

Abstract
Type I (T1) diabetes is an autoimmune and metabolic disease associated with bone loss. Previous studies demonstrate that T1-diabetes decreases osteoblast activity and viability. Bisphosphonate therapy, commonly used to treat osteoporosis, is demonstrated to inhibit osteoclast activity as well as osteoblast apoptosis. Therefore, we examined the effect of weekly alendronate treatments on T1-diabetes induced osteoblast apoptosis and bone loss. Bone TUNEL assays identified that alendronate therapy prevents the diabetes-induced osteoblast death observed during early stages of diabetes development. Consistent with this, alendronate treatment for 40 days was able to prevent diabetes-induced trabecular bone loss. Alendronate was also able to reduce marrow adiposity in both control diabetic mice compared to untreated mice. Mechanical testing indicated that 40 days of alendronate treatment increased bone stiffness but decreased the work required for fracture in T1-diabetic and alendronate treated mice. Of concern at this later time point, bone formation rate and osteoblast markers, which were already decreased in diabetic mice, were further suppressed in alendronate treated diabetic mice. Taken together, our results suggest that short term alendronate treatment can prevent T1-diabetes-induced bone loss in mice, possibly in part by inhibiting diabetes onset associated osteoblast death, while longer treatment enhanced bone density but at the cost of further suppressing bone formation in diabetic mice.
Keywords: Bone, Diabetes, Alendronate, Death, Osteoblast, Bone formation
INTRODUCTION
Type I (T1) diabetes is both an autoimmune and metabolic disease characterized by little to no insulin production and hyperglycemia. Medical advances to maintain euglycemia are increasing patient lifespan, however, even today proper maintenance of glucose level remains difficult. Prolonged exposure to hyperglycemia results in increased occurrence of secondary complications such as neuropathy, nephropathy, retinopathy and osteoporosis [Beisswenger et al., 2005; Duby et al., 2004; Goralski and Sinal, 2007; McCabe, 2007; Susztak et al., 2006; Valeri et al., 2004]. Osteoporosis is defined as 2.5 standard deviations below average bone density in humans. A decrease in bone density by even 1 standard deviation results in increased fracture risk by roughly 2.4 times. Bone loss and increased fracture risk are evident in the hip [Alexopoulou et al., 2006; Forsen et al., 1999; Hamilton et al., 2009; Meyer et al., 1993; Miao et al., 2005; Munoz-Torres et al., 1996; Schwartz et al., 2001] and spine [Alexopoulou et al., 2006; Hamilton et al., 2009; Munoz-Torres et al., 1996] of T1-diabetic patients and are independent of gender and age. Spontaneous and pharmacologic-induced rodent models of T1-diabetes display comparable decreases in bone density to T1-diabetic patients [Botolin and McCabe, 2006a; Coe et al., 2012].
Analysis of bone parameters in diabetic patients and mouse models indicate that bone formation is reduced [Bain et al., 1997; Botolin et al., 2005; Botolin and McCabe, 2007; Bouillon et al., 1995; Cakatay et al., 1998; Glajchen et al., 1988; Lumachi et al., 2009; McCabe et al., 2011; McCabe, 2007; Motyl and McCabe, 2009b; Motyl et al., 2009; Motyl and McCabe, 2009c]. Bone formation is regulated by numerous factors and can be suppressed through multiple processes including increasing mesenchymal cell progression to adipocyte over osteoblast lineage selection as well as by increasing osteoblast death. Elevated marrow adiposity is associated with diabetic bone loss [Botolin and McCabe, 2007; McCabe, 2007; Motyl and McCabe, 2009a]. However, inhibition of marrow adiposity does not prevent diabetes-induced bone loss [Botolin and McCabe, 2006b; Martin and McCabe, 2007; Motyl and McCabe, 2009c] suggesting that additional mechanisms are required. Recently, we reported elevated osteoblast apoptosis during diabetes onset when the first detectable decrease in osteocalcin expression is observed [Coe et al., 2011; Motyl et al., 2009]. Osteoblast death has also been shown to be elevated in bacteria-challenged type 2 diabetic animals [He et al., 2004] and reduced by treatment with TNFα-specific inhibitors [Alblowi et al., 2009; Kayal et al., 2010]. Bone marrow cells from diabetic mice contribute to the increased osteoblast death seen in T1-diabetes [Coe et al., 2011].
To date, no perfect therapy exists for treating diabetes-induced bone loss. The most commonly used preventive therapy for decreased bone density is bisphosphonates [Bushardt et al., 2006; Cole et al., 2008; McClung, 2003a; McClung, 2003b]. These drugs selectivity integrate into bone mineral surfaces where they inhibit osteoclast-mediated resorption, recruitment and differentiation [Black, 1996; Devogelaer, 1996; Devogelaer et al., 1996; Giannini et al., 1993; Hosking et al., 1998; Liberman et al., 1995; McClung et al., 1998] and prevent both vertebral and non-vertebral fractures [Hughes et al., 1989; Russell and Rogers, 1999; Sahni et al., 1993; Sato et al., 1991]. Previous studies suggest that bisphosphonates not only inhibit osteoclast-mediated resorption but they also inhibit osteoblast and osteocyte death [Plotkin et al., 2006; Plotkin et al., 1999], thus making them a possible beneficial therapy for treating diabetic patients. In vitro studies report that bisphosphonates protect rodent-derived osteoblasts from advanced glycation end products (AGE)-induced death (previously shown elevated in diabetic bone) [Gangoiti et al., 2008]. Clinically, type 2 diabetic and non-diabetic women taking bisphosphonates displayed a benefit in overall bone health [Keegan et al., 2004]. The goal of this study was to determine if weekly alendronate therapy, a commonly used bisphosphonate, would prevent diabetes-associated osteoporosis. Our findings indicate that short-term alendronate treatment reduced diabetes-induced osteoblast death and prevented diabetes-induced bone loss, but at 6 weeks the consistent diabetes-induced reduction of bone formation markers was further suppressed by alendronate treatment.
METHODS
Diabetic Mouse Models
Diabetes was induced (starting at experimental day 0) in adult (16 week old) male C57BL/6 mice by daily intraperitoneal injections of streptozotocin (50 mg/kg body weight in 0.1 M citrate buffer, pH 4.5) for 5 consecutive days. Controls were given citrate buffer alone. Alendronate was delivered (beginning on day 0) by weekly subcutaneous injections in control (n=8) and diabetic (n=8) mice at a concentration of 2 mg/kg/week, a dose consistent with past mouse studies [Nijenhuis et al., 2008]. Mice were maintained on a 12-hour light, 12-hour dark cycle at 23°C, given standard lab chow and water ad libitum. Diabetes was confirmed 12 days after initial injection using an Accu-Check compact glucometer (Roche Diagnostics Corporation, Indianapolis, IN) with a drop of blood from the saphenous vein. Mice were euthanized at either 7 or 40 days after the start of diabetes induction. At this time general parameters were measured, including blood glucose levels and total body, tibialis anterior and subcutaneous femoral fat pad mass. Animal procedures were approved by the Michigan State University Institutional Animal Care and Use Committee.
RNA Analysis
Immediately after euthanasia, one tibia and femur were cleared of soft tissue and snap frozen in liquid nitrogen and stored at −80°C. Frozen tibias were crushed under liquid nitrogen conditions, homogenized, and placed in Tri Reagent (Molecular Research Center, Inc., Cincinnati, OH). RNA integrity was determined through formaldehyde-agarose gel electrophoresis. cDNA was synthesized through a reverse transcriptase reaction utilizing Superscript II Reverse Transcriptase Kit and oligo dT (12–18) primers (Invitrogen, Carlsbad, CA), amplified by quantitative real time PCR with iQ SYBR Green Supermix (Bio-Rad, Hercules, CA), and gene-specific primers. Forward and reverse primers for aP2, Bax, HPRT, osteocalcin and cathepsin K have been previously described [Botolin et al., 2005; Harris et al., 2009]. Expression of HPRT is constant in diabetic and control mouse bones and therefore was used as a housekeeping gene. Real time PCR was carried out for 40 cycles, each cycle consisting of 95°C for 15 seconds, 60°C for 30 seconds (except osteocalcin which has an annealing temperature of 65°C) and 72°C for 30 seconds. RNA-free samples were used as a negative control and did not produce amplicons. PCR products were separated on 1.5% agarose gel electrophoresis and sequenced to verify the desired gene is being amplified.
Micro-computed Tomography (μCT) analysis
Fixed femurs in 70% ethanol were scanned using the GE Explore μCT system at a voxel resolution of 20um from 720 views with a beam strength of 80kvp and 450uA. Integration time for each scan was 2000ms. Scans included bones from each condition and a phantom bone to standardize the grayscale values and maintain consistency between runs. Using the systems auto-threshold (800) and isosurface analysis confirmation, trabecular bone densities (bone volume fraction (BV/TV), bone mineral density (BMD) and content (BMC), and trabecular thickness (tb. th), number (tb. n) and spacing (tb. sp)) were measured in the trabecular region defined at 0.17mm under the growth plate of the femur extending 2mm toward to diaphysis, and excluding the outer cortical shell. Cortical bone measurements are determined with a 2-mm3 region of interest (ROI) in the mid-diaphysis. Cortical thickness, moment of inertia, cortical area, marrow area, total area, inner perimeter and outer perimeter as well as all trabecular parameters were computed using the GE Microview Software for visualization and analysis of volumetric image data.
