

Abstract
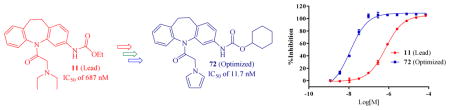
Constitutive androstane receptor (CAR, NR1I3) and pregnane X receptor (PXR, NR1I2) are master regulators of endobiotic and xenobiotic metabolism and disposition. Because CAR is constitutively active in certain cellular contexts, inhibiting CAR might reduce drug-induced hepatotoxicity and resensitize drug-resistant cancer cells to chemotherapeutic drugs. We recently reported a novel CAR inhibitor/inverse agonist CINPA1 (11). Here, we have obtained or designed 54 analogs of CINPA1 and used a time-resolved fluorescence resonance energy transfer (TR-FRET) assay to evaluate their CAR inhibition potency. Many of the 54 analogs showed CAR inverse agonistic activities higher than those of CINPA1, which has an IC50 value of 687 nM. Among them, 72 has an IC50 value of 11.7 nM, which is about 59-fold more potent than CINPA1 and over 10-fold more potent than clotrimazole (an IC50 value of 126.9 nM), the most potent CAR inverse agonist in a biochemical assay previously reported by others. Docking studies provide a molecular explanation of the structure-activity relationship (SAR) observed experimentally. To our knowledge, this effort is the first chemistry endeavor in designing and identifying potent CAR inverse agonists based on a novel chemical scaffold, leading to 72 as the most potent CAR inverse agonist so far. The 54 chemicals presented are novel and unique tools for characterizing CAR’s function, and the SAR information gained from these 54 analogs could guide future efforts to develop improved CAR inverse agonists.
Keywords: constitutive androstane receptor, CINPA1, inverse agonist, TR-FRET
Graphical abstract
1. Introduction
Constitutive androstane receptor (CAR, NR1I3) and pregnane X receptor (PXR, NR1I2) are members of the nuclear receptor (NR) superfamily and are master regulators of endobiotic and xenobiotic metabolism and disposition [1–3]. CAR and PXR are highly expressed in the liver, where most drug metabolism and clearance occurs [4]. CAR regulates the transactivation of many P450 enzymes and transporters, including CYP2Bs, CYP2Cs, CYP3As, multi-drug resistance protein 1 (MDR1), and multidrug resistance-associated proteins (MRPs) [5].
Depending on the cellular context, CAR may be constitutively active or activated by agonists [6]. CAR activation contributes to drug-induced hepatotoxicity [7], drug resistance [8], liver hypertrophy and liver tumor development in animals [9–11]. Therefore, small molecule inhibitors of CAR, either function as inverse agonists that decrease the constitutive activity, or as antagonists in the presence of agonists, or as deactivators that decrease the levels of CAR, will provide beneficial pharmacological effects by reducing the activity of CAR.
CAR has two main structural domains, the DNA-binding domain (DBD) and the ligand-binding domain (LBD) [12], which are common structural features of the NR family [13]. CAR recognizes a wide range of structurally diverse ligands because of its relatively large ligand binding pocket [14]. There are multiple splice variants of human CAR, with human CAR1 (wild-type CAR) being the most abundant and constitutively active isoform [15], and the target of our studies discussed here.
A panel of structurally diverse small molecules (Figure 1) has been reported as being CAR inverse agonists, inhibitors, deactivators, or antagonists. Androstanol (1), clotrimazole (2), and PK11195 (3) have been demonstrated to be CAR inverse agonists in a biochemical CAR-mediated PGC1α coactivator recruitment/repression assay, with IC50s of 1.2 μM, 80 nM, and 390 nM, respectively [16]. In another report, clotrimazole displayed similar CAR inverse agonistic activity, with an IC50 of 100 nM in a similar biochemical coactivator recruitment/repression assay (using SRC-1 instead of PGC1α), and a Ki of ca. 100 nM in a Scintillation Proximity Binding Assay [17], however in a cell-based CAR inverse agonistic assay it exhibited a higher IC50 value of 0.69 μM. It was believed that cell permeability or metabolism of clotrimazole contributed to the activity discrepancy between the biochemical and cell-based assays. Among all reported CAR inverse agonists tested using various assay systems [18], clotrimazole was consistently the most potent CAR inverse agonist in the biochemical CAR-mediated coregulator recruitment/repression assays. However, the activity of clotrimazole against CAR in cell-based assays has varied from being inverse agonist [16, 17, 19] to agonist [16, 20, 21], most likely because of different cellular contexts, such as the levels of co-regulators in the different systems used [22]. Because of the complexity of cell-based assays, biochemical assays are more appropriate for structure-activity relationship (SAR) analysis. Meclizine (4) was reported to be another CAR inverse agonist [23], but it failed to act as an effective hCAR inverse agonist or antagonist in cultured human hepatocytes [24]. As an estrogen receptor agonist, 17α-ethynyl-3,17β-estradiol (EE2, 5) was shown to possess modest CAR inverse agonistic activity with an IC50 of ca. 3 μM in a cell-based assay [20]. S07662 (6) [25], TO901317 (T0901317, 7) [26], Nigramide J (8) [27] and LY2090314 (9) [28] were also shown to be CAR inverse agonists with PXR agonistic activity. Although most CAR inverse agonists also act as PXR agonists, allyl isothiocyanate (AITC, 10) was found to have inhibitory effects against both CAR and PXR [29], but only when very high concentrations (i.e., at 20 or 40 μM) were used. Because of the relatively weak CAR inverse agonistic activities, no IC50 value is available for other reported CAR inverse agonists. Recently, we discovered CINPA1 (11) [6] to be a potent CAR inverse agonist having a novel chemical scaffold. Uniquely, it does not activate PXR. Here, we present our efforts to develop CINPA1 analogs and evaluate their activities as inverse agonists of CAR.
Figure 1.


Chemical structures of a panel of representative CAR inverse agonists
Our efforts to identify more potent CAR inverse agonists are based on CINPA1 and were implemented in three stages. First, we obtained 10 close analogs of CINPA1 (11) that are commercially available and evaluated their CAR inverse agonistic activities and those of the lead compound 11 (CINPA1) in a biochemical CAR-mediated fluorescein-PGC1α recruitment/repression assay [6, 16]. We chose to use the biochemical CAR-mediated fluorescent coactivator recruitment/repression assay because it provides consistent potency, regardless of whether SRC1 or PGC1α peptide is used [16, 17]. Second, on the basis of the preliminary SAR information gained from these 11 compounds (i.e., CINPA1 and its 10 close analogs), we designed 23 novel analogs during our first-round chemistry effort to improve CAR inverse agonistic activities, which were evaluated by using the same biochemical CAR-mediated fluorescein-PGC1α recruitment/repression assay. Third, the SAR information of the 23 novel analogs directed us to design 20 additional novel analogs and evaluate their activities. In summary, we have evaluated 54 chemicals (CINPA1, 10 commercially available analogs, and 43 novel analogs) and found that many of them have more potent CAR inverse agonistic activities than the lead chemical CINPA1 (11) does. Among them, the most potent analog 72 has an IC50 value of 11.7 nM which is about 59-fold as potent as the lead compound CINPA1 (an IC50 value of 687 nM) and it is over 10-fold as potent as the most potent and previously reported inverse agonist clotrimazole (with an IC50 value of 126.9 nM from the same biochemical assay). To our knowledge, our effort is the first chemistry endeavor to develop and identify potent CAR inverse agonists based on a novel chemical scaffold identified from a compound screening campaign. The set of 54 compounds is a novel and useful tool for the study of CAR function.
2. Results and Discussion
2.1. CAR inverse agonistic activities and preliminary structure-activity relationship of CINPA1 and its 10 commercially available analogs
2.1.1. CAR inverse agonistic activities of CINPA1 and its 10 commercially available analogs
On the basis of the chemical scaffold of CINPA1, 3-amino-10,11-dihydro-5H-dibenz[b,f]azepine or saturated 3-amino-dibezapine (Figure 2), we searched for commercially available close analogs of CINPA1 by using SciFinder® (provided by Chemical Abstracts Service) and identified and obtained 10 CINPA1 close analogs. We then evaluated their CAR inverse agonistic activities by using a biochemical CAR-mediated fluorescent PGC1α coactivator recruitment/repression assay as previously reported [6, 16], with clotrimazole (positive control) and DMSO (negative control) used as controls. The chemical structures of CINPA1 and the 10 analogs, together with their CAR inverse agonistic activities, are summarized in Table 1. In our assay, clotrimazole had an IC50 of 126.9 ± 8.0 nM (Table 1), which is consistent with published results [16, 17]. CINPA1 (11) had an IC50 of 687 ± 71 nM.
Figure 2.
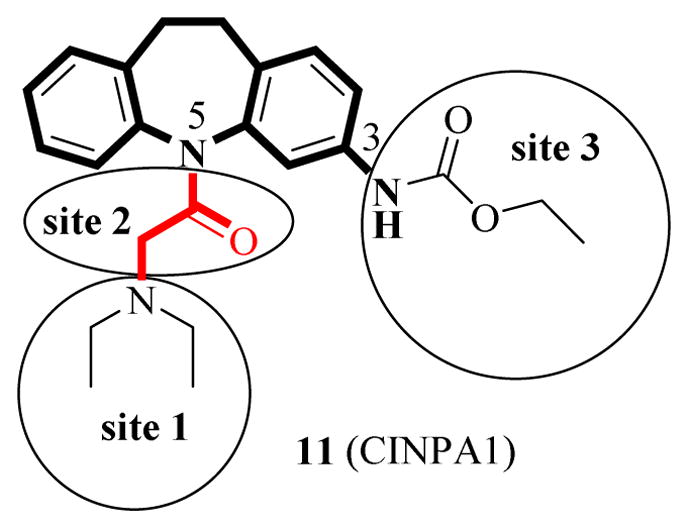
Structural features of 11 (CINPA1)
Table 1.
CAR inverse agonistic activities of CINPA1 and 10 commercially available analogs
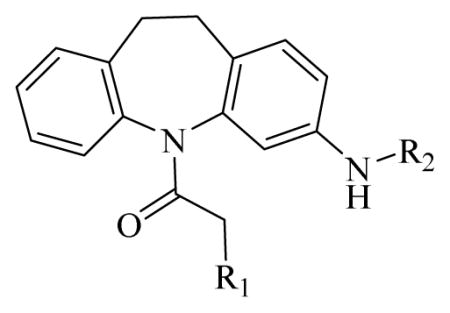
| |||
|---|---|---|---|
| Compound | R1 | R2 | IC50 (nM) |
| 2 (clotrimazole) | NA | NA | 126.9 ± 8.0 |
| 11 (CINPA1) |
|
|
687 ± 71 |
| 12 |
|
(12 to 18) |
235 ± 20 |
| 13 |
|
38,755 ± 5,451 | |
| 14 |
|
4,078 ± 239 | |
| 15 | NH2 | > 70,000 | |
| 16 |
|
3,631 ± 187 | |
| 17 |
|
320 ± 37 | |
| 18 |
|
5,010 ± 84 | |
| 19 |
|
|
288 ± 13 |
| 20 |
|
 (20 and 21) |
1,792 ± 87 |
| 21 |
|
> 70,000 | |
NA: not applicable
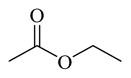
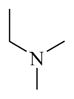
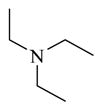
CINPA1 has a chemical scaffold of 3-amino-10,11-dihydro-5H-dibenz[b,f]azepine or saturated 3-amino-dibezapine (Figure 2, with scaffold highlighted in bold and black) with a diethylamino substitute to the 5-nitrogen on the saturated dibezapine ring through a methylene carbonyl linker and an ethyl carbamate modification at the 3-amino group of the scaffold. These structure features provide CINPA1 with 3 major sites for chemical modifications (if the core scaffold remains unchanged): 1) substitutes (specified as site 1) attached to the 5-nitrogen on the saturated dibezapine ring by the methylene carbonyl linker; 2) the methylene carbonyl linker (specified as site 2); and 3) modifications on the 3-amino group site (specified as site 3) at the scaffold.
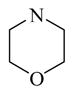
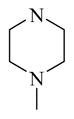
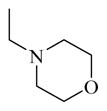
CINPA1 and the 10 commercially available analogs (Table 1) have varied modifications at all 3 major modifiable sites. CINPA1 (11) and chemicals 12 to 18 all have an ethyl carbamate group at the 3-amino position (site 3), but have different modifications at site 1 and site 2. However, chemical 19 has a methyl carbamate group at site 3, and chemicals 20 and 21 both have an isopropyl carbamate group at site 3. Although both CINPA1 (11) and chemical 12 have an ethyl carbamate modification at the 3-amino position, chemical 12 has a morpholino structure feature at site 1 instead of the diethylamino group presented in chemical 11 (CINPA1). This modification enhances the CAR inverse agonistic activity of chemical 12 (IC50 of 235 nM) by about 3-fold over that of lead chemical 11 (IC50 of 687 nM). The Morpholino group is similar in size to the diethylamino group but has reduced rotation flexibility because the diethylamino group in chemical 11 is locked in the form of a 6-member morpholino ring in chemical 12. However, locking the diethylamino group conformation of chemical 11 in the form of an N-methyl-piperazino group resulted in a significantly weaker chemical 13 with a 56-fold higher IC50 value (IC50 of 38,755 nM) than that of chemical 11. Compared to the Morpholino group, the N-methyl-piperazino group is bigger; an additional aliphatic tertiary amino group may allow chemical 13 to be protonated at a greater degree under assay conditions (pH 7.5), which may adversely affect ligand receptor interaction.
Chemicals 14 (IC50 of 4,078 nM) and 15 (IC50 > 70,000 nM) are much weaker CAR inverse agonists. Both have smaller substitutes at the position of the diethylamino group in chemical 11 (IC50 of 687 nM), with a dimethyl amino group in chemical 14 and an amino group in chemical 15. Therefore, substitutions with a smaller group at the position of the diethylamino group in chemical 12 contributed negatively to the CAR inverse agonistic activities.
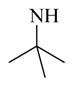
Chemical 16 (IC50 of 3,631 nM) has a secondary amino group (tert-butylamino) at the position of diethylamino group in chemical 11 (IC50 of 687 nM), which reduced its CAR inverse agonistic activity (IC50 increases by 5-fold). A secondary amino group such as tert-butylamino in chemical 16 at the position of diethylamino group in chemical 11 has a free proton on the nitrogen, which may contribute as a hydrogen bond donor. A hydrogen bond donor at this position may affect the CAR inverse agonistic activity negatively.
In comparison to chemical 12 (IC50 of 235 nM) and chemical 14 (IC50 of 4,078 nM), chemical 17 (IC50 of 320 nM) and chemical 18 (IC50 of 5,010 nM), which have an ethylene carbonyl linker, had slightly decreased CAR inverse agonistic activities, suggesting that a longer linker at site 2 may negatively affect the CAR inverse agonistic activity of the analogs. The only difference between chemical 17 (IC50 of 320 nM) and chemical 19 (IC50 of 288 nM) is at site 3, with an ethyl carbamate at 3-amino group in chemical 17 and a methyl carbamate in chemical 19. Here a slightly increased CAR inverse agonistic activity was observed with a methyl carbamate rather than an ethyl carbamate at the 3-amino group. However, a methyl carbamate at the 3-amino group position was not taken into consideration in further development because the activity increase was marginal, and the configuration is less enzymatically and metabolically stable than an ethyl carbamate. More importantly, site 3 with a larger group such as an isopropyl carbamate in chemical 20, endowed the analog with increased CAR inverse agonistic activity.
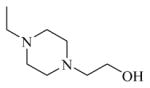
Chemical 18 (IC50 of 5,010 nM) and chemical 20 (IC50 of 1,792nM) have a difference only at site 3, with an ethyl carbamate in chemical 18 and an isopropyl carbamate in chemical 20. The isopropyl carbamate at site 3 increased the potency of chemical 20 by about 3-fold compared to chemical 18. Chemical 21 (IC50 > 70,000 nM) has an even larger, protonable, and maybe more hydrophilic substitute (2-hydroxyethylpiperazino group) at site 1 and an ethylene carbonyl linker at site 2 which rendered it inactive as a CAR inverse agonist even though its isopropyl carbamate feature at site 3 could increase its CAR inverse agonistic activity.
2.1.2. Preliminary structure-activity relationship summary based on the CINPA1 and 10 commercially available analogs
Based on the CAR inverse agonistic activities gained from CINPA1 and 10 commercially available analogs, a preliminary SAR for the CINPA1 analogs was summarized (schematic CINPA1 structural features in Figure 2):
Scaffold: 3-amino-10,11-dihydro-5H-dibenz[b,f]azepine (saturated 3-amino-dibezapine) (bold and black in Figure 2);
Site 1: Medium constrained substitutes to the 5-nitrogen position through the methylene carbonyl linker are favorable; less charged or less protonated favorable; H-bond donor unfavorable;
Site 2: A methylene carbonyl linker between 5-nitrgen position of the saturated 3-amino-dibezapine and additional substitutes is preferred over an ethylene carbonyl linker (bold and red in Figure 2);
Site 3: Proper modification at the 3-amino group on the saturated dibezapine ring: isopropyl carbamate > ethyl carbamate ≥ methyl carbamate.
2.2. Design, syntheses and activity evaluation of 23 CINPA1 analogs in the round 1 chemistry effort
2.2.1. Design of the 23 CINPA1 analogs in the round 1 chemistry effort
By using preliminary SAR gained from CINPA1 and 10 commercially available analogs as CAR inverse agonists, novel analogs were designed in a stepwise approach. Modifications were focused on site 1 (the substitutes to 5-nitrogen position through the methylene carbonyl linker), site 2 (the methylene carbonyl linker), and site 3 (the modifications at the 3-amino group at the saturated dibezapine ring), with the saturated 3-amino-dibezapine scaffold unchanged.
In the first-round chemistry effort, 23 analogs were designed, with CINPA1 as the lead compound; their structures are summarized in Table 2. Chemicals 22 to 30 have a variety of di-substituted amino group modifications at site 1: open rings, closed rings, aliphatic rings, or aromatic rings. Chemicals 31 and 32 have an ethylene carbonyl linker at site 2 to further explore and confirm the effect of the additional methylene insert within site 2. Chemicals 33 and 34 have a secondary amino group feature that was designed to further explore and confirm the hydrogen donor property at the site 1 position. Chemical 35 has a tert-butoxy group at the site 1 position instead of a regular tertiary amino group. A tert-butoxy group could eliminate the protonation possibility at site 1 under physiological conditions. Chemicals 36 to 44 have modifications on site 3 with various structure features of amide, carbamates, ureas, sulfamide, and carbamate heterocyclic isostere.
Table 2.
