

Abstract
Ischemic preconditioning (IPC) and remote ischemic precondition (RIPC) are resistance to ischemia-reperfusion (IR) injury. They have common protective mechanism. Cyclooxygenase (COX)-2 participate in the mechanism of IPC. So, the purpose of this study was to determine whether RIPC protects endothelial function of radial artery in human against IR and whether COX-2 involves in this effect. Endothelial IR injury was induced by arm ischemia (20 min) and reperfusion. Flow-mediated dilation (FMD) of the radial artery was measured before and after IR. RIPC (three 5-min cycles of ischemia of the contralateral arm) was applied immediately and 24 h before IR. All volunteers received the COX-2 inhibitor celecoxib (200 mg orally twice daily) for 5 days. On day 6, all subjects experienced the same studies as described. FMD was reduced by IR without administration of RIPC (P<0.0001). RIPC prevent this impairment of FMD immediately (P=NS) and at 24 h (P=NS). Nevertheless, the COX-2 inhibiter abolished protective effect of RIPC at 24 h (P=NS), but not immediately (P=0.001). After administration of the COX-2 inhibiter, post-IR FMD after RIPC performed immediately had significant increase than after RIPC performed at 24 h (P=0.001) and without administration of RIPC (P=0.003). The COX-2 inhibiter made post-IR FMD evidently decrease after RIPC performed at 24 h (P=0.002). RIPC prevents radial artery endothelial dysfunction induced by IR. This protective effect of RIPC in the late phase is mediated by a COX-2-dependent mechanism.
Keywords: Remote ischemic preconditioning, endothelial function, ischemia and reperfusion, cyclooxygenase-2
Introduction
Reperfusion measures using either thrombolytic therapy or primary percutaneous coronary intervention (PPCI), timely and effectively reopening the infarct-related coronary artery, limit the infarct size and improve the prognosis in patients with ST-elevation myocardial infarction (STEMI). However, these myocardial reperfusion can themself induce further cardiomyocyte injury, known as ischemia and reperfusion (IR) injury contribute to attenuate the full benefits of myocardial reperfusion [1,2]. Ischemic precondition (IPC) reduces the susceptibility of myocardium and vascular endothelium for IR injury and decreases infart size in STEMI [3,4]. Nevertheless, it is difficult for IPC to be appied in unexpected cardiovascular events in high-risk patients. Remote ischemic precondition (RIPC) also can protect vascular endothelial function and reduce infarct size against IR injury in STEMI. In particular, the RIPC induced by limbs is easy and simple in clinical application [5,6]. Moreover, RIPC resembles IPC that its protection has two phases [5-8]: the early one and the late one, sharing common triggers and signal transduction pathway [9]. Thus, IPC and RIPC may have the same mechanism in protecting myocardium and vascular endothelial function against IR injury, but the protective mechanism of RIPC is not fully elucidated at present. The late protection of IPC partly depends on activating celecoxib-2 (COX-2). Whether COX-2 invovle in the mechanism of RIPC remain uninvestigated.
Methods
Subjects
There were twenty healthy nonsmoking volunteers (32.7 ± 8.3 age) to be enrolled at random. All subjects gave informed consent. The local research ethics committee approved this study. All studies were performed in a temperature-controlled laboratory (24°C to 26°C). On admission, sitting blood pressure was measured and peripheral venous blood was obtained for baseline lipid analysis in all subjects. All subjects participated in all studies and repeated in studies were at least seven days apart.
Experimental design
All subjects were divided into baseline group, RIPC group, RIPC 24 h group, Celecoxib + baseline group, Celecoxib + RIPC group, and Celecoxib + RIPC 24 h group according to administration of IR injury before RIPC or after RIPC at immediate or 24 h moment as well as whether or not administering COX-2 inhibitor. IR injury was performed and flow-mediated dilation (FMD) before and after IR injury was measured in subjects in every group. Subjects in Celecoxib + baseline group, Celecoxib + RIPC group, and Celecoxib + RIPC 24 h group were administered celecoxib, a selective COX-2 inhibitor, 200 mg twice daily for 5 days, then experiencing the measurement of FMD before and after IR injury (Figure 1).