Mechanical Strength Testing
Mouse femurs were subjected to three-point bending to determine their mechanical properties [Isaksson et al., 2009a; Isaksson et al., 2009b; Jamsa et al., 1998; Phillips et al., 2008; Schriefer et al., 2005]. Specimens were thawed at room temperature prior to testing and kept wet in saline solution. The bones were placed on the apparatus support with the medial surface facing up and loaded using an MTS Insight at 0.05 mm/s until failure. The stiffness was determined by calculating the slope of the load versus displacement curve. The elastic modulus was calculated using elementary beam theory (Equation 1) [Jamsa et al., 1998].
| (1) |
Where P is the load, L is the span length of the three-point bend support (10 mm), I is the moment of inertia, and δ is the deflection at the center of the bone. The load and displacement values were determined using the linear portion of the load versus displacement curve. The ultimate stress was calculated using Equation 2.
| (2) |
In equation 2, P is the ultimate load, L is the span length, c is half the bone diameter along the loading axis, and I is the moment of inertia. To calculate the moment of inertia, the mouse bone was modeled as a hollow ellipsoid. The thickness of the bone was averaged from MicroCT data for each specimen. The moment of inertia for a hollow ellipsoid can be calculated using equation 3 where B and D represent the outer major and minor diameters (respectively) and b and d represent the inner major and minor diameters (respectively).
| [Harris et al.] |
Bone Histology and Histomorphometry
Bones were fixed in 10% buffered formalin, transferred to 70% alcohol after 24 hours and then processed for dehydration, clearing and infiltration on a routine overnight processing schedule. Samples were then paraffin embedded on a Sakura Tissue Tek II embedding center and 5 μm sections prepared. Osteoclasts were identified, by staining for tartrate resistant acid phosphatase (TRAP) (Sigma). The number or surface area of osteoclasts, osteoblasts and adipocytes was determined in the trabecular region of the femur (defined at 0.17mm under the growth plate of the femur extending 2mm toward to diaphysis). Osteoclast and osteoblast surface was measured in three trabecular regions for each mouse and expressed as a percent of total trabecular surface. Visible adipocytes, greater than 30 μm, were counted in the same trabecular region.
Scanning Electron Microscopy
Bone specimens SEM micrographs were obtained using a JEOL 6400V SEM (Japan Electron Optics Laboratories, Tokyo, Japan) with a LaB6 emitter (Noran EDS, Noran Instruments Inc., Middleton, WI) at an accelerating voltage of 15 kV and magnification of 25,000×.
Cell death
Cell death was determined using a TACS•XL® Basic In Situ Apoptosis Detection Kit (TUNEL, Trevigen Inc., Gaithersburg, MD) on the L1–3 vertebrae or femur sections. Osteoblasts with positive nuclei were counted and expressed as a percentage of total osteoblasts counted per bone. Positive controls included slides incubated with nuclease. Five trabecular regions were examined for each mouse. Total osteoblasts counted ranged between 20 and 100 per bone.
Dynamic bone formation
Mice were injected intraperitoneally with 200μl of 10mg/ml calcein (Sigma, St. Louis, MO, USA) dissolved in saline at 7 and 2 days prior to harvest. L3–L4 vertebrae were fixed in formalin at time of harvest then transferred to 70% ethanol 24 hours later. Vertebrae were then embedded, sectioned and examined under UV light. Five images were taken and the distance between the calcein lines and their length along the bone surface was measured and used to calculate MAR. Mineralizing surface was calculated by measuring total calcein labeling per total trabecular surface. Bone formation rate was calculated by the equation BFR=mineralizing surface × mineral apposition rate as previously stated [Bennett et al., 2007; Schriefer et al., 2005].
Serum Measurements
Blood was collected at the time of harvest, allowed to clot at room temperature for 5 minutes, then centrifuged at 4,000 rpm for 10 minutes. Serum was removed and stored at −80°C. Serum went through no more than two freeze/thaw cycles. Serum TRAP5b and Osteocalcin were measured using a Mouse TRAP and OC assay kits (SB-TR103, Immunodiagnostic Systems Inc., Fountain Hills, AZ, USA and BT-470, Biomedical Technologies Inc., Stoughton, MA, USA respectively) according to the manufacturer’s protocol.
Statistical Analysis
All data is presented as mean ± standard error. Statistical significance (p-value < 0.05) of main effects (treatment or diabetes) as well as treatment × diabetes interaction (which determines if diabetes alters the treatment effect or vise versa) was determined using 2-way factorial analysis of variance (ANOVA) where noted, by using SPSS statistical software (Chicago, IL). When diabetes and treatment were found to have a significant effect, a Fisher post hoc test was performed to determine significant differences between treatment groups. When 2-way ANOVA analysis did not show significance we used the less stringent 1-way ANOVA with a Fisher post hoc test to determine significant differences between groups. Specific statistical analyses are noted in the figure legends.
RESULTS
Previously, we demonstrated an increase in osteoblast death in T1-diabetic mice during early stages of disease induction [Coe et al., 2011; Motyl et al., 2012]. Given that past reports indicate a role for bisphosphonates in suppressing osteoblast apoptosis, we examined if alendronate treatment could suppress diabetes induced osteoblast apoptosis at this early time point. Diabetes was induced by streptozotocin injection in 16-week-old adult male C57BL/6 mice, and the mice were divided into two subgroups: alendronate treated and non-treated mice. Mouse femur sections were examined for TUNEL positive osteoblasts seven days after the start of the study, a time point where osteoblast death is readily detectable [Coe et al., 2011]. Consistent with past reports, the untreated diabetic mouse bones exhibited a significant (Fisher post-hoc test) increase in TUNEL positive osteoblasts (elevated nearly 4-fold). Two-way ANOVA analysis indicated that there was treatment × disease effect indicative of alendronate not affecting controls but affecting diabetic mice. In parallel, expression of the pro-apoptotic factor Bax was elevated by 40% compared to control mice (Figure 1B). Consistent with our hypothesis, alendronate-treated T1-diabetic mice did not display an increase in the number of TUNEL positive osteoblasts or in Bax mRNA levels in bone (Figure 1).
Figure 1. Alendronate suppresses early stage diabetes-induced osteoblast death.
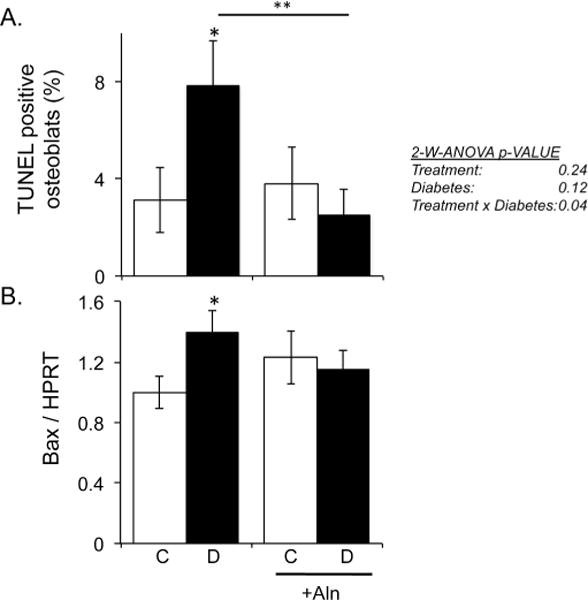
Mouse bones were examined during early stage diabetes development, 7 days post-diabetes induction with or without alendronate treatment. Bars represent the average value ± SE (n≥5 per group). A) Percentage of TUNEL positive stained osteoblasts in femur bone. Data was analyzed by 2-way ANOVA (charts on right) followed by Fisher post hoc analyses that determined significant differences * p< 0.05 (control vs diabetic), ** p<0.01 (untreated vs treated diabetic). B) Bax mRNA expression from control (white bars) and diabetic (black bars) non-treated and alendronate treated (+ Aln) mice. Bax expression is calculated relative to the housekeeping gene HPRT. Bars represent the average value ± SE (n≥5 per group). *denotes a significant difference (p<0.05) compared to control as determined by one-way ANOVA.
Because of the beneficial effects of alendronate on early stage (7 day) T1D diabetic bone changes, we examined if there were benefits to general body and bone parameters with 40 days of bisphosphonate treatment in control and diabetic. Analysis of general mouse parameters indicated that 40 days alendronate treatment decreased both femoral and visceral fat pad mass by 12.5% and 42%, respectively, compared to controls (table I), but did not prevent any of the diabetes-induced physiological changes. Specifically, diabetic mice with or without alendronate treatment displayed comparable elevations in blood glucose levels when compared to their respective controls (averaging 511 mg/dl and 494 mg/dl, respectively) (Table I). Both alendronate treated and untreated diabetic mouse groups displayed decreases in total body mass (14% and 13%, respectively), femoral fat pad mass (62% and 61%, respectively), tibialis anterior mass (24% and 25%, respectively), and visceral fat mass (94% and 96%, respectively) when compared to their respective treatment controls.
Table I.