CAR inverse agonistic activities of CINPA1 and its 23 analogs from the first-round chemistry effort
|
| |||
|---|---|---|---|
| Compound | R1 | R2 | IC50 (nM) |
| 11 (CINPA1) |
|
|
687 ± 71 |
| 22 |
|
210 ± 10 | |
| 23 |
|
22 to 35 |
3,050 ± 125 |
| 24 |
|
1,580 ± 88 | |
| 25 |
|
620 ± 29 | |
| 26 |
|
4,080 ± 162 | |
| 27 |
|
1,410 ± 41 | |
| 28 |
|
39,810 ± 4,384 | |
| 29 |
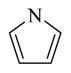
|
37.0 ± 1.5 | |
| 30 |
|
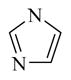
6,890 ± 350 | |
| 31 |
|
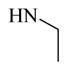
24,490 ± 845 | |
| 32 |
|
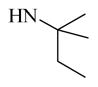
2,060 ± 163 | |
| 33 |
|
2,430 ± 114 | |
| 34 |
|
3,180 ± 131 | |
| 35 |
|
70.8 ± 1.0 | |
| 36 |
36 to 44 |
|
2,320 ± 95 |
| 37 |
|
680 ± 31 | |
| 38 |
|
1,030 ± 38 | |
| 39 |
|
14,420 ± 742 | |
| 40 |
|
4,930 ± 146 | |
| 41 |
|
12,970 ± 183 | |
| 42 |
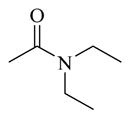
|
13,990 ± 318 | |
| 43 |
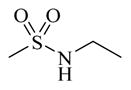
|
19,090 ± 1,397 | |
| 44 |
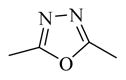
|
18,790 ± 1,585 | |
2.2.2. Syntheses of the 23 novel CINPA1 analogs in the first-round chemistry effort
2.2.2.1 Syntheses of starting materials of 11, 46, 47, and 48 for the novel CINPA1 analogs
In preparing novel CINPA1 analogs for the 2 runs of chemistry efforts, compounds 46, 47, 48, and 11 (CINPA1) —starting materials for various CINPA1 (11) analogs— were first synthesized according to published methods [30–32]. The procedures in preparing these 4 chemicals are summarized in Scheme 1. Briefly, compound 46 [30, 31] was subjected to subsequent reactions of ethoxycarbonylation to compound 47, chloroacetylation to compound 48, and amination to compound 11 [32].
Scheme 1. Syntheses of intermediate chemicals 46, 47, 48, and 11.

Reagents and conditions: (a) ethyl chloroformate, EtOH, 5°C to 7°C; (b): Na2CO3/H2O, 2 h; (c): 2-chloroacetyl chloride, anhydrous toluene, reflux, 1 h; (d): Et2NH, anhydrous toluene, reflux, 4 h.
In the first-run chemistry effort to prepare chemicals 22 to 44, chemical 48 was used as the common starting material to prepare chemicals 22 to 32 (Scheme 2); chemical 47 was used as the common starting material to prepare chemicals 33 to 35 (Scheme 3); and chemical 11 was used as the common starting material to prepare chemicals 36 to 44 (Scheme 4). In the second-run chemistry effort to prepare chemicals 53 to 72, chemical 46 was used as the common starting material to prepare chemicals 53 to 64 (Scheme 5) and 65 to 72 (Scheme 6).
Scheme 2. Syntheses of CINAP1 analog compounds 22 to 32.

Reagents and conditions: (a) R1H, NaH, DMF, microwave, 80°C, 0.5 h; (b): R1H, NaH, DMF, 0°C, 1.5 h; (c): NaI, acetone, N2, 25°C, 12 h; (d): 2,2,6,6-tetramethylpiperidine, toluene, 120°C, 16 h; (e): NH3.H2O, MeOH, 120°C, 12 h; (f): furan-2,5-dione, AcOH, 100°C, 12 h.
*This intermediate chemical was prepared (purified or unpurified) and used as raw starting material for the next step without structural characterization.
Scheme 3. Syntheses of CINPA1 analog compounds 33 to 35.

Reagents and conditions: (a) R1CH2COOH, T3P, reflux, 12 h; (b): 2-tert-butoxyacetic acid, T3P, 55°C, 12 h.
Scheme 4. Syntheses of CINPA1 analog compounds 36 to 44.

Reagents and conditions: (a) H2SO4, AcOH, 120°C, 4 h; (b): R2Cl, Et3N, dichloromethane, 10°C to 35°C, 16 h; (c): Boc2O, Et3N, dichloromethane, RT, 24 h; (d): CDI, Et3N, dichloromethane, RT, 2 h; (e): corresponding primary or secondary amine, DMAP, Et3N, dichloromethane, RT, 12 h; (f): EtNHSO2Cl, dichloromethane, Et3N, 15°C to 25°C, 1 h; (g): 1-(2-oxopyridine-1-carbothioyl)pyridin-2-one, dichloromethane, 20°C, 16 h; (h): acetohydrazide, THF, 20°C, 16 h; (i): EDC, Et3N, DMF, 20°C, 16 h.
#Purified or unpurified intermediate with partial structural characterization (1H NMR, MS, or both).
*This intermediate chemical was prepared (purified or unpurified) and used as raw starting material for the next step without structural characterization.
Scheme 5. Syntheses of CINPA1 analogs: chemicals 53 to 64.

Reagents and conditions: (a) isopropyl chloroformate, EtOH, 0°C, 20 min; (b) Na2CO3, H2O, 15°C to 25°C, 1 h; (c) 3-chloropropionyl chloride, toluene, 100°C, 3 h; (d) diethyl amine, toluene, 100°C, 10 h; (e): 2-chloroacetyl chloride, toluene, 100°C, 3 h; (f) R1H, DMF, NaH, 25°C, 3 h; (g) NaI, acetone, 25°C, 12 h; (h) R1H, NaHMDS, N2, THF, 0°C to 25°C, 1.5 h.
#Purified or unpurified intermediate, with partial structural characterization (1H NMR, MS, or both).
*An intermediate chemical prepared, purified or unpurified, and used as raw starting material for the next step without structural characterization.
Scheme 6. Syntheses of CINPA1 analogs: chemicals 65 to 72.

Reagents and conditions: (a) benzyl chloroformate, EtOH, 0°C, 1 h; (b) Na2CO3, H2O, 15°C, 1 h; (c) 2-chloroacetyl chloride, toluene, 110°C, 3 h; (d) R1H, toluene, 110°C, 6 h; (e) 10% Pd/C, MeOH, H2 (15 psi), 25°C, 12 h; (f) (Boc)2O, MeOH, 25°C, 3 h; (g) 2-propanol, Na, 90°C → 60°C, 30 min; (h) Boc2O, Pd/C, MeOH, H2 (15 psi), 25°C, 2 h; (i) pyrrole, DMF, NaH, N2, 0°C to 25°C, 12 h; (j) Pd/C, H2 (15 psi), EtOAc, 25°C, 4 h; (k) alkyl chloroformate, THF, Et3N, 0°C to 25°C, 2 h.
#Purified or unpurified intermediate, with partial structural characterization (1H NMR, MS, or both).
*An intermediate chemical prepared, purified or unpurified, and used as raw starting material for the next step without structural characterization.
2.2.2.2 Syntheses of the 23 CINPA1 analogs in the round 1 chemistry effort from chemicals 11, 47, and 48
Chemicals 22 to 32 (Scheme 2) were prepared by using chemical 48 as the common starting material. In the presence of sodium hydride, alkylations of corresponding amines with the chloride chemical 48 at microwave-heated 80°C [33] or 0°C [34] yielded chemicals 22, 23, 27, 31, and 32 or 24, 25, 29, and 30, respectively. To prepare chemical 26, the chloride chemical 48 was first converted to the more reactive species of iodide chemical 49 with sodium iodide [35] and then 2,2,6,6-tetramethylpiperidine was alkylated with the iodide chemical 49 to chemical 26 at 120°C in toluene [36]. To prepare chemical 28, the chloride chemical 48 was first converted to the amino chemical 50 with ammonium hydroxide solution in methanol at 120°C under sealed conditions [37]; the amino chemical 50 was subsequently converted to chemical 28 by reacting with furan-2,5-dione in acetic acid at 100°C [38].
Chemicals 33 to 34 were prepared from chemical 47 (Scheme 3) with corresponding carboxylic acids in the presence of propylphosphonic anhydride at reflux or 55°C reaction conditions [39].
In the syntheses of chemicals 36 to 44 with site 3 modifications (Scheme 4), the chemical 11 was first converted to chemical 51 by acidic hydrolysis to remove the ethoxy carbonyl group [40]. Chemical 51 was further converted to chemicals 36 and 37 by reacting with acyl chlorides of butyryl chloride and isopropyl chloroformate, respectively, in methylene chloride in the presence of triethylamine at 10°C to 35°C [41] and then converted to chemical 38 by reacting with di-tert-butyl dicarbonate under similar conditions [42]. To prepare chemicals 39 to 42, chemical 51 was first activated into the form of imidazole carboxamide 52 with carbonyldiimidazole [43] in methylene chloride in the presence of triethylamine, and the chemical 52 was then converted to chemicals 39 to 42 by reacting with corresponding amines in methylene chloride in the presence of triethylamine and 4-dimethylaminopyridine [44]. To prepare chemical 43, the chemical 51 was reacted with freshly prepared N-ethyl-sulfamoyl chloride in methylene chloride in the presence of triethylamine at 15°C to 25°C [45]. To prepare chemical 44, the chemical 51 was subsequently reacted with reagents of 1-(2-oxopyridine-1-carbothioyl)pyridin-2-one, acetohydrazide, and EDC [46].
2.2.3. CAR inverse agonistic activities of the 23 CINPA1 analogs in the first-round chemistry effort
After we synthesized the 23 CINPA1 analogs, we determined their CAR inverse agonistic activities by using the biochemical CAR-mediated fluorescent PGC1α coactivator recruitment/repression assay. The chemical structures and CAR inverse agonistic activities of the 23 analogs are summarized in Table 2 along with those of the lead chemical 11 (CINPA1).
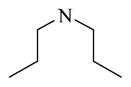
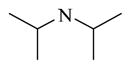
Among the 23 analogs prepared in the first-round chemistry effort, chemicals 22 to 32 have modifications only at the site 1 position of the lead chemical CINPA1 (11)’s structure. Chemical 22 (IC50 of 210 nM) has a dipropylamino group at the site 1 position of CINPA1. The introduction of this dipropylamino group increased the CAR inverse agonistic activity of chemical 22 to 3-fold more than that of lead chemical 11, which has a diethylamino group at this position. However, a branching, disubstituted amino group is not favorable for CAR inverse agonistic activity, as chemical 23 (IC50 of 3,050 nM), which has a diisopropyl amino group at site 1, is much weaker, with an IC50 value more than 4-fold higher than that of lead chemical 11.
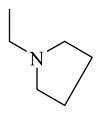
The preliminary SAR data from the 10 commercially available analogs suggested that reduced side chain flexibility at site 1 might improve the CAR inverse agonistic activity, as seen in chemical 12. To further explore the effect of reducing side chain flexibility, chemicals 24 to 30 were designed to have ring structure features, which could reduce side chain rotation flexibility at site 1. Chemical 24 (IC50 of 1,580 nM) and chemical 25 (IC50 of 620 nM) were designed and prepared by directly constraining the diethylamino group in chemical 11 (IC50 of 687 nM) in the form of a pyrrolidinyl group or piperidinyl group, respectively. Compared to the CAR inverse agonistic potency of lead chemical 11, that of chemical 24 decreased (IC50 increased by more than 2-fold) whereas that of chemical 25 was marginally increased. Therefore, constraint at site 1 may not always increase CAR inverse agonistic activities of the analogs. However, reasonable size at site 1 might be important as the piperidinyl group in chemical 25 is larger than the pyrrolidinyl group in chemical 24, and chemical 25 exhibited higher CAR inverse agonistic potency than did chemical 24.
An additional hydrogen-bond acceptor at the farthest end of the site 1 position may also give analogs improved CAR inverse agonistic activities. For example, chemical 12 (IC50 of 235 nM) has an additional oxygen atom as a hydrogen-bond acceptor at the very end of site 1 within its morpholino group, whereas chemical 25 (IC50 of 620 nM) has a similar 6-member ring structure to chemical 12 (IC50 of 235 nM) at site 1 but without a hydrogen-bond acceptor built in, and it is less active as a CAR inverse agonist. Chemical 26 (IC50 of 4,080 nM) has a 2,2,6,6-tetramethylpiperidinyl group at site 1: this group is branched at positions next to the nitrogen and is bulkier than the similar 6-member ring feature at this site in chemical 25 (IC50 of 620 nM). This difference in site 1 rendered chemical 26 significantly less active than chemical 25 (The IC50 of chemical 26 is over 6-fold more than that of chemical 25).
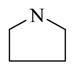
Chemical 27 (IC50 of 1,410 nM) has a (1s,4s)-7-azabicyclo[2.2.1]heptanyl group at the site 1 position: this group branches at positions next to the nitrogen, with constraint in the form of an additional 5-member ring, and is slightly bulkier than the single 5-member ring in chemical 24 (IC50 of 1,580 nM). Chemical 27 is slightly more active than chemical 24, suggesting that chemical groups at site 1 with appropriate bulkiness are desirable for improving the CAR inverse agonistic activities of analogs. Chemical 28 (IC50 of 39,810 nM) has a 2,5-dioxo-2,5-dihydro-1H-pyrrolyl substitute at site 1, making it branched at α-positions of the nitrogen atom but with a hydrogen-bond acceptor property. This structural feature rendered chemical 28 less active than chemical 24 as a CAR inverse agonist (the IC50 of chemical 28 is 25-fold lower than that of chemical 24) even though a similar 5-member ring structure was maintained at site 1 in both chemical 24 and 28, which may indicate hydrogen-bond acceptor property at α-positions of the nitrogen atom is not desirable for CAR inverse agonistic activities. Chemicals 29 (IC50 of 37 nM) and 30 (IC50 of 6,890 nM) have a similar 5-member aromatic ring structure feature at site 1 of the lead compound CINPA1, with a pyrrolyl group in chemical 29 and an imidazolyl group in chemical 30. Chemical 29 is the aromatic version of chemical 24 at the site 1 position but is 43- and 19-fold more potent than chemicals 24 and 11 (CINPA1), respectively. The aromatic feature at site 1 has the most significant positive impact for analogs as a CAR inverse agonist. However, when an additional heteroatom in the form of nitrogen was added to the 5-member aromatic ring in chemical 29 to make chemical 30 (which now has an imidazolyl group), a dramatic loss of CAR inverse agonistic activity was observed (the IC50 of chemical 30 is 186-fold less than that of chemical 29). This observation further confirms that protonable groups at the site 1 position are indeed undesirable for CAR inverse agonistic activity because an imidazolyl group can be easily protonated under assay conditions (pH 7.5).
Chemical 31 (IC50 of 24,490 nM) has a mono-substituted amino group (ethylamino group) at site 1, with a proton attached to the nitrogen that can function as a hydrogen-bond donor. The extremely weak activity of chemical 31 further confirms that a hydrogen-bond donor feature at site 1 is undesirable (chemical 31 has an IC50 that is over 35-fold less than that of the lead chemical 11) even though a smaller size at site 1 in chemical 31 may also partially contribute to the activity loss. However, chemical 32 (IC50 of 2,060 nM), which has a similar mono-substituted amino group (tert-pentylamino group) and reasonable bulkiness, only slightly lost its CAR inverse agonistic activity (IC50 is 3-fold less than that of the lead chemical 11). In this example, having a hydrogen-bond donor feature at site 1 is still undesirable, but the suitable group size maintained at site 1 might have compensated for the negative effect of the hydrogen-bond donor and helped retain reasonable CAR inverse agonistic activity for chemical 32.
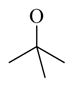
Chemical 33 (IC50 of 2,430 nM) has an ethylene carbonyl linker at site 2 to replace the methylene carbonyl linker at the same site in lead chemical 11 (IC50 of 687 nM). This modification decreased the activity of chemical 33 (a 3.5-fold increase in the IC50 value over that of lead chemical 11). Chemical 34 (IC50 of 3,180 nM) has a further modification at site 1, with a constrained pyrrolidinyl group to replace the diethylamino group in lead chemical 11 in addition to the ethylene carbonyl linker modification at site 2 as in chemical 33. This constrained modification without enough bulkiness at site 1 caused a slight decrease in CAR inverse agonistic activity (IC50 of 2,430 nM for chemical 33 vs. IC50 of 3,180 nM for chemical 34), which is consistent with the observed difference between chemical 11 (IC50 of 687 nM) and chemical 24 (IC50 of 1,580 nM). Chemical 35 (IC50 of 70.8 nM) has a tert-butoxy group at the site 1 position of lead chemical 11 (IC50 of 687 nM). This modification in chemical 35 increased its CAR inverse agonistic activity by 9.7-fold over that of lead chemical 11, demonstrating that a non-protonable group with reasonable size at site 1 could improve CAR inverse agonistic activity. However the tert-butoxy group is not protonable under assay conditions (pH 7.5) but has reasonable size, and the diethylamino group in lead chemical 11 is protonable under similar conditions. The success of chemical 35 as a substantially more potent CAR inverse agonist than lead chemical 11 led us to focus on site 1 modifications with the tert-butoxy and diethylamino functional groups, as reflected in 9 of 20 novel chemicals in the second-round chemistry effort (discussed in section 2.3).
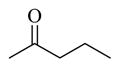
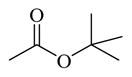
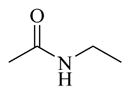
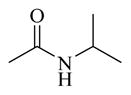
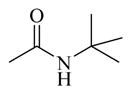
Chemicals 36 to 44 have modifications on site 3 of lead chemical 11, with structural features of amide in chemical 36, carbamates in chemicals 37 and 38, ureas in chemicals 39 to 42, sulfamide in chemical 43, and carbamate heterocyclic isostere in chemical 44. Chemical 36 (IC50 of 2,320 nM) has a butyramide at site 3 instead of the ethyl carbamate of lead chemical 11 (IC50 of 687 nM) at the same position. Butyramide and ethyl carbamate are similar in size and shape; the only difference between them is that the ethoxy oxygen (non-carbonyl oxygen) in lead chemical 11 was replaced by a methylene group in chemical 44. The change of the ethoxy oxygen in chemical 11 to a methylene group in chemical 44 decreased the CAR inverse agonistic activity (IC50 increased by about 3.4-fold). Therefore, it appears that the ethoxy oxygen in chemical 11 is beneficial for its CAR inverse agonistic activity, possibly because oxygen may serve as a hydrogen bond acceptor. Chemicals 37 and 38 have an isopropyl carbamate and a tert-butyl carbamate, respectively, at site 3 of lead chemical 11 (IC50 of 687 nM), which has an ethyl carbamate at this site. The isopropyl carbamate modification maintains the CAR inverse agonistic activity for chemical 37 (IC50 of 680 nM), which is consistent with the observed behavior of chemical 20.