Figure 1.

Trial profile. Flow-mediated dilation (FMD) of the radial artery was assessed before 15 min of arm ischemia reperfusion (IR) and at 15 min of reperfusion (A~F). Remote ischemic preconditioning (RIPC) was applied immediately (B, D) and 24 h (C, F) before IR. The COX-2 inhibitor celecoxib (200 mg orally twice daily) was administered for 5 days (D~F).
Induction of IR and RIPC
A pneumatic cuff placed above the antecubital fossa in the nondominant forearm was inflated to 200 mmHg for 20 min to induce IR injury and then deflated for 15 min of reperfusion before FMD was measured again.
RIPC was induced by three cycles that inflated a pneumatic cuff placed above the antecubital fossa in the contralateral arm to 200 mmHg for 5 min (ischemia) and followed by a 5 min deflation.
Assessment of radial artery function
It was required for subjects to rest for at least 10 minutes in the suprine position before measurements were started. FMD was measured before and after IR at 15 minutes, as previously described [10,11].
Radial artery images were taken with an Acuson Sequoia 512 with a 7- to 12-MHz linear array transducer. Longitudinal, electrocardiogram-gated, end-diastolic B-mode images of radial artery were acquired at 5-second for offline analysis. Baseline radial artery diameter was averaged from 6 separate images taken. Subsequently, a pneumatic cuff placed at the level that was distal to the site of radial artery measurement in the wrist was inflated to 250 mmHg for 4 minutes, 30 seconds. after wrist-cuff deflation, FMD was calculated according to percent maximum increase of radial artery diameter measured starting at 30 seconds and ending at 3 minutes, 30 seconds. Radial artery diameter was calculated semiautomatically using a modified version of the Image J software and custom-designed software.
Baseline blood flow and postischemic blood flow (reactive hyperemia) in radial artery were measured using pulsed-wave Doppler as average velocity-time integral for the first 5 cardiac cycles after cuff deflation [11].
Calculations and statistics
All data are presented as mean ± SE unless otherwise stated. Radial artery diameter was measured in millimeters and dilation expressed as percentage increase from baseline diameter. Data were compared using the Student paired t test, as appropriate. In all cases, P<0.05 was considered statistically significant. SPSS version 17.0 (SPSS Inc, Chicago, IL) was used for all statistical analyses.
Results
Baseline characteristics
All subjects tolerated the procedures without any complications. There was no significant differences in resting radial artery diameter before and after IR injury among groups (Table 1). The change of radial diameter between pre-IR and post-IR have significant difference in baseline group (0.145 ± 0.033 vs. 0.102 ± 0.027, P<0.0001), Celecoxib + Baseline group (0.145 ± 0.026 vs. 0.108 ± 0.034, P<0.0001), and Celecoxib + RIPC 24 h group (0.167 ± 0.038 vs. 0.113 ± 0.038, P<0.0001). Compared with baseline group, there were distinct difference in RIPC 24 h group (0.102 ± 0.027 vs. 0.136 ± 0.028, P<0.0001), Celecoxib + RIPC group (0.102 ± 0.027 vs. 0.143 ± 0.032, P<0.0001) and RIPC group (0.102 ± 0.027 vs. 0.139 ± 0.035, P=0.001) for the change of radial diameter after IR injury. Likewise, compared with Celecoxib + baseline group, there were evident difference in RIPC group (0.108 ± 0.034 vs. 0.139 ± 0.035, P=0.008), RIPC 24 h group (0.108 ± 0.034 vs. 0.136 ± 0.028, P=0.008), and Celecoxib + RIPC group (0.108 ± 0.034 vs. 0.143 ± 0.032, P=0.002) for the change of radial diameter after IR injury. In addition, the change of radial diameter after IR injury significantly increased in RIPC group (0.113 ± 0.038 vs. 0.139 ± 0.035, P=0.033), RIPC 24 h group (0.113 ± 0.038 vs. 0.136 ± 0.028, P=0.039), and Celecoxib + RIPC group (0.113 ± 0.038 vs. 0.143 ± 0.032, P=0.011) than Celecoxib + RIPC 24 h group (Table 1).