Body parameters in control and diabetic with or without alendronate.
| Alendronate
|
||||
|---|---|---|---|---|
| Control (n=7) |
Diabetic (n=7) |
Control (n=8) |
Diabetic (n=8) |
|
| Body Mass (g) | 28.4 ± 0.9 | 24.6 ± 1.0* (−13%) | 27.4 ± 0.6 | 23.7 ± 0.5* (−14%) |
| Blood Glucose (mg/dl) | 156 ± 7 | 494 ± 29* (217%) | 166 ± 7 | 511 ± 34* (208%) |
| Fem Fat Mass (mg) | 15.8 ± 1.6 | 6.1 ± 0.7* (−61%) | 12.5 ± 1.1ˆ (−12.5%) | 4.8 ± 0.7* (−62%) |
| Tibialis Mass(mg) | 3.2 ± 0.2 | 2.4 ± 0.3* (−25%) | 3.3 ± 0.8 | 2.5 ± 0.3* (−24%) |
| Vis Fat Mass (mg) | 6.88 ± 1.7 | 0.17 ± 0.01* (−96%) | 4.01 ± 0.61ˆ (−42%) | 0.26 ± 0.17* (−94%) |
Abbreviations: Fem. Fat, femoral fat pad mass, Vis. Fat, visceral fat pad mass. Statistics:
p<0.05 compared to respective control.
p<0.05 compared to untreated controls. (%) change indicated compared to control littermates and vehicle treated control.
Next, we examined bone density parameters at 40 days post-diabetes induction using micro-computed tomography quantification and three-dimensional isosurface imaging of the distal femur trabecular region (Figure 2A). As expected and consistent with past studies, diabetes reduced trabecular bone volume fraction (BV/TV) and bone mineral density (BMD), by 31% and 27%, respectively (Figure 2B). Changes in trabecular parameters of the diabetic mice were characterized by decreases in trabecular thickness and number and increases in trabecular spacing (Table II). In contrast, alendronate therapy significantly increased both BV/TV and BMD by 54% and 27%, respectively, beyond control mouse levels. Alendronate treatment also increased trabecular number by 25% compared to non-treated controls. Even more importantly, alendronate therapy maintained high bone density and volume in the diabetic mice. Consistent with previous reports, T1-diabetes decreased femur diaphyseal cortical thickness, cortical area and the percent cortical area/total area compared to control bones (Figure 3, Table II). In contrast, T1-diabetic mice treated with alendronate displayed cortical bone parameters that were similar to control mice (Figure 3, table II). Two-way ANOVA analysis did not reveal an interaction between diabetes and alendronate treatment for bone volume or cortical parameters.
Figure 2. Long-term treatment with alendronate (40 days) prevents diabetes-induced trabecular bone loss.
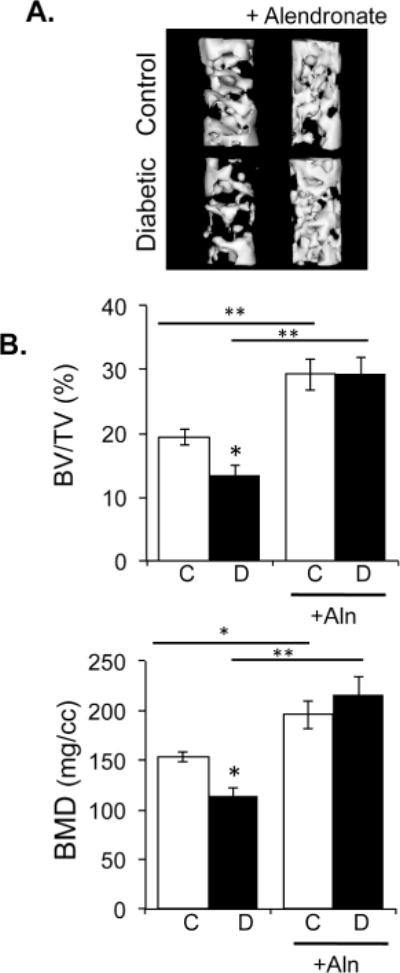
A) Representative microcomputed tomography isosurface images of trabecular bone volume/total volume (BV/TV) from distal femurs were obtained from control and diabetic mice treated with or without alendronate at 40 days post-diabetes induction. B) Average BV/TV and bone mineral density (BMD) in control (white bars) and diabetic (black bars), non-alendronate treated and alendronate treated mice. Bars represent the average value ± SE (n=7–8 per group). * p<0.05, ** p<0.001 as determined by one-way ANOVA.
Table II.
pCT femur parameters in control and diabetic vehicle and alendronate treatment littermates.
| Alendronate | ||||
|---|---|---|---|---|
|
|
||||
| Trabecular | Control | Diabetic | Control | Diabetic |
| BMC (mg) | 0.48 ± 0.02 | 0.34 ± 0.02* | 0.60 ± 0.05* | 0.65 ± 0.05*ˆ |
| BMD (mg/cc) | 154 ±5 | 113 ±9* | 195±14* | 216 ± 18*ˆ |
| BV/TV (%) | 19.4 ± 1.2 | 13.4 ±1.6* | 29.8 ±4.1* | 28.5±2.1*ˆ |
| Tb. Th (μm) | 40.0 ± 1.2 | 36.4 ±2.0* | 48.7 ± 5.1$ | 54.9 ± 7.1 *ˆ |
| Tb. N (1/mm) | 4.79 ± 0.26 | 3.56 ± 0.27* | 5.97 ± 0.22* | 5.96±0.19*ˆ |
| Tb. Sp (mm) | 0.17 ±0.01 | 0.26 ± 0.03* | 0.12±0.01* | 0.11 ±0.01*ˆ |
| Cortical | ||||
|
| ||||
| Ct. Th (mm) | 0.220 ± 0.003 | 0.206 ± 0.005* | 0.230 ± 0.006 | 0.230 ±0.011ˆ |
| Imax(mm4) | 0.20 ± 0.03 | 0.16 ±0.01 | 0.19 ±0.02 | 0.18 ±0.02 |
| Ec. Pm (mm) | 3.68 ± 0.07 | 3.79 ± 0.07 | 3.53 ± 0.06* | 3.57 ± 0.06ˆ |
| Ps. Pm (mm) | 5.09 ± 0.08 | 5.01 ±0.07 | 5.00 ± 0.07 | 5.01 ±0.06 |
| Ma. Ar (mm2) | 0.93 ± 0.03 | 0.96 ± 0.04 | 0.86 ± 0.03 | 0.89 ±0.03 |
| Ct. Ar (mm2) | 0.92 ± 0.02 | 0.78 ± 0.03* | 0.93 ± 0.03 | 0.92 ± 0.04ˆ |
| Tt.Ar (mm2) | 1.85 ±0.06 | 1.76 ±0.05 | 1.79 ±0.04 | 1.81 ±0.04 |
| Ct. Ar/ Tt. Ar (%) | 49.9 ± 0.44 | 44.6 ± 1.56* | 52.0 ± 1.09 | 50.8 ± 1.58ˆ |
| BMD (mg/cc) | 919 + 27 | 961 ± 26 | 914± 17 | 973 ± 24* |
Significance:
p<0.05 compared to non-treated control
p<0.05 compared to non-treated diabetic
p<0.07 compared to non-treated control.
Abbreviations: BMC, bone mineral content, BMD, bone mineral density, BV/TV, bone volume fraction, Tb. Th, trabecular thickness, Tb. Sp, trabecular spacing, Tb. N, trabecular number, Imax, maximum moment of inertia, Ec. Pm, endocortical perimeter, Ps. Pm, periosteal perimeter, outer perimeter, Ma. Ar, Marrow area, Ct. Ar, cortical area, Tt. Ar, total area.
Figure 3. Decreased cortical thickness in diabetic mice.
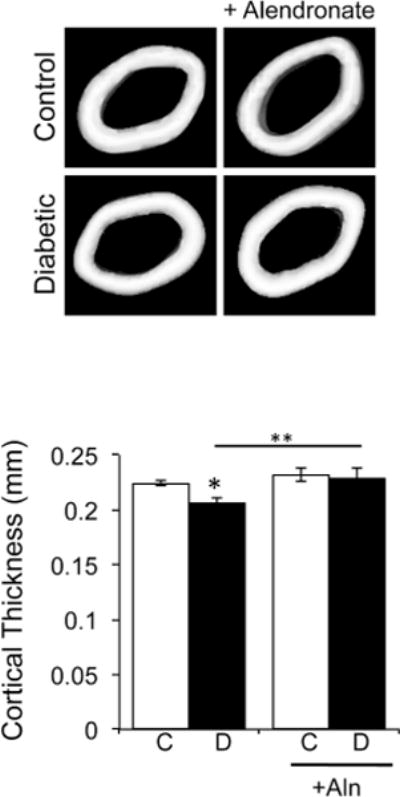
Representative microcomputed tomography isosurface images of cortical thickness from the mid-diaphysis region of femurs were obtained from control and diabetic mice treated with or without alendronate. Graphical representation of cortical thickness in millimeters is displayed from control (white bars) and diabetic (black bars), non-alendronate treated and alendronate treated mice. Bars represent the average value ± SE (n=7–8 per group). * p<0.05 compared to respective control. ** p<0.05 compared to untreated diabetic bone as determined by one-way ANOVA.
Consistent with alendronate’s benefit to bone density, it significantly reduce the overall marrow adipocyte number per total marrow area in both control and diabetic mice resulting in diabetic-treated mice having significantly less adipocytes than untreated diabetic mice (Figure 4). In contrast, bone aP2 expression, an adipocyte marker, was elevated in diabetic mice but levels were not suppressed by alendronate treatment in control or diabetic mice. The discordance between alendronate treatment reducing basal adipocyte numbers but not aP2 levels in mice could result from the presence of smaller adipocytes present in alendronate treated mice but not included in our counts (hence the basal adipocyte number is actually higher), altered adipocyte maturation (greater aP2 expression per adipocyte, so fewer adipocytes would still express the same level of aP2 as untreated mice) or changes in macrophage numbers in the marrow of alendronate treated mice (leading to increased overall levels of aP2). Of interest, the specific T1-diabetes induced bone marrow adiposity effect [Botolin and McCabe, 2007; Martin and McCabe, 2007; McCabe, 2007; Motyl and McCabe, 2009c] was seen in both untreated and treated mice (Figure 4). Specifically, T1-diabetes induced a significant greater than 2-fold increase in bone marrow adipocyte number per total marrow area and a 60% increase in bone aP2 mRNA levels (a mature adipocyte marker) in both untreated and treated mice.