Therefore, an isopropyl carbamate at site 3 was further confirmed as being a more favorable structural feature than ethyl carbamate at the same site. An isopropyl group is slightly larger than an ethyl group, suggesting that a slightly larger chemical group at site 3 may be favored for CAR inverse agonistic activity. In addition, isopropyl carbamate should provide analogs with higher enzymatic and metabolic stability than ethyl carbamate does. Further increase in size at site 3 to a tert-butyl carbamate group in chemical 38 actually slightly decreased its activity (IC50 of 1,030 nM) from that of lead chemical 11 (IC50 of 687 nM). Compared to lead chemical 11 (IC50 of 687 nM), urea, sulfamide, and carbamate heterocyclic isostere modifications at site 3 yielded chemicals 39 (IC50 of 14,420 nM), 40 (IC50 of 4,930 nM), 41 (IC50 of 12,970 nM), 42 (IC50 of 13,990 nM), 43 (IC50 of 19,090 nM), and 44 (IC50 of 18,790 nM), all with dramatically reduced CAR inverse agonistic activity. Therefore, these modifications at site 3 are unfavorable for CAR inverse agonistic activity, confirming that an alkoxy oxygen is optimal at site 3 to maintain the CAR inverse agonistic activity.
In addition, ureas in chemicals 39, 40, and 41 and sulfamide in chemical 43 both have a proton attached to the nitrogen atom, which could serve as a hydrogen-bond donor. The observation that a hydrogen-bond donor at site 3 may be unfavorable for CAR inverse agonistic activity is again seen in the chemical pair of 39 (IC50 of 14,420 nM, with proton on nitrogen) and 42 (IC50 of 13,990 nM, without a proton on nitrogen) in which chemical 42 is marginally more active than chemical 39. However, a diethyl substitution in chemical 42 instead of a mono-ethyl substitution in chemical 39 might also contribute to the marginal difference. Chemical 44, which has the non-carbonyl oxygen but the carbonyl oxygen is replaced with nitrogen, had dramatically lower activity (IC50 of 18,790 nM) than did lead chemical 11 (IC50 of 687 nM), indicating that a carbamate structure with both carbonyl oxygen and non-carbonyl oxygen (alkoxy oxygen) is required for optimal CAR inverse agonistic activity.
2.2.4. Structure-activity relationship summary from CINPA1, 10 commercial analogs and the 23 novel CINPA1 analogs prepared from the first-round chemistry effort
Based on the overall CAR inverse agonistic activities from the 33 CINPA1 analogs, a brief SAR report of analogs with CAR inverse agonistic activities at least comparable to CINPA1 was summarized (structural features shown in Figures 2 and 3)
Figure 3.
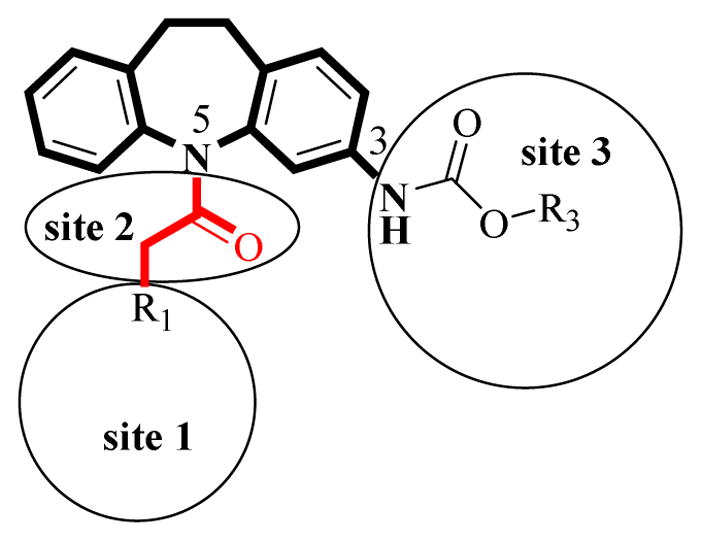
Structural features of CINPA1 analogs as potent CAR inverse agonists
Scaffold of 3-amino-10,11-dihydro-5H-dibenz[b,f]azepine (saturated 3-amino-dibezapine).
Site 1: An unbranched disubstituted amino group is favorable; protonability is very undesirable; a suitably bulky group is desirable; an oxygen-contained group in the form of an alkoxy group may be better than a nitrogen-containing group in the form of an aliphatic tertiary amino group because an aliphatic amino group is susceptible to protonation under assay conditions (pH 7.5), which may be unfavorable for CAR inverse agonistic activity; a mono-nitrogen–containing aromatic ring is favorable because it is not as susceptible to protonation under assay conditions (pH 7.5) as an aliphatic tertiary amino group is.
Site 2: A methylene carbonyl linker is better than an ethylene carbonyl linker.
Site 3: A carbamate structure is favorable; a hydrogen-bond donor is unfavorable; medium-sized substitutes, such as isopropyl carbamate, are favorable.
2.3. Design, syntheses, and activity evaluation of 20 CINPA1 analogs in the second-round chemistry effort
2.3.1. Design of 20 novel CINPA1 analogs in the second-round chemistry effort
The SAR obtained from CINPA1, the 23 CINPA1 analogs of the first-round of chemistry, and the 10 analogs from commercial sources demonstrated that isopropyl carbamate at site 3 is desirable for CAR inverse agonistic activity. We, therefore, designed 12 novel analogs with isopropyl carbamate at site 3. In addition, 4 novel analogs with tert-butyl carbamate at site 3 were designed for comparison. Among these 16 novel analogs, 9 were given various alkoxy groups at site 1 because SAR from the 23 analogs in round 1 suggested that an alkoxy group at this site could improve CAR inverse agonistic activity. The other 7 analogs were given several site 1 and site 2 groups that are similar to those explored in the first-round chemistry effort in combination with either isopropyl carbamate or tert-butyl carbamate at site 3 to gain additional insight about the functional preference at site 3. To identify optimal groups at site 3, we designed 4 additional chemicals having novel carbamates at site 3 and the optimal pyrrolyl group at site 1. In total, 20 novel CINPA1 analogs were designed in the second-round chemistry effort.
2.3.2. Syntheses of the 20 novel CINPA1 analogs in the second-round chemistry effort
Chemical 46 was the common starting material to prepare all the analogs in the second-round chemistry effort. The syntheses of chemicals 53 to 64 are summarized in Scheme 5. To prepare chemical 53 [32], chemical 46 was first converted to chemical 73 by selectively acylating the primary 3-amino group at the saturated 3-amino-dibezapine ring in chemical 46 to form the isopropyl carbamate in chemical 73 with isopropyl chloroformate because this 3-primary amino group is more electronically and sterically reactive than is the secondary amino group (the 5-position nitrogen) on the saturated dibezapine ring. Chemical 73 was then subjected to further acylation with 3-chloropropionyl chloride in toluene at 100°C to yield the chloride chemical 74, which was then converted to chemical 53 by alkylating diethyl amine in toluene at 100°C. Chemical 73 was also converted to the chloride chemical 75 with 2-chloroacetyl chloride in toluene at 100°C [33]. Amines of dipropylamine, piperidine, pyrrole were alkylated with the chloride chemical 75 in dimethylformamide (DMF) in the presence of sodium hydride at 25°C to form the corresponding products of chemicals 54, 55, and 56 [34]. In addition, the chloride chemical 75 was converted to a more reactive alkylation chemical—the iodide chemical 76—with sodium iodide in acetone at room temperature [35]. In the presence of sodium bis(trimethylsilyl)amide (NaHMDS) and under nitrogen atmosphere, respective alcohols were then alkylated with the iodide chemical 76 in tetrahydrofuran (THF) at 0°C to 25°C to yield corresponding chemicals 57 to 64 [47].
In the preparation of chemicals 65 to 72 (Scheme 6), chemical 46 was first converted to chemical 77 with benzyl chloroformate in ethanol at 0°C; chemical 77 was then further acylated into the chloride chemical 78 with 2-chloroacetyl chloride in toluene at 110°C. The chloride chemical 78 was further converted to chemicals 65 – 72 by using different synthetic approaches. To prepare chemicals 65 and 66, corresponding dipropylamine and piperidine groups were first alkylated with the chloride chemical 78 to form the intermediate chemicals 79 and 80 [32], which were then subjected to de-carboxybenzyl (de-Cbz) [48] with 10% Pd-C catalyzed hydrogenolysis in methanol at room temperature and acylation with di-tert-butyl dicarbonate [42] in methanol at room temperature to give respective target chemicals. To prepare chemical 67, 2-propanol was first deprotonated with sodium at temperatures from 90°C to 60°C to form sodium isopropoxide, which was alkylated with the chloride chemical 78 to form the intermediate chemical 81 [49]. The intermediate chemical 81 was then converted to chemical 67 [42] with di-tert-butyl dicarbonate in methanol at room temperature under 10% Pd-C-mediated catalytic hydrogenolysis conditions. During the preparation of chemicals 68 to 72, pyrrole was de-protonated with sodium hydride in dimethylformamide at 0°C to 25°C under nitrogen atmosphere and then alkylated with the chloride chemical 78 to yield chemical 82 [50], which was then subjected to 10% Pd-C-mediated catalytic hydrogenolysis in ethyl acetate at room temperature to form chemical 83 [48], with the 3-amino group at the saturated dibezapine being available for further carbamation. Chemicals 68 to 72 were then obtained by carbamation of the 3-amino group in chemical 83 with the corresponding alkyl chloroformates in tetrahydrofuran (THF) in the presence of triethylamine at temperatures of 0°C to 25°C [51].
2.3.3. CAR inverse agonistic activities of the 20 novel CINPA1 analogs in the second-round chemistry effort
We synthesized and evaluated the CAR inverse agonistic activities of these 20 novel CINPA1 analogs using the biochemical CAR-mediated fluorescent PGC1α coactivator recruitment/repression assay. Their structures and CAR inverse agonistic activities are summarized in Table 3 along with those of lead chemical 11 (CINPA1).
Table 3.
CAR inverse agonistic activities of CINPA1 and its novel 20 analogs from the second-round chemistry effort
|
| |||
|---|---|---|---|
| Compound | R1 | R2 | IC50 (nM) |
| 11 (CINPA1) |
|
|
687 ± 71 |
| 53 |
|
6,400 ± 124 | |
| 54 |
|
53 to 64 |
116.4 ± 8.6 |
| 55 |
|
249.0 ± 16.6 | |
| 56 |
|
20.9 ± 2.6 | |
| 57 |
|
11,350 ± 670 | |
| 58 |
|
60.1 ± 3.4 | |
| 59 |
|
35.3 ± 4.8 | |
| 60 |
|
21.2 ± 1.9 | |
| 61 |
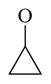
|
81.5 ± 7.4 | |
| 62 |
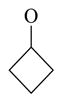
|
37.6 ± 2.8 | |
| 63 |
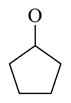
|
19.5 ± 1.5 | |
| 64 |
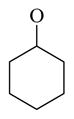
|
24.1 ± 1.4 | |
| 65 |
|
65 to 68 |
152.9 ± 11.1 |
| 66 |
|
216.2 ± 15.6 | |
| 67 |
|
116.0 ± 7.3 | |
| 68 |
|
31.9 ± 2.2 | |
| 69 |
69 to 72 |
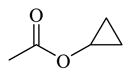
|
22.1 ± 1.3 |
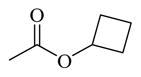
| 70 |
|
20.1 ± 2.8 | |
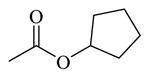
| 71 |
|
13.9 ± 0.9 | |
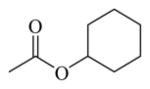
| 72 |
|
11.7 ± 0.8 | |
Among the 20 novel CINPA1 analogs from the round 2 chemistry, chemical 53 is the only one with an ethylene carbonyl group modification and an isopropyl carbamate at site 3. Chemical 53 (IC50 of 6,400 nM) is substantially less active than is lead chemical 11 (IC50 of 687 nM) even though the isopropyl carbamate group at site 3 is expected to increase the CAR inverse agonistic activity as observed in other analogs with the isopropyl carbamate structural feature at site 3. The ethylene carbonyl linker at site 2 is again proven to be a less desirable group than the methylene carbonyl linker group.
Chemicals 22 (IC50 of 210 nM), 54 (IC50 of 116.4 nM), and 65 (IC50 of 152.9 nM) are only different at site 3, with a corresponding ethyl, isopropyl, or tert-butyl carbamate group. Among this group of 3 chemicals, chemical 54, which has an isopropyl carbamate at site 3, has the highest CAR inverse agonistic activity. This slight positive effect of a site 3 isopropyl carbamate on the CAR inverse agonistic activity was also observed among chemicals 29 (IC50 of 37.0 nM), 56 (IC50 of 20.9 nM), and 68 (IC50 of 31.9 nM), with the only difference being their respective ethyl, isopropyl, or tert-butyl carbamate groups at site 3. In another group of chemicals —25 (IC50 of 620 nM), 55 (IC50 of 249.0 nM), and 66 (IC50 of 216.2 nM) — that had a corresponding ethyl, isopropyl, or tert-butyl carbamate group at site 3, 66 (with a tert-butyl carbamate) was only slightly more active than 55 (with an isopropyl carbamate), but both were substantially more active than 25, which has an ethyl carbamate. Overall, an isopropyl carbamate is similar to a tert-butyl carbamate, but both are better than an ethyl carbamate at site 3 for the CAR inverse agonistic activity. Having an IC50 of 20.9 nM, chemical 56 was the most active CAR inverse agonist and is 32.8-fold more potent than lead chemical 11 (IC50 687 nM).
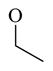
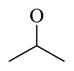
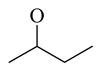
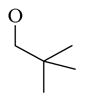
Chemicals 57 to 64 and 67 have alkoxy groups at site 1 and an isopropyl or tert-butyl carbamate at site 3. These structural features improve the CAR inverse agonistic activities of the analogs compared to that of lead chemical 11, with the exception of chemical 57 (IC50 value 11,350 nM), which has an ethoxy group at site 1 that might be too small to maintain a high CAR inverse agonistic activity. Chemicals 58 (IC50 60.1 nM) and 67 (IC50 116.0 nM) both have an isopropoxy group at site 1 and an isopropyl or a tert-butyl carbamate, respectively, at site 3. Both 58 and 67 are substantially more active than lead chemical 11 (IC50 of 687 nM). Here, the isopropyl carbamate at site 3 in chemical 58 again endowed the analog with higher CAR inverse agonistic activity than did the tert-butyl carbamate at the same site in chemical 67. Increasing the size of the alkoxy group at site 1 from an ethoxy group in chemical 57 (IC50 of 11,350 nM) and an isopropoxy group in chemical 58 (IC50 of 60.1 nM) to a sec-butoxy group in chemical 59 (IC50 of 35.3 nM) and a neopentyloxy group in chemical 60 (IC50 of 21.2 nM) increases CAR inverse agonistic activity. Similar observations were made in the cyclic alkoxy group series at site 1: from chemical 61 (IC50 of 81.5 nM) with a cyclopropoxy group to chemical 62 (IC50 of 37.5 nM) with a cyclobutoxy group, and to chemical 63 (IC50 of 19.5 nM) with a cyclopentyloxy group. However, a slight activity decrease was observed when the size of site 1 was further increased by incorporating a cyclohexyloxy group, as in chemical 64 (IC50 of 24.1 nM). The highest CAR inverse agonistic activities were observed when a branched 5-carbon alkoxy group was at site 1, as in chemicals 60 and 63.
Chemicals 69 to 72 were designed to further explore the effect of cyclic carbamates at site 3 on CAR inverse agonistic activities. Chemicals 69 (IC50 of 22.1 nM), 70 (IC50 of 20.1 nM), 71 (IC50 of 13.9 nM), and 72 (IC50 of 11.7 nM), which respectively have cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl carbamates at site 3 and the optimal pyrrolyl functional group at site 1, had substantially higher CAR inverse agonistic activities than did lead chemical 11 (IC50 of 687 nM). These results demonstrate the significant contribution of cyclohexyl carbamates at site 3 to high CAR inverse agonistic activity. Chemical 72 is the most active CAR inverse agonist among chemicals 69 to 72: having an IC50 value of 11.7 nM, it is approximately 59-fold more potent than the lead compound 11 (CINPA1) and is the most active analog among all 54 analogs evaluated in this study.
2.3.4. Structure-activity relationship summary of all 54 CINPA1 analogs evaluated as CAR inverse agonists
Based on the overall CAR inverse agonistic activities of the 54 CINPA1 analogs, this brief summary of the SAR of modifications that affect CAR inverse agonistic activities (structural features in Figure 3) is provided:
Scaffold: 3-amino-10,11-dihydro-5H-dibenz[b,f]azepine (saturated 3-amino-dibezapine).
Site 1: medium size alkoxy groups or tertiary amino groups in aromatic rings with no additional substitute at α-position to the nitrogen; hydrogen-bond donor is highly unfavorable; protonability is very undesirable at this site.
Site 2: a methylene carbonyl linker is better than an ethylene carbonyl linker.
Site 3: a carbamate is better than an amide, urea, sulfamide, or carbamate heterocyclic isostere structure; a medium-sized aliphatic cyclic carbamate structure is favorable; a hydrogen-bond donor is unfavorable.
2.3.5. Representative molecular modeling study of compound 11 (CINPA1) and compound 72 (the most potent analog) to hCAR-LBD
In order to predict the binding mode of CINPA1 (11) and the most potent analog (72) to the hCAR-LBD, we conducted docking studies using AutoDock Vina, which showed the ligands positioned in the binding pocket (Figure 4). The top ligand-poses were selected based on the superposition of the scaffold for both ligands and comparisons with the SAR data. In this model, the ethyl carbamate (position 3) of compound 11 (Figure 4A) and the cyclohexyl carbamate of compound 72 (Figure 4B) have the potential to form hydrogen bonding with Asn-165 or His-203. Because hydrogen bonds impart strong ligand-protein interactions, it is plausible that the postulated hydrogen bonding accounts for the necessity of the ethoxy and carbonyl oxygens.
Figure 4.

Docking of CINPA1 (11) and compound 72 to hCAR-LBD depicting interactions of site 1 of compound 11 (A), site 1 of compound 72 (B), site 3 of compound 11 (C) and site 3 of compound 72 (D). The ligands are illustrated as sticks surrounded by protein residues (surface representation). Carbon, oxygen, nitrogen and sulfur atoms are shown in grey, red, blue and yellow, respectively. The carbon atoms of Asn-165 and His-203 are represented in pink, while carbon atoms of Phe-217 are shown in green. Potential hydrogen bonding is indicated with dash lines.
In relation to site 1, the models agree with the SAR data with regard to the need of hydrophobic groups without moieties containing hydrogen bond donors or are incapable of protonation, because site 1 occupies a highly hydrophobic pocket in the protein ligand cavity. Aromatic groups such as the 5-member ring in compound 72 would potentially benefit of potential aromatic stacking with residues such as Phe-217 (Figures 4A and 4B).
Site 3 also occupies a very hydrophobic pocket with enough space to accommodate the cyclohexyl ring of compound 72 (Figure 4D), where groups containing hydrogen bond donors would negatively affect binding as observed in the SAR studies. In the same way as with site 1, larger groups at site 3 would provide stronger hydrophobic contacts with the protein, presenting an explanation for the stronger potency of compound 72 compared to compound 11 (Figures 4C).
