

Abstract
Purpose: We aim to report a genetic testing and fertility guidance for the deaf through analyzing pedigree and molecular genetic characteristics of the couple who have non-syndromic sensorineural hearing loss (NSHL). Methods: One of hospitalized congenial deaf couple and family members were included in this study. The wife was twin pregnant woman and her gestational age was 31+5 pregnant weeks. The DNA was extracted from peripheral blood and umbilical vein blood, respectively. Mutation screening of common deafness genes was performed in pregnant women and other family members. Nine common mutations in four major deafness genes, GJB2 (35delG, 176del16, 235delC, 299delAT), GjB3 (C538T), SLC26A4 (IVS7-2A>G, A2168G) and Mitochondrial 12S rRNA (A1555G, C1494T), were detected simultaneously with a microarray based method. SLC26A4 whole genome sequencing was carried out for the results of the DNA microarray. According to the test results, the couple chose abortion termination of pregnancy twins, and after one year obtained singleton pregnancy by artificial insemination by donor (AID). In week 16 of pregnancy, amniocentesis had been done to collect fetal somatic cell and extract DNA, and then the above tests had been repeated. Results: The couple had SLC26A4 combined heterozygous mutation. Both parents had SLC26A4 single heterozygous mutation. Twin fetuses had SLC26A4 combined heterozygous mutation. The probability of naturally being pregnant and bearing deaf children for the pregnant women was 100%. Fetus obtained by AID had SLC26A4 single heterozygous mutation. After the birth of the baby, her hearing has been normal. Conclusions: To reduce children with congenital deafness, screening high mutation sites by microarray, combined with pedigree analysis and gene sequencing is effective, and should be used as a routine inspection item for the deaf before marriage and pregnancy. On the basis of genetic testing for the couple with hearing loss, human assisted reproductive technology is a viable option to avoid the birth of infant with hereditary deafness.
Keywords: Hereditary deafness, SLC26A4 gene, gene diagnosis, prenatal diagnosis, assisted reproductive technology
Introduction
Congenital deafness is one of the most common birth defects and its incidence rate is about 1‰~3‰, the data were further increasing trend in certain areas [1-3]; Above fifty percent of deaf neonates are caused by the genetic factors [4,5]. The lately national population census data show that about 27,800,000 persons in China are troubled by hearing disability and the prevalence rate of hearing disability is 2.11% [6], the rate of non-syndromic hearing impairment(NSHL) is 70% of the genetic deafness. It is worth noticed that about 77% of NSHL belong to autosomal recessive inheritances [7]. Extensive studies of deafness molecular epidemiology in China have shown that quite a number of NSHL are caused by only several mutated genes such as gap junction protein beta-2 gene (GJB2 gene), SLC 26A4 gene (PDS gene) and mitochondrial gene (mtDNA). Pathological mutations of these genes lead to high incidence of the hereditary deafness [8-13]. This provides a basis for screening deafness gene generally. One typical case of family gene of non-syndromic deafness was studied using gene chip and sequencing analysis to reveal the significance of detecting deafness genes in normal and deaf population.
Materials and methods
Study objects
The 28-year-old primigravida with congenital deafness, who carried twin and in 31+5 weeks of pregnancy, was given the pregnant health examination in the obstetric clinic of Yuhuangding hospital in Yantai in September of 2012. On the basis of pedigree analysis, all subjects including the deaf couple, their parents, other family members, were given the clinical historical survey, imaging examination, general physical examination and routine examination of ear-nose-throat department including pure tone test, acoustic immittance and auditory brainstem response (ABR). Both ears of the couple were found non-syndromic sensorineural deafness, and thin slice temporal bone CT pictures and the auditory brainstem response binaural 100dB nHL were not elicit reaction. 40 Hz auditory event-related potential (40 Hz AERP) displayed binaural 1 kHz 100dB nHL. Temporal bone CT and body checked showed no abnormal in the others. After one year induction of labor was performed, hysterosalpingography showed that uterine was normal in size and shape, and bilateral tubal patency was unobstructed. The deaf couple accepted artificial insemination by donor (AID) to get pregnancy again.
Informed consent and ethical oversight
The study had been approved by Ethics Committee of Qingdao University Affiliated Yantai Yuhuangding Hospital and all the subjects signed informed consent.
