

Abstract
Background
Several epidemiological cross-sectional studies have found positive associations between serum concentrations of lipids and perfluorooctanoic acid (PFOA, or C8). A longitudinal study should be less susceptible to biases from uncontrolled confounding or reverse causality.
Methods
We investigated the association between within-individual changes in serum PFOA and perfluorooctanesulfonic acid (PFOS) and changes in serum lipid levels (low-density lipoprotein [LDL] cholesterol, high-density lipoprotein cholesterol, total cholesterol, and triglycerides) over a 4.4-year period. The study population consisted of 560 adults living in parts of Ohio and West Virginia where public drinking water had been contaminated with PFOA. They had participated in a cross-sectional study in 2005–2006, and were followed up in 2010, by which time exposure to PFOA had been substantially reduced.
Results
Overall serum concentrations of PFOA and PFOS fell by half from initial geometric means of 74.8 and 18.5 ng/mL, respectively, with little corresponding change in LDL cholesterol (mean increase 1.8%, standard deviation 26.6%). However, there was a tendency for people with greater declines in serum PFOA or PFOS to have greater LDL decrease. For a person whose serum PFOA fell by half, the predicted fall in LDL cholesterol was 3.6% (95% confidence interval = 1.5–5.7%). The association with a decline in PFOS was even stronger, with a 5% decrease in LDL (2.5–7.4%).
Conclusions
Our findings from this longitudinal study support previous evidence from cross-sectional studies of positive associations between PFOA and PFOS in serum and LDL cholesterol.
Perfluorooctanoic acid (PFOA; also known as C8), a fluorinated eight-carbon member of the perfluoroalkyl acid (PFAA) family, has been used as a processing aid to manufacture fluoropolymers, which are used in products for nonstick, heat and chemical resistance applications. In rodents PFOA causes peroxisome proliferation, developmental toxicity, immunotoxicity, and hepatotoxicity.1,2 PFOA activates the α-isoform of the peroxisome proliferation-activated receptors (PPARα), a nuclear receptor important in lipid regulation.3,4 In animals, PFOA reduces serum triglyceride and cholesterol levels, as well as lipid accumulation in the liver, similar to the response in humans to treatment with fibrates, which act via PPARα.1,5,6 PFOA does not break down in the environment and persists in humans, with half-life estimated at 2.3 to 3.8 years.7,8 Still, its effects on human health are not clear.9,10 Studies in human populations have reported positive associations between PFOA and low-density lipoprotein (LDL) and total cholesterol, but not usually high-density lipoprotein (HDL) cholesterol or triglycerides.11–17 However, a recent study in 55 highly exposed Chinese workers (PFOA geometric mean 1,272 ng/mL) found an inverse association between HDL cholesterol and PFOA.18 Some of the studies on PFOA also investigated associations between serum concentrations of lipids and perfluoroctanesulfonic acid (PFOS), another eight-carbon PFAA. PFOS has been found to be independently associated with serum lipids, after adjusting for PFOA.17
Most of the positive associations between PFOA or PFOS and total or LDL cholesterol are from cross-sectional studies; thus there are limitations to causal inference. Cross-sectional associations could arise if, for example, PFOA uptake or excretion is affected by lipid levels, or if some other factor is responsible for variation in both PFOA and lipids. For this study, we had measurements on individuals at two time points 4 to 5 years apart, and we may thus consider the association between change in serum PFAAs and change lipids with potentially less bias. This study design means that parameter estimates are not biased by confounding variables that do not change over time, and whose effects do not change (for instance, genetic factors that affect the excretion rate of PFAAs). We investigated the association between changes in both serum PFOA and PFOS and changes in serum lipids (LDL cholesterol, HDL cholesterol, total cholesterol, and triglycerides), in a population exposed to high levels of PFOA via contaminated drinking water.
METHODS
Study Design and Participants
The study sample was derived from The C8 Short-Term Follow-up Study, one of the C8 Science Panel studies, based on a population with high exposure to PFOA via contaminated drinking water. Those studies are described at http://www.c8sciencepanel.org. Details are provided in the eAppendix (http://links.lww.com/EDE/A675).
Plant emissions of PFOA peaked in 2000 and decreased between 2001 and 2004 to much lower levels.19 However, contamination remained in the local environment, and exposure to PFOA in drinking water was further reduced by filtration of the public water supplies between March 2006 and September 2008. Serum concentrations of PFOS fell during the period of this study, in a pattern fairly similar to that observed in the US population between 1999 and 2008.20 PFOA and PFOS were measured in serum samples collected at the original survey and follow-up. At baseline, concentrations were determined using protein precipitation followed by reversed-phase high-performance liquid chromatography/tandem mass spectrometry.21 At follow-up, the approach followed an online solid phase extraction coupled with reversed-phase high-performance liquid chromatography separation and detection by isotope-dilution tandem mass spectrometry.22 The two techniques are known to produce equivalent results for the analysis of serum PFAAs.23 Furthermore, both laboratories participated in an interlaboratory study that reported a reasonable agreement, particularly for PFOS and PFOA.24 Therefore, any differences in serum concentrations of PFAAs between baseline and follow-up should not be attributable to differences in the analytical methods. Further details are provided in the eAppendix (http://links.lww.com/EDE/A675).