Table 1.
Radial Arterial Diameter (mm)
| Pre-IR | Post-IR | |||
|---|---|---|---|---|
|
| ||||
| Baseline | Change in Diameter | Baseline | Change in Diameter | |
|
|
|
|||
| After Wrist Cuff Deflation | After Wrist Cuff Deflation | |||
| Baseline | 2.36 ± .35 | .145 ± .033 | 2.39 ± .30 | .102 ± .027* |
| RIPC | 2.37 ± .34 | .147 ± .028 | 2.37 ± .28 | .139 ± .035†,‡ |
| RIPC 24 h | 2.35 ± .31 | .145 ± .026 | 2.38 ± .29 | .136 ± .028†,‡ |
| Celecoxib + Baseline | 2.39 ± .31 | .157 ± .036 | 2.38 ± .29 | .108 ± .034* |
| Celecoxib + RIPC | 2.42 ± .31 | .164 ± .040 | 2.40 ± .30 | .143 ± .032†,‡ |
| Celecoxib + RIPC 24 h | 2.38 ± .32 | .167 ± .038 | 2.40 ± .31 | .113 ± .038* |
P<0.0001 corresponding value in the baseline group.
P<0.01 versus corresponding value in the Celecoxib + Baseline group, p value in the baseline group versus corresponding value in the RIPC group;
P<0.05 versus corresponding value post-IR in the Celecoxib + RIPC 24 h group.
There were no significant differences in baseline blood flow and reactive hyperemia before and after IR injury among groups (Table 2).
Table 2.
Radial blood flow
| Blood Flow (ml/min) | Pre-IR | Post-IR | ||
|---|---|---|---|---|
|
|
|
|||
| Baseline | Reactive Hyperemia (%) | Baseline | Reactive Hyperemia (%) | |
| Baseline | 33.6 ± 15.0 | 481 ± 162 | 35.3 ± 17.8 | 468 ± 223 |
| RIPC | 36.4 ± 21.0 | 472 ± 156 | 38.2 ± 19.6 | 488 ± 220 |
| RIPC 24 h | 34.3 ± 16.9 | 478 ± 160 | 37.6 ± 19.4 | 500 ± 214 |
| Celecoxib + Baseline | 40.8 ± 15.6 | 463 ± 156 | 39.2 ± 19.4 | 480 ± 200 |
| Celecoxib + RIPC | 36.5 ± 15.5 | 480 ± 130 | 38.9 ± 17.8 | 474 ± 112 |
| Celecoxib + RIPC 24 h | 38.6 ± 14.7 | 466 ± 121 | 37.2 ± 16.7 | 486 ± 100 |
Effect of RIPC on endothelial function induced by IR injury
IR injury significantly blunted FMD in the baseline group (pre-IR: 6.8 ± 1.3%; post-IR: 4.7 ± 2.1%, P<0.0001) (Figure 2). On contrary, RIPC protected endothelial function immediately (FMD pre-IR: 7.4 ± 1.7%; post-IR: 6.7 ± 1.9%, P>0.05) and at 24 h (FMD pre-IR: 7.3 ± 1.6%; post-IR: 6.8 ± 1.7%, P>0.05) (Figure 2). FMD in the baseline was lower than that in the RIPC group (post-IR: 4.7 ± 2.1%; post-IR: 6.7 ± 1.9%, P=-0.003) and in the RIPC 24 h group (post-IR: 4.7 ± 2.1%; post-IR: 6.8 ± 1.7%, P=0.001).
Figure 2.
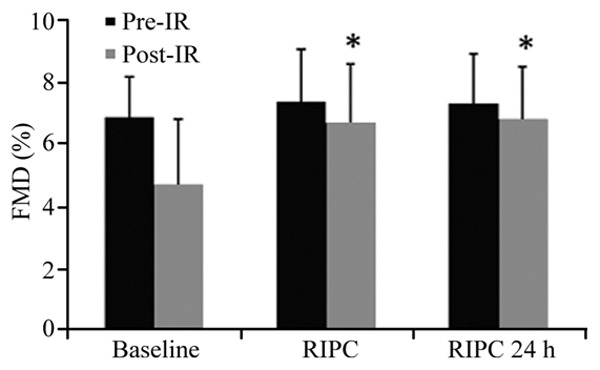
Effect of remote ischemic preconditioning (RIPC) on ischemia-reperfusion (IR) induced endothelial dysfunction. In the Baseline group, Flow-mediated dilation (FMD) was significantly blunted post-IR. This effect was prevented by RIPC applied immediately and 24 h. *P<0.01 versus FMD post-IR in the baseline group.