Figure 4. Diabetes induces marrow adiposity and alendronate therapy decreases marrow adiposity.
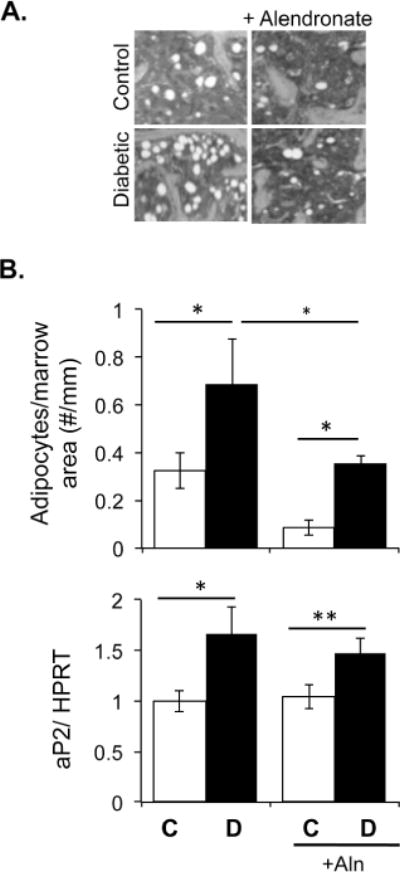
Representative images of bone marrow adiposity from distal femurs were obtained from control and diabetic mice treated with or without alendronate for 40 days. Graphical representations of adipocyte number per total marrow area and aP2 mRNA expression are displayed from control (white bars) and diabetic (black bars), non-alendronate treated and alendronate treated mice. Bars represent the average value ± SE (n=7–8 per group). RNA levels are calculated relative to the housekeeping gene HPRT. Data analyzed by one-way ANOVA, * p<0.05, **p<0.01.
To test if the observed changes in bone density translated into stronger bone, we examined femur diaphyseal bone strength parameters by three-point bending following 40-days of alendronate treatment. Both diabetes and alendronate showed limited effects on the ultimate stress and elastic modulus of the specimens (Figure 5). The bones did, however, show a change in stiffness. Bones from alendronate treated mice showed a higher stiffness (~20%) compared to control specimens. T1-diabetic mouse bones showed a decrease in stiffness (~20%) and alendronate significantly increased the stiffness (~30%) (Figure 5). However, compared to control mice, the work to fracture was significantly decreased in both diabetic (45%) and alendronate treated (~30%) mice, while alendronate treated diabetic mice displayed a minor trend to decrease (~10%).
Figure 5. Alendronate increases bone stiffness while diabetes and alendronate reduce force to fracture.
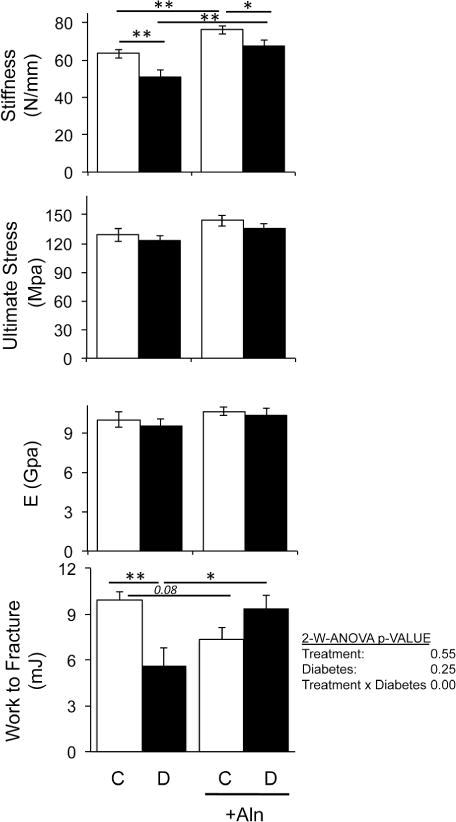
Three-point bending tests were performed on femurs from control (white bars) and diabetic (black bars) non-treated and alendronate treated mice. Stiffness, ultimate stress, energy and work to fracture (area under the stress strain curve) were calculated and represented as average value ± SE (n≥7–8 per group). Work to fracture was analyzed by 2-way ANOVA to determine the effects of bisphosphonate treatment, diabetes and bisphosphonate treatment × diabetes on femur strength parameters. Fisher post hoc test was used to compare differences between groups. The rest of the graphs were analyzed by one-way ANOVA followed by Fisher post hoc tests to compare differences between groups, *p<0.05, **p<0.01.
Following fracture, the cross sectional images of the femurs were viewed using scanning electron microscopy (SEM) to further assess bone quality. Representative images (Figure 6) demonstrate the structural differences between bones from control compared to bones from diabetic and alendronate treated mice, which displayed evidence of circumferential cracks running parallel to the bone surface (as indicated in Figure 6 by the white arrows) on the compressive portion of the bones. The presence of cracks is consistent with the calculated work to fracture (Figure 7) that decreases in the bones from the untreated-diabetic mice (p<0.05), the alendronate treated control mice (p<0.05), and the treated-diabetic mice bone (trend) when compared to the bones from the untreated control mice. Interestingly, the largest cracks appear in the samples having the lowest work to fracture (untreated-diabetic and the alendronate treated control) in comparison to the smaller cracks in the treated-diabetic mice bone. The functional significance of these cracks is not fully known, since we did not control for variations in the bone matrix, pore size and pore distribution, but is an area of future investigation.
Figure 6. Representative scanning electron microscopy (SEM) images of fracture surfaces of bones obtained from alendronate treated control and diabetic mice.
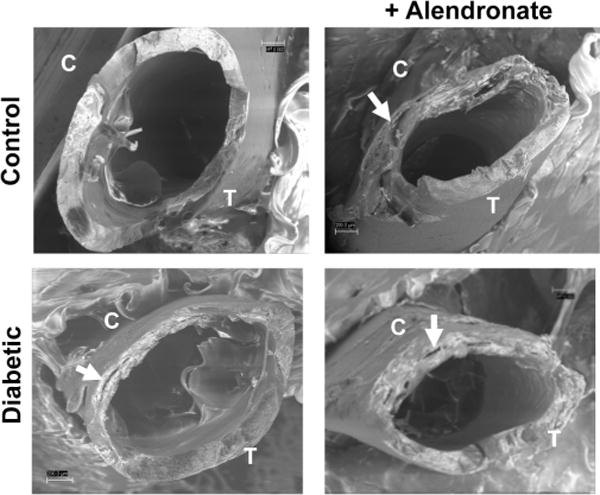
After strength testing, the bone fracture surfaces were examined. Noted are the tensile (T) and compressive fracture areas (C). White arrows point to circumferential cracks that run parallel to the bone surface.
Figure 7. Alendronate effects on bone resorption markers.
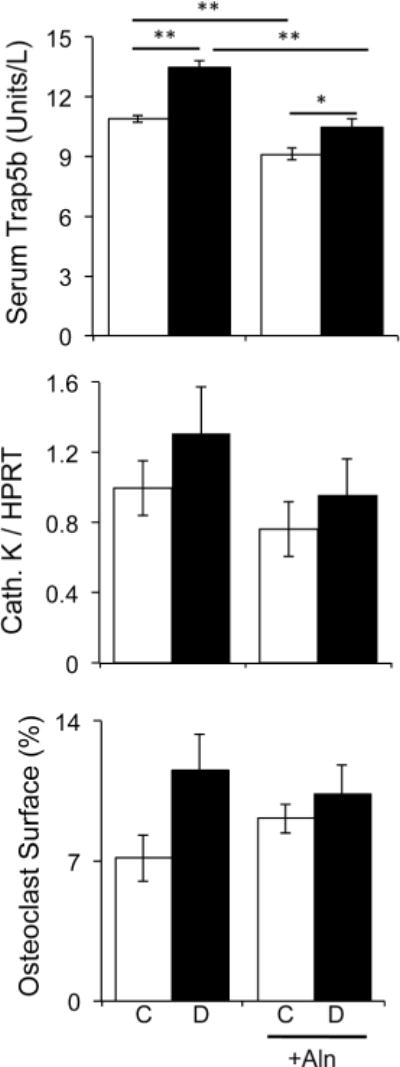
Serum TRAP5 concentration, cathepsin K mRNA levels, and osteoclast surface were analyzed in non-treated and alendronate treated in control (white bars) and diabetic (black bars) mice at the 40 day time point. Bars represent the average value ± SE (n=7–8 per group). Data was analyzed by one-way ANOVA followed by Fisher post hoc tests to compare differences between groups, *p<0.05, **p<0.01.
To assess the mechanism of alendronate treatment effects on diabetic bone we examined osteoclast and osteoblast markers. Bisphosphonates are known suppressors of bone resorption and accordingly we observed that alendronate therapy decreased serum active Trap5b levels in both the control and diabetic mice (Figure 7). However, we did not observe significant reductions in other osteoclast markers (cathepsin K expression or percent osteoclast surface), this is likely due to the time point that they were measured at (40 days) and variability among mice. Similarly, T1-diabetes induced changes were variable, but did show an increase in serum Trap5 levels in both untreated and treated mice.