Specimen collection and processing
The vein blood and umbilical vein blood from previous twin fetus were collected in test tubes with contained EDTA anticoagulant. Amniotic fluid was punctured in week 16 after the deaf couple accepted Artificial insemination by donor (AID) for second pregnancy. Fetal cells in amniotic fluid culture were cultured for one week until cell number reached to 107. The DNA from blood and fetal cells were extracted with QIAGEN DNA extraction kit (Catalog number: DP319-01, Germany). Using Thermo NANODROP 2000C model spectrophotometer, the isolated DNA was carried out quantitative purity detection to ensure that the concentration was 100-200 ng/ul and the purity OD260/280 was 1.7-2.0.
PCR reaction
The primers of nine groups with nine mutation sites were divided into 3MIX and 1MIX. The two reaction systems were carried out multiplex PCR, respectively. The mixture of PCR amplification primers in the kit and the amplification reagent mixture were thoroughly mixed (A tube and B tube). Each 17 μl reaction system was added 3 μL DNA sample, which was recognized as PCR amplification template. The PCR program was denaturation at 37°C for 10 min, 95°C for 15 min and 96°C for 1 min, amplification at 94°C for 30 s, 55°C for 30 s and 70°C for 45 s, 32 cycles at 60°C for 10 min. During the amplification procedure, the parameter was set to make the temperature be cooled from 94°C to 55°C at a speed of 0.4°C/s and then be warmed from 55°C to 70°C at a speed of 0.2°C/s. Bio-Rad PCR machine (USA) was used for PCR production.
Hybridization
Heredity Hearing Loss Array Detection Kit was purchased from Capital Bio Corporation (Beijing, China). It includes nine common mutations in four major deafness genes, GJB2 (35delG, 176del16, 235delC, 299delAT), GjB3 (C538T), SLC26A4 (IVS7-2A>G, A2168G) and Mitochondrial 12S rRNA (A1555G, C1494T), and they were detected simultaneously by a microarray. After denaturation at 95°C for 5 min, the PCR products were immediately immersed in ice water mixture for an ice bath for 3 min. The PCR products were respectively extracted 2.5 μl from two different amplification systems and were then added into the 10 μl hybridization buffer tube to be mixed well and transiently centrifuged. The mixture was putted through the sample hole of a coverslip to be added to sampling region of the chip. Finally, the hybrid box was closed and then placed in a 50°C preheated hybridization box for insulation for 1 hour.
Washing and scanning
The chips were uniformly shaken in 42°C washing liquid I for 2 min by 80 r/min shaker. Then they were quickly put into 42°C washing liquid II to be shook 1 min for two times. Finally put chip into microarray chip centrifuge tube to be centrifuged at 1000 r/min for 2 min for drying. Crystal core LuxScanTM 10K-B microarray scanner and the corresponding genetic deafness gene chip detection discrimination system were used for signal reading and judging. Test results were automatically interpreted by genetic deafness gene chip interpretation system. Deafness microarray dot arrangement is shown in Figure 1.
Figure 1.

Lattice arrangement figure of deafness gene chip. QC: surface chemical Quality control probe; PC: hybridize positive control probe; NC: hybridize negative control probe; IC: PCR Internal control probe; BC: blank control; site examine probe W: wild type probe; site examine probe; M: mutant type probe.
Detection principle
For different mutation sites, Design two kinds of the upstream probe with different labels and a public primer with CY3 fluorescently labeled downstream. Firstly, specific primers were extended on multiplex alleles in the liquid phase (PCR). The PCR amplification products were brought a tag sequence and marked by monochromatic fluorescence. After denaturation of the products, they were hybridized with chips fixed with tags complementary oligonucleotide sequence. The mutation points were detected using laser scanner. The chips results are shown in Figure 2.
Figure 2.

The deafness gene chip. A: Woman’s chip: IVS7-2A>G carrier; B: Husband’s chip: IVS7-2A>G carrier; C, D: Both mothers’ chips: IVS7-2A>G carriers; E, F: Both father’s chips: completely norma.; G: Chip of woman’s older sister: completely normal.; H: Chip of husband’s young sister :IVS7-2A>G carrier; I, J: The twin fetus’s chips: IVS7-2A>G carriers; K: Chip of Amniotic fluid by the second cycle AID: IVS7-2A>G carrier.