Effect of celecoxib on FMD after RIPC
Likewise, after administering celecoxib, IR injury significantly reduced FMD in the baseline + baseline group (pre-IR: 7.1 ± 1.4%; post-IR: 4.9 ± 2.5%, P=0.002) (Figure 3). RIPC also protected endothelial function immediately (FMD pre-IR: 7.2 ± 1.6%; post-IR: 7.0 ± 1.6%, P>0.05), but didn’t do endothelial function at 24h (FMD pre-IR: 7.0 ± 1.4%; post-IR: 4.7 ± 2.3%, P=0.001) (Figure 3). In comparison with Celecoxib + baseline group (4.9 ± 2.5% vs. 7.0 ± 1.6%, P=0.003) and Celecoxib + RIPC 24 h group (4.7 ± 2.3% vs. 7.0 ± 1.6%, P=0.001), Celecoxib + RIPC group deceased in FMD post-IR (Figure 3).
Figure 3.
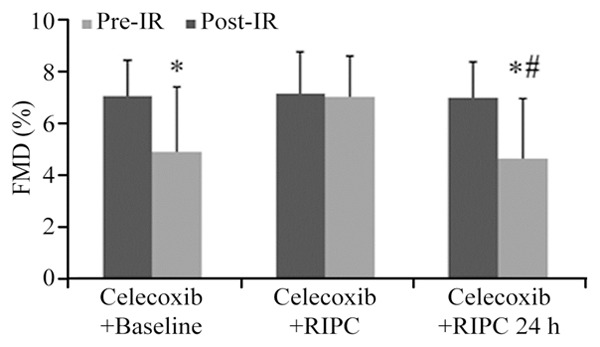
Effect of Celecoxib on FMD after remote ischemic preconditioning (RIPC). In the Celecoxib + baseline and Celecoxib + RIPC24 h groups, Flowmediated dilation (FMD) were significant decrease in post-IR, but was’nt in the Celecoxib + RIPC group. *P<0.01 versus FMD post-IR in the Celecoxib + RIPC group. #P=0.002 versus FMD post-IR in the RIPC group.
After IR injury, FMD in Celecoxib + RIPC 24 h group had more significant decrease than in RIPC 24 h group (4.7 ± 2.3% vs. 6.8 ± 1.7%, P=0.002) (Figure 3).
Discussion
The present study demonstrates that COX-2 participate in the mechanism that RIPC protect endothelial function of radial artery free from IR injury. This COX-2-dependent mechanism occurs in the late phase of RIPC protection.
At present, although the process of myocardial reperfusion in STEMI has maken great progresses, due to myocardial IR injury in STEMI causing damage to the endothelium, exacertion of myocardial reperfusion [12] and worse clinical outcomes [13] the benefit obtained by these process is limited in patients with STEMI.
Human study [5] demonstrates that RIPC prevents vascular endothelial dysfunction induced by IR. This protective role has an early phase of protection lasting four hours after the RIPC stimulus, and followed 24 h later by a late phase of protection lasting for up to 48 h. In the present study, RIPC also protects vascular endothelial function against IR injury, which has an early phase of protection and a late phase of that, resembling the above mentioned study. In some recent studies, RIPC not only provide perioperative myocardial protection and a decrease in all-cause mortality in patients undergoing elective coronary artery bypass graft (CABG) surgery [14], but significantly improves endothelial function [15] increases myocardial salvage [6], and reduces major adverse cardiac and cerebrovascular events (MACCE) in patients with STEMI experiencing primary PCI [16]. In addition, in the CRISP stent trial, RIPC reduced the MACCE rate at 6 years after elective PCI [17]. Therefore, RIPC can protect endothelial function and improve long-term clinical prognosis by preventing or attenuating IR injury.