From the molecular modeling study, several important features were identified in terms of compounds 11 (CINPA1) and 72 interacting with the hCAR-LBD. The ligands reside at the binding pocket of hCAR-LBD, where site 1 and site 3 of the ligands occupy a highly hydrophobic environment, with potential for hydrogen bonding with Asn-165 or His-203. The larger moieties at sites 1 and 3 of compound 72 would correlate with its higher potency in comparison to compound 11 because of the stronger hydrophobic interactions with the protein. The models provide a molecular explanation of the SAR observed experimentally.
3. Conclusions
In this study, the following 54 CINPA1 analogs were obtained and evaluated for their CAR inverse agonistic activities in a SAR-guided stepwise approach: the lead compound and 10 commercial analogs; 23 novel analogs in the first-round chemistry effort; and 20 novel analogs in the second-round chemistry effort. Many analogs having improved CAR inverse agonistic activities were discovered. Among them, chemical 72 was the most active analog, with an IC50 of 11.7 nM, which is approximately 59-fold more potent than the lead compound CINPA1 (an IC50 value of 687 nM) and over 10-fold more potent than the most potent inverse agonist previously reported by others, clotrimazole (an IC50 value of 126.9 nM in our assay). This set of 54 chemicals is a novel tool for investigating the function of CAR. The SAR data gained from these 54 analogs provides a basis to guide future efforts to develop additional CAR inverse agonists to study the function of CAR. To our knowledge, our effort is the first chemistry endeavor in developing highly active CAR inverse agonists based on a novel chemical scaffold discovered in a compound-screening campaign.
4. Experimental section
4.1. Chemistry
General procedures: Organic reagents were purchased from commercial suppliers unless otherwise noted and were used without further purification. All solvents were analytical- or reagent-grade. All reactions were carried out in flame-dried glassware under argon or nitrogen. Flash column chromatography was performed by using Sigma-Aldrich silica gel 60 (200–400 mesh) and carried out under moderate pressure, with columns of an appropriate size packed and eluted with appropriate eluents. All reactions were monitored by performing thin-layer chromatography (TLC) on precoated plates (silica gel HLF). TLC spots were visualized either by exposure to iodine vapor or by irradiation with UV light. Organic solvents were removed under vacuum by a rotary evaporator. The reactions, purities, or identities of final compounds were monitored or determined by performing TLC or using a Waters Acquity UPLC MS system with a C18 column in a 2-min gradient (H2O + 0.1% formic acid → Acetonitrile + 0.1% formic acid) and detectors of PDA (215 – 400 nm), ELSD, and Acquity SQD ESI Positive MS. Preparative TLC separation was performed by using self-casted preparative TLC plates with Sigma-Aldrich silica gel 60 (200–400 mesh) on 20-cm × 20-cm glass plates. The purifications of reaction products were performed by using a Dionex APS 3000 dual purification/analytical LC/PDA/MS system with a C18 column in a 15-min gradient (H2O with 0.05% NH3•H2O → Acetonitrile) and ESI Positive MS. High-resolution mass spectra were determined by using a Waters Acquity UPLC system with a C18 column (H2O + 0.1% formic acid → acetonitrile + 0.1% formic acid gradient over 2.5 min) under Xevo G2Q-TOF ESI in positive, resolution mode. Compounds were internally normalized to leucine-enkephalin lock solution, with a calculated error of < 3 ppm. All 1H NMR spectra were recorded on a Bruker ULTRASHIELD 400 plus NMR spectrometer and all 13C NMR spectra were recorded on a Bruker Ascend 126 MHz Fourier transform (FT) NMR spectrometer at room temperature. The chemical shift values are expressed in parts per million (ppm) relative to tetramethylsilane as the internal standard. Coupling constants (J) are reported in hertz (Hz).
4.1.1
Compounds 46 [30, 31], 47 [32], 48 [32] and 11 [32] were prepared by following published methods.
4.1.2. Ethyl (5-(dipropylglycyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (22)
Dipropylamine (68 mg, 670 μmol) in DMF (2 mL) was added to compound 48 (200 mg, 550 μmol) in DMF (1 mL) in a microwavable reaction vessel. The vessel was sealed and heated to 80°C in a microwave for 0.5 hour, and LC-MS showed that starting material 48 was depleted. The mixture was extracted with ethyl acetate (20 mL × 2) and washed with brine (10 mL × 2). The organic layer was dried with anhydrous Na2SO4 and concentrated to give a residue. The residue was purified by preparative HPLC to give compound 22 (115.9 mg, 274 μmol, 49.8% yield, 99.4% purity). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.33 (br. s., 1H), 8.15 (s, 1H), 7.48 (s, 1H), 7.18–7.33 (m, 5H), 7.14 (d, J = 8.28 Hz, 1H), 4.14 (q, J = 7.15 Hz, 2H), 3.22 (br. s, 2H), 2.73–2.80 (m, 2H), 2.46 (t, J = 7.28 Hz, 4H), 1.21 – 1.36 (m, 7H), 0.73–0.85 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 169.63, 163.37, 153.50, 141.25, 141.12, 140.82, 140.71, 137.98, 137.35, 137.11, 135.11, 130.46, 130.27, 130.06, 128.57, 128.34, 127.69, 127.27, 127.08, 126.25, 117.99, 117.42, 60.24, 60.13, 55.56, 54.96, 54.81, 30.06, 29.79, 29.43, 29.19, 20.31, 20.09, 20.01, 14.51, 11.68, 11.62. ESI-TOF HRMS: m/z 424.2604 (C25H33N3O3 + H+ requires 424.2602).
4.1.3. Ethyl (5-(diisopropylglycyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (23)
Diisopropylamine (68 mg, 670 μmol) in DMF (2 mL) was added to compound 48 (200 mg, 550 μmol) in DMF (1 mL) in a microwavable reaction vessel. The reaction vessel was sealed and heated to 80°C in a microwave for 0.5 hour, and LC-MS showed that starting material 48 was depleted. The mixture was extracted with ethyl acetate (20 mL × 2) and washed with brine (10 mL × 2). The organic layer was dried with anhydrous Na2SO4 and concentrated to give a residue. The residue was purified by preparative HPLC to give compound 23 (45.2 mg, 106 μmol, 19.4% yield, 98.7% purity). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.31 (br. s., 1H), 8.14 (s, 1H), 7.46–7.53 (m, 1H), 7.17–7.35 (m, 5H), 7.13 (d, J = 8.41 Hz, 1H), 4.13 (q, J = 7.11 Hz, 2H), 3.21–3.28 (m, 4H), 2.99–3.04 (m, 2H), 2.72–2.80 (m, 2H), 1.25 (t, J = 7.03 Hz, 3H), 0.82–1.03 (m, 12H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 170.86, 163.44, 153.51, 141.16, 130.54, 130.29, 128.58, 128.35, 60.21, 48.99, 48.37, 47.60, 46.73, 29.96, 29.82, 29.31, 20.63, 20.48, 19.68, 17.56, 14.50. ESI-TOF HRMS: m/z 424.2602 (C25H33N3O3 + H+ requires 424.2602).
4.1.4. Ethyl (5-(2-(pyrrolidin-1-yl)acetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (24)
NaH (33.44 mg, 1.39 mmol) was added to a solution of pyrrolidine (50 mg, 703 μmol) in DMF (2 mL) at 0°C. The mixture was stirred at 0°C for 0.5 hour. Compound 48 (250 mg, 697 μmol) in DMF (1 mL) was then added dropwise. The mixture was stirred at 0°C for 1 hour, and LC-MS showed that starting material 48 was depleted. The reaction was quenched with aqueous NH4Cl solution, extracted with ethyl acetate (20 mL × 3), and washed with brine (20 mL × 2). The organic layer was dried with anhydrous Na2SO4 and concentrated to give a residue. The residue was dissolved in MeOH (3 mL) and purified by preparative HPLC to give compound 24 (42.7 mg, 108 μmol, 15.5% yield, 99.2% purity). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.29 (s, 1H), 8.18 (br. s., 1H), 7.49 (d, J = 2.13 Hz, 1H), 7.20–7.34 (m, 5H), 7.14 (d, J = 8.28 Hz, 1H), 4.14 (q, J = 7.03 Hz, 2H), 3.24–3.32 (m, 4H), 2.71–2.81 (m, 2H), 2.46–2.50 (m, 4H), 1.60–1.69 (m, 4H), 1.22–1.31 (m, 3H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 168.70, 163.67, 153.49, 141.20, 140.54, 140.38, 138.03, 137.35, 135.03, 130.79, 130.44, 130.11, 129.97, 128.63, 128.49, 127.60, 127.31, 127.12, 126.26, 118.36, 118.00, 117.48, 60.15, 56.61, 56.46, 53.37, 30.18, 29.67, 29.49, 29.00, 23.35, 14.51. ESI-TOF HRMS: m/z 394.2133 (C23H27N3O3 + H+ requires 394.2132).
4.1.5. Ethyl (5-(2-(piperidin-1-yl)acetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (25)
NaH (33.44 mg, 1.39 mmol) was added to a solution of piperidine (60 mg, 704 μmol) in DMF (2 mL) at 0°C. The mixture was stirred at 0°C for 0.5 hour. Compound 48 (250 mg, 697 μmol) in DMF (1 mL) was then added dropwise. The mixture was stirred at 0°C for 1 hour, and LC-MS showed that starting material 48 was depleted. The reaction was quenched with aqueous NH4Cl solution, extracted with ethyl acetate (20 mL × 3), and washed with brine (20 mL × 2). The organic layer was dried with anhydrous Na2SO4 and concentrated to give a residue. The residue was dissolved in MeOH (3 mL) and purified by preparative HPLC to give compound 25 (85.4 mg, 209 μmol, 30.1% yield, 97.6% purity). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.22 (br. s., 1H), 8.16 (s, 1H), 7.48 (d, J = 2.07 Hz, 1H), 7.17–7.36 (m, 5H), 7.13 (d, J = 8.48 Hz, 1H), 4.14 (q, J = 7.10 Hz, 2H), 3.28 (br. s., 2H), 2.72–2.84 (m, 4H), 2.26–2.33 (m, 4H), 1.31–1.49 (m, 6H), 1.26 (t, J = 7.06 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 169.10, 163.87, 154.00, 153.94, 141.87, 141.18, 141.05, 138.27, 137.85, 135.62, 131.30, 130.83, 130.44, 130.31, 129.12, 129.04, 128.71, 127.81, 127.36, 126.71, 118.64, 118.44, 117.96, 117.65, 60.70, 60.62, 54.26, 30.51, 30.40, 29.86, 29.75, 25.78, 23.88, 14.98. ESI-TOF HRMS: m/z 408.2287 (C24H29N3O3 + H+ requires 408.2289).
4.1.6. Ethyl (5-(2-(2,2,6,6-tetramethylpiperidin-1-yl)acetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (26)
NaI (376 mg, 2.51 mmol) was added to compound 48 (300 mg, 836 μmol) in acetone (5 mL) in one portion at 25°C under N2. The mixture was stirred at 25°C for 10 hours, and TLC showed that the reaction was completed. The mixture was filtered, and the filtrate was concentrated under vacuum to afford compound 49 (325 mg, 721.8 μmol) as a yellow solid. A mixture of compound 49 (150 mg, 333.2 μmol) and 2,2,6,6-tetramethylpiperidine (2.00 g, 14.16 mmol) in toluene (10 mL) was heated to 120°C and stirred for 16 hours. LC-MS showed that starting material 49 was depleted. The mixture was cooled to 25°C and concentrated under reduced pressure at 50°C. The residue was purified by preparative HPLC to afford compound 26 (10.00 mg, 12.94 μmol, 6.48% yield, 95.6% purity) as a yellow solid. 1H NMR (400 MHz, chloroform-d) δ (ppm) 1.12–1.51 (m, 15 H), 1.68 (d, J = 5.77 Hz, 2 H), 2.78–2.93 (m, 3 H), 3.07–3.33 (m, 4 H), 3.56 (d, J = 17.82 Hz, 1 H), 4.19 (ddt, J = 10.54, 7.03, 3.64, 3.64 Hz, 3 H), 5.05 (d, J = 17.82 Hz, 1 H), 7.16–7.32 (m, 5 H), 7.36–7.49 (m, 1 H), 8.00 (d, J = 7.78 Hz, 1 H), 8.17 (br. s., 1 H), 9.27–9.52 (m, 1 H). ESI-TOF HRMS: m/z 464.2918 (C28H37N3O3 + H+ requires 464.2915).
4.1.7. Ethyl (5-(2-((1s,4s)-7-azabicyclo[2.2.1]heptan-7-yl)acetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (27)
7-Azabicyclo[2.2.1]heptane (65 mg, 669 μmol) in DMF (2 mL) was added to compound 48 (200 mg, 550 μmol) in DMF (1 mL) in a microwavable reaction vessel. The vessel was sealed and heated to 80°C in a microwave for 0.5 h, and LC-MS showed that starting material 48 was depleted. The mixture was extracted with ethyl acetate (20 mL × 2) and washed with brine (10 mL × 2). The organic layer was dried with anhydrous Na2SO4 and concentrated to give a residue. The residue was purified by preparative HPLC to give compound 27 (75.7 mg, 180 μmol, 32.8% yield, 98.9% purity). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.32 (br. s., 1H), 8.18 (br. s., 1H), 7.49 (s, 1H), 7.17–7.37 (m, 5H), 7.12 (d, J = 8.28 Hz, 1H), 4.06–4.18 (m, 2H), 3.20–3.29 (m, 2H), 3.02–3.15 (m, 4H), 2.65–2.83 (m, 2H), 1.44–1.62 (m, 4H), 1.13–1.30 (m, 7H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 169.65, 164.07, 153.98, 141.81, 141.18, 141.03, 138.30, 138.00, 137.82, 135.72, 131.48, 130.81, 130.49, 130.36, 129.24, 129.03, 128.82, 127.98, 127.80, 127.43, 126.70, 118.42, 117.97, 60.61, 60.45, 49.58, 30.49, 30.39, 29.83, 28.25, 27.98, 14.98. ESI-TOF HRMS: m/z 420.2291 (C25H29N3O3 + H+ requires 420.2289).
4.1.8. Ethyl (5-(2-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)acetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (28)
NH3.H2O (10 mL) was added to compound 48 (200 mg, 557 μmol) in MeOH (3 mL) in one portion at 25°C in a sealable reaction tube. The reaction mixture was sealed and stirred at 120°C for 12 hours. TLC showed that the reaction was completed. The mixture was concentrated under vacuum-to afford compound 50 (185.00 mg, 545.10 μmol, 97.80% yield) as a yellow solid. Furan-2,5-dione (260 mg, 2.65 mmol) was added to compound 50 (180 mg, 530 μmol) in AcOH (5 mL) in one portion at 25°C. The mixture was stirred at 100°C for 12 hours, and TLC showed that the reaction was completed. The mixture was cooled to 25°C and concentrated under reduced pressure. The residue was poured into saturated NaHCO3 solution (5 mL) and stirred for 3 min. The aqueous phase was extracted with ethyl acetate (3 mL × 3). The combined organic phase was washed with saturated brine (3 mL), dried with anhydrous Na2SO4, filtered, and concentrated under vacuum. The residue was purified by performing silica gel chromatography (Petroleum ether/ethyl acetate = 3/1 to 1/1) to afford the compound 28 (14.00 mg, 33.38 μmol, 6.29% yield, 96.7% purity) as an off-white solid. 1H NMR(400 MHz, chloroform-d) δ (ppm) 1.23–1.42 (m, 3 H), 2.79–2.93 (m, 2 H), 3.29–3.43 (m, 1 H), 3.48–3.61 (m, 1 H), 3.82–3.99 (m, 1 H), 4.17–4.31 (m, 2 H), 4.35–4.57 (m, 1 H), 6.50–6.69 (m, 1 H), 6.76 (s, 2 H), 7.07–7.23 (m, 2 H), 7.25–7.39 (m, 7 H), 7.43 (d, J = 7.53 Hz, 1 H), 7.58 (s, 1 H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 171.00, 170.97, 166.09, 165.98, 154.02, 153.89, 140.67, 140.32, 140.28, 140.25, 138.90, 137.93, 137.88, 135.57, 135.43, 131.23, 131.05, 130.88, 130.85, 129.62, 128.85, 128.71, 128.20, 128.14, 128.09, 126.91, 60.82, 60.62, 30.44, 29.91, 29.87, 29.42, 14.98, 14.96. ESI-TOF HRMS: m/z 420.1566 (C23H21N3O5 + H+ requires 420.1561).
4.1.9. Ethyl (5-(2-(1H-pyrrol-1-yl)acetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (29)
NaH (33.5 mg, 1.39 mmol) was added to a solution of pyrrole (47 mg, 700 μmol) in DMF (2 mL) at 0°C. The mixture was stirred at 0°C for 0.5 hour. Compound 48 (250 mg, 696 μmol) in DMF (1 mL) was then added dropwise. The mixture was stirred at 0°C for 1 hour, and LC-MS showed that starting material 48 was depleted. The reaction was quenched with aqueous NH4Cl solution, extracted with ethyl acetate (20 mL × 3), and washed with brine (20 mL × 2). The organic layer was dried with anhydrous Na2SO4 and concentrated to give a residue. The residue was dissolved in MeOH (3 mL) and purified by preparative HPLC to give compound 29 (81.4 mg, 209 μmol, 30.0% yield, 99.5% purity). 1H NMR (400 MHz, chloroform-d) δ (ppm) 7.53 (br. s., 1H), 7.27–7.43 (m, 3H), 6.99–7.22 (m, 3H), 6.62 (d, J = 14.43 Hz, 1H), 6.47 (d, J = 9.79 Hz, 2H), 6.11 (t, J = 1.94 Hz, 2H), 4.47–4.72 (m, 2H), 4.16–4.29 (m, 2H), 3.16 (br. s., 2H), 2.73 (d, J = 9.41 Hz, 2H), 1.27–1.37 (m, 3H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 167.65, 154.08, 153.91, 140.79, 140.66, 138.66, 138.02, 137.89, 135.62, 131.52, 130.95, 130.84, 129.35, 129.09, 128.83, 128.21, 128.01, 127.84, 126.80, 122.37, 119.47, 118.15, 108.06, 108.00, 60.78, 60.61, 51.03, 50.83, 30.48, 30.22, 29.84, 29.59, 14.98. ESI-TOF HRMS: m/z 390.1823 (C23H23N3O3 + H+ requires 390.1819).