Examination of osteoblast markers indicated that T1-diabetes decreased serum osteocalcin levels and osteocalcin mRNA levels by 30% and 37%, respectively, similar to past studies (Figure 8). Alendronate therapy alone decreased serum and tibia osteocalcin levels while alendronate treated diabetic mice displayed the lowest osteocalcin levels of the groups (Figure 8). Consistent with this finding, diabetes alone and alendronate therapy alone or in combination (diabetic mice treated with alendronate) decreased dynamic bone formation rate. Specifically, bisphosphonate treatment resulted in an ~80% reduction in bone formation rate, while diabetes only decreased BFR by ~40%. One-way ANOVA determined that alendronate treatment caused further suppression (by more than 66% compared to untreated diabetic mice, p<0.001) of an already low BFR in diabetic mice. Interestingly, we observed the circumferential cracks in the three groups with low osteoblast marker levels.
Figure 8. Bone formation is decreased at 40 day time point as a result of both diabetes and bisphosphonate therapy.
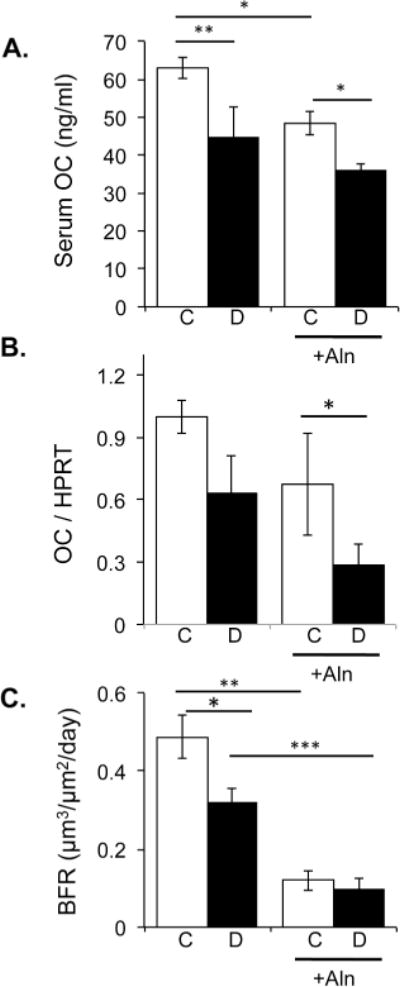
A & B) Serum osteocalcin concentration and tibia mRNA osteocalcin expression was analyzed in non-treated and alendronate treated in control (white bars) and diabetic (black bars) mice. Bars represent the average value ± SE (n=7–8 per group). RNA levels are calculated relative to the housekeeping gene HPRT. C) Mice were injected with calcein dissolved in saline 7 and 2 days prior to harvest. L3–L4 vertebrae sections were photographed under UV light and the distance between the calcein lines were measured. Bone formation rate is graphed and bars represent the average value ± SE (n=7–8 per group). One-way ANOVA followed by Fishers post hoc tests determined differences among the groups. * p<0.05 compared to respective control, ** p<0.01, ***p<0.001.
DISCUSSION
Bisphosphonate therapy is the first line of pharmacological defense against osteoporosis. Previous studies have demonstrated that bisphosphonates selectively integrate into bone mineral surfaces and increase bone mineral density [Black, 1996; Devogelaer, 1996; Devogelaer et al., 1996; Giannini et al., 1993; Hosking et al., 1998; Liberman et al., 1995; McClung et al., 1998] by inhibiting osteoclast-mediated resorption, recruitment and differentiation and by stimulating osteoclast apoptosis. Previous studies suggest that bisphosphonates not only inhibit osteoclast-mediated resorption, but they also inhibit osteoblast and osteocyte death [Plotkin et al., 2006; Plotkin et al., 1999]. Previously, we demonstrated that T1-diabetes results in elevated osteoblast apoptosis and increased Bax expression in bone [Coe et al., 2011]. Here, we demonstrate that alendronate prevents osteoblast death and elevated Bax expression during early stages of diabetes development and more importantly prevents diabetes-induced bone loss at 6 weeks post-diabetes induction. However, the longer treatment time led to decreased osteoblast marker levels and a lower work to fracture, both of which can increase fracture risk and bone loss with extended treatment. This is a major concern considering that diabetes is associated with altered matrix properties that can already weaken the bone [McCabe et al., 2011; Saito et al., 2014].
Only a few studies have addressed the impact of bisphosphonate therapy on diabetic bone and all of these studies have been directed at T2-diabetic changes. One clinical study involving patients on long-term (4 years) bisphosphonate treatment indicated that the treatment did not prevent bone loss in postmenopausal women with type 2 diabetes [Dagdelen et al., 2007]. In contrast, a study performed on 13 post-menopausal Japanese women with type 2 diabetes taking bisphosphonates for 6 or 12 months demonstrated elevated bone density in the lumbar spine at both time points [Kanazawa et al., 2010]. Similarly, Keegan et al. found that non-diabetic and type 2 diabetic women benefitted in overall bone health from bisphosphonate therapy [Keegan et al., 2004]. Interestingly, in this study more diabetic women reported taking estrogen supplements compared to non-diabetic women. This is important to note since estrogen would contribute to bone health due to its positive impact on bone formation.
Bisphosphonates, while thought to predominantly affect osteoclast activity, can also affect osteoblasts. Of benefit is their ability to suppress osteoblast and osteocyte apoptosis [Plotkin et al., 1999], similar to what we observed. Osteocyte apoptosis is also increased in the STZ-induced diabetes model [Portal-Nunez et al., 2010] and could also contribute to the increase we observe in Bax mRNA levels. Of additional interest to the diabetic condition, Gangoiti, et al. reported that mouse and rat osteoblasts treated with advanced glycation end products to induce cell death were protected from death at low bisphosphonate concentrations [Gangoiti et al., 2008].
Although inhibition of osteoblast and osteocyte apoptosis is beneficial for T1-diabetic bone, bisphosphonates can also suppress bone formation and osteoblast differentiation [Giuliani et al., 1998; Reinholz et al., 2000], similar to what we observed. The anti-resorptive properties of bisphosphonates likely reduce overall bone turnover and can be problematic under T1-diabetic conditions where bone turnover and anabolic properties are already significantly suppressed. Many studies support the notion that T1-diabetic bone loss is due primarily to decreased bone formation with little to no change in bone resorption ([Bonfanti et al., 1997; Cakatay et al., 1998; Kemink et al., 2000: Botolin and McCabe, 2006b; Motyl and McCabe, 2009c: Botolin and McCabe, 2007; Martin and McCabe, 2007). Thus giving an anti-resorptive drug to patients that do not have an osteoclast defect could be counterproductive. Therefore, bisphosphonate variants that do not affect osteoclast activity or bone turnover but still inhibit osteoblast and osteocyte apoptosis could be beneficial [Takeuchi et al., 1999].
It should also be noted the bisphosphonate dose could influence the outcome of the response. In this study we used a weekly subcutaneous injection of 2 mg/kg/week. While this is a common mid-range dose in mouse studies [Nijenhuis et al., 2008], it is given by injection and not orally (the way patients would receive it). Thus the bioavailability is likely greater in our model. However, consequences of high dose alendronate treatment, such as increased osteoblast apoptosis [Gangoiti et al., 2013; Gangoiti et al., 2008], were not observed in our study, but we did observe a decrease in bone toughness in treated controls. Clearly more work is needed to understand the mechanisms of the bisphosphonate effects on diabetic bone.
Bisphosphonates are known for their ability to suppress bone resorption markers [Liberman et al., 1995; Orwoll et al., 2000; Saag et al., 1998] and induce osteoclast apoptosis in vitro and in vivo [Plotkin et al., 2006; Plotkin et al., 1999; Takeuchi et al., 1999]. While we observed a decrease in serum active Trap5b levels we did not observe changes in other osteoclast markers. The majority of clinical studies, however, support the premise that resorption is decreased or not altered in T1-diabetic patients while formation is clearly decreased [Bonfanti et al., 1997; Cakatay et al., 1998; Kemink et al., 2000], consistent with our overall findings in this study.
Elevated marrow adiposity is often (but not always [Botolin and McCabe, 2006b; Motyl and McCabe, 2009c]) inversely related to diabetes-induced bone loss [Botolin and McCabe, 2007; Martin and McCabe, 2007; Slade et al., 2012]. Here, we show that alendronate treatment did not affect the diabetes-induced increase in marrow adiposity as determined by elevated adipocyte numbers and increased aP2 expression compared to controls. Interestingly, alendronate treatment of control and diabetic mice did result in and overall decrease in adipocyte number per total marrow area compared to corresponding untreated conditions. Consistent with our data, a recent clinical study indicated that bisphosphonate therapy suppressed marrow adipocyte number by as much as 20% in post-menopausal women [Duque et al., 2010]. Additionally, a clinical study that examined over 2400 postmenopausal women treated with risedronate (another bisphosphoante) demonstrated significant decreases in adiposity while increasing BMD levels and reducing the incidence of vertebral and non-vertebral fractures [Harris et al., 1999]. In our study, the lack of an observed decrease in aP2 expression in control mice treated with alendronate could be due to several factors including altered adipocyte size and/or the influence of other cells present in the bone marrow microenvironment that express aP2. One study reported that hematopoietic cells, specifically monocytes, express both early and late adipocyte markers, PPARγ and aP2, respectively [Pelton et al., 1999]. Thus, elevated basal levels of monocytes in the control mice treated with alendronate could account for the lack of effect of alendronate on aP2 expression.