Whole genome sequencing of SLC26A4
The PCR products were purified using ExoSAP-IT enzyme. The excess free PCR primers and substrates dNTPs were removed by adding 3 μl ExoSAP-IT in each reaction tube. After mixed by low-speed centrifugation, they were placed in a PCR instrument maintaining at 37°C for 30 min, then maintaining at 80°C for 15 min and then maintaining at 4°C. The products from purification treatment were used as a sequencing reaction template. The 10 μl reaction system was used for sequencing reaction. It contained 8 μl sequencing primers and 2 μl PCR product treated by ExoSAP-IT. The reaction was 25 cycles at 96°C for 20 s, 50°C for 30 s, 60°C for 2 min and 4°C. The sequencing reaction products were precipitated with sodium acetate/ethanol method. Adding 15 μl formamide (Hi-Di Formamide), they were denatured in the PCR instrument at 95°C for 3 min. The PCR products were analyzed by dideoxy chain termination method using a sequencing machine (Applied Bio systems 3130, USA). The sequencing results were compared with standard sequence. Assign-SBT 3.5+ (Conexio Genomics, Western Australian) software was used to analyze SLC26A4 allele type of subjects. The SLC26A4 gene sequencing ratings and the mean of the tetranucleotide peak were calculated. The sequencing results are shown in Figure 3.
Figure 3.
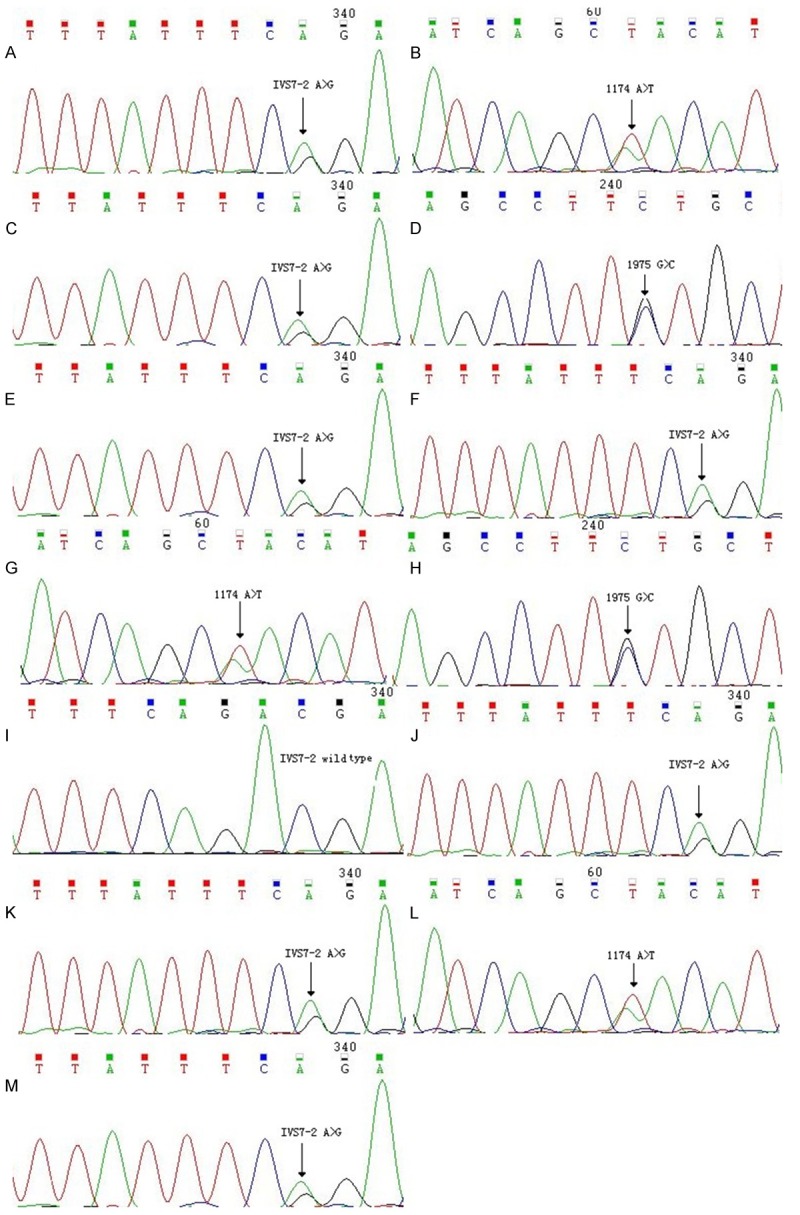
The SLC26A4 gene sequencing. A, B: The sequencing of pregnant woman were SLC26A4 1174 A>T and IVS7-2A>G two-hybrid mutation. C, D: The sequencing of the husband were SLC26A4 1975G>C and IVS7-2A>G two-hybrid mutation. E, F: The sequencing of two sides mothers were SLC26A4 IVS7-2A>G single heterozygous mutation. G: The sequencing of the woman’s father was SLC26A4 1174 A>T single heterozygous mutation. H: The sequencing of the husband’s father was SLC26A4 1975 G>C single heterozygous mutation. I: The sequencing of the woman’s older sister was SLC26A4 wild-type. J: The sequencing of the husband’s the young sister was SLC26A4 IVS7-2A>G single heterozygous mutation. K, L: The sequencing of the twin fetus were SLC26A4 1174 A>T and IVS7-2 A>G two-hybrid mutation. M: The sequencing of f Amniotic fluid by the second cycle AID was SLC26A4 IVS7-2A>G single heterozygous mutation.
Results