The blood samples collected at the initial survey and follow-up were also used to determine lipid profiles. Participants were not required to fast before blood draw, but fasting status was recorded and participants classified as fasting if the time since their last meal was more than 6 hours. Serum was separated from red cells, put in transport tubes and refrigerated before being shipped to an accredited commercial laboratory. Lipids were measured enzymatically. LDL cholesterol was calculated by the Friedewald equation when triglycerides were lower than 400 mg/dL.25 Serum creatinine was also measured, and we used this to calculate the glomerular filtration rate (GFR; mL/min), a measure of the rate of flow of filtered fluid through the kidneys, using the equation developed by the Modification of Diet in Renal Disease study: 186*creatinine−1.154* age−0.203*(7.43 if female)*(1.21 if black).26
For the analysis of change in serum lipids, we excluded 195 participants in the C8 Short-Term Follow-up Study (n = 755) who had reported using lipid-lowering drugs at baseline or follow-up. Excluding those pharmacologically treated for high cholesterol left 560 eligible participants, all of whom had 2 measurements of total cholesterol, HDL cholesterol, and triglycerides. However, 33 at baseline and 20 at follow-up had no LDL cholesterol calculated because their triglycerides were greater than 400 mg/dL, leaving 521 people for the LDL analysis.
Statistical Analyses
A model for the cross-sectional association between serum lipids yit and PFAA in serum xit in individual i at times t = 1 (C8 Health Project 2005/6) and t = 2 (follow-up 2010) may be given as yit = αt + βxit + γ′wi + δ′tzit + εit, where w represents time-invariant confounding variables and z represents confounding variables that change (or whose effects change) over time; we have assumed that the association between serum lipids and PFAA does not change. A simple model for change in serum lipids versus change in PFAA may be derived by subtraction of the cross-sectional model at baseline from that at follow-up. A strength of this approach is that bias due to time-invariant confounding (w) is eliminated by subtraction. Further detail on the properties of models of this type is discussed by Liker et al.27
In the cross-sectional analysis of the C8 Health Project data, a linear model for LDL in relation to PFOA, in which measurements of both were logarithmically transformed, provided a better fit than a linear model for untransformed values.17 We also found this in a cross-sectional analysis of the follow-up study. We based this conclusion on inspection of residual plots (not shown) and on comparison of global tests of model assumptions.28 At baseline, the test statistic for the hypothesis that four linear model assumptions were satisfied was 8.61 for logarithmically transformed changes, whereas without transformation the test statistic was 59.38. These may be compared with a chi-square distribution with four degrees of freedom and provide evidence that model assumptions are satisfied with log-transformed changes but not with untransformed changes.
To examine the relationships between changes in PFOA and PFOS and changes in serum lipids, we first fit linear regression models for the logarithm (base 10) of ratio change in each serum lipid measurement in relation to the logarithm of ratio change in PFOA and PFOS separately. This is equivalent to the difference between baseline and follow-up log(lipids) being a linear function of the difference in log(PFAA). The change-versus-change model is given by
That is,
where (αr, βr, δr) are the parameters for the model, zi may vary across time, and εi is the difference between error terms in the two cross-sectional models.
By design, this model eliminates (by subtraction) confounding by factors that do not change (or whose effects do not change) over time. This design should also eliminate most biases that might occur due to regression to the mean (people with extreme values of lipids at the first time tending to have values closer to the mean at the second time). We confirmed this by simulation, and further details are provided in the eAppendix (http://links.lww.com/EDE/A675). Also, it is known that when the true change in response is independent of the initial response, regression to the mean may bias the estimate of interest if the initial value of the response is included as a predictor; we did not include the initial value of the response in our models.29
A further advantage of this design is that the parameter estimate of interest is robust to systematic lab drift in the measurement of either predictor or response. For instance, if the first or second measurement were consistently over- or underestimated by a constant, the intercept of the change-versus-change model would alter but the slope would remain the same. (However, the average change in the predictor would be altered by a systematic change in measurement, and therefore, the relationship between the average change in response and average change in predictor would be affected.) Furthermore, if there were more variability in measurements conducted by one laboratory than the other, this would affect variance estimates but would not bias the parameter estimates. However, the geometric standard deviations (SDs) for serum PFOA in the 755 blood samples were similar, 3.22 at baseline, and 3.54 at follow-up.
These models were adjusted for age at baseline in four 10-year categories, continuous time between measurements, sex, and fasting status in four categories (fasting at both times, fasting then nonfasting, nonfasting then fasting, and nonfasting at both times). Age, sex, and time interval are possible confounders of the relationship between change in serum lipids and change in PFOA/PFOS. Fasting status, which is related to time of day, can affect lipid levels, and proportions of people travelling from differentially exposed areas to the centers for blood testing varied during the day; hence people who had to travel further were more likely to be nonfasting when they gave their second samples, and change in fasting status may confound the relationship between changes in serum PFOA and certain lipids. We had no a priori expectation that other measured variables would confound the relationship between changes in serum lipid measurements and changes in PFOA or PFOS, as factors that do not change (and whose effect does not change over time) are eliminated by design. However, as a sensitivity analysis, we calculated the parameter estimates after adjusting for baseline body mass index (BMI) (follow-up BMI was not reported), years of schooling, change in smoking, and baseline and change in GFR.