Nevertheless, up to now, the protective mechanism of RIPC has not been expounded clearly. RIPC protects vascular endothelium and myocardial by triggers (including adenosine, bradykinin, and opioids.) and upregulation of a spectrum of established prosurvival kinases (including the e-isoform of protein kinase C, extracellular signal-related kinase, Janus kinase, and phosphatidylinositol 3-kinase/protein kinase B) [9], which of these triggers and kinases also are activated in the protective effect of IPC [18]. Moreover, some studies demonstrated that the late protection of RIPC and IPC have common signaling pathway and transcriptional factors, including ERK 1/2, PI3K/Akt, JAK-STAT, and Akt/eNOS pathway as well as NF-κB and iNOS [19-26]. So, RIPC and IPC may have an identical mechanism in protenting myocardium and vascular endothelium.
In animal model study, the cardioprotective effects in the late phase of IPC resulted in increase in COX-2 protein expression and in the myocardial content of prostaglandin (PG) E2 and PGI2. Whereas, the increase in myocardial tissue levels of PGE2 and PGF2 was abolished, while COX-2 selective inhibitor completely blocked the cardioprotective effects of late IPC [27]. Guo et al. demonstrated that targeted disruption of the COX-2 gene completely abrogates the cardioprotection of the late IPC, which provide unequivocal molecular genetic evidence for a crucial role of the COX-2/PGI2 receptor axis in the cardioprotection of the late IPC [28]. In addition, late IPC upregulate COX-2 via protein kinase C (PKC)-epsilon in the cardioprotection [19]. Furthermore, COX-2 is located downstream of iNOS in the protective pathway of late IPC, which is modulated by iNOS via cGMP-independent mechanisms [26]. The upregulation of COX-2 also requires upregulated iNOS and iNOS-derived NO via a JAK1/2-STAT1/3 pathway [29]. Moreover, COX-2 achieves the regulation in the protection of late IPC through PGE2 and PGI2 [27]. In another study, PGI2 is evidently upregulated in the cardioprotection of late IPC and COX-2 inhibitor can completely abolish the protective effect of late IPC [30]. Therefore, COX-2 and PGI2 might be the most main participants in the mechanism of the cardioprotection of late IPC. Meanwhile, many studies have confirmed that IPC and RIPC share with common mechanism in their late protection [19-26]. Likewise, COX-2 might involve the mechanism in the late protection of RIPC. In present study, COX-2 inhibitor suppressed the late protection of RIPC in the radial artery, which show for the first time that COX-2 participate in the late protection of RIPC. Hence, COX-2 might involve in the late protection of RIPC.
In a recent study, the specific prostaglandin receptors (IP), downstream of the COX-2/prostanoid pathway, is a key mediator of the late IPC [28]. STAT3 plays an obligatory role by increasing the expression of cardioprotective (COX-2 and Heme Oxygenase-1) and anti-apoptotic proteins in the cardioprotection of the late IPC [31]. But, IP mediates STAT via stimulation of STAT3 Tyr (705) and Ser (727) phosphorylations [32]. Therefore, IP not only may adjust the signal transduction for COX-2, but play the part of promotor for feedback enhancement in multiple pathways mediating the late IPC. Nevertheless, the detailed mechanism of COX-2 in the late phase of RIPC may be analogous to the role of that in the late phase of IPC. However, the study about this aspect hitherto is little reported, and that is also the direction of study in future. If these key role of IP in protection of late RIPC are verified, the receptor of IP will become the most important determinant for achieving the protection of late RIPC. Thus, the selective and specific agonist of IP would be an appealing pharmacological approach to mimic the late phase of RIPC. If this targeted drug is screened or found, it would be applied to clinical treatment by simulating pharmacologic cardioprotection of late RIPC.
In our study, there are some limitations. Firstly, the data of the present study were obtained in healthy volunteers and in radial artery, the latter is different from the coronary circulation and needs to be validated. Secondly, the assessment of IR-induced changement in endothelial function was implemented in radial artery and not in the distal microcirculation that is crucial in clinical IR injury. Thirdly, IR injury only resulted in endothelial function stuning and not in endothelial function necrosis. Additionally, COX-2 protein expression and the correlative trigger and kinase of COX-2 in the cardioprotection of late RIPC also aren’t investigated. So, these result need to be further verified in animal study.