The test results of deafness gene chips were found that the couples, both mothers and the man’s younger sister and twin fetus were IVS7-2A>G carriers, indicating that they were obliged to sequence SLC26A4. However, their fathers and the woman’s older sister were normal in the test results of deafness gene chips.
The sequencing results of pregnant woman were SLC26A4 1174 A>T and IVS7-2A>G two-hybrid mutation. The sequencing results of the husband were SLC26A4 1975G>C and IVS7-2A>G two-hybrid mutation. The sequencing results of two sides mothers were SLC26A4 IVS7-2A>G single heterozygous mutation. The sequencing result of the woman’s father was SLC26A4 1174 A>T single heterozygous mutation. The sequencing result of the husband’s father was SLC26A4 1975 G>C single heterozygous mutation. The sequencing result of the woman’s older sister was SLC26A4 wild-type. The sequencing result of the husband’s the young sister was SLC26A4 IVS7-2A>G single heterozygous mutation.
Because amniotic fluid was punctured for prenatal diagnosis is likely to cause the risk of preterm delivery in pregnant women in late pregnancy (approximately 31 weeks), and we understood study objects genotypes and pedigree analysis information from the two families, so the prenatal diagnosis of the twin fetus were not done, but we had clearly inferred that the twin fetus were congenital deafness. We had proved the twin fetuses after the termination of pregnancy by gene chips and whole genome sequencing of SLC26A4. The sequencing results of the twin fetus were SLC26A4 1174 A>T and IVS7-2A>G two-hybrid mutation.
The woman did not be pregnant in the first cycle AID, and the second cycle she obtained Single births. In week 16 of pregnancy, amniotic fluid was punctured after the deaf couple accepted AID. The results of SLC26A4 gene chips and sequencing confirmed that the fetus was a single heterozygous mutation. A baby girl was born in full-term. The result of audiometry confirmed that her hearing had been normal on the third day and the thirtieth day after birth.
The couple family tree genetic spectrum of SLC26A4 gene is shown in Figure 4. The SLC26A4 gene is autosomal recessive, the deafness gene of the couple is inherited from both parents carrying SLC26A4 mutation gene by pedigree analysis.
Figure 4.