We fit analogous models with PFOS as the exposure of interest. Finally we fit joint models including changes in PFOA and PFOS, first with the logarithm of PFOA ratio and a spline smooth for PFOS ratio, and then vice versa. We did this by fitting generalized additive models, with spline smooths chosen by generalized cross-validation.30
From the estimated regression coefficients we calculated the predicted percentage change and 95% confidence interval (CI) in the response with a 50% drop in PFOA or PFOS serum concentrations. Models were fit using R Version 2.15.1.31
RESULTS
The population comprised more women (54%) than men, and ages were evenly distributed across the range (20–60 years at baseline), although there was a slightly larger proportion in the 30–50 years range. Mean BMI was 28.0, and 19% were smokers at the time of the initial survey. The mean interval between surveys was 4.4 years (SD = 11.7 weeks). There were 262 (47%) people who gave two nonfasting samples and 53 (10%) who gave two fasting samples, 164 (29%) who were fasting for the first sample but not the second, and 78 (14%) who were not fasting for the first sample but were for the second.
The serum concentrations of lipids, PFOA, and PFOS at the time of survey and follow-up are summarized in Table 1. The mean serum PFOA and PFOS both decreased by approximately one half between baseline and follow-up. The mean changes in lipids were very small, but there was large variability among individuals. For instance, mean serum LDL cholesterol increased 1.8% (from 112.4 to 113.8 mg/dL) with a standard deviation of 27%.
TABLE 1.
Relative and Absolute Differences in Serum Concentrations of PFOA, PFOS, and Lipids at Baseline (2005/6) and Follow-up (2010)
| Minimum | 33rd Percentile | 66th Percentile | Maximum | Geometric Mean | Arithmetic Mean | SD | |
|---|---|---|---|---|---|---|---|
| PFOA (ng/mL) | |||||||
| Baseline | 1.0 | 44.4 | 118.6 | 2,495 | 74.8 | 140.1 | 208.7 |
| Follow-up | 0.25 | 16.3 | 54.2 | 2,140 | 30.8 | 68.2 | 143.9 |
| Ratio (follow-up/baseline) | 0.0004 | 0.36 | 0.51 | 7.86 | 0.41 | 0.49 | 0.49 |
| Difference (follow-up − baseline) | −1,710 | −62.9 | −22.6 | 1,819 | −71.9 | 149.9 | |
| PFOS (ng/mL) | |||||||
| Baseline | 0.25 | 15.8 | 24.7 | 90.5 | 18.5 | 22.6 | 13.5 |
| Follow-up | 0.1 | 6.6 | 12.2 | 61 | 8.2 | 10.5 | 7.1 |
| Ratio (follow-up/baseline) | 0.002 | 0.39 | 0.53 | 7.2 | 0.44 | 0.51 | 0.47 |
| Difference (follow-up − baseline) | −60.5 | −13.5 | −7.3 | 8.9 | −12.1 | 9.3 | |
| LDL cholesterol (mg/dL) | |||||||
| Baseline | 42 | 96 | 123 | 249 | 107.8 | 112.4 | 35.6 |
| Follow-up | 30 | 98.7 | 123 | 250 | 109.2 | 113.8 | 32.3 |
| Ratio (follow-up/baseline) | 0.26 | 0.94 | 1.1 | 2.98 | 1.02 | 1.05 | 0.26 |
| Difference (follow-up − baseline) | −99 | −7 | 11 | 116 | 1.9 | 25.0 | |
| Total cholesterol (mg/dL) | |||||||
| Baseline | 100 | 178 | 209 | 324 | 192.5 | 196 | 36.7 |
| Follow-up | 95 | 177 | 210.7 | 337 | 192.8 | 196.3 | 37.5 |
| Ratio (follow-up/baseline) | 0.59 | 0.95 | 1.05 | 1.62 | 1 | 1.09 | 0.15 |
| Difference (follow-up − baseline) | −116 | −10 | 10 | 11 | 0.35 | 9.0 | |
| HDL cholesterol (mg/dL) | |||||||
| Baseline | 19 | 43 | 55 | 117 | 48.6 | 50.5 | 14.5 |
| Follow-up | 16 | 42 | 55 | 99 | 47.2 | 49.2 | 14.2 |
| Ratio (follow-up/baseline) | 0.55 | 0.9 | 1.04 | 1.65 | 0.97 | 0.99 | 0.17 |
| Difference (follow-up − baseline) | −37 | −5 | 2 | 30 | −1.3 | 28.9 | |
| Triglycerides (mg/dL) | |||||||
| Baseline | 30 | 111 | 183.7 | 1,130 | 144.1 | 177.2 | 138.7 |
| Follow-up | 32 | 111.3 | 186.7 | 1,500 | 146.9 | 173.9 | 120.5 |
| Ratio (follow-up/baseline) | 0.14 | 0.85 | 1.25 | 4.93 | 1.02 | 1.15 | 0.62 |
| Difference (follow-up − baseline) | −792 | −23 | 30 | 561 | −3.2 | 104.4 |
Figures 1A and 2A illustrate the change in LDL ratio by tertile of PFOA and PFOS, respectively. Ratios are the baseline value divided by the follow-up value, and PFAA tertiles are ordered by magnitude of decrease. Figure 1A shows that people whose PFOA fell by less than one half had a slight increase in LDL on average. In contrast, those whose PFOA fell by more than 64% had very little change in LDL cholesterol. Plots for total cholesterol show a similar pattern to LDL (Figures 1C and 2C). There is slight suggestion of associations for HDL cholesterol (Figures 1B and 2B) but no suggestion of associations for triglycerides (Figures 1D and 2D).
FIGURE 1.