4.1.10. Ethyl (5-(2-(1H-imidazol-1-yl)acetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (30)
NaH (33.5 mg, 1.39 mmol) was added to a solution of imidazole (48 mg, 705 μmol) in DMF (2 mL) at 0°C. The mixture was stirred at 0°C for 0.5 hour. Compound 48 (250 mg, 697 μmol) in DMF (1 mL) was then added dropwise. The mixture was stirred at 0°C for 1 hour, and LC-MS showed that starting material 48 was depleted. The reaction was quenched with aqueous NH4Cl solution, extracted with ethyl acetate (20 mL × 3), and washed with brine (20 mL × 2). The organic layer was dried with anhydrous Na2SO4 and concentrated to give a residue. The residue was dissolved in MeOH (3 mL) and purified by preparative HPLC to give compound 30 (91.5 mg, 234 μmol, 33.7% yield, 99.1% purity). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.27 (br. s., 1H), 7.59 (s, 1H), 7.45 (s, 2H), 7.25–7.33 (m, 4H), 7.18 (d, J = 8.29 Hz, 1H), 7.03 (s, 1H), 6.85 (s, 1H), 4.79 (d, J = 9.80 Hz, 2H), 4.15 (q, J = 6.97 Hz, 2H), 3.20–3.31 (m, 2H), 2.72–2.81 (m, 2H), 1.26 (t, J = 7.06 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 166.99, 154.08, 153.91, 140.58, 140.52, 140.49, 140.40, 138.84, 138.71, 138.12, 137.90, 135.50, 131.70, 130.99, 130.88, 129.48, 129.04, 128.82, 128.18, 128.12, 128.05, 127.89, 126.81, 121.29, 121.26, 119.67, 118.12, 60.78, 60.61, 60.23, 48.44, 48.29, 30.60, 30.21, 29.90, 29.54, 21.24, 14.98, 14.56. ESI-TOF HRMS: m/z 391.1775 (C22H22N4O3 + H+ requires 391.1772).
4.1.11. Ethyl (5-(ethylglycyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (31)
Ethylamine (31 mg, 687 μmol) in DMF (2 mL) was added to compound 48 (200 mg, 550 μmol) in DMF (1 mL) in a microwavable vessel. The vessel was sealed and heated to 80°C in a microwave for 0.5 hour, and LC-MS showed that starting material 48 was depleted. The mixture was extracted with ethyl acetate (20 mL × 2) and washed with brine (10 mL × 2). The organic layer was dried with anhydrous Na2SO4 and concentrated to yield a residue. The residue was purified by preparative HPLC to yield compound 31 (104.5 mg, 284 μmol, 51.7% yield, 99.1% purity). 1H NMR (400 MHz, DMSO-d6) δ 9.29 (br. s., 1H), 8.17 (br. s., 1H), 7.48 (s, 1H), 7.18–7.35 (m, 5H), 7.15 (d, J = 8.48 Hz, 1H), 4.14 (q, J = 6.97 Hz, 2H), 3.20–3.21 (m, 2H), 2.78 (d, J = 9.42 Hz, 2H), 2.51–2.59 (m, 4H), 2.09 (s, 1H), 1.25 (t, J = 7.06 Hz, 3H), 0.96 (t, J = 7.06 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 169.70, 169.49, 164.83, 154.00, 153.93, 140.77, 140.56, 140.50, 138.65, 137.93, 137.85, 135.49, 131.24, 131.03, 130.96, 130.88, 130.70, 129.34, 128.91, 128.01, 127.83, 126.84, 119.19, 118.19, 60.76, 60.62, 49.96, 49.89, 43.16, 30.62, 30.01, 29.95, 29.39, 14.97, 14.43, 14.27. ESI-TOF HRMS: m/z 368.1980 (C21H25N3O3 + H+ requires 368.1976).
4.1.12. Ethyl (5-(tert-pentylglycyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (32)
Tert-Amylamine (59 mg, 677 μmol) in DMF (2 mL) was added to compound 48 (200 mg, 550 μmol) in DMF (1 mL) in a microwavable vessel. The vessel was sealed and heated to 80°C in a microwave for 0.5 hour, and LC-MS showed that starting material 48 was depleted. The mixture was extracted with ethyl acetate (20 mL × 2) and washed with brine (10 mL × 2). The organic layer was dried with anhydrous Na2SO4 and concentrated to yield a residue. The residue was purified by preparative HPLC to yield compound 32 (96.3 mg, 235 μmol, 42.8% yield, 95.0% purity). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.35 (br. s., 1H), 8.18 (s, 1H), 7.50 (s, 1H), 7.22–7.34 (m, 5H), 7.15 (d, J = 8.28 Hz, 1H), 4.10–4.16 (m, 2H), 3.18–3.27 (m, 4H), 2.71–2.81 (m, 2H), 1.20–1.27 (m, 5H), 0.95 (s, 1H), 0.86 (s, 6H), 0.73–0.80 (m, 3H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 171.08, 170.95, 164.40, 154.00, 153.93, 141.09, 140.94, 140.68, 138.55, 137.87, 135.54, 131.17, 131.00, 130.87, 130.60, 129.24, 128.93, 127.97, 127.89, 127.75, 126.80, 119.04, 118.31, 118.06, 117.66, 60.75, 60.67, 60.61, 52.81, 52.44, 44.13, 44.04, 33.07, 33.02, 30.59, 29.99, 29.44, 26.37, 26.20, 26.14, 26.08, 22.62, 14.97, 8.64, 8.58, 8.33. ESI-TOF HRMS: m/z 410.2455 (C24H31N3O3 + H+ requires 410.2445).
4.1.13. Ethyl (5-(3-(diethylamino)propanoyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (33)
A mixture of 3-(diethylamino)propionic acid (124 mg, 854 μmol) and compound 47 (200 mg, 708 μmol) in T3P (5 mL) was stirred at reflux for 12 hours, and LC-MS showed that starting material 47 was depleted. The mixture was concentrated to yield a residue that was then purified by preparative HPLC to yield compound 33 (147.3 mg, 360 μmol, 50.8% yield, 97.7% purity) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.32 (br. s., 1H), 8.16 (s, 1H), 7.50 (br. s., 1H), 7.19–7.35 (m, 5H), 7.15 (d, J = 8.28 Hz, 1H), 4.14 (q, J = 7.15 Hz, 2H), 3.19–3.28 (m, 2H), 2.69–2.81 (m, 4H), 2.32–2.43 (m, 6H), 1.26 (t, J = 7.09 Hz, 3H), 0.85–0.94 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 171.09, 171.06, 164.37, 153.99, 153.92, 141.90, 141.88, 141.21, 141.07, 138.58, 137.84, 137.65, 135.60, 131.08, 130.76, 130.73, 130.63, 129.12, 128.99, 128.95, 128.33, 127.78, 127.67, 126.71, 118.84, 118.41, 117.98, 117.82, 60.73, 60.58, 48.53, 48.48, 46.66, 46.63, 31.13, 31.10, 30.53, 30.23, 29.86, 29.59, 14.99, 14.96, 11.36, 11.33. ESI-TOF HRMS: m/z 410.2446 (C24H31N3O3 + H+ requires 410.2445).
4.1.14. Ethyl (5-(3-(pyrrolidin-1-yl)propanoyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (34)
A mixture of 1-pyrrolidinepropanoic acid (122 mg, 852 μmol) and compound 47 (200 mg, 708 μmol) in T3P (5 mL) was stirred at reflux for 12 hours, and LC-MS showed that starting material 47 was depleted. The mixture was concentrated to yield a residue. The residue was purified by preparative HPLC to yield compound 34 (197.4 mg, 485 μmol, 68.5% yield, 99.0% purity) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.25 (br. s., 1H), 8.14 (s, 1H), 7.47 (s, 1H), 7.10–7.30 (m, 5H), 4.13 (q, J = 6.97 Hz, 2H), 3.03–3.15 (m, 2H), 2.70 (t, J = 6.97 Hz, 4H), 2.20–2.44 (m, 6H), 1.65 (td, J = 3.30, 6.59 Hz, 4H), 1.25 (t, J = 7.06 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 170.81, 170.78, 164.27, 154.00, 153.92, 141.78, 141.74, 141.21, 141.09, 138.54, 137.83, 137.69, 135.74, 131.11, 130.81, 130.73, 130.68, 130.66, 129.23, 128.95, 128.33, 127.81, 127.68, 126.71, 118.86, 118.38, 117.98, 117.86, 60.74, 60.59, 53.83, 51.62, 51.54, 32.70, 32.63, 30.44, 30.27, 29.80, 29.65, 23.41, 14.99, 14.97. ESI-TOF HRMS: m/z 408.2290 (C24H29N3O3 + H+ requires 408.2289).
4.1.15. Ethyl (5-(2-(tert-butoxy)acetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (35)
A mixture of compound 47 (150 mg, 530 μmol) and 2-tert-butoxyacetic acid (105 mg, 796 μmol) in propylphosphonic acid (T3P) (5 mL) was stirred at 55°C for 12 hours and, LC-MS showed that starting material 47 was depleted. The mixture was washed with NaHCO3 aqueous solution (20 mL × 3). The organic layer was dried with anhydrous Na2SO4 and concentrated under vacuum to yield give a residue. The residue was purified by preparative HPLC to give compound 35 (50.00 mg, 126.11 μmol, 23.74% yield, 99.5% purity) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.63 (br. s., 1 H), 7.03–7.61 (m, 7 H), 4.04–4.22 (m, 2 H), 3.87 (d, J = 8.91 Hz, 2 H), 3.10–3.32 (m, 2 H), 2.63–2.87 (m, 2 H), 1.23 (t, J = 7.09 Hz, 3 H), 0.92 (br. s., 9 H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 169.08, 153.96, 141.68, 140.95, 138.35, 138.17, 137.81, 130.90, 130.48, 130.26, 128.95, 127.82, 127.40, 126.73, 118.37, 118.01, 73.79, 62.07, 60.65, 30.44, 30.17, 29.82, 29.60, 27.16, 14.97. ESI-TOF HRMS: m/z 419.1952 (C23H28N2O4 + Na+ requires 419.1949).
4.1.16. 1-(3-amino-10,11-dihydro-5H-dibenzo[b,f]azepin-5-yl)-2-(diethylamino)ethan-1-one (51)
Compound 11 (2.5 g, 6.32 mmol) in H2SO4/AcOH (40 mL) was stirred at 120°C for 4 hours, and LC-MS showed that starting material 11 was depleted. The mixture was basified to pH 7 with Na2CO3 aqueous solution and extracted with ethyl acetate (50 mL × 3). The organic layer was dried by anhydrous Na2SO4 and concentrated to give compound 51 (2.0 g, 97.8% yield) as a light yellow oil. LCMS: MS+1=324.2. 1H NMR (400 MHz, DMSO-d6) δ (ppm) 7.17–7.36 (m, 4H), 6.76–6.99 (m, 1H), 6.36–6.59 (m, 2H), 4.92–5.15 (m, 2H), 3.02–3.22 (m, 4H), 2.51–2.81 (m, 4H), 2.44 (br. s., 2H), 0.75–0.87 (m, 6H).
4.1.17. N-(5-(diethylglycyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)butyramide (36)
A mixture of 51 (180 mg, 556 μmol), butyryl chloride (118.6 mg, 1.11 mmol) and Et3N (141 mg, 1.39 mmol) in dichloromethane (3 mL) was stirred at 10°C to 35°C for 16 hours, and LC-MS showed that starting material 51 was depleted. The mixture was concentrated to give a residue. The residue was purified by preparative HPLC to give compound 36 (95.2 mg, 242 μmol, 43.4% yield, 98.8% purity). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.55 (br. s., 1H), 8.15 (s, 1H), 7.61 (d, J = 1.88 Hz, 1H), 7.40 (dd, J = 2.07, 8.29 Hz, 1H), 7.18–7.36 (m, 4H), 7.14 (d, J = 8.29 Hz, 1H), 3.21 (d, J = 1.13 Hz, 4H), 2.72–2.84 (m, 2H), 2.52–2.59 (m, 4H), 2.28 (t, J = 7.25 Hz, 2H), 1.58–1.70 (m, 2H), 0.94 (t, J = 7.44 Hz, 3H), 0.81–0.89 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 206.58, 171.62, 171.47, 170.99, 169.88, 163.86, 141.83, 141.57, 141.13, 141.03, 138.43, 137.90, 137.78, 135.55, 131.80, 130.79, 130.52, 130.40, 129.65, 129.03, 128.82, 128.08, 127.75, 127.50, 126.70, 119.40, 118.72, 61.49, 54.63, 47.19, 42.78, 38.69, 38.65, 30.52, 30.19, 30.01, 29.70, 21.44, 18.99, 16.97, 14.07, 13.93, 12.25, 12.22. ESI-TOF HRMS: m/z 394.2501 (C24H31N3O2 + H+ requires 394.2496).
4.1.18. Isopropyl (5-(diethylglycyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (37)
A mixture of 51 (180 mg, 556 μmol), isopropyl chloroformate (118.6 mg, 1.11 mmol), and Et3N (1401 mg, 1.4 mmol) in dichloromethane (3 mL) was stirred at 10°C to 35°C for 16 hours, and LC-MS showed that starting material 51 was depleted. The mixture was concentrated to give a residue. The residue was purified by preparative HPLC to give 37 (61.1 mg, 149 μmol, 26.8% yield, 97.9% purity). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.16 (br. s., 1H), 8.15 (br. s., 1H), 7.49 (s, 1H), 7.17–7.35 (m, 5H), 7.12 (d, J = 8.48 Hz, 1H), 4.91 (td, J = 6.22, 12.43 Hz, 1H), 3.18–3.27 (m, 4H), 2.71–2.80 (m, 2H), 2.51–2.58 (m, 4H), 1.27 (dd, J = 1.41, 6.31 Hz, 6H), 0.85 (t, J = 7.16 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 169.90, 164.06, 153.56, 141.80, 141.66, 141.28, 141.17, 138.48, 137.91, 137.71, 135.60, 131.00, 130.77, 130.61, 130.45, 129.00, 128.78, 128.11, 127.74, 127.48, 126.71, 118.65, 118.39, 117.89, 68.04, 67.92, 54.72, 54.50, 47.18, 30.51, 30.26, 29.87, 29.65, 22.42, 12.30, 12.21. ESI-TOF HRMS: m/z 410.2444 (C24H31N3O3 + H+ requires 410.2445).
4.1.19. tert-Butyl (5-(diethylglycyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (38)
A mixture of compound 51 (100 mg, 310 μmol), Boc2O (135 mg, 619 μmol), and Et3N (78 mg, 772 μmol) in dichloromethane (2 mL) was stirred at 20°C to 25°C for 24 hours, and LC-MS showed that starting material 51 was depleted. The mixture was concentrated to give a residue. The residue was purified by preparative HPLC to give compound 38 (50 mg, 118.05 μmol, 38.18% yield, 99.6% purity) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ (ppm) 8.95 (br. s., 1H), 8.16 (br. s., 1H), 7.49 (d, J = 1.70 Hz, 1H), 7.16–7.36 (m, 5H), 7.11 (d, J = 8.48 Hz, 1H), 3.20 (s, 4H), 2.71–2.84 (m, 2H), 2.51–2.58 (m, 4H), 1.49 (s, 9H), 0.86 (t, J = 7.06 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 169.85, 163.91, 153.16, 141.80, 141.60, 141.27, 141.15, 138.70, 138.13, 137.67, 135.58, 130.76, 130.65, 130.49, 129.04, 128.75, 128.12, 127.72, 127.47, 126.69, 118.64, 118.27, 117.91, 117.70, 79.68, 79.49, 54.70, 54.45, 47.19, 30.56, 30.29, 29.84, 29.62, 28.58, 12.29, 12.20. ESI-TOF HRMS: m/z 424.2603 (C25H33N3O3 + H+ requires 424.2602).
4.1.20. N-(5-(diethylglycyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)-1H-imidazole-1-carboxamide (52)
A mixture of compound 51 (500 mg, 1.55 mmol), CDI (250.6 mg, 1.55 mmol), and Et3N (312.9 mg, 3.09 mmol) in dichloromethane (5 mL) was stirred at 20°C to 25°C for 2 hours, and LC-MS showed that starting material 51 was depleted. The mixture was used directly for the next step without further purification.
4.1.21. 1-(5-(Diethylglycyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)-3-ethylurea (39)
A mixture of ethylamine (26 mg, 577 μmol), compound 52 (120 mg, 287 μmol), DMAP (17.6 mg, 143 μmol), and Et3N (29.1 mg, 287.42 μmol) in dichloromethane (2 mL) was stirred at 20°C to 25°C for 12 hours, and LC-MS showed that starting material 52 was depleted. The mixture was concentrated to give a residue. The residue was purified by preparative HPLC to give compound 39 (82.3 mg, 208 μmol, 72.6% yield, 97.6% purity) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ (ppm) 8.22 (br. s., 1H), 8.14 (s, 1H), 7.44 (br. s., 1H), 7.13–7.36 (m, 5H), 7.06 (d, J = 8.41 Hz, 1H), 5.95 (br. s., 1H), 3.07–3.18 (m, 6H), 2.69–2.80 (m, 2H), 2.50–2.58 (m, 4H), 0.99–1.14 (m, 3H), 0.77–0.88 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 169.76, 169.62, 163.96, 155.51, 141.86, 141.53, 141.20, 141.08, 139.91, 139.25, 137.83, 135.63, 130.77, 130.68, 130.43, 129.46, 129.00, 128.77, 128.06, 127.69, 127.45, 126.65, 118.10, 117.81, 117.38, 117.22, 54.62, 54.35, 47.29, 47.24, 34.40, 34.28, 30.65, 30.31, 29.85, 29.56, 15.93, 12.25, 12.14. ESI-TOF HRMS: m/z 395.2450 (C23H30N4O2 + H+ requires 395.2449).
4.1.22. 1-(5-(Diethylglycyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)-3-isopropylurea (40)
A mixture of iso-propylamine (34 mg, 575 μmol), compound 52 (120.00 mg, 287 μmol), DMAP (17.6 mg, 143 μmol), and Et3N (29.08 mg, 287.42 μmol) in dichloromethane (2 mL) was stirred at 20°C to 25°C for 12 hours, and LC-MS showed that starting material 52 was depleted. The mixture was concentrated to give a residue. The residue was purified by preparative HPLC to give compound 40 (52.7 mg, 128 μmol, 44.9% yield, 99.7% purity) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ (ppm) 8.07 (s, 1H), 7.44 (d, J = 1.88 Hz, 1H), 7.12–7.35 (m, 5H), 7.03–7.09 (m, 1H), 5.77 (d, J = 7.35 Hz, 1H), 3.78 (qd, J = 6.59, 13.59 Hz, 1H), 3.16–3.34 (m, 4H), 2.69–2.82 (m, 2H), 2.51–2.56 (m, 4H), 1.04–1.19 (m, 6H), 0.86 (t, J = 7.06 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 170.10, 169.99, 154.87, 142.04, 141.73, 141.30, 141.20, 139.78, 139.12, 137.82, 135.64, 130.71, 130.42, 129.50, 129.05, 128.69, 128.09, 127.65, 127.43, 126.63, 117.95, 117.82, 117.27, 117.16, 54.75, 54.45, 47.14, 47.09, 41.39, 41.29, 30.65, 30.31, 29.87, 29.57, 23.44, 23.41, 12.45, 12.34. ESI-TOF HRMS: m/z 409.2607 (C24H32N4O2 + H+ requires 409.2605).