It is important also to note that studies investigating diabetic patients taking bisphosphonate therapy indicate an increased (albeit rare) incidence of osteonecrosis of the jaw at high doses commonly used to treat cancer patients compared to non-diabetic patients [Khamaisi et al., 2007]. Additionally, there is the link between bisphosphonate therapy and increases in rare femur fractures [Black et al., 2010; Kim et al., 2010]. Given that diabetes and bisphosphonates both reduce bone turnover, there is the potential for additive or synergistic effects with long-term treatment that could result in an increase in brittle bone. However, our bone strength measurements indicate that alendronate treatment increased stiffness and yield strength in control mice. However, examination of the fractured bone suggests increased brittleness.
Taken together, the benefits of bisphosphonate therapy in combating osteoporosis are well known for the majority of healthy patients. Our results suggest that short-term bisphosphonate therapy can benefit diabetic bone by increasing osteoblast viability and bone density. However, given that T1-diabetes suppresses bone anabolic activity and that bisphosphonate treatment causes yet a further reduction of osteoblast markers, our data suggests caution is needed with regard to long-term treatment protocols for T1-diabetic bone loss. Future studies examining the chronic effects of alendronate on T1-diabetic bone health are needed. Understanding the positives and negatives of such a treatment on T1-diabetic bone health is critical for identifying new and improved bisphosphonate therapies.
Acknowledgments
The authors thank Regina Irwin for her technical expertise and critical review of the manuscript, Jeffery Dylan Denison for histomorphometry analysis of adipocytes, and Jing Zhang and Christopher Giles for their helpful suggestions. We thank the Investigative Histology Laboratory in the Department of Physiology, Division of Human Pathology at Michigan State University for their assistance with histological analyses. The authors have no conflict of interests to disclose. These studies were supported by funding from the American Diabetes Association (7-07-RA-105) and NIH (DK101050) to LRM.
Footnotes
Authors have nothing to disclose.
References
- Alblowi J, Kayal RA, Siqueira M, McKenzie E, Krothapalli N, McLean J, Conn J, Nikolajczyk B, Einhorn TA, Gerstenfeld L, Graves DT. High levels of tumor necrosis factor-alpha contribute to accelerated loss of cartilage in diabetic fracture healing. Am J Pathol. 2009;175:1574–85. doi: 10.2353/ajpath.2009.090148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexopoulou O, Jamart J, Devogelaer JP, Brichard S, de Nayer P, Buysschaert M. Bone density and markers of bone remodeling in type 1 male diabetic patients. Diabetes Metab. 2006;32:453–8. doi: 10.1016/s1262-3636(07)70303-8. [DOI] [PubMed] [Google Scholar]
- Bain S, Ramamurthy NS, Impeduglia T, Scolman S, Golub LM, Rubin C. Tetracycline prevents cancellous bone loss and maintains near-normal rates of bone formation in streptozotocin diabetic rats. Bone. 1997;21:147–53. doi: 10.1016/s8756-3282(97)00104-x. [DOI] [PubMed] [Google Scholar]
- Beisswenger PJ, Drummond KS, Nelson RG, Howell SK, Szwergold BS, Mauer M. Susceptibility to diabetic nephropathy is related to dicarbonyl and oxidative stress. Diabetes. 2005;54:3274–81. doi: 10.2337/diabetes.54.11.3274. [DOI] [PubMed] [Google Scholar]
- Bennett CN, Ouyang H, Ma YL, Zeng Q, Gerin I, Sousa KM, Lane TF, Krishnan V, Hankenson KD, MacDougald OA. Wnt10b increases postnatal bone formation by enhancing osteoblast differentiation. J Bone Miner Res. 2007;22:1924–32. doi: 10.1359/jbmr.070810. [DOI] [PubMed] [Google Scholar]
- Black DM. Screening and treatment in the elderly to reduce osteoporotic fracture risk. Br J Obstet Gynaecol. 1996;103(Suppl 13):2–7. discussion 7–8. [PubMed] [Google Scholar]
- Black DM, Kelly MP, Genant HK, Palermo L, Eastell R, Bucci-Rechtweg C, Cauley J, Leung PC, Boonen S, Santora A, de Papp A, Bauer DC. Bisphosphonates and fractures of the subtrochanteric or diaphyseal femur. N Engl J Med. 2010;362:1761–71. doi: 10.1056/NEJMoa1001086. [DOI] [PubMed] [Google Scholar]
- Bonfanti R, Mora S, Prinster C, Bognetti E, Meschi F, Puzzovio M, Proverbio MC, Chiumello G. Bone modeling indexes at onset and during the first year of follow-Up in insulin-dependent diabetic children. Calcif Tissue Int. 1997;60:397–400. doi: 10.1007/s002239900251. [DOI] [PubMed] [Google Scholar]
- Botolin S, Faugere MC, Malluche H, Orth M, Meyer R, McCabe LR. Increased bone adiposity and peroxisomal proliferator-activated receptor-gamma2 expression in type I diabetic mice. Endocrinology. 2005;146:3622–31. doi: 10.1210/en.2004-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botolin S, McCabe LR. Bone Loss and Increased Bone Adiposity in Spontaneous and Pharmacologically Induced Diabetic Mice. Endocrinology. 2006a;148:198–205. doi: 10.1210/en.2006-1006. [DOI] [PubMed] [Google Scholar]
- Botolin S, McCabe LR. Inhibition of PPARgamma prevents type I diabetic bone marrow adiposity but not bone loss. J Cell Physiol. 2006b;209:967–76. doi: 10.1002/jcp.20804. [DOI] [PubMed] [Google Scholar]
- Botolin S, McCabe LR. Bone loss and increased bone adiposity in spontaneous and pharmacologically induced diabetic mice. Endocrinology. 2007;148:198–205. doi: 10.1210/en.2006-1006. [DOI] [PubMed] [Google Scholar]
- Bouillon R, Bex M, Van Herck E, Laureys J, Dooms L, Lesaffre E, Ravussin E. Influence of age, sex, and insulin on osteoblast function: osteoblast dysfunction in diabetes mellitus. J Clin Endocrinol Metab. 1995;80:1194–202. doi: 10.1210/jcem.80.4.7714089. [DOI] [PubMed] [Google Scholar]
- Bushardt RL, Turner JL, Ragucci KR, Askins DG., Jr Non-estrogen treatments for osteoporosis: an evidence-based review. JAAPA. 2006;19:25–30. doi: 10.1097/01720610-200612000-00006. [DOI] [PubMed] [Google Scholar]
- Cakatay U, Telci A, Kayali R, Akcay T, Sivas A, Aral F. Changes in bone turnover on deoxypyridinoline levels in diabetic patients. Diabetes Res Clin Pract. 1998;40:75–9. doi: 10.1016/s0168-8227(98)00025-4. [DOI] [PubMed] [Google Scholar]
- Coe LM, Irwin R, Lippner D, McCabe LR. The bone marrow microenvironment contributes to type I diabetes induced osteoblast death. Journal of cellular physiology. 2011;226:477–83. doi: 10.1002/jcp.22357. [DOI] [PubMed] [Google Scholar]
- Coe LM, Zhang J, McCabe LR. Both spontaneous Ins2(+/−) and streptozotocin-induced type I diabetes cause bone loss in young mice. Journal of Cellular Physiology. 2012 doi: 10.1002/jcp.24177. [DOI] [PubMed] [Google Scholar]
- Cole Z, Dennison E, Cooper C. Update on the treatment of post-menopausal osteoporosis. Br Med Bull. 2008;86:129–43. doi: 10.1093/bmb/ldn017. [DOI] [PubMed] [Google Scholar]
- Dagdelen S, Sener D, Bayraktar M. Influence of type 2 diabetes mellitus on bone mineral density response to bisphosphonates in late postmenopausal osteoporosis. Adv Ther. 2007;24:1314–20. doi: 10.1007/BF02877778. [DOI] [PubMed] [Google Scholar]
- Devogelaer JP. Clinical use of bisphosphonates. Curr Opin Rheumatol. 1996;8:384–91. doi: 10.1097/00002281-199607000-00018. [DOI] [PubMed] [Google Scholar]
- Devogelaer JP, Broll H, Correa-Rotter R, Cumming DC, De Deuxchaisnes CN, Geusens P, Hosking D, Jaeger P, Kaufman JM, Leite M, Leon J, Liberman U, Menkes CJ, Meunier PJ, Reid I, Rodriguez J, Romanowicz A, Seeman E, Vermeulen A, Hirsch LJ, Lombardi A, Plezia K, Santora AC, Yates AJ, Yuan W. Oral alendronate induces progressive increases in bone mass of the spine, hip, and total body over 3 years in postmenopausal women with osteoporosis. Bone. 1996;18:141–50. doi: 10.1016/8756-3282(95)00436-x. [DOI] [PubMed] [Google Scholar]