Conclusions
In present study, RIPC protected endothedial function of radial artery against IR injury. COX-2 inhibitor completely abrogated the protective effect of late RIPC, evidence that showed that activation of COX-2 mediated the protection of RIPC in the late phase.
Acknowledgements
The authors of this manuscript have demonstrated that they abide by Principles of Ethical Publishing in the International Journal of Cardiology.
Disclosure of conflict of interest
None.
References
- 1.Abela CB, Homer-Vanniasinkham S. Clinical implications of ischaemia-reperfusion injury. Pathophysiology. 2003;9:229–240. doi: 10.1016/s0928-4680(03)00025-7. [DOI] [PubMed] [Google Scholar]
- 2.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 3.Kharbanda RK, Peters M, Walton B, Kattenhorn M, Mullen M, Klein N, Vallance P, Deanfield J, MacAllister R. Ischemic preconditioning prevents endothelial injury and systemic neutrophil activation during ischemia-reperfusion in humans in vivo. Circulation. 2001;103:1624–1630. doi: 10.1161/01.cir.103.12.1624. [DOI] [PubMed] [Google Scholar]
- 4.Sanada S, Komuro I, Kitakaze M. Pathophysiology of myocardial reperfusion injury: preconditioning, postconditioning, and translational aspects of protective measures. Am J Physiol Heart Circ Physiol. 2011;301:H1723–1741. doi: 10.1152/ajpheart.00553.2011. [DOI] [PubMed] [Google Scholar]
- 5.Loukogeorgakis SP, Panagiotidou AT, Broadhead MW, Donald A, Deanfield JE, MacAllister RJ. Remote ischemic peconditioning provides early and late protection against endothelial ischemia-reperfusion injury in humans. J Am Coll Cardiol. 2005;46:450–456. doi: 10.1016/j.jacc.2005.04.044. [DOI] [PubMed] [Google Scholar]
- 6.Bøtker HE, Kharbanda R, Schmidt MR, Bøttcher M, Kaltoft AK, Terkelsen CJ, Munk K, Andersen NH, Hansen TM, Trautner S, Lassen JF, Christiansen EH, Krusell LR, Kristensen SD, Thuesen L, Nielsen SS, Rehling M, Sørensen HT, Redington AN, Nielsen TT. Remote ischaemic conditioning before hospital admission, as a complement to angioplasty, and effect on myocardial salvage in patients with acute myocardial infarction: a randomised trial. Lancet. 2010;375:727–734. doi: 10.1016/S0140-6736(09)62001-8. [DOI] [PubMed] [Google Scholar]
- 7.Marber MS, Latchman DS, Walker JM, Yellon DM. Cardiac stress protein elevation 24 hours after brief ischemia or heat stress is associated with resistance to myocardial infarction. Circulation. 1993;88:1264–1272. doi: 10.1161/01.cir.88.3.1264. [DOI] [PubMed] [Google Scholar]
- 8.Baxter GF, Goma FM, Yellon DM. Characterisation of the infarct-limiting effect of delayed preconditioning: time course and dose dependency studies in rabbit myocardium. Basic Res Cardiol. 1997;92:159–167. doi: 10.1007/BF00788633. [DOI] [PubMed] [Google Scholar]
- 9.Przyklenk K, Whittaker P. Remote ischemic preconditioning: current knowledge, unresolved questions, and future priorities. J Cardiovasc Pharmacol Ther. 2011;16:255–259. doi: 10.1177/1074248411409040. [DOI] [PubMed] [Google Scholar]
- 10.Bartoli G, Menegaz G, Lisi M, Di Stolfo G, Dragoni S, Gori T. Model-based analysis of flow-mediated dilation and intima-media thickness. Int J Biomed Imaging. 2008;2008:738545. doi: 10.1155/2008/738545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gori T, Sicuro S, Dragoni S, Donati G, Forconi S, Parker JD. Sildenafil prevents endothelial dysfunction induced by ischemia and reperfusion via opening of adenosine triphosphatesensitive potassium channels: a human in vivo study. Circulation. 2005;111:742–746. doi: 10.1161/01.CIR.0000155252.23933.2D. [DOI] [PubMed] [Google Scholar]