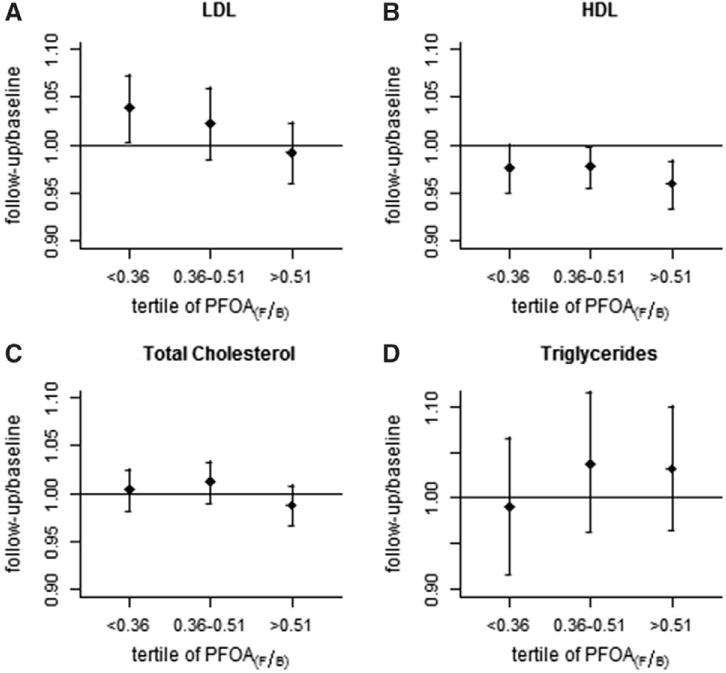
Geometric means and 95% CIs for follow-up/baseline change in each lipid measurement (mg/dL), according to tertile of followup/baseline change in PFOA (ng/mL).
FIGURE 2.
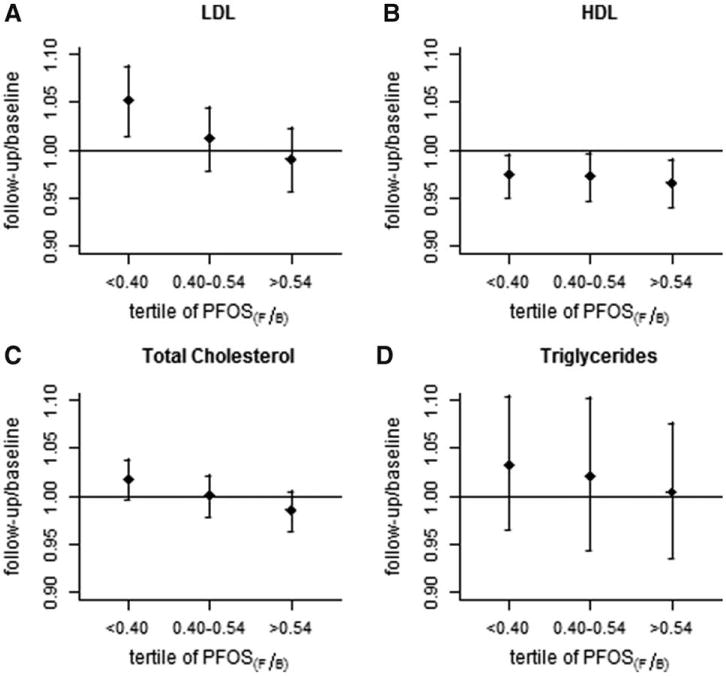
Geometric means and 95% CIs for follow-up/baseline change in each lipid measurement (mg/dL), according to tertile of followup/baseline change in PFOS (ng/mL).
These figures describe the broad picture. Regression models used change as a continuum, expressed as the logarithm of the ratio between final and initial values. Results are summarized in Table 2. The estimates from the adjusted models (Model 2) were slightly larger than the estimates from the unadjusted models (Model 1) for LDL, HDL, and total cholesterol. From Model 2, for change in LDL ratio versus change in PFOA ratio, adjusted for age, sex, fasting status, and interval between measurements, we found that for persons whose PFOA halved over the study time, LDL decreased an average of 3.6% (95% CI = 1.5–5.7%). We also found that halving PFOA predicted a total cholesterol decrease of 1.7% (95% CI = 0.3–3.0%). There was some suggestion of a similar association between change in HDL cholesterol and change in PFOA, but no evidence for an association between change in triglycerides and change in PFOA.
TABLE 2.
Predicted Percentage Decrease (95% CI) in Each Serum Lipid per Halving PFOA or PFOS in Serum
| Predictor | Response | Model 1a | Model 2b | Model 3c | Model 2 R2 | Model 2 Partial R2d |
|---|---|---|---|---|---|---|
| PFOA | LDL cholesterol | 3.08 (0.98 to 5.14) | 3.58 (1.47 to 5.66) | 2.92 (0.71 to 5.09) | 0.06 | 0.02 |
| Total cholesterol | 1.31 (0.01 to 2.60) | 1.65 (0.32 to 2.97) | 0.63 (−0.88 to 2.12) | 0.03 | 0.01 | |
| HDL cholesterol | 0.91 (−0.61 to 2.41) | 1.33 (−0.21 to 2.85) | 1.24 (−0.34 to 2.79) | 0.04 | 0.005 | |
| Triglycerides | −0.86 (−5.44 to 3.52) | −0.78 (−5.34 to 3.58) | −1.16 (−5.85 to 3.33) | 0.08 | 0.0002 | |
| PFOS | LDL cholesterol | 4.58 (2.12 to 6.97) | 4.99 (2.46 to 7.44) | 4.59 (1.97 to 7.14) | 0.07 | 0.03 |
| Total cholesterol | 2.75 (1.22 to 4.25) | 3.20 (1.63 to 4.76) | 3.21 (1.59 to 4.81) | 0.04 | 0.03 | |
| HDL cholesterol | 0.87 (−0.95 to 2.66) | 1.28 (−0.59 to 3.12) | 0.82 (−1.11 to 2.70) | 0.04 | 0.003 | |
| Triglycerides | 2.12 (−3.23 to 7.19) | 2.49 (−2.88 to 7.57) | 3.24 (−2.2 to 8.42) | 0.08 | 0.001 |
Model 1 is unadjusted.