4.1.23. 1-(tert-Butyl)-3-(5-(diethylglycyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)urea (41)
A mixture of tert-butylamine (42 mg, 574 μmol), compound 52 (120.00 mg, 287 μmol), DMAP (17.6 mg, 143 μmol), and Et3N (29 mg, 287 μmol) in dichloromethane (2 mL) was stirred at 20°C to 25°C for 12 hours, and LC-MS showed that starting material 52 was depleted. The mixture was concentrated to give a residue. The residue was purified by preparative HPLC to give 41 (71.7 mg, 170 μmol, 59.2% yield, 99.8% purity) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ (ppm) 8.17 (s, 1H), 7.98 (br. s., 1H), 7.49 (s, 1H), 7.17-7.39 (m, 5H), 7.09 (d, J = 8.16 Hz, 1H), 3.25-3.42 (m, 8H), 2.68-2.82 (m, 2H), 2.52-2.57 (m, 4H), 0.99-1.31 (m, 6H), 0.75-0.93 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 169.77, 169.63, 163.95, 154.76, 141.88, 141.59, 141.18, 139.92, 139.25, 137.78, 135.60, 130.77, 130.65, 130.44, 129.37, 129.06, 128.75, 128.10, 127.68, 127.45, 127.27, 126.64, 117.88, 117.56, 117.10, 116.93, 54.63, 54.31, 49.86, 47.28, 47.23, 30.68, 30.33, 29.84, 29.48, 12.25, 12.16. ESI-TOF HRMS: m/z 423.2762 (C25H34N4O2 + H+ requires 423.2762).
4.1.24. 3-(5-(Diethylglycyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)-1,1-diethylurea (42)
A mixture of diethylamine (42 mg, 575 μmol), compound 52 (120 mg, 287.42 μmol), DMAP (17.6 mg, 143.8 μmol), and Et3N (29 mg, 287.4 μmol) in dichloromethane (2 mL) was stirred at 20°C to 25°C for 12 hours, and LC-MS showed that starting material 52 was depleted. The mixture was concentrated to give a residue. The residue was purified by preparative HPLC to give 42 (102.7 mg, 243 μmol, 84.8% yield, 95.2% purity) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ (ppm) 8.05-8.23 (m, 2H), 7.44 (br. s., 1H), 7.32 (br. s., 1H), 7.16-7.27 (m, 3H), 7.00-7.12 (m, 2H), 5.81 (br. s., 1H), 3.26-3.37 (m, 4H), 2.67-2.80 (m, 2H), 2.51-2.56 (m, 4H), 1.29 (s, 9H), 0.77-0.90 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 169.89, 169.74, 164.11, 154.75, 141.86, 141.20, 140.84, 139.98, 139.40, 137.74, 135.62, 130.76, 130.48, 130.12, 129.95, 129.02, 128.74, 128.09, 127.66, 127.44, 126.62, 120.16, 120.04, 119.75, 119.47, 54.54, 54.45, 47.25, 47.18, 30.66, 30.36, 29.83, 29.59, 14.35, 12.30, 12.22, 11.53. ESI-TOF HRMS: m/z 423.2769 (C25H34N4O2 + H+ requires 423.2762).
4.1.25. 1-(5-(Diethylglycyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)-3-ethylsulfamide (43)
Ethanamine (10 g, 222 mmol) in dichloromethane (30 mL) was added dropwise to a suspension of sulfuryl chloride (100 g, 741 mmol) at 0°C to 10°C. After addition, the mixture was heated to 75°C for 15 hours and then cooled to 20°C. The mixture was concentrated to dryness to yield compound N-ethyl-sulfamoyl chloride (50 g, crude), which was freshly used directly in the next step. N-Ethyl-sulfamoyl chloride (532.7 mg, 3.71 mmol) was added to a mixture of compound 51 (200 mg, 618.4 μmol) in dichloromethane (6 mL), and Et3N (18.8 mg, 185.52 μmol) was added dropwise. The mixture was stirred at 15°C to 25°C for 1 hour, and LC-MS showed that starting material 51 was depleted. The mixture was concentrated, dried, and purified by preparative HPLC to give compound 43 (76.6 mg, 178 μmol, 28.8% yield, 94.4% purity) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.62 (br. s., 1H), 6.94-7.57 (m, 8H), 3.03-3.28 (m, 4H), 2.64-2.93 (m, 4H), 2.44 (br. s., 4H), 0.96 (t, J = 7.22 Hz, 3H), 0.79 (br. s., 6H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 170.23, 170.07, 141.93, 141.38, 141.29, 138.55, 137.82, 137.62, 135.67, 131.06, 130.83, 130.72, 130.48, 130.38, 129.18, 128.98, 128.77, 128.13, 127.71, 127.48, 126.70, 119.00, 118.14, 116.78, 54.82, 47.06, 37.51, 30.47, 30.20, 29.90, 29.63, 15.01, 12.30. ESI-TOF HRMS: m/z 431.2120 (C22H30N4O3S + H+ requires 431.2119).
4.1.26. 2-(Diethylamino)-1-(3-((5-methyl-1,3,4-oxadiazol-2-yl)amino)-10,11-dihydro-5H-dibenzo[b,f]azepin-5-yl)ethan-1-one (44)
1-(2-Oxopyridine-1-carbothioyl)pyridin-2-one (344.7 mg, 1.48 mmol) in dichloromethane (0.5 mL) was added dropwise to a solution of compound 51 (320 mg, 989.39 μmol) in dichloromethane (1 mL) at 0°C. The mixture was stirred at 20°C for 16 hours and then concentrated and purified by performing silica gel chromatography (dichloromethane: MeOH = 30:1). A mixture of acetohydrazide (146.6 mg, 1.98 mmol) and purified product in THF (1 mL) was stirred at 20°C for 16 hours and then concentrated; EDC (307.19 mg, 1.98 mmol), TEA (200.23 mg, 1.98 mmol), and DMF (1 mL) were then added, and the reaction was stirred at 20°C for 16 hours. The residue was further purified by preparative HPLC to give compound 44 (39.8 mg, 98 μmol, 9.9% yield, 94.1% purity) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ (ppm) 10.28 (br. s., 1H), 9.68 (br. s., 1H), 7.80 - 7.20 (m, 7H), 4.32 (br. s., 1H), 3.94 - 3.47 (m, 2H), 3.26 (br. s., 5H), 2.92 - 2.75 (m, 2H), 2.40 (s, 3H), 1.24 (br. s., 6H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 160.21, 159.94, 157.68, 157.51, 138.59, 137.82, 137.70, 135.61, 131.55, 131.42, 131.11, 131.01, 130.34, 129.82, 128.53, 128.10, 128.04, 127.87, 127.02, 117.37, 116.35, 116.28, 49.03, 45.73, 30.45, 30.23, 29.79, 29.58, 10.86, 9.78, 8.86. ESI-TOF HRMS: m/z 406.2247 (C23H27N5O2 + H+ requires 406.2245).
4.1.27. Isopropyl (10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (73)
Isopropyl carbonochloridate (4.20 g, 34.24 mmol) was added dropwise to a solution of compound 46 (3.60 g, 17.12 mmol) in ethanol (36 mL) at 0°C for approximately 20 min. A solution of sodium carbonate (1.81 g, 17.12 mmol) in water (36 mL) was then added dropwise while the temperature was maintained below 15°C. The reaction was stirred at 25°C for 1 hour. TLC showed that starting material 46 was depleted. The reaction solution was poured into water and filtered yielding pure compound 73 (5.20 g, crude) as a green solid. LC-MS: MS + 1 = 297.15. 1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.35 (s, 1H), 8.28 (s, 1H), 7.24 (s, 1H), 7.01-6.96 (m, 3H), 6.87- 6.85(d, J = 8.0 Hz, 1H), 6.66-6.64 (m, 2H), 4.91-4.85 (m, 1H), 2.93-2.84 (m, 4H), 1.26-1.25 (d, J = 4.0 Hz, 6H).
4.1.28. Isopropyl (5-(3-(diethylamino)propanoyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (53)
A mixture of compound 73 (200 mg, 674.85 μmol) and 3-chloropropionyl chloride (128.5 mg, 1.01 mmol) in toluene (2 mL) was stirred at 100°C for 3 hours. TLC showed that the reaction was complete. The mixture was concentrated to afford compound 74 as gray gum, which was directly used in the next step. A mixture of compound 74 (261 mg, 675 μmol) and diethyl amine (247 mg, 3.38 mmol) in toluene (2 mL) was stirred at 100°C for 10 hours. TLC showed that the reaction was complete. The solvent was removed, and the residue was first purified by flash column chromatography (petroleum ether/EtOAc = 10/1 to 100% EtOAc) and then further purified by preparative HPLC to afford compound 53 (60 mg, 382.48 μmol, 56.66% yield, 96.4% purity) as a colorless oil. 1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.50-9.79 (d, J = 34 Hz, 1 H) 8.93 (br, 1 H) 7.73-7.07 (m, 7 H) 4.81-4.95 (m, 1 H) 3.15-3.36 (m, 5 H) 3.05-3.15 (m, 4 H) 2.86-2.99 (m, 1 H) 2.71-2.85 (m, 2 H) 2.33-2.45 (m, 1 H) 1.24 (d., J = 5.8 Hz, 6 H) 1.09-1.20 (m, 6 H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 169.36, 169.35, 158.73, 158.47, 158.22, 157.96, 153.66, 153.52, 141.39, 141.37, 140.55, 140.25, 138.81, 137.95, 137.76, 135.34, 131.35, 131.04, 130.95, 130.79, 130.76, 129.28, 128.94, 128.87, 128.16, 127.98, 127.91, 126.73, 119.36, 118.57, 118.21, 117.81, 116.19, 68.17, 67.97, 48.03, 47.42, 47.39, 47.26, 47.14, 47.11, 30.72, 30.04, 29.94, 29.32, 22.43, 22.41, 9.04, 8.98, 8.94, 8.91, 8.87. ESI-TOF HRMS: m/z 424.2604 (C25H33N3O3 + H+ requires 424.2602).
4.1.29. Isopropyl (5-(2-chloroacetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (75)
A mixture of compound 73 (5.2 g, 17.55 mmol) and 2-chloroacetyl chloride (2.97 g, 26.32 mmol) in toluene (52 mL) was stirred at 100°C for 3 hours. TLC showed that the reaction was complete. The mixture was concentrated to afford compound 75 (5.20 g, 13.95 mmol, 79.47% yield) as a green solid. LC-MS: MS + 1 = 373.12. 1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.62-9.59 (d, J = 12.0 Hz, 1H), 7.63-7.11 (m, 7H), 4.91-4.85 (m, 1H), 4.34-4.09 (m, 2H), 3.21-3.02 (m, 2H), 2.77-2.68 (m, 2H), 1.25-1.24 (m, 6H).
4.1.30. Isopropyl (5-(dipropylglycyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (54)
NaH (12 mg 500 μmol) was added to a mixture of dipropylamine (60 mg, 592 μmol) in DMF (2 mL) at 25°C. The mixture was stirred at 25°C for 0.5 hour. Compound 75 (100 mg, 268 μmol) in DMF (1 mL) was then added dropwise. The mixture was stirred at 25°C for 2.5 hours, and LC-MS showed that starting material 75 was mostly depleted. The reaction was quenched with aqueous NH4Cl solution. The product was extracted with EtOAc (50 mL × 3), and the combined organic layer was washed with brine (20 mL × 2), dried with anhydrous Na2SO4, filtered, and concentrated to give a residue. The residue was dissolved in acetonitrile (3 mL), purified by preparative HPLC to give the compound 54 (52 mg, 119 μmol, 44.4% yield, 98.6% purity). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.68 (s, 1H), 7.61-7.32 (m, 7H), 4.97 (s, 1H), 3.54-3.30 (m, 8H), 2.83 (s, 2H), 1.33(s, 10 H), 0.85 (s, 6H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 170.16, 153.56, 141.76, 141.58, 141.31, 141.20, 138.53, 137.91, 137.55, 135.58, 130.97, 130.73, 130.52, 129.04, 128.78, 128.17, 127.72, 127.53, 126.71, 118.67, 118.43, 117.87, 68.06, 67.91, 56.03, 55.78, 55.46, 55.28, 30.53, 30.28, 29.89, 29.66, 22.41, 20.78, 20.61, 20.52, 12.16, 12.09. ESI-TOF HRMS: m/z 438.2759 (C26H35N3O3 + H+ requires 438.2758).
4.1.31. Isopropyl (5-(2-(piperidin-1-yl)acetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (55)
NaH (12 mg, 500 μmol) was added to a mixture of piperidine (50 mg, 587 μmol) in DMF (2 mL) at 25°C. The reaction was stirred at 25°C for 0.5 hour. Compound 75 (100 mg, 268 μmol) in DMF (1 mL) was then added dropwise. The mixture was stirred at 25°C for 2.5 hours, and LC-MS showed that starting material 75 was mostly depleted. The reaction was quenched with aqueous NH4Cl solution. The product was extracted with EtOAc (50 mL × 3), and the combined organic layer was washed with brine (20 mL × 2), dried with anhydrous Na2SO4, filtered, and concentrated to give a residue. The residue was dissolved in acetonitrile (3 mL) and purified by preparative HPLC to give the compound 55 (83.6 mg, 198 μmol, 73.8% yield, 99.1% purity). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.62-9.59 (m, 1H), 7.60-7.10 (m, 7H), 4.89-4.86 (m, 1H), 3.34-3.16 (m, 6H), 2.77-2.74 (m, 4 H), 1.50(s, 4 H), 1.35(s, 2 H), 1.26 -1.24 (dd, J1 = 4.0 Hz, J2 = 4.0 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 162.76, 153.59, 153.53, 138.46, 137.97, 137.86, 135.55, 131.26, 130.88, 130.50, 128.96, 127.81, 127.49, 126.75, 118.14, 117.62, 68.06, 67.94, 54.12, 36.25, 31.24, 30.54, 30.34, 29.84, 29.65, 25.36, 22.42, 1.63. ESI-TOF HRMS: m/z 422.2452 (C25H31N3O3 + H+ requires 422.2445).
4.1.32. Isopropyl (5-(2-(1H-pyrrol-1-yl)acetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (56)
NaH (12 mg, 500 μmol) was added to a mixture of pyrrole (40 mg, 596 μmol) in DMF (2 mL) at 25°C. The mixture was stirred at 25°C for 0.5 hour. Compound 75 (100 mg, 268 μmol) in DMF (1 mL) was then added dropwise. The mixture was stirred at 25°C for 2.5 hour, and LC-MS showed that starting material 75 was mostly depleted. The reaction was quenched with aqueous NH4Cl solution. The product was extracted with EtOAc (50 mL × 3), and the combined organic layer was washed with brine (20 mL × 2), dried by anhydrous Na2SO4, filtered, and concentrated to give a residue. The residue was dissolved in acetonitrile (3 mL), purified by preparative HPLC to give the compound 56 (50 mg, 124 μmol, 46.3% yield, 99.4% purity). 1H NMR (400 MHz, Methanol-d4) δ (ppm) 7.67 (s, 1H), 7.50 (s, 1H), 7.36-7.31 (m, 2H), 7.24 (s, 2H), 7.13-7.11 (d, J = 8.0 Hz, 1H), 6.52-6.49 (d, J = 12.0 Hz, 2H), 6.03 (s, 2H), 4.76-4.59 (m, 3H), 3.24-3.20 (m, 2H), 2.79-2.77(d, J = 8.0 Hz, 2 H), 1.33 -1.29 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 167.65, 153.67, 153.52, 140.79, 140.67, 138.74, 138.00, 135.61, 131.43, 130.97, 130.85, 129.34, 129.01, 128.83, 128.22, 128.00, 127.83, 126.80, 122.38, 119.43, 118.12, 108.05, 108.00, 68.14, 67.94, 55.39, 51.03, 50.83, 30.49, 30.23, 29.83, 29.58, 22.42. ESI-TOF HRMS: m/z 404.1978 (C24H25N3O3 + H+ requires 404.1976).
4.1.33. Isopropyl (5-(2-iodoacetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (76)
Sodium iodide (1.21 g, 8.05 mmol) was added to a mixture of compound 75 (3.00 g, 8.05 mmol) in acetone (30 mL) in one portion at 25°C. The mixture was stirred at 25°C for 12 hours, and LC-MS showed that starting material 75 was depleted. The mixture was concentrated, and water (80 mL) was added to the residue. The aqueous phase was extracted with ethyl acetate (200 mL × 3). The combined organic phase was washed with saturated brine (200 mL × 2), dried with anhydrous sodium sulfate, filtered, and vacuum concentrated to afford compound 76 (3.00 g, 6.46 mmol, 80.25% yield) as a gray solid. LC-MS: MS + 1 = 465.1.
4.1.34. Isopropyl (5-(2-ethoxyacetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (57)
Ethanol (90 mg, 1.95 mmol) was added to NaHMDS (1 M, 2.07 mL, 2.07 mmol) at 0°C under nitrogen. The mixture was stirred at 0°C for 0.5 hour. Compound 76 (300 mg, 646 μmol) in THF (3 mL) was then added dropwise. The mixture was stirred at 25°C for 1 hour. TLC showed that starting material 76 was depleted. The reaction was quenched with aqueous saturated ammonium chloride solution, extracted with ethyl acetate (20 mL × 3), and washed with brine (20 mL × 2). The organic layer was dried with anhydrous sodium sulfate and concentrated to give a residue. The residue was dissolved in acetonitrile (3 mL) and purified by preparative HPLC to give compound 57 (51 mg, 133 μmol, 20.6% yield, 99.5% purity). 1H NMR (400 MHz, chloroform-d) δ (ppm) 7.08-7.54 (m, 7H), 6.71 (s, 1H), 5.01 (br, s, 1H), 4.16-4.30 (m, 1H), 3.76-3.08 (m, 1H), 3.54 (q, J = 8.0 Hz, 2H), 3.35 (br, s, 2H), 2.77-2.82 (m, 2H), 1.30 (d, J = 8.0 Hz, 6H), 1.20 (t, J = 4.0 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 168.73, 153.57, 140.70, 137.94, 130.89, 130.56, 129.15, 128.99, 127.90, 127.65, 126.78, 118.34, 118.06, 68.72, 67.99, 66.40, 30.61, 30.10, 29.95, 29.48, 22.42, 15.40. ESI-TOF HRMS: m/z 383.1970 (C22H26N2O4 + H+ requires 383.1973).