- 12.Hombach V, Grebe O, Merkle N, Waldenmaier S, Höher M, Kochs M, Wöhrle J, Kestler HA. Sequelae of acute myocardial infarction regarding cardiac structure and function and their prognostic significance as assessed by magnetic resonance imaging. Eur Heart J. 2005;26:549–557. doi: 10.1093/eurheartj/ehi147. [DOI] [PubMed] [Google Scholar]
- 13.Ganame J, Messalli G, Dymarkowski S, Rademakers FE, Desmet W, Van de Werf F, Bogaert J. Impact of myocardial haemorrhage on left ventricular function and remodelling in patients with reperfused acute myocardial infarction. Eur Heart J. 2009;30:1440–1449. doi: 10.1093/eurheartj/ehp093. [DOI] [PubMed] [Google Scholar]
- 14.Thielmann M, Kottenberg E, Kleinbongard P, Wendt D, Gedik N, Pasa S, Price V, Tsagakis K, Neuhäuser M, Peters J, Jakob H, Heusch G. Cardioprotective and prognostic effects of remote ischaemic preconditioning in patients undergoing coronary artery bypass surgery: a single-centre randomised, double-blind, controlled trial. Lancet. 2013;382:597–604. doi: 10.1016/S0140-6736(13)61450-6. [DOI] [PubMed] [Google Scholar]
- 15.Manchurov V, Ryazankina N, Khmara T, Skrypnik D, Reztsov R, Vasilieva E, Shpektor A. Remote ischemic preconditioning and endothelial function in patients with acute myocardial infarction and primary PCI. Am J Med. 2014;127:670–673. doi: 10.1016/j.amjmed.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 16.Sloth AD, Schmidt MR, Munk K, Kharbanda RK, Redington AN, Schmidt M, Pedersen L, Sørensen HT, Bøtker HE CONDI Investigators. Improved long-term clinical outcomes in patients with ST-elevation myocardial infarction undergoing remote ischaemic conditioning as an adjunct to primary percutaneous coronary intervention. Eur Heart J. 2014;35:168–175. doi: 10.1093/eurheartj/eht369. [DOI] [PubMed] [Google Scholar]
- 17.Davies WR, Brown AJ, Watson W, McCormick LM, West NE, Dutka DP, Hoole SP. Remote ischemic preconditioning improves outcome at 6 years after elective percutaneous coronary intervention: the CRISP stent trial long-term follow-up. Circ Cardiovasc Interv. 2013;6:246–251. doi: 10.1161/CIRCINTERVENTIONS.112.000184. [DOI] [PubMed] [Google Scholar]
- 18.Minamino T. Cardioprotection from ischemia/reperfusion injury: basic and translational research. Circ J. 2012;76:1074–1082. doi: 10.1253/circj.cj-12-0132. [DOI] [PubMed] [Google Scholar]
- 19.Cai ZP, Parajuli N, Zheng X, Becker L. Remote ischemic preconditioning confers late protection against myocardial ischemia-reperfusion injury in mice by upregulating interleukin-10. Basic Res Cardiol. 2012;107:277. doi: 10.1007/s00395-012-0277-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xuan YT, Guo Y, Zhu Y, Wang OL, Rokosh G, Bolli R. Endothelial nitric oxide synthase plays an obligatory role in the late phase of ischemic preconditioning by activating the protein kinase C epsilon p44/42 mitogen-activated protein kinase pSer-signal transducers and activators of transcription 1/3 pathway. Circulation. 2007;116:535–544. doi: 10.1161/CIRCULATIONAHA.107.689471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li G, Labruto F, Sirsjö A, Chen F, Vaage J, Valen G. Myocardial protection by remote preconditioning: the role of nuclear factor kappa-B p105 and inducible nitric oxide synthase. Eur J Cardiothorac Surg. 2004;26:968–973. doi: 10.1016/j.ejcts.