Model 2 is adjusted for age, sex, interval between measurements, and fasting status.
Model 3 is joint model for PFOA with PFOS as a spline smooth or vice versa. Models are also adjusted for age, sex, interval, and fasting status.
Partial R2 is for the change in PFOA or PFOS.
The second part of Table 2 shows results for PFOS. The results are similar, although the magnitude (and variability) of decreases in lipids predicted by PFOS is slightly larger. We found an adjusted decrease in LDL cholesterol of 5.0% (95% CI = 2.5–7.4%) and an adjusted decrease in total cholesterol of 3.2% (95% CI = 1.6–4.8%), predicted by a 50% decline in PFOS. We found no evidence of association between changes in HDL or triglycerides and changes in PFOS.
The R2 and partial R2 values for the change in each PFAA separately for Model 2 are also shown in Table 2. Many other factors influence variations in cholesterol, and these R2 values are small. For both PFAAs, the partial R2 values for the LDL and total cholesterol responses make up a substantial proportion of the overall R2.
Decreases in serum PFOA and PFOS were quite highly correlated (Spearman’s ρ = 0.51). When, as a sensitivity analysis (Model 3), we included both PFOA and PFOS ratio changes in the model, with the control variable included as a smooth term, most of the associations weakened. However, the associations of LDL cholesterol with PFOA and PFOS, and of total cholesterol with PFOS, remained after mutual adjustment.
The sensitivity analysis in which we included in Model 2 variables that we considered would not change (or whose effects would not change over time) gave parameter estimates that were very close to the estimated effect of change in PFOA on change in LDL from Model 2. We included years of schooling, baseline BMI, and change in smoking status in Model 2 for LDL versus PFOA, but these had very little impact on the parameter estimate. The largest effect was after adding a smooth term for BMI, giving an estimated decrease in LDL per halving PFOA of 3.6% (95% CI = 1.4–5.6%)—very close to the estimate without adjustment for BMI. Baseline GFR was associated with change in PFOA, predicting 0.50% (95% CI = 0.14–0.86%) more decrease in PFOA per unit increase in baseline GFR, but including baseline GFR had very little effect on the parameter estimate for change in PFOA. Including change in GFR as a smooth term had little effect on the parameter estimate, the magnitude being similar to what we observed upon inclusion of BMI (the estimated decrease in LDL per halving PFOA was 3.6% with 95% CI [1.5–5.7%]).
DISCUSSION
These change-versus-change models are equivalent to repeated measures models with fixed effects for subjects. We decided against using repeated measures models with random effects for subjects, as change-versus-change models are not subject to bias due to time-invariant confounding. We have found that LDL cholesterol was positively related to both PFOA and PFOS in serum. This supports previous evidence of a positive association between serum LDL cholesterol and PFOA and adds to evidence for a causal effect of PFOA or PFOS on lipids.
During the study period, overall serum concentrations of both PFAAs decreased by approximately one half, and there was little change in LDL cholesterol, with mean increase 1.8% (SD 26.6%). Some evidence from previous studies suggests a slight overall increase in total cholesterol over a few years in untreated people, mainly due to increases in younger age groups.32 In clinical trials, there is little evidence of change in cholesterol among placebo groups over this length of time.33 These appear to be consistent with our study, where overall there is little evidence of change in cholesterol.
We found a tendency for study participants with a larger PFOA decrease to have a larger LDL decrease, such that halving of PFOA predicted a 3.6% (1.5–5.7%) fall in LDL cholesterol, adjusted for possible confounders. We also found a greater estimated decrease in LDL cholesterol of 5.0% (2.5–7.4%) when PFOS was halved. We found similar results for total cholesterol. There was some suggestion of association between change in HDL cholesterol and change in PFOA, but no evidence of association between change in triglycerides and change in either PFOA or PFOS.
The joint models including both PFOA and PFOS should be interpreted with caution because PFOA and PFOS are highly correlated. Nevertheless, despite increased standard errors in these models, which made identification of associations more challenging, associations of LDL with both PFOA and PFOS remained.
As a sensitivity analysis of the logarithmic transformation, and for comparison with previous studies, we fit models for the untransformed difference in response versus the difference in PFOA and PFOS, adjusted for possible time-varying confounders, and we discuss this analysis in the eAppendix (http://links.lww.com/EDE/A675). We do not find these results inconsistent with the conclusions regarding our primary hypothesis.
Steenland and colleagues,17 using data from the C8 Health Project, found an average difference in LDL of 0.015 (standard error = 0.001) for a one unit change in PFOA on the natural logarithm scale—that is, a predicted decrease of 1.0% (0.9%, 1.2%) in LDL for a halving in PFOA. This estimate is smaller than our longitudinal estimate of 3.6%. This may be due to biases operating differently in the two study designs.