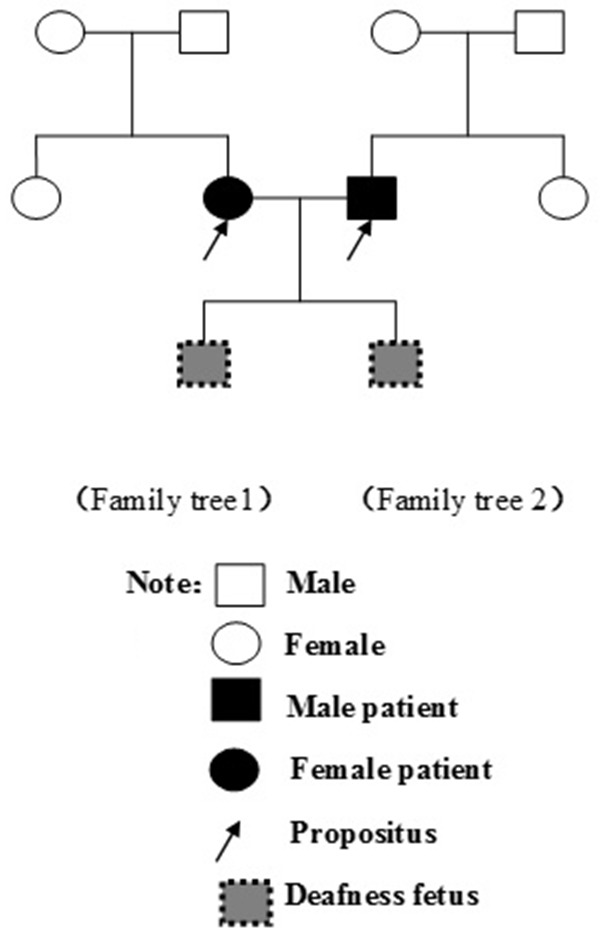
The couple Family trees genetic spectrum SLC26A4 (Family tree 1, 2).
Discussions
Congenital deafness is the most common neurosensory disorder. It can be divided into two types: genetic deafness and non-genetic deafness. The genetic deafness has been thought to be a highly complex problem due to genetic heterogeneity. It also can be divided into syndromic hearing loss (SHL) and non-syndromic hearing loss (NSHL) which accounts for more than 70% [5]. Until now, more than 100 mutations leading to NSHL have been located and 68 pathogenic genes have been cloned including 40 autosomal recessive genes, 25 autosomal dominant genes and 3 X-linked genes. Each gene contains multiple deafness-associated mutation sites (http://hereditaryhearingloss.org/, updated in March 9th, 2012). In Chinese population, molecular epidemiological investigation related to hearing loss showed that four genes including GJB2, GJB3, SLC26A4 and mitochondrial 12S rRNA gene (A1555G and C1494T) are the most common ones in NSHL patients [14].
In this study, nine mutation hot spots of the four common deafness-related genes were detected using the deafness gene chip technology, including GJB2 (35delG, 176del16, 235delC and 299delAT), GJB3 (C538T), SLC26A4 (IVS7-2A>G, A2168G) and mitochondrial 12S rRNA (A1555G and C1494T). Furthermore, the whole gene sequencing analysis of SLC26A4 gene was carried out according to the results of microarray-based genetic testing. There is IVS7-2A>G mutation in 8 of 11 samples from the two pedigrees in this report, showing that the mutation rate of this locus is much higher. Therefore, the IVS7-2A>G mutation is a hot-spot mutation inducing deafness in Chinese population, which is in consistence with the report of Dai et al. [15].
The IVS7-2A>G is a splicing mutation located in the splice point of the 3’ end of intron 7. When the IVS7-2A>G mutation occurs, this splice point disappears causing exon 8 (the transcription product) to be sheared entirely and exon 7 directly to be connected with exon 9. Thereby the consequence of this transformation affects the normal production of Pendrin [16]. It suggests that the universal screening of the IVS7-2A>G mutation for childbearing age population can significantly reduce the birth rate of infant with large vestibular aqueduct. Because the SLC26A4 gene is autosomal recessive, the deafness gene of the couple is inherited from both parents carrying SLC26A4 gene by pedigree analysis. In this report, 1174 A>T single heterozygous mutation of the SLC26A4 gene in pregnant woman’s father and 1975 G>C single heterozygous mutation in pregnant woman’s father-in-law are also mutation sites leading to hearing loss. The mutation frequency at two mutation sites is 2.82% and 0.56%, respectively [17,18]. Consequently, the genotype of fetal can be one of four types, namely IVS7-2A>G and 1174 A>T dual (compound) heterozygous mutation, IVS7-2A>G and 1975 G>C dual (compound) heterozygous mutation, IVS7-2A>G homozygous mutation, and 1174 A>T and 1975 G>C dual (compound) heterozygous mutation. Because any kind of four genotypes can cause fetal deafness, induction of labor had been performed for pregnant woman under the circumstance of informed consent. The result of molecular diagnosis confirmed that the genotypes of the twins were the same as IVS7-2A>G and 1174 A>T dual (compound) heterozygous mutations.