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 22.Bell RM, Clark JE, Hearse DJ, Shattock MJ. Reperfusion kinase phosphorylation is essential but not sufficient in the mediation of pharmacological preconditioning: characterisation in the biphasic profile of early and late protection. Cardiovasc Res. 2007;73:153–163. doi: 10.1016/j.cardiores.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 23.Xuan YT, Guo Y, Zhu Y, Wang OL, Rokosh G, Messing RO, Bolli R. Role of the protein kinase C-epsilon-Raf-1-MEK-1/2-p44/42 MAPK signaling cascade in the activation of signal transducers and activators of transcription 1 and 3 and induction of cyclooxygenase-2 after ischemic preconditioning. Circulation. 2005;112:1971–1978. doi: 10.1161/CIRCULATIONAHA.105.561522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Kudo M, Xu M, Ayub A, Ashraf M. Mitochondrial K (ATP) channel as an end effector of cardioprotection during late preconditioning: triggering role of nitric oxide. J Mol Cell Cardiol. 2001;33:2037–2046. doi: 10.1006/jmcc.2001.1468. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Kodani E, Wang J, Zhang SX, Takano H, Tang XL, Bolli R. Cardioprotection during the final stage of the late phase of ischemic preconditioning is mediated by neuronal NO synthase in concert with cyclooxygenase-2. Circ Res. 2004;95:84–91. doi: 10.1161/01.RES.0000133679.38825.a6. [DOI] [PubMed] [Google Scholar]
- 26.Shinmura K, Xuan YT, Tang XL, Kodani E, Han H, Zhu Y, Bolli R. Inducible nitric oxide synthase modulates cyclooxygenase-2 activity in the heart of conscious rabbits during the late phase of ischemic preconditioning. Circ Res. 2002;90:602–608. doi: 10.1161/01.res.0000012202.52809.40. [DOI] [PubMed] [Google Scholar]
- 27.Shinmura K, Tang XL, Wang Y, Xuan YT, Liu SQ, Takano H, Bhatnagar A, Bolli R. Cyclooxygenase-2 mediates the cardioprotective effects of the late phase of ischemic preconditioning in conscious rabbits. Proc Natl Acad Sci U S A. 2000;97:10197–10202. doi: 10.1073/pnas.97.18.10197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo Y, Tukaye DN, Wu WJ, Zhu X, Book M, Tan W, Jones SP, Rokosh G, Narumiya S, Li Q, Bolli R. The COX-2/PGI2 receptor axis plays an obligatory role in mediating the cardioprotection conferred by the late phase of ischemic preconditioning. PLoS One. 2012;7:e41178. doi: 10.1371/journal.pone.0041178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xuan YT, Guo Y, Zhu Y, Han H, Langenbach R, Dawn B, Bolli R. Mechanism of cyclooxygenase-2 upregulation in late preconditioning. J Mol Cell Cardiol. 2003;35:525–537. doi: 10.1016/s0022-2828(03)00076-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kodani E, Xuan YT, Shinmura K, Takano H, Tang XL, Bolli R. Delta-opioid receptor-induced late preconditioning is mediated by cyclooxygenase-2 in conscious rabbits. Am J Physiol Heart Circ Physiol. 2002;283:H1943–1957. doi: 10.1152/ajpheart.00150.2002. [DOI] [PubMed] [Google Scholar]
- 31.Bolli R, Stein AB, Guo Y, Wang OL, Rokosh G, Dawn B, Molkentin JD, Sanganalmath SK, Zhu Y, Xuan YT. A murine model of inducible, cardiac-specific deletion of STAT3: its use to determine the role of STAT3 in the upregulation of cardioprotective proteins by ischemic preconditioning. J Mol Cell Cardiol. 2011;50:589–597. doi: 10.1016/j.yjmcc.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 32.Lo RK, Liu AM, Wise H, Wong YH. Prostacyclin receptor-induced STAT3 phosphorylation in human erythroleukemia cells is mediated via Galpha(s) and Galpha(16) hybrid signaling. Cell Signal. 2008;20:2095–2106. doi: 10.1016/j.cellsig.2008.08.003. [DOI] [PubMed] [Google Scholar]