4.1.35. Isopropyl (5-(2-isopropoxyacetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (58)
Isopropanol (117 mg, 1.94 mmol) was added to NaHMDS (1 M, 2.07 mL, 2.07 mmol) at 0°C under nitrogen. The mixture was stirred at 0°C for 0.5 hour. Compound 76 (300 mg, 646 μmol) in THF (3 mL) was then added dropwise. The mixture was stirred at 25°C for 1 hour. TLC showed that starting material 76 was depleted. The reaction was quenched with aqueous saturated ammonium chloride solution, extracted with ethyl acetate (20 mL × 3) and washed with brine (20 mL × 2). The organic layer was dried with anhydrous sodium sulfate and concentrated to give a residue. The residue was dissolved in acetonitrile (3 mL) and purified by preparative HPLC to give compound 58 (52 mg, 131 μmol, 20.3% yield, 99.1% purity). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.58 (s, 1H), 7.52-7.11 (m, 7H), 4.91-4.86 (m, 1H), 4.07-3.78 (m, 3H), 3.24 (m, 2H), 2.75 (m, 2H), 1.26-1.24 (d, J = 8.0 Hz, 6H), 0.99-0.98(d, J = 4.0 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 168.92, 153.56, 140.82, 137.90, 130.86, 130.66, 130.46, 129.01, 127.84, 127.57, 126.74, 118.32, 118.02, 71.76, 67.97, 66.61, 33.40, 30.56, 30.13, 29.92, 29.52, 22.42, 22.07. ESI-TOF HRMS: m/z 397.2129 (C23H28N2O4 + H+ requires 397.2129).
4.1.36. Isopropyl (5-(2-(sec-butoxy)acetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (59)
2-Butanol (144 mg, 1.94 mmol) was added to NaHMDS (1 M, 2.07 mL, 2.07 mmol) at 0°C under nitrogen. The mixture was stirred at 0°C for 0.5 h. Compound 76 (300 mg, 646 μmol) in THF (3 mL) was then added dropwise. The mixture was stirred at 25°C for 1 hour. TLC showed that starting material 76 was depleted. The reaction was quenched with aqueous saturated ammonium chloride solution, extracted with ethyl acetate (20 mL × 3), and washed with brine (20 mL × 2). The organic layer was dried with anhydrous sodium sulfate and concentrated to give a residue. The residue was dissolved in acetonitrile (3 mL) and purified by preparative HPLC to give compound 59 (17 mg, 41.4 μmol, 6.4% yield, 95.0% purity). 1H NMR (400 MHz, chloroform-d) δ (ppm) 7.62-6.96 (m, 6H), 6.66 (br. s., 1H), 5.10-4.88 (m, 1H), 4.39-3.65 (m, 2H), 3.54-3.18 (m, 3H), 2.93-2.66 (m, 2H), 1.61-1.48 (m, 1H), 1.46-1.34 (m, 1H), 1.30 (d, J = 6.3 Hz, 6H), 1.07 (br. s., 3H), 0.87 (t, J = 7.5 Hz, 3H). ESI-TOF HRMS: m/z 411.2287 (C24H30N2O4 + H+ requires 411.2286).
4.1.37. isopropyl (5-(2-(neopentyloxy)acetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (60)
2,2-Dimethyl-1-Propanol (171 mg, 1.94 mmol) was added to NaHMDS (1 M, 2.07 mL, 2.07 mmol) at 0°C under nitrogen. The mixture was stirred at 0°C for 0.5 hour. Compound 76 (300 mg, 646 μmol) in THF (3 mL) was then added dropwise. The mixture was stirred at 25°C for 1 hour. TLC showed that starting material 76 was depleted. The reaction was quenched with aqueous saturated ammonium chloride solution, extracted with ethyl acetate (20 mL × 3), and washed with brine (20 mL × 2). The organic layer was dried with anhydrous sodium sulfate and concentrated to give a residue. The residue was dissolved in acetonitrile (3 mL), purified by preparative HPLC to give compound 60 (102.8 mg, 242 μmol, 37.5% yield, 99.4% purity). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.59 (s, 1H), 7.53 (s, 1H), 7.45-7.10 (m, 6H), 4.91-4.85 (m, 1H), 4.19-4.16 (d, J = 12.0 Hz, 1H), 3.83-3.72 (m, 1H), 3.23 (s, 2H), 2.80-2.75(m, 2 H), 1.26-1.25 (d, J = 4.0 Hz, 6H), 0.82 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 168.70, 153.57, 140.85, 140.66, 137.88, 131.18, 130.88, 130.55, 129.08, 128.87, 128.78, 127.88, 127.64, 127.03, 126.73, 118.38, 118.02, 81.56, 69.73, 67.96, 32.22, 30.63, 30.54, 30.17, 29.99, 29.56, 27.41, 26.96, 22.42. ESI-TOF HRMS: m/z 425.2451 (C25H32N2O4 + H+ requires 425.2442).
4.1.38. Isopropyl (5-(2-cyclopropoxyacetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (61)
Cyclopropanol (113 mg, 1.94 mmol) was added to NaHMDS (1 M, 2.07 mL, 2.07 mmol) at 0°C under nitrogen. The mixture was stirred at 0°C for 0.5 hour. Compound 76 (300 mg, 646 μmol) in THF (3 mL) was then added dropwise. The mixture was stirred at 25°C for 1 hour. TLC showed that starting material 76 was depleted. The reaction was quenched with aqueous saturated ammonium chloride solution, extracted with ethyl acetate (20 mL × 3), and washed with brine (20 mL × 2). The organic layer was dried with anhydrous sodium sulfate and concentrated to give a residue. The residue was dissolved in acetonitrile (3 mL) and purified by preparative HPLC to give compound 61 (82 mg, 208 μmol, 32.2% yield, 98.6% purity). 1H NMR (400 MHz, chloroform-d) δ (ppm) 7.38–7.86 (m, 7H), 5.32 (br, s, 1H), 4.51–4.65 (m, 1H), 4.13–4.24 (m, 1H), 3.68–3.84 (m, 4H), 3.08–3.15 (m, 2H), 1.61 (d, J = 4.0 Hz, 6H), 0.88 (br, s, 2H), 0.74–0.75 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 168.34, 153.56, 140.90, 140.77, 140.59, 137.94, 135.48, 131.21, 130.97, 130.75, 130.55, 129.25, 128.99, 128.83, 127.92, 127.71, 126.78, 119.09, 118.30, 118.08, 117.55, 68.82, 68.11, 67.95, 53.75, 30.61, 30.02, 29.95, 29.39, 22.41, 5.79, 5.76. ESI-TOF HRMS: m/z 395.1974 (C23H26N2O4 + H+ requires 395.1973).
4.1.39. Isopropyl (5-(2-cyclobutoxyacetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (62)
Cyclobutanol (140 mg, 1.94 mmol) was added to NaHMDS (1 M, 2.07 mL, 2.07 mmol) at 0°C under nitrogen. The mixture was stirred at 0°C for 0.5 hour. Compound 76 (300 mg, 646 μmol) in THF (3 mL) was then added dropwise. The mixture was stirred at 25°C for 1 hour. TLC showed that starting material 76 was depleted. The reaction was quenched with aqueous saturated ammonium chloride solution, extracted with ethyl acetate (20 mL × 3) and washed with brine (20 mL × 2). The organic layer was dried with anhydrous sodium sulfate and concentrated to give a residue. The residue was dissolved in acetonitrile (3 mL) and purified by preparative HPLC to give compound 62 (52 mg, 127 μmol, 19.7% yield, 98.5% purity). 1H NMR (400 MHz, chloroform-d) δ (ppm) 7.08–7.52 (m, 7H), 6.69 (s, 1H), 5.01 (br, s, 1H), 3.69–4.18 (m, 3H), 2.07 (br, s, 2H), 2.77–2.82 (m, 2H), 2.14–2.23 (m, 2H), 1.90 (t, J = 8.0 Hz, 2H), 1.61–1.69 (m, 1H), 1.37–1.49 (m, 1H), 1.30 (d, J = 8.0 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 168.49, 153.56, 140.96, 140.74, 137.92, 131.18, 130.92, 130.70, 130.52, 129.16, 128.96, 127.90, 127.64, 126.76, 118.29, 118.05, 73.12, 67.96, 65.97, 65.93, 30.60, 30.13, 30.00, 29.43, 22.41, 22.32, 12.36. ESI-TOF HRMS: m/z 409.2138 (C24H28N2O4 + H+ requires 409.2129).
4.1.40. Isopropyl (5-(2-(cyclopentyloxy)acetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (63)
Cyclopentanol (167 mg, 1.94 mmol) was added to NaHMDS (1 M, 2.07 mL, 2.07 mmol) at 0°C under nitrogen. The mixture was stirred at 0°C for 0.5 hour. Compound 76 (300 mg, 646 μmol) in THF (3 mL) was then added dropwise. The mixture was stirred at 25°C for 1 hour. TLC showed that starting material 76 was depleted. The reaction was quenched with aqueous saturated ammonium chloride solution, extracted with ethyl acetate (20 mL × 3), and washed with brine (20 mL × 2). The organic layer was dried with anhydrous sodium sulfate and concentrated to give a residue. The residue was dissolved in acetonitrile (3 mL) and purified by preparative HPLC to give compound 63 (80.5 mg, 190.6 μmol, 29.5% yield, 99.3% purity). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.59 (s, 1H), 7.52-7.10 (m, 7H), 4.91-4.85 (m, 1H), 3.99 (s, 1H), 3.78 (s, 2H), 3.24-3.18(m, 2H), 2.75(m, 2 H), 1.51-1.41 (d, J = 40.0 Hz, 8H), 1.26-1.24 (d, J = 8.0 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 168.70, 153.56, 141.20, 141.03, 140.76, 138.53, 137.92, 131.19, 130.89, 130.62, 130.43, 129.01, 127.83, 127.55, 126.74, 118.93, 118.33, 118.03, 117.55, 81.62, 67.97, 67.38, 31.93, 30.57, 30.12, 29.92, 29.50, 23.48, 22.41. ESI-TOF HRMS: m/z 423.2288 (C25H30N2O4 + H+ requires 423.2286).
4.1.41. Isopropyl (5-(2-(cyclohexyloxy)acetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (64)
Cyclohexanol (194 mg, 1.94 mmol) was added to NaHMDS (1 M, 2.07 mL, 2.07 mmol) at 0°C under nitrogen. The mixture was stirred at 0°C for 0.5 hour. Compound 76 (300 mg, 646 μmol) in THF (3 mL) was then added dropwise. The mixture was stirred at 25°C for 1 hour. TLC showed that starting material 76 was depleted. The reaction was quenched with aqueous saturated ammonium chloride solution, extracted with ethyl acetate (20 mL × 3), and washed with brine (20 mL × 2). The organic layer was dried with anhydrous sodium sulfate and concentrated to give a residue. The residue was dissolved in acetonitrile (3 mL) and purified by preparative HPLC to give compound 64 (28 mg, 64.2 μmol, 9.9% yield, 99.3% purity). 1H NMR (400 MHz, chloroform-d) δ (ppm) 7.09–7.52 (m, 7H), 6.60 (s, 1H), 5.01 (d, J = 4.0 Hz, 1H), 3.84–4.25 (m, 2H), 3.28–3.30 (m, 3H), 2.79–2.82 (m, 2H), 1.86–1.88 (m, 3H), 1.68 (br, s, 2H), 1.51 (br, s, 1H), 1.17–1.31 (m, 10H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 168.99, 153.56, 141.14, 140.94, 140.77, 137.90, 131.17, 130.88, 130.65, 130.45, 129.02, 127.84, 127.58, 126.74, 118.34, 118.01, 77.34, 67.95, 66.41, 31.78, 30.58, 30.14, 29.93, 29.52, 25.73, 23.75, 22.41. ESI-TOF HRMS: m/z 437.2445 (C26H32N2O4 + H+ requires 437.2442).
4.1.42. Benzyl (10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (77)
Benzyl chloroformate (16.23 g, 95.12 mmol) was added dropwise to a solution of compound 46 (10 g, 47.56 mmol) in EtOH (100 mL) at 0°C over a period of 1 hour. A solution of Na2CO3 (5.04 g, 47.56 mmol) in H2O (50 mL) was then added dropwise while the reaction temperature was maintained below 15°C. The reaction was stirred for another hour until TLC demonstrated that starting material 46 was depleted. The reaction solution was poured into water (500 mL) and filtered to give the crude product. The crude product was triturated with petroleum ether/ethyl acetate = 100/1, filtered, and concentrated to give compound 77 (15.00 g, crude) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ (ppm) 2.91 (d, J = 5.77 Hz, 4 H), 5.15 (s, 2 H), 6.61 - 6.71 (m, 2 H), 6.87 (d, J = 8.03 Hz, 1 H), 6.95 - 7.03 (m, 2 H), 7.25 (br. s., 1 H), 7.31 - 7.49 (m, 6 H), 8.30 (s, 1 H), 9.57 (s, 1 H).
4.1.43. Benzyl (5-(2-chloroacetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (78)
A mixture of compound 77 (15 g, 43.55 mmol) and 2-chloroacetyl chloride (7.38 g, 65.33 mmol) in toluene (150 mL) was stirred at 110°C for 3 hours, and LCMS showed that compound 77 was depleted. The mixture was concentrated, and the residue was purified by re-crystallization with petroleum ether and EtOAc (500 mL, 10/1), filtered, and concentrated to give compound 78 (14.00 g, crude) as a white solid. LCMS: MS + 1 = 420.12.
4.1.44. tert-Butyl (5-(dipropylglycyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (65)
A mixture of compound 78 (500 mg, 1.19 mmol) and dipropylamine (120 mg, 1.19 mmol) in toluene (1.8 mL) was stirred at 110°C for 6 hours, and LCMS showed that starting material 78 was depleted. The mixture was concentrated and purified by pre-HPLC (FA) to give compound 79. Then, 10% Pd/C (65.00 mg) was added to the solution of compound 79 that was just prepared in MeOH (5 mL), and the mixture was stirred at room temperature under H2 (15psi) for 12 hours. TLC showed that compound 79 was depleted. (Boc)2O (260 mg, 1.19 mmol) was then added at 25°C, and the mixture was stirred at 25°C for 3 hours; LCMS showed that the reaction was complete. The mixture was filtered and concentrated to give crude product that was further purified by preparative HPLC to give compound 65 (116 mg, 257 μmol, 21.6% yield, 99.5% purity). 1H NMR (400 MHz, chloroform-d) δ (ppm) 0.826 (t, J = 7.2 Hz, 6 H), 1.36 (s, 4 H), 1.51 (s, 9 H), 2.53–2.55 (m, 3 H), 2.74–2.80 (m, 2 H), 3.11–3.45(m, 4 H), 6.47 (s, 1 H), 7.06–7.49 (m, 7 H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 170.16, 153.18, 153.15, 141.77, 141.55, 141.32, 141.19, 138.75, 138.13, 137.51, 135.57, 130.73, 130.62, 130.60, 129.08, 128.74, 128.20, 127.70, 127.51, 126.69, 118.66, 118.31, 117.88, 117.73, 79.68, 79.48, 56.04, 55.45, 55.23, 30.58, 30.31, 29.87, 29.64, 28.57, 20.63, 20.53, 12.16. ESI-TOF HRMS: m/z 452.2914 (C27H37N3O3 + H+ requires 452.2915).
4.1.45. tert-Butyl (5-(2-(piperidin-1-yl)acetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (66)
A mixture of compound 78 (500 mg, 1.19 mmol) and piperidine (101 mg, 1.19 mmol) in toluene (1.8 mL) was stirred at 110°C for 6 hours, and LCMS showed that starting material compound 78 was depleted. The mixture was concentrated and purified by pre-HPLC (FA) to give compound 80. Then, 10% Pd/C (65.00 mg) was added to the solution of compound 80 that was just prepared in MeOH (5 mL), and the mixture was stirred at room temperature under H2 (15psi) for 12 hours. TLC showed that compound 80 was depleted. (Boc)2O (260 mg, 1.19 mmol) was then added at 25°C, and the mixture was stirred at 25°C for 3 hours; LCMS showed that the reaction was complete. The mixture was filtered and concentrated to give crude product that was further purified by preparative HPLC to give compound 66 (100 mg, 229.7 μmol, 19.3% yield, 99.6% purity). 1H NMR (400 MHz, chloroform-d) δ (ppm) 1.30 - 1.91 (m, 15 H), 2.11 - 2.90 (m, 5 H), 2.96 - 3.73 (m, 4 H), 6.28 - 6.58 (m, 1 H), 6.95 - 7.69 (m, 7 H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 169.23, 153.16, 141.20, 141.04, 138.58, 138.13, 137.83, 135.58, 131.01, 130.82, 130.67, 130.28, 129.08, 128.80, 128.64, 127.83, 127.72, 127.33, 126.68, 118.57, 118.31, 117.92, 117.56, 79.65, 79.49, 60.80, 54.28, 30.59, 30.44, 29.84, 29.68, 28.58, 25.86, 23.94. ESI-TOF HRMS: m/z 436.2604 (C26H33N3O3 + H+ requires 436.2602).
4.1.46. tert-Butyl (5-(2-isopropoxyacetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (67)
Na (0.5 g, 21.7 mmol) was added to 2-propanol (20 mL), and the reaction mixture was heated at 90°C until Na was completely consumed. The reaction mixture (6 mL) was then added to a solution of compound 78 (2.0 g, 4.75 mmol) in 2-propanol (20 mL) at 60°C. After 30 min, TLC showed that compound 78 was depleted, and the desired compound 81 was detected by LCMS. Water (20 mL) was added, and the reaction mixture was extracted with EtOAc (20 mL × 3). The combined organic phase was washed with brine, dried with anhydrous Na2SO4, and concentrated to give a brown oil that was purified by prep-HPLC (TFA) to give compound 81 (400 mg, 18.9 % yield) as a white solid. A mixture of compound 81 (400 mg, 0.90 mmol), Boc2O (237 mg, 1.09 mmol), and 10% Pd/C (40 mg) in MeOH (10 mL) was stirred under H2 atmosphere (15 psi) at 25°C for 2 hours. TLC showed that compound 81 was depleted, and the desired compound 67 was detected by LCMS. The reaction mixture was filtered through a Celite pad, and the filtrate was concentrated to give a crude product that was purified by silica gel chromatography (Petroleum ether/Ethyl acetate = 10/1 to 3/1) to afford compound 67 (103 mg, 27.9 % yield, 95.6% purity) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.39 (br. s., 1 H), 7.02–7.67 (m, 6 H), 3.97–4.21 (m, 1 H), 3.70–3.89 (m, 1 H), 3.38–3.52 (m, 1 H), 3.21 (d, J = 11.80 Hz, 2 H), 2.75 (br. s., 2 H), 1.47 (s, 9 H), 0.99 (d, J = 5.52 Hz, 6 H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 170.80, 168.90, 153.16, 141.14, 140.78, 138.14, 137.87, 130.80, 130.54, 129.03, 128.63, 128.14, 127.99, 127.84, 127.56, 126.72, 118.92, 118.21, 118.00, 117.53, 79.54, 71.74, 71.65, 66.64, 60.22, 30.61, 30.15, 29.92, 29.50, 28.58, 22.42, 22.07, 21.24, 14.56. ESI-TOF HRMS: m/z 411.2289 (C24H30N2O4 + H+ requires 411.2286).