For all kinds of people, the combination of gene microarray technology and sequencing may be a feasible method for universal screening and clinical diagnosis of hereditary deafness. Deafness gene microarrays used in this experiment has many advantages such as high-throughput, simplified testing procedures and high accuracy. So it is very suitable for rapid screening of clinical specimens to detect common deafness gene mutation hotspots. The principle of gene chip technology is that human genomic DNA is used as a template, the known gene locus-specific primers is applied for amplification and fluorescent marker of the gene fragment and is then hybridized with a specific nucleotide sequence. The result is interpreted using fluorescent labeling technique, but other non-hot spot mutations cannot be detected. Fully sequencing can present a variety of gene mutation sites directly and draw the mutation spectrum of all kinds of people properly. However, it is not be feasible for large-scale population’s screening of clinical specimens due to time and effort consumption and high cost. Therefore, preliminary screening with deafness gene microarray should be carried out, and then the result of preliminary screening can supply the basis of selective sequencing for detecting different deafness-associated gene mutations to offer the molecular diagnosis basis of NSHL.
The results of this study suggest that it is essential of genetic counseling before marriage among sufferers with hearing impairment, pre-pregnancy screening after marriage and early detection in pregnant women. Due to the particularity of mutual exchanges among sufferers with hearing loss, they are married each other at high rate. Thus the incidence of genetic deafness children is greatly increased. The main aim of extensive genetic screening is to avoid sufferers with identical deafness-associated gene mutation sites to marry each other and reduce the birth rate of offspring suffering from genetic deafness. For child-bearing age population, especially for the deaf population, deaf genetic detection should be used as a conventional item of pre-pregnancy screening. Deaf couple who do not receive pre-marital and pre-pregnancy fertility guidance should be strictly detected the fetal genetic mutations in early pregnancy to prevent induction of labor in the third trimester of pregnancy from doing physiological and psychological harm to pregnant women. Consequently, it is necessary for scientific propaganda among deaf persons and assessment of the risk of offspring suffering from genetic deafness to reduce the birth rate of neonate attacked by predictable genetic deafness. The deaf couple in this study obtained a infant with normal hearing. According to the results of genetic diagnosis, human assisted reproductive technology is a viable option to avoid the birth of fetus with genetic disorders.
Conclusions
To reduce children with congenital deafness, screening high mutation sites by microarray, combined with pedigree analysis and gene sequencing, is effective, and should be used as a routine inspection item for the deaf before marriage and pregnancy. On the basis of results of genetic testing for the couple with hearing loss, human assisted reproductive technology is a viable option to avoid the birth of infant with hereditary deafness.
Acknowledgements
This study was supported by a grant from the development of science and technology plan projects in yantai (grant no. 2012089).
Disclosure of conflict of interest
None.