There have been only three other longitudinal studies of the relationship between cholesterol and PFOA, all smaller than the current study. A study34 of highly exposed workers (n = 175) reported positive associations between serum PFOA and total cholesterol and triglycerides, but not HDL; the authors found no associations between cholesterol and PFOS. Another longitudinal study,15 on 454 exposed workers, found a positive association between PFOA and total cholesterol but no association with LDL, HDL, or triglycerides. A recent study35 of 179 workers exposed to high levels of PFOA found little evidence of association between PFOA and total, HDL, or non-HDL cholesterol but did not examine LDL cholesterol or triglycerides. This study was limited in time of follow-up (mean 5.5 months). The main analysis was for untransformed differences in response versus predictor (the authors also fit a model in a subset with the predictor log-transformed but not the response); a linear model for untransformed changes in total cholesterol versus PFOA gave an adjusted parameter estimate of −1.86 with 95% CI (−4.25 to 1.15), which overlaps our estimate of −0.72 (−2.34, 0.90) shown in Table 3 in the eAppendix (http://links.lww.com/EDE/A675). With PFOS as the predictor, the parameter estimate was 0.59 (−3.75, 4.91), which also overlaps our estimate of 0.41 (0.15, 0.67). Lower power might explain failure of these studies to identify some of the associations found in this study. Also, the first two studies described did not control for use of lipid-lowering drugs.
In the change-versus-change analysis, confounding is less of a problem than in a cross-sectional analyses because many risk factors varying between people (such as GFR, diet, exercise, BMI, and other lifestyle factors) are relatively constant within people over time, or at least not likely to vary with changes in serum PFOA and PFOS. We took account of possible confounding due to changes in use of lipid-lowering drugs by exclusion, also allowing for possible nonidentification of treatment (see eAppendix, http://links.lww.com/EDE/A675). We also considered the possibility that our estimates were affected by regression to the mean, but neither analytic considerations nor simulations indicated that this was the case. Moreover, we observed little evidence of associations for triglycerides or HDL cholesterol, which would also likely give rise to spurious associations if regression to the mean were an issue. Also, the change in exposure to PFOA was due to a specific intervention—the ending of contamination of public water supplies. This “natural experiment” further reduces likelihood of reverse causation and thus strengthens a causal interpretation of the association we observed between changes in serum concentrations of PFAAs and cholesterol.
This study has certain limitations. We had only two time points from which to estimate the relationship between change in serum cholesterol and change in PFOA and are therefore unable to distinguish an association more complex than linear. Another limitation is the uncertainty about a possible lag in the response to changes in PFOA and PFOS; however, the potential to investigate this using our data is limited, having measurements at only two times. Also, this study does not allow us to detect a possible irreversible effect of PFOA/PFOS on serum lipids. If PFOA/PFOS causes an increase in cholesterol due to a wholly or partly irreversible mechanism, we would not expect to see any effect on cholesterol with a fall in PFOA. A logistic regression model for the association between changing from high (>100 mg/dL) to normal LDL cholesterol and the change in PFOA was limited due to power considerations; there were only 56 people who changed from high to normal LDL cholesterol, and we would ideally require more than 100 cases to detect an association.36,37 Nevertheless, an unadjusted logistic model for the association between changing from high to normal LDL cholesterol and change in (log-transformed) PFOA predicted an odds of 0.96 (95% CI = 0.92–1.00) for a 50% decrease in PFOA, which is consistent with the results of the linear models. This suggests the effect may be of clinical relevance.
The exact mechanism by which PFAAs might elevate serum lipids in humans is unknown, and this limits a causal interpretation. However, both PFOA and PFOS are ligands for the PPARα.3,4 The natural ligands for PPARα include saturated and unsaturated fatty acids,38 as well as eicosanoids derived from n-3 and n-6 polyunsaturated fatty acids.39 Ligand activation of PPARα is associated with transcriptional upregulation of a wide range of genes that code for proteins associated with fatty acid oxidation and lipoprotein metabolism, such as acyl coenzyme A cholesterol acyltransferase, carnine palmitoyl transferase 1, lipoprotein lipase, and apo CIII.40 Thus PFOA and PFOS may interfere with lipid regulation by altering normal PPARα activity.
The estimated decrease of 3% to 5% in LDL cholesterol for a 50% fall in PFOA or PFOS is modest, although at a population level such a shift could have public health impact. Caution is necessary in extrapolating the results for PFOA from this study (based on a highly exposed population) to the general population, with much lower concentrations of PFOA. Still, it appears the impact on LDL of reducing PFOA is greater in those with lower serum PFOA concentrations. For serum PFOS, concentrations in the study population and the rate of decline are comparable with those measured in the industrialized world.
In summary, our findings from this longitudinal study provide further evidence of positive associations between LDL cholesterol and PFOA and PFOS in serum.
Supplementary Material
Acknowledgments
We thank the participants for their contributions to this study. We also acknowledge the technical assistance of Brian Basden and Tao Jia in measuring the serum concentrations of PFOS and PFOA. The LSHTM Ethics Committee approved this study.
Supported by The C8 Class Action Settlement Agreement.
Footnotes
Supplemental digital content is available through direct URL citations in the HTML and PDF versions of this article (www.epidem.com). This content is not peer-reviewed or copy-edited; it is the sole responsibility of the author.
Editors’ note: Related papers appear on pages 577 and 580.