- Duby JJ, Campbell RK, Setter SM, White JR, Rasmussen KA. Diabetic neuropathy: an intensive review. Am J Health Syst Pharm. 2004;61:160–73. doi: 10.1093/ajhp/61.2.160. quiz 175–6. [DOI] [PubMed] [Google Scholar]
- Duque G, Li W, Adams M, Xu S, Phipps R. Effects of risedronate on bone marrow adipocytes in postmenopausal women. Osteoporos Int. 2010 doi: 10.1007/s00198-010-1353-8. [DOI] [PubMed] [Google Scholar]
- Forsen L, Meyer HE, Midthjell K, Edna TH. Diabetes mellitus and the incidence of hip fracture: results from the Nord-Trondelag Health Survey. Diabetologia. 1999;42:920–5. doi: 10.1007/s001250051248. [DOI] [PubMed] [Google Scholar]
- Gangoiti MV, Anbinder PS, Cortizo AM, McCarthy AD. Morphological changes induced by advanced glycation endproducts in osteoblastic cells: effects of co-incubation with alendronate. Acta Histochem. 2013;115:649–57. doi: 10.1016/j.acthis.2013.01.004. [DOI] [PubMed] [Google Scholar]
- Gangoiti MV, Cortizo AM, Arnol V, Felice JI, McCarthy AD. Opposing effects of bisphosphonates and advanced glycation end-products on osteoblastic cells. Eur J Pharmacol. 2008;600:140–7. doi: 10.1016/j.ejphar.2008.10.031. [DOI] [PubMed] [Google Scholar]
- Giannini S, D’Angelo A, Malvasi L, Castrignano R, Pati T, Tronca R, Liberto L, Nobile M, Crepaldi G. Effects of one-year cyclical treatment with clodronate on postmenopausal bone loss. Bone. 1993;14:137–41. doi: 10.1016/8756-3282(93)90240-b. [DOI] [PubMed] [Google Scholar]
- Giuliani N, Pedrazzoni M, Negri G, Passeri G, Impicciatore M, Girasole G. Bisphosphonates stimulate formation of osteoblast precursors and mineralized nodules in murine and human bone marrow cultures in vitro and promote early osteoblastogenesis in young and aged mice in vivo. Bone. 1998;22:455–61. doi: 10.1016/s8756-3282(98)00033-7. [DOI] [PubMed] [Google Scholar]
- Glajchen N, Epstein S, Ismail F, Thomas S, Fallon M, Chakrabarti S. Bone mineral metabolism in experimental diabetes mellitus: osteocalcin as a measure of bone remodeling. Endocrinology. 1988;123:290–5. doi: 10.1210/endo-123-1-290. [DOI] [PubMed] [Google Scholar]
- Goralski KB, Sinal CJ. Type 2 diabetes and cardiovascular disease: getting to the fat of the matter. Can J Physiol Pharmacol. 2007;85:113–32. doi: 10.1139/y06-092. [DOI] [PubMed] [Google Scholar]
- Hamilton EJ, Rakic V, Davis WA, Chubb SA, Kamber N, Prince RL, Davis TM. Prevalence and predictors of osteopenia and osteoporosis in adults with Type 1 diabetes. Diabet Med. 2009;26:45–52. doi: 10.1111/j.1464-5491.2008.02608.x. [DOI] [PubMed] [Google Scholar]
- Harris L, Senagore P, Young VB, McCabe LR. Inflammatory bowel disease causes reversible suppression of osteoblast and chondrocyte function in mice. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1020–9. doi: 10.1152/ajpgi.90696.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris ST, Watts NB, Genant HK, McKeever CD, Hangartner T, Keller M, Chesnut CH, 3rd, Brown J, Eriksen EF, Hoseyni MS, Axelrod DW, Miller PD. Effects of risedronate treatment on vertebral and nonvertebral fractures in women with postmenopausal osteoporosis: a randomized controlled trial. Vertebral Efficacy With Risedronate Therapy (VERT) Study Group. JAMA. 1999;282:1344–52. doi: 10.1001/jama.282.14.1344. [DOI] [PubMed] [Google Scholar]
- He H, Liu R, Desta T, Leone C, Gerstenfeld LC, Graves DT. Diabetes causes decreased osteoclastogenesis, reduced bone formation, and enhanced apoptosis of osteoblastic cells in bacteria stimulated bone loss. Endocrinology. 2004;145:447–52. doi: 10.1210/en.2003-1239. [DOI] [PubMed] [Google Scholar]
- Hosking D, Chilvers CE, Christiansen C, Ravn P, Wasnich R, Ross P, McClung M, Balske A, Thompson D, Daley M, Yates AJ. Prevention of bone loss with alendronate in postmenopausal women under 60 years of age. Early Postmenopausal Intervention Cohort Study Group. N Engl J Med. 1998;338:485–92. doi: 10.1056/NEJM199802193380801. [DOI] [PubMed] [Google Scholar]
- Hughes DE, MacDonald BR, Russell RG, Gowen M. Inhibition of osteoclast-like cell formation by bisphosphonates in long-term cultures of human bone marrow. J Clin Invest. 1989;83:1930–5. doi: 10.1172/JCI114100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaksson H, Tolvanen V, Finnila MA, Iivarinen J, Turunen A, Silvast TS, Tuukkanen J, Seppanen K, Arokoski JP, Brama PA, Jurvelin JS, Helminen HJ. Long-term voluntary exercise of male mice induces more beneficial effects on cancellous and cortical bone than on the collagenous matrix. Exp Gerontol. 2009a;44:708–17. doi: 10.1016/j.exger.2009.08.005. [DOI] [PubMed] [Google Scholar]
- Isaksson H, Tolvanen V, Finnila MA, Iivarinen J, Tuukkanen J, Seppanen K, Arokoski JP, Brama PA, Jurvelin JS, Helminen HJ. Physical exercise improves properties of bone and its collagen network in growing and maturing mice. Calcif Tissue Int. 2009b;85:247–56. doi: 10.1007/s00223-009-9273-3. [DOI] [PubMed] [Google Scholar]
- Jamsa T, Jalovaara P, Peng Z, Vaananen HK, Tuukkanen J. Comparison of three-point bending test and peripheral quantitative computed tomography analysis in the evaluation of the strength of mouse femur and tibia. Bone. 1998;23:155–61. doi: 10.1016/s8756-3282(98)00076-3. [DOI] [PubMed] [Google Scholar]
- Kanazawa I, Yamaguchi T, Hayashi K, Takase H, Shimizu T, Sugimoto T. Effects of treatment with risedronate and alfacalcidol on progression of atherosclerosis in postmenopausal women with type 2 diabetes mellitus accompanied with osteoporosis. Am J Med Sci. 2010;339:519–24. doi: 10.1097/MAJ.0b013e3181db6dfe. [DOI] [PubMed] [Google Scholar]
- Kayal RA, Siqueira M, Alblowi J, McLean J, Krothapalli N, Faibish D, Einhorn TA, Gerstenfeld LC, Graves DT. TNF-alpha mediates diabetes-enhanced chondrocyte apoptosis during fracture healing and stimulates chondrocyte apoptosis through FOXO1. J Bone Miner Res. 2010;25:1604–15. doi: 10.1002/jbmr.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan TH, Schwartz AV, Bauer DC, Sellmeyer DE, Kelsey JL. Effect of alendronate on bone mineral density and biochemical markers of bone turnover in type 2 diabetic women: the fracture intervention trial. Diabetes Care. 2004;27:1547–53. doi: 10.2337/diacare.27.7.1547. [DOI] [PubMed] [Google Scholar]
- Kemink SA, Hermus AR, Swinkels LM, Lutterman JA, Smals AG. Osteopenia in insulin-dependent diabetes mellitus; prevalence and aspects of pathophysiology. J Endocrinol Invest. 2000;23:295–303. doi: 10.1007/BF03343726. [DOI] [PubMed] [Google Scholar]
- Khamaisi M, Regev E, Yarom N, Avni B, Leitersdorf E, Raz I, Elad S. Possible association between diabetes and bisphosphonate-related jaw osteonecrosis. J Clin Endocrinol Metab. 2007;92:1172–5. doi: 10.1210/jc.2006-2036. [DOI] [PubMed] [Google Scholar]
- Kim SY, Schneeweiss S, Katz JN, Levin R, Solomon DH. Oral bisphosphonates and risk of subtrochanteric or diaphyseal femur fractures in a population-based cohort. J Bone Miner Res. 2010 doi: 10.1002/jbmr.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman UA, Weiss SR, Broll J, Minne HW, Quan H, Bell NH, Rodriguez-Portales J, Downs RW, Jr, Dequeker J, Favus M. Effect of oral alendronate on bone mineral density and the incidence of fractures in postmenopausal osteoporosis. The Alendronate Phase III Osteoporosis Treatment Study Group. N Engl J Med. 1995;333:1437–43. doi: 10.1056/NEJM199511303332201. [DOI] [PubMed] [Google Scholar]
- Lumachi F, Camozzi V, Tombolan V, Luisetto G. Bone mineral density, osteocalcin, and bone-specific alkaline phosphatase in patients with insulin-dependent diabetes mellitus. Ann N Y Acad Sci. 2009;1173(Suppl 1):E64–7. doi: 10.1111/j.1749-6632.2009.04955.x. [DOI] [PubMed] [Google Scholar]
- Martin LM, McCabe LR. Type I diabetic bone phenotype is location but not gender dependent. Histochem Cell Biol. 2007;128:125–33. doi: 10.1007/s00418-007-0308-4. [DOI] [PubMed] [Google Scholar]