References
- 1.Nadol JB Jr. Hearing loss. N Engl J Med. 1993;329:1092–1102. doi: 10.1056/NEJM199310073291507. [DOI] [PubMed] [Google Scholar]
- 2.Khabori MA, Patton MA. Consanguinity and deafness in Omani children. Int J Audiol. 2008;47:30–33. doi: 10.1080/14992020701703539. [DOI] [PubMed] [Google Scholar]
- 3.Sajjad M, Khattak AA, Bunn JE, Mackenzie I. Causes of childhood deafness in Pukhtoonkhwa Province of Pakistan and the role of consanguinity. J Laryngol Otol. 2008;122:1057–1063. doi: 10.1017/S0022215108002235. [DOI] [PubMed] [Google Scholar]
- 4.Morton CC, Nance WE. Newborn hearing screening--a silent revolution. N Engl J Med. 2006;354:2151–2164. doi: 10.1056/NEJMra050700. [DOI] [PubMed] [Google Scholar]
- 5.Morton NE. Genetic epidemiology of hearing impairment. Ann N Y Acad Sci. 1991;630:16–31. doi: 10.1111/j.1749-6632.1991.tb19572.x. [DOI] [PubMed] [Google Scholar]
- 6.White KR. Early hearing detection and intervention programs: opportunities for genetic services. Am J Med Genet A. 2004;130A:29–36. doi: 10.1002/ajmg.a.30048. [DOI] [PubMed] [Google Scholar]
- 7.Sun XB, Wei ZY, Yu LM, Wang Q, Liang W. [Prevalence and etiology of people with hearing impairment in China] . Zhonghua Liu Xing Bing Xue Za Zhi. 2008;29:643–646. [PubMed] [Google Scholar]
- 8.Yuan Y, You Y, Huang D, Cui J, Wang Y, Wang Q, Yu F, Kang D, Yuan H, Han D, Dai P. Comprehensive molecular etiology analysis of nonsyndromic hearing impairment from typical areas in China. J Transl Med. 2009;7:79. doi: 10.1186/1479-5876-7-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y, Sun J. Journal of Translational Medicine Genetic mutations in non-syndromic deafness patients of Uyghur and Han Chinese ethnicities in Xinjiang, China: A comparative study. 2011;154:154. doi: 10.1186/1479-5876-9-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu X, Liang F, Zhao M, Gong A, Berry ER, Shi Y, Wang Y, Chen Y, Liu A, Qu C. Mutational analysis of the SLC26A4 gene in Chinese sporadic nonsyndromic hearing-impaired children. Int J Pediatr Otorhinolaryngol. 2012;76:1474–1480. doi: 10.1016/j.ijporl.2012.06.027. [DOI] [PubMed] [Google Scholar]
- 11.Yao G, Li S, Chen D, Wang H, Zhang J, Feng Z, Guo L, Yang Z, Yang S, Sun C, Zhang X, Ma D. Compound heterozygous mutations of SLC26A4 in 4 Chinese families with enlarged vestibular aqueduct. Int J Pediatr Otorhinolaryngol. 2013;77:544–549. doi: 10.1016/j.ijporl.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 12.Pourova R, Janousek P, Jurovcik M, Dvorakova M, Malikova M, Raskova D, Bendova O, Leonardi E, Murgia A, Kabelka Z, Astl J, Seeman P. Spectrum and frequency of SLC26A4 mutations among Czech patients with early hearing loss with and without Enlarged Vestibular Aqueduct (EVA) Ann Hum Genet. 2010;74:299–307. doi: 10.1111/j.1469-1809.2010.00581.x. [DOI] [PubMed] [Google Scholar]
- 13.Wang GJ, Han DY. Application of DNA microarray in rapid genetic diagnosis of non-syndromic hearing loss. Chinese Journal of Otology. 2008;6:61–66. [Google Scholar]
- 14.Han M. Progress in clinical application of gene diagnosis in China. Beijing Medical Journal. 2011;33:419–421. [Google Scholar]
- 15.Dai P, Feng B. Genetic testing for the enlarged vestibular aqueduct syndrome and mutation analysis of the SLC26A4 gene. Chinese Archives of Otolaryngology-Head and Neck Surgery. 2006:13. [Google Scholar]
- 16.Yang JJ, Tsai CC, Hsu HM, Shiao JY, Su CC, Li SY. Hearing loss associated with enlarged vestibular aqueduct and Mondini dysplasia is caused by splice-site mutation in the PDS gene. Hear Res. 2005;199:22–30. doi: 10.1016/j.heares.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 17.Park HJ, Shaukat S, Liu XZ, Hahn SH, Naz S, Ghosh M, Kim HN, Moon SK, Abe S, Tukamoto K, Riazuddin S, Kabra M, Erdenetungalag R, Radnaabazar J, Khan S, Pandya A, Usami SI, Nance WE, Wilcox ER, Riazuddin S, Griffith AJ. Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: global implications for the epidemiology of deafness. J Med Genet. 2003;40:242–248. doi: 10.1136/jmg.40.4.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang QJ, Zhao YL, Rao SQ, Guo YF, Yuan H, Zong L, Guan J, Xu BC, Wang DY, Han MK, Lan L, Zhai SQ, Shen Y. A distinct spectrum of SLC26A4 mutations in patients with enlarged vestibular aqueduct in China. Clin Genet. 2007;72:245–254. doi: 10.1111/j.1399-0004.2007.00862.x. [DOI] [PubMed] [Google Scholar]