References
- 1.Kennedy GL, Jr, Butenhoff JL, Olsen GW, et al. The toxicology of perfluorooctanoate. Crit Rev Toxicol. 2004;34:351–384. doi: 10.1080/10408440490464705. [DOI] [PubMed] [Google Scholar]
- 2.Lau C, Anitole K, Hodes C, Lai D, Pfahles-Hutchens A, Seed J. Perfluoroalkyl acids: a review of monitoring and toxicological findings. Toxicol Sci. 2007;99:366–394. doi: 10.1093/toxsci/kfm128. [DOI] [PubMed] [Google Scholar]
- 3.Vanden Heuvel JP, Thompson JT, Frame S, Gillies PJ. Differential activation of nuclear receptors by perfluorinated fatty acid analogs and natural fatty acids: a comparison of human, mouse, and rat peroxisome proliferator-activated receptor-α, -β, and -γ, liver X receptor-β, and retinoid X receptor-α. Toxicol Sci. 2006;92:476–489. doi: 10.1093/toxsci/kfl014. [DOI] [PubMed] [Google Scholar]
- 4.Takacs ML, Abbott BD. Activation of mouse and human peroxisome proliferator-activated receptors (α, β/δ, γ) by perfluorooctanoic acid and perfluorooctane sulfonate. Toxicol Sci. 2009;95:108–117. doi: 10.1093/toxsci/kfl135. [DOI] [PubMed] [Google Scholar]
- 5.Maloney EK, Waxman DJ. trans-Activation of PPARalpha and PPARgamma by structurally diverse environmental chemicals. Toxicol Appl Pharmacol. 1999;161:209–218. doi: 10.1006/taap.1999.8809. [DOI] [PubMed] [Google Scholar]
- 6.Xie Y, Yang Q, Nelson BD, DePierre JW. The relationship between liver peroxisome proliferation and adipose tissue atrophy induced by peroxisome proliferator exposure and withdrawal in mice. Biochem Pharmacol. 2003;66:749–756. doi: 10.1016/s0006-2952(03)00386-1. [DOI] [PubMed] [Google Scholar]
- 7.Olsen GW, Burris JM, Ehresman DJ, et al. Half-life of serum elimination of perfluorooctanesulfonate,perfluorohexanesulfonate, and perfluorooctanoate in retired fluorochemical production workers. Environ Health Perspect. 2007;115:1298–1305. doi: 10.1289/ehp.10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bartell SM, Calafat AM, Lyu C, Kato K, Ryan PB, Steenland K. Rate of decline in serum PFOA concentrations after granular activated carbon filtration at two public water systems in Ohio and West Virginia. Environ Health Perspect. 2010;118:222–228. doi: 10.1289/ehp.0901252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Betts KS. Perfluoroalkyl acids: what is the evidence telling us? Environ Health Perspect. 2007;115:A250–A256. doi: 10.1289/ehp.115-a250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steenland K, Fletcher T, Savitz DA. Epidemiologic evidence on the health effects of perfluorooctanoic acid (PFOA) Environ Health Perspect. 2010;118:1100–1108. doi: 10.1289/ehp.0901827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olsen GW, Burris JM, Burlew MM, Mandel JH. Plasma cholecystokinin and hepatic enzymes, cholesterol and lipoproteins in ammonium perfluorooctanoate production workers. Drug Chem Toxicol. 2000;23:603–620. doi: 10.1081/dct-100101973. [DOI] [PubMed] [Google Scholar]
- 12.Emmett EA, Zhang H, Shofer FS, et al. Community exposure to perfluorooctanoate: relationships between serum levels and certain health parameters. J Occup Environ Med. 2006;48:771–779. doi: 10.1097/01.jom.0000233380.13087.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olsen GW, Zobel LR. Assessment of lipid, hepatic, and thyroid parameters with serum perfluorooctanoate (PFOA) concentrations in fluorochemical production workers. Int Arch Occup Environ Health. 2007;81:231–246. doi: 10.1007/s00420-007-0213-0. [DOI] [PubMed] [Google Scholar]
- 14.Sakr CJ, Kreckmann KH, Green JW, Gillies PJ, Reynolds JL, Leonard RC. Cross-sectional study of lipids and liver enzymes related to a serum biomarker of exposure (ammonium perfluorooctanoate or APFO) as part of a general health survey in a cohort of occupationally exposed workers. J Occup Environ Med. 2007;49:1086–1096. doi: 10.1097/JOM.0b013e318156eca3. [DOI] [PubMed] [Google Scholar]
- 15.Sakr CJ, Leonard RC, Kreckmann KH, Slade MD, Cullen MR. Longitudinal study of serum lipids and liver enzymes in workers with occupational exposure to ammonium perfluorooctanoate. J Occup Environ Med. 2007;49:872–879. doi: 10.1097/JOM.0b013e318124a93f. [DOI] [PubMed] [Google Scholar]
- 16.Costa G, Sartori S, Consonni D. Thirty years of medical surveillance in perfluooctanoic acid production workers. J Occup Environ Med. 2009;51:364–372. doi: 10.1097/JOM.0b013e3181965d80. [DOI] [PubMed] [Google Scholar]
- 17.Steenland K, Tinker S, Frisbee S, Ducatman A, Vaccarino V. Association of perfluorooctanoic acid and perfluorooctane sulfonate with serum lipids among adults living near a chemical plant. Am J Epidemiol. 2009;170:1268–1278. doi: 10.1093/aje/kwp279. [DOI] [PubMed] [Google Scholar]
- 18.Wang J, Zhang Y, Zhang W, Jin Y, Dai J. Association of perfluorooctanoic acid with HDL cholesterol and circulating miR-26b and miR-199-3p in workers of a fluorochemical plant and nearby residents. Environ Sci Technol. 2012;46:9274–9281. doi: 10.1021/es300906q. [DOI] [PubMed] [Google Scholar]