4.1.47. Benzyl (5-(2-(1H-pyrrol-1-yl)acetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (82)
NaH (836.4 mg, 20.91 mmol) was added to a mixture of pyrrole (1.4 g, 20.91 mmol) in DMF (30 mL) in portions at 0°C under N2. The mixture was stirred at 0°C for 1 hour, then a solution of compound 78 (8.0 g, 19.01 mmol) in DMF (70 mL) was added dropwise at 0°C. The mixture was allowed to warm to room temperature (25°C) and stirred for 12 hours. LCMS showed that the reaction was complete. The mixture was diluted with water and extracted with EtOAc (50 mL × 3). The combined organic layers were washed twice with water, dried with anhydrous Na2SO4, and concentrated to afford crude product that was purified by silica gel chromatography (Petroleum ether/Ethyl acetate = 4/1 to 3/2) to afford compound 82 (2.30 g, 5.1 mmol, 26.8% yield) as a yellow solid. LCMS: MS + 1 = 452.1. 1H NMR (400 MHz, chloroform-d) δ (ppm) 7.63-7.01 (m, 11H), 6.81 (d, J = 17.7 Hz, 1H), 6.47 (d, J = 8.9 Hz, 2H), 6.12 (t, J=2.0 Hz, 2H), 5.21 (d, J=16.9 Hz, 2H), 4.74-4.41 (m, 2H), 3.26-3.07 (m, 2H), 2.86-2.65 (m, 2H).
4.1.48. 1-(3-Amino-10,11-dihydro-5H-dibenzo[b,f]azepin-5-yl)-2-(1H-pyrrol-1-yl)ethan-1-one (83)
First, 10% Pd/C (200.00 mg) was added to a solution of compound 82 (2.30 g, 5.1 mmol) in EtOAc (50 mL) at 25°C. The mixture was degassed under vacuum and purged with H2. The mixture was then stirred under H2 (15 psi) for 4 hours. TLC (Petroleum ether/Ethyl acetate = 2/1) showed that the reaction was complete. After the solution was filtered over celite pad, the filtrate was concentrated to afford compound 83 (1.20 g, crude) as a yellow solid. LCMS: MS+1= 318.1.
4.1.49. tert-Butyl (5-(2-(1H-pyrrol-1-yl)acetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (68)
A solution of tert-butoxycarbonyl chloride (65 mg, 475 μmol) in THF (0.5 mL) was added dropwise to a solution of compound 83 (150 mg, 475 μmol) in THF (0.5 mL) at 0°C. A solution of Et3N (48 mg, 475 μmol) in THF (1 mL) was then added dropwise at 0°C. The mixture was allowed to warm to room temperature (25°C) and stirred for 2 hours. The mixture was filtered, and the filtrate was concentrated. The residue was purified by preparative TLC (petroleum ether/ethyl acetate = 2/1) to give compound 68 (103 mg, 247 μmol, 52.0% yield, 99.4% purity) as a white solid. 1H NMR (400 MHz, chloroform-d) δ (ppm) 7.64-6.98 (m, 6H), 6.62-6.37 (m, 3H), 6.12 (s, 2H), 4.75-4.51 (m, 2H), 3.24-3.08 (m, 2H), 2.82-2.67 (m, 2H), 1.53 (d, J = 12.4 Hz, 9H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 167.63, 153.24, 153.12, 140.83, 140.77, 140.63, 138.97, 138.20, 138.00, 135.57, 131.24, 130.84, 130.76, 129.31, 128.92, 128.87, 128.74, 128.23, 127.98, 127.82, 126.78, 122.38, 119.41, 118.11, 117.98, 108.04, 107.98, 79.77, 79.52, 51.04, 50.82, 30.55, 30.24, 29.83, 29.53, 28.59. ESI-TOF HRMS: m/z 440.1960 (C25H27N3O3 + Na+ requires 440.1952).
4.1.50. Cyclopropyl (5-(2-(1H-pyrrol-1-yl)acetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (69)
A solution of cyclopropyl chloroformate (58 mg, 475 μmol) in THF (0.5 mL) was added dropwise to a solution of compound 83 (150 mg, 475 μmol) in THF (0.5 mL) at 0°C. A solution of Et3N (48 mg, 475 μmol) in THF (1 mL) was then added dropwise at 0°C. The mixture was allowed to warm to room temperature (25°C) and stirred for 2 hours. The mixture was filtered, and the filtrate was concentrated. The residue was purified by preparative TLC (petroleum ether/ethyl acetate = 2/1) to give compound 69 (58 mg, 144.5 μmol, 30.4% yield, 95.1% purity) as a white solid. 1H NMR (400 MHz, chloroform-d) δ (ppm) 7.69 - 6.98 (m, 6H), 6.62 (br. s., 1H), 6.48 (d, J=7.5 Hz, 2H), 6.12 (s, 2H), 4.71-4.51 (m, 2H), 4.21-4.06 (m, 1H), 3.23-3.09 (m, 2H), 2.81-2.66 (m, 2H), 0.76 (d, J=12.4 Hz, 4H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 167.65, 154.43, 154.25, 140.77, 138.41, 138.03, 137.64, 135.62, 131.68, 130.99, 130.83, 129.36, 129.26, 128.82, 128.20, 128.02, 127.85, 126.81, 122.38, 119.50, 118.20, 108.05, 108.00, 55.39, 51.02, 50.85, 49.36, 49.23, 30.45, 30.19, 29.85, 29.60, 5.28. ESI-TOF HRMS: m/z 402.1821 (C24H23N3O3 + H+ requires 402.1819).
4.1.51. Cyclobutyl (5-(2-(1H-pyrrol-1-yl)acetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (70)
A solution of cyclobutyl chloroformate (64 mg, 475 μmol) in THF (0.5 mL) was added dropwise to a solution of compound 83 (150 mg, 475 μmol) in THF (0.5 mL) at 0°C. A solution of Et3N (48 mg, 475 μmol) in THF (1 mL) was then added dropwise at 0°C. The mixture was allowed to warm to room temperature (25°C) and stirred for 2 hours. The mixture was filtered, and the filtrate was concentrated. The residue was purified by preparative TLC (petroleum ether/ethyl acetate = 2/1) to give compound 70 (65 mg, 156.5 μmol, 32.9% yield, 96.1% purity) as a white solid. 1H NMR (400 MHz, chloroform-d) δ (ppm) 7.62-6.95 (m, 6H), 6.68-6.40 (m, 3H), 6.19-6.04 (m, 2H), 5.16-4.94 (m, 1H), 4.78-4.47 (m, 2H), 3.24-3.07 (m, 2H), 2.84-2.65 (m, 2H), 2.40 (br. s., 2H), 2.22-2.05 (m, 2H), 1.92-1.76 (m, 1H), 1.73-1.63 (m, 1H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 167.64, 153.29, 153.11, 140.77, 138.56, 138.02, 137.79, 135.60, 131.57, 130.94, 130.83, 129.35, 129.10, 128.92, 128.83, 128.21, 128.00, 127.84, 126.80, 122.38, 118.13, 108.04, 107.99, 68.60, 51.02, 50.84, 30.51, 30.20, 29.85, 29.58, 13.40. ESI-TOF HRMS: m/z 416.1978 (C25H25N3O3 + H+ requires 416.1976).
4.1.52. Cyclopentyl (5-(2-(1H-pyrrol-1-yl)acetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (71)
A solution of cyclopentyl chloroformate (71 mg, 475 μmol) in THF (0.5 mL) was added dropwise to a solution of compound 83 (150 mg, 475 μmol) in THF (0.5 mL) at 0°C. A solution of Et3N (48 mg, 475 μmol) in THF (1 mL) was then added dropwise at 0°C. The mixture was allowed to warm to room temperature (25°C) and stirred for 2 hours. The mixture was filtered, and the filtrate was concentrated. The residue was purified by preparative TLC (petroleum ether/ethyl acetate = 2/1) to give compound 71 (87 mg, 202.7 μmol, 42.7% yield, 97.8% purity) as a white solid. 1H NMR (400 MHz, chloroform-d) δ (ppm) 7.72-6.97 (m, 6H), 6.69-6.40 (m, 3H), 6.12 (s, 2H), 5.21 (d, J = 15.8 Hz, 1H), 4.79-4.47 (m, 2H), 3.27-3.07 (m, 2H), 2.82-2.64 (m, 2H), 2.04-1.64 (m, 8H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 167.64, 153.88, 153.74, 140.79, 138.73, 138.01, 135.61, 131.41, 130.95, 130.84, 129.33, 128.98, 128.92, 128.82, 128.20, 128.00, 127.83, 126.80, 122.37, 119.42, 118.10, 108.05, 107.99, 77.37, 77.21, 55.39, 51.03, 50.83, 32.77, 30.49, 30.22, 29.83, 29.58, 23.73. ESI-TOF HRMS: m/z 430.2137 (C26H27N3O3 + H+ requires 430.2132).
4.1.53. Cyclohexyl (5-(2-(1H-pyrrol-1-yl)acetyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate (72)
A solution of cyclohexyl chloroformate (77 mg, 475 μmol) in THF (0.5 mL) was added dropwise to a solution of compound 83 (150 mg, 475 μmol) in THF (0.5 mL) at 0°C. A solution of Et3N (48 mg, 475 μmol) in THF (1 mL) was then added dropwise at 0°C. The mixture was allowed to warm to room temperature (25°C) and stirred for 2 hours. The mixture was filtered, and the filtrate was concentrated. The residue was purified by preparative TLC (petroleum ether/ethyl acetate = 2/1) to give compound 72 (134 mg, 302.3 μmol, 63.6% yield, 98.0% purity) as a white solid. 1H NMR (400 MHz, chloroform-d) δ (ppm) 7.67-6.95 (m, 6H), 6.67-6.39 (m, 3H), 6.12 (s, 2H), 4.82-4.49 (m, 3H), 3.27-3.03 (m, 2H), 2.85-2.63 (m, 2H), 2.01-1.86 (m, 2H), 1.83-1.71 (m, 2H), 1.55-1.28 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 165.25, 151.21, 151.06, 138.39, 138.32, 136.34, 135.64, 135.58, 133.24, 129.02, 128.59, 128.51, 128.45, 126.95, 126.61, 126.41, 125.81, 125.62, 125.44, 124.41, 119.98, 117.06, 115.76, 105.66, 105.61, 70.59, 70.43, 48.63, 48.45, 29.67, 28.09, 27.83, 27.45, 27.20, 22.98, 21.49. ESI-TOF HRMS: m/z 444.2290 (C27H29N3O3 + H+ requires 444.2289).
4.2. Biology
Materials: Tb-anti-GST antibody, GST-hCAR-LBD, Fluorescein-PGC1α coactivator peptide, TR-FRET coregulator buffer G, and 1 M DTT were purchased from Invitrogen (Carlsbad, CA, USA). Clotrimazole was purchased from Sigma (St. Louis, MO, USA). Dimethyl sulfoxide (DMSO) was purchased from Fisher Scientific (Pittsburgh, PA, USA). Black 384-well low-volume assay plates were purchased from Corning (Tewksbury, MA, USA). Chemicals 10, 13, 14, 15, 19, and 20 were purchased from ChemDiv (San Diego, CA, USA). Chemicals 11, 12, 16, 17, and 18 were purchased from ChemBridge (San Diego, CA, USA).
4.2.1. CAR-mediated fluorescent PGC1α coactivator recruitment/repression assay
In black 384-well low-volume assay plates, titrations of chemicals (final concentrations in assay: clotrimazole, 1-to-3 dilutions from 42 μM to 0.71 nM for 11 concentration levels; other compounds, 1-to-3 dilutions from 70 μM to 1.18 nM for 11 concentration levels; DMSO, 0.7%) were mixed with fluorescein-PGC1α coactivator peptide (125 nM), GST-hCAR-LBD (5 nM), and Tb-anti-GST antibody (5 nM) in TR-FRET coregulator buffer G supplemented with 5 mM DTT at 20 μl per well. In addition, DMSO (final concentration: 0.7%) and clotrimazole (final concentration: 42 μM with 0.7% DMSO) were included in each plate and served as negative control (0% Inhibition) and positive control (100% Inhibition), respectively. The assay plates were then briefly spun down and incubated for 1 hour at room temperature (25°C). The TR-FRET signals for individual wells were collected by using a PHERAstar FS plate reader (BMG Labtech; Durham, NC, USA) with a 340-nm excitation filter, 100-μs delay time, and 200-μs integration time to measure the fluorescence emission ratio (10,000 × 520 nm/490 nm). The data were then normalized to positive control (42 μM clotrimazole, 100% inhibition) and negative control (DMSO, 0% inhibition) values by using Equation 1 to derive the %Inhibition for individual chemicals at respective concentrations.
| Equation 1 |
Where applicable, the data were fit into a sigmoidal dose-response equation to derive IC50 values by using the graphic software GraphPad Prism 5.04 (GraphPad Software, La Jolla, CA, USA).
4.3. Molecular Modeling
The hCAR-LBD protein crystal structure was obtained from the RCSB Protein Data Bank (http://www.rcsb.org) in PDB format (PDB code 1XVP) [52]. All water molecules and ligand from the protein structure were removed using the molecular graphics systems Pymol (http://www.pymol.org). The 3D-ligand structures with energy-minimization were generated in ChemBio3D (CambridgeSoft Corporation). The PDBQT files for docking (protein and ligands) were created in AutoDockTools (ADT) version 1.5.6 (http://mgltools.scripps.edu). The protein search space with XYZ dimensions 22 Å × 22 Å × 22 Å was centered at coordinates 34.231 (x), 59.568 (y), 78.066 (z). Docking of the ligands to hCAR-LBD was carried out with the program AutoDock Vina version 1.1.1 [53], with the exhaustiveness value set to 300. The docking results were analyzed and the figures created in Pymol with assistance of Ligplot+ [54].
Supplementary Material
Highlights.
We study a novel scaffold in CINPA1 to discover better CAR inverse agonists
We have obtained, or designed and synthesized 54 analogs of CINPA1
We have evaluated the CAR inverse agonistic activity of all analogs
Chemical 72 is the most potent CAR inverse agonist so far (IC50 = 11.7 nM)
This is the first chemical endeavor in systematically exploring CAR inverse agonism
Acknowledgments
We thank WuXi AppTec for technical assistance in chemical synthesis; Cherise Guess, PhD, ELS, for editing the manuscript; and members of the Chen laboratory for valuable discussions. This work was supported by the American Lebanese Syrian Associated Charities (ALSAC) and the National Institutes of Health [Grants RO1 GM086415, RO1 GM110034, and P30 CA21765].
Abbreviations
- CAR
constitutive androstane receptor
- PXR
pregnane X receptor
- NR
nuclear receptor
- CYP2Bs
cytochrome P450, family 2, subfamily B proteins
- CYP2Cs
cytochrome P450, family 2, subfamily C proteins
- CYP3As
cytochrome P450, family 3, subfamily A proteins
- MDR1
multi-drug resistance protein 1
- MRPs
multi-drug resistance–associated proteins
- TCPOBOP
1,4-bis-[2-(3,5-dichloropyridyloxy)]benzene
- DBD
DNA-binding domain
- LBD
ligand-binding domain
- PK11195
1-(2-Chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinolinecarboxamide
- IC50
half maximal inhibitory concentration
- Ki
binding affinity of an inhibitor
- EE2
17α-ethynyl-3,17β-estradiol
- S07662
N-[(2-Methyl-3-benzofuranyl)methyl]-N′-(2-thienylmethyl)urea
- TO901317 (T0901317)
N-(2,2,2-trifluoroethyl)-N-[4-[2,2,2-trifluoro-1-hydroxy-1-(trifluoromethyl)ethyl]phenyl]-benzenesulfonamide
- LY2090314
3-(9-fluoro-2-(piperidine-1-carbonyl)-1,2,3,4-tetrahydro-[1,4]diazepino[6,7,1-hi]indol-7-yl)-4-(imidazo[1,2-a]pyridin-3-yl)-1H-pyrrole-2,5-dione
- AITC
allyl isothiocyanate
- CINPA1
ethyl (5-(diethylglycyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate
- T3P
propylphosphonic anhydride
- DMF
dimethylformamide
- NaHMDS
sodium bis(trimethylsilyl)amide
- THF
tetrahydrofuran
- SRC1
nuclear receptor coactivator 1
- PGC1α
peroxisome proliferator-activated receptor gamma, coactivator 1 alpha
- SAR
structure-activity relationship
- DMSO
dimethyl sulfoxide
- GST
glutathione S-transferase
- TR-FRET
time-resolved fluorescence resonance energy transfer
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Banerjee M, Robbins D, Chen T. Targeting xenobiotic receptors PXR and CAR in human diseases. Drug Discov Today. 2015;20:618–628. doi: 10.1016/j.drudis.2014.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Banerjee M, Robbins D, Chen T. Modulation of xenobiotic receptors by steroids. Molecules. 2013;18:7389–7406. doi: 10.3390/molecules18077389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chai X, Zeng S, Xie W. Nuclear receptors PXR and CAR: implications for drug metabolism regulation, pharmacogenomics and beyond. Expert Opin Drug Metab Toxicol. 2013;9:253–266. doi: 10.1517/17425255.2013.754010. [DOI] [PubMed] [Google Scholar]
- 4.Wang YM, Ong SS, Chai SC, Chen T. Role of CAR and PXR in xenobiotic sensing and metabolism. Expert Opin Drug Metab Toxicol. 2012;8:803–817. doi: 10.1517/17425255.2012.685237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lamba JK. Pharmacogenetics of the constitutive androstane receptor. Pharmacogenomics. 2008;9:71–83. doi: 10.2217/14622416.9.1.71. [DOI] [PubMed] [Google Scholar]
- 6.Cherian MT, Lin W, Wu J, Chen T. CINPA1 Is an Inhibitor of Constitutive Androstane Receptor (CAR) that Does Not Activate Pregnane X Receptor (PXR) Mol Pharmacol. 2015;87:878–889. doi: 10.1124/mol.115.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang J, Huang W, Chua SS, Wei P, Moore DD. Modulation of acetaminophen-induced hepatotoxicity by the xenobiotic receptor CAR. Science. 2002;298:422–424. doi: 10.1126/science.1073502. [DOI] [PubMed] [Google Scholar]
- 8.Takwi AA, Wang YM, Wu J, Michaelis M, Cinatl J, Chen T. miR-137 regulates the constitutive androstane receptor and modulates doxorubicin sensitivity in parental and doxorubicin-resistant neuroblastoma cells. Oncogene. 2014;33:3717–3729. doi: 10.1038/onc.2013.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tamura K, Inoue K, Takahashi M, Matsuo S, Irie K, Kodama Y, Gamo T, Ozawa S, Yoshida M. Involvement of constitutive androstane receptor in liver hypertrophy and liver tumor development induced by triazole fungicides. Food Chem Toxicol. 2015;78C:86–95. doi: 10.1016/j.fct.2015.01.021. [DOI] [PubMed] [Google Scholar]
- 10.Sakamoto Y, Inoue K, Takahashi M, Taketa Y, Kodama Y, Nemoto K, Degawa M, Gamou T, Ozawa S, Nishikawa A, Yoshida M. Different pathways of constitutive androstane receptor-mediated liver hypertrophy and hepatocarcinogenesis in