- McCabe L, Zhang J, Raehtz S. Understanding the skeletal pathology of type 1 and 2 diabetes mellitus. Critical reviews in eukaryotic gene expression. 2011;21:187–206. doi: 10.1615/critreveukargeneexpr.v21.i2.70. [DOI] [PubMed] [Google Scholar]
- McCabe LR. Understanding the pathology and mechanisms of type I diabetic bone loss. J Cell Biochem. 2007;102:1343–57. doi: 10.1002/jcb.21573. [DOI] [PubMed] [Google Scholar]
- McClung M. Use of highly potent bisphosphonates in the treatment of osteoporosis. Curr Osteoporos Rep. 2003a;1:116–22. doi: 10.1007/s11914-996-0006-5. [DOI] [PubMed] [Google Scholar]
- McClung M, Clemmesen B, Daifotis A, Gilchrist NL, Eisman J, Weinstein RS, Fuleihan Ge-H, Reda C, Yates AJ, Ravn P. Alendronate prevents postmenopausal bone loss in women without osteoporosis. A double-blind, randomized, controlled trial. Alendronate Osteoporosis Prevention Study Group. Ann Intern Med. 1998;128:253–61. doi: 10.7326/0003-4819-128-4-199802150-00001. [DOI] [PubMed] [Google Scholar]
- McClung MR. Bisphosphonates. Endocrinol Metab Clin North Am. 2003b;32:253–71. doi: 10.1016/s0889-8529(02)00079-8. [DOI] [PubMed] [Google Scholar]
- Meyer HE, Tverdal A, Falch JA. Risk factors for hip fracture in middle-aged Norwegian women and men. Am J Epidemiol. 1993;137:1203–11. doi: 10.1093/oxfordjournals.aje.a116622. [DOI] [PubMed] [Google Scholar]
- Miao J, Brismar K, Nyren O, Ugarph-Morawski A, Ye W. Elevated hip fracture risk in type 1 diabetic patients: a population-based cohort study in Sweden. Diabetes Care. 2005;28:2850–5. doi: 10.2337/diacare.28.12.2850. [DOI] [PubMed] [Google Scholar]
- Motyl K, McCabe LR. Streptozotocin, Type I Diabetes Severity and Bone. Biol Proced Online. 2009a doi: 10.1007/s12575-009-9000-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motyl K, McCabe LR. Streptozotocin, Type I Diabetes Severity and Bone. Biol Proced Online. 2009b;11:296–315. doi: 10.1007/s12575-009-9000-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motyl KJ, Botolin S, Irwin R, Appledorn DM, Kadakia T, Amalfitano A, Schwartz RC, McCabe LR. Bone inflammation and altered gene expression with type I diabetes early onset. J Cell Physiol. 2009;218:575–83. doi: 10.1002/jcp.21626. [DOI] [PubMed] [Google Scholar]
- Motyl KJ, McCabe LR. Leptin treatment prevents type I diabetic marrow adiposity but not bone loss in mice. J Cell Physiol. 2009c;218:376–84. doi: 10.1002/jcp.21608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motyl KJ, McCauley LK, McCabe LR. Amelioration of type I diabetes-induced osteoporosis by parathyroid hormone is associated with improved osteoblast survival. Journal of Cellular Physiology. 2012;227:1326–34. doi: 10.1002/jcp.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Torres M, Jodar E, Escobar-Jimenez F, Lopez-Ibarra PJ, Luna JD. Bone mineral density measured by dual X-ray absorptiometry in Spanish patients with insulin-dependent diabetes mellitus. Calcif Tissue Int. 1996;58:316–9. doi: 10.1007/BF02509378. [DOI] [PubMed] [Google Scholar]
- Nijenhuis T, van der Eerden BC, Hoenderop JG, Weinans H, van Leeuwen JP, Bindels RJ. Bone resorption inhibitor alendronate normalizes the reduced bone thickness of TRPV5(−/−) mice. J Bone Miner Res. 2008;23:1815–24. doi: 10.1359/jbmr.080613. [DOI] [PubMed] [Google Scholar]
- Orwoll E, Ettinger M, Weiss S, Miller P, Kendler D, Graham J, Adami S, Weber K, Lorenc R, Pietschmann P, Vandormael K, Lombardi A. Alendronate for the treatment of osteoporosis in men. N Engl J Med. 2000;343:604–10. doi: 10.1056/NEJM200008313430902. [DOI] [PubMed] [Google Scholar]
- Pelton PD, Zhou L, Demarest KT, Burris TP. PPARgamma activation induces the expression of the adipocyte fatty acid binding protein gene in human monocytes. Biochem Biophys Res Commun. 1999;261:456–8. doi: 10.1006/bbrc.1999.1071. [DOI] [PubMed] [Google Scholar]
- Phillips JA, Almeida EA, Hill EL, Aguirre JI, Rivera MF, Nachbandi I, Wronski TJ, van der Meulen MC, Globus RK. Role for beta1 integrins in cortical osteocytes during acute musculoskeletal disuse. Matrix Biol. 2008;27:609–18. doi: 10.1016/j.matbio.2008.05.003. [DOI] [PubMed] [Google Scholar]
- Plotkin LI, Manolagas SC, Bellido T. Dissociation of the pro-apoptotic effects of bisphosphonates on osteoclasts from their anti-apoptotic effects on osteoblasts/osteocytes with novel analogs. Bone. 2006;39:443–52. doi: 10.1016/j.bone.2006.02.060. [DOI] [PubMed] [Google Scholar]
- Plotkin LI, Weinstein RS, Parfitt AM, Roberson PK, Manolagas SC, Bellido T. Prevention of osteocyte and osteoblast apoptosis by bisphosphonates and calcitonin. J Clin Invest. 1999;104:1363–74. doi: 10.1172/JCI6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portal-Nunez S, Lozano D, de Castro LF, de Gortazar AR, Nogues X, Esbrit P. Alterations of the Wnt/beta-catenin pathway and its target genes for the N- and C-terminal domains of parathyroid hormone-related protein in bone from diabetic mice. FEBS Lett. 2010;584:3095–100. doi: 10.1016/j.febslet.2010.05.047. [DOI] [PubMed] [Google Scholar]
- Reinholz GG, Getz B, Pederson L, Sanders ES, Subramaniam M, Ingle JN, Spelsberg TC. Bisphosphonates directly regulate cell proliferation, differentiation, and gene expression in human osteoblasts. Cancer Res. 2000;60:6001–7. [PubMed] [Google Scholar]
- Russell RG, Rogers MJ. Bisphosphonates: from the laboratory to the clinic and back again. Bone. 1999;25:97–106. doi: 10.1016/s8756-3282(99)00116-7. [DOI] [PubMed] [Google Scholar]
- Saag KG, Emkey R, Schnitzer TJ, Brown JP, Hawkins F, Goemaere S, Thamsborg G, Liberman UA, Delmas PD, Malice MP, Czachur M, Daifotis AG. Alendronate for the prevention and treatment of glucocorticoid-induced osteoporosis. Glucocorticoid-Induced Osteoporosis Intervention Study Group. N Engl J Med. 1998;339:292–9. doi: 10.1056/NEJM199807303390502. [DOI] [PubMed] [Google Scholar]
- Sahni M, Guenther HL, Fleisch H, Collin P, Martin TJ. Bisphosphonates act on rat bone resorption through the mediation of osteoblasts. J Clin Invest. 1993;91:2004–11. doi: 10.1172/JCI116422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M, Kida Y, Kato S, Marumo K. Diabetes, collagen, and bone quality. Curr Osteoporos Rep. 2014;12:181–8. doi: 10.1007/s11914-014-0202-7. [DOI] [PubMed] [Google Scholar]
- Sato M, Grasser W, Endo N, Akins R, Simmons H, Thompson DD, Golub E, Rodan GA. Bisphosphonate action. Alendronate localization in rat bone and effects on osteoclast ultrastructure. J Clin Invest. 1991;88:2095–105. doi: 10.1172/JCI115539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schriefer JL, Robling AG, Warden SJ, Fournier AJ, Mason JJ, Turner CH. A comparison of mechanical properties derived from multiple skeletal sites in mice. J Biomech. 2005;38:467–75. doi: 10.1016/j.jbiomech.2004.04.020. [DOI] [PubMed] [Google Scholar]
- Schwartz AV, Sellmeyer DE, Ensrud KE, Cauley JA, Tabor HK, Schreiner PJ, Jamal SA, Black DM, Cummings SR. Older women with diabetes have an increased risk of fracture: a prospective study. J Clin Endocrinol Metab. 2001;86:32–8. doi: 10.1210/jcem.86.1.7139. [DOI] [PubMed] [Google Scholar]
- Slade JM, Coe LM, Meyer RA, McCabe LR. Human bone marrow adiposity is linked with serum lipid levels not T1-diabetes. Journal of diabetes and its complications. 2012;26:1–9. doi: 10.1016/j.jdiacomp.2011.11.001. [DOI] [PubMed] [Google Scholar]
- Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55:225–33. [PubMed] [Google Scholar]
- Takeuchi K, Ukawa H, Furukawa O, Kawauchi S, Araki H, Sugimoto Y, Ishikawa A, Ushikubi F, Narumiya S. Prostaglandin E receptor subtypes involved in stimulation of gastroduodenal bicarbonate secretion in rats and mice. J Physiol Pharmacol. 1999;50:155–67. [PubMed] [Google Scholar]
- Valeri C, Pozzilli P, Leslie D. Glucose control in diabetes. Diabetes Metab Res Rev. 2004;20(Suppl 2):S1–8. doi: 10.1002/dmrr.512. [DOI] [PubMed] [Google Scholar]