- 19.Shin HM, Viera VM, Ryan PB, et al. Environmental fate and transport modelling for perfluouroocatannoic acid emitted from the Washington works factory in West Virginia. Environ Sci Technol. 2011;45:1435–1442. doi: 10.1021/es102769t. [DOI] [PubMed] [Google Scholar]
- 20.Kato K, Wong LY, Jia LT, Kuklenyik Z, Calafat AM. Trends in exposure to polyfluoroalkyl chemicals in the U.S. Population: 1999–2008. Environ Sci Technol. 2011;45:8037–8045. doi: 10.1021/es1043613. [DOI] [PubMed] [Google Scholar]
- 21.Frisbee SJ, Brooks AP, Jr, Maher A, et al. The C8 health project: design, methods, and participants. Environ Health Perspect. 2009;117:1873–1882. doi: 10.1289/ehp.0800379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kato K, Basden BJ, Needham LL, Calafat AM. Improved selectivity for the analysis of maternal serum and cord serum for polyfluoroalkyl chemicals. J Chromatogr A. 2011;1218:2133–2137. doi: 10.1016/j.chroma.2010.10.051. [DOI] [PubMed] [Google Scholar]
- 23.Keller JM, Calafat AM, Kato K, et al. Determination of perfluorinated alkyl acid concentrations in human serum and milk standard reference materials. Anal Bioanal Chem. 2010;397:439–451. doi: 10.1007/s00216-009-3222-x. [DOI] [PubMed] [Google Scholar]
- 24.Van Leeuwen SJP, Karrmen A, Van Bavel B, De Boer J, Lindstropm G. Struggle for quality of determination of perfluorinated contaminants in human and environmental samples. Environ Sci Technol. 2006;40:7854–7860. doi: 10.1021/es061052c. [DOI] [PubMed] [Google Scholar]
- 25.Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18:499–502. [PubMed] [Google Scholar]
- 26.Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med. 1999;130:461–470. doi: 10.7326/0003-4819-130-6-199903160-00002. [DOI] [PubMed] [Google Scholar]
- 27.Liker JK, Augustiniak S, Duncan GJ. Panel data and models of change: a comparison of first difference and conventional two-wave models. Soc Sci Res. 1985;14:80–101. [Google Scholar]
- 28.Peña EA, Slate EH. Global validation of linear model assumptions. J Am Stat Assoc. 2006;101:341. doi: 10.1198/016214505000000637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Glymour MM, Weuve J, Berkman LF, Kawachi I, Robins JM. When is baseline adjustment useful in analyses of change? Am J Epidemiol. 2005;162:267–278. doi: 10.1093/aje/kwi187. [DOI] [PubMed] [Google Scholar]
- 30.Wood SN. Generalized Additive Models: An Introduction with R. Boca Raton, FL: Chapman and Hall/CRC; 2006. [Google Scholar]
- 31.R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna: R foundation for Statistical Computing; 2011. [Google Scholar]
- 32.Arnett DK, Jacobs DR, Jr, Luepker RV, Blackburn H, Armstrong C, Claas SA. Twenty-year trends in serum cholesterol, hypercholesterolemia, and cholesterol medication use: the Minnesota Heart Survey, 1980–1982 to 2000–2002. Circulation. 2005;112:3884–3891. doi: 10.1161/CIRCULATIONAHA.105.549857. [DOI] [PubMed] [Google Scholar]
- 33.Taylor F, Ward K, Moore TH, et al. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2011;(1):CD004816. doi: 10.1002/14651858.CD004816.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olsen GW, Burris JM, Burlew MM, Mandel JH. Epidemiologic assessment of worker serum perfluorooctanesulfonate (PFOS) and perfluorooctanoate (PFOA) concentrations and medical surveillance examinations. J Occup Environ Med. 2003;45:260–270. doi: 10.1097/01.jom.0000052958.59271.10. [DOI] [PubMed] [Google Scholar]
- 35.Olsen GW, Ehresman DJ, Buehrer BD, Gibson BA, Butenhoff JL, Zobel LR. Longitudinal assessment of lipid and hepatic clinical parameters in workers involved with the demolition of perfluoroalkyl manufacturing facilities. J Occup Environ Med. 2012;54:974–983. doi: 10.1097/JOM.0b013e31825461d2. [DOI] [PubMed] [Google Scholar]
- 36.Peduzzi P, Concato J, Kemper E, Holford TR, Feinstein AR. A simulation study of the number of events per variable in logistic regression analysis. J Clin Epidemiol. 1996;49:1373–1379. doi: 10.1016/s0895-4356(96)00236-3. [DOI] [PubMed] [Google Scholar]
- 37.Long JS. Regression Models for Categorical and Limited Dependent Variables. Thousand Oaks, CA: Sage Publications; 1997. [Google Scholar]
- 38.Rigamonti E, Chinetti-Gbaguidi G, Staels B. Regulation of macrophage functions by PPARa, PPARg, and LXRs in mice and men. Arterioscler Thromb Vasc Biol. 2008;28:1050–1059. doi: 10.1161/ATVBAHA.107.158998. [DOI] [PubMed] [Google Scholar]
- 39.Krey G, Braissant O, L’Horset F, et al. Fatty acids, eicosanoids, and hypolipidemic agents identified as ligands of peroxisome proliferator-activated receptors by coactivator-dependent receptor ligand assay. Mol Endocrinol. 1997;11:779–791. doi: 10.1210/mend.11.6.0007. [DOI] [PubMed] [Google Scholar]
- 40.Brown JD, Plutzky J. Peroxisome proliferator-activated receptors as transcriptional nodal points and therapeutic targets. Circulation. 2007;115:518–533. doi: 10.1161/CIRCULATIONAHA.104.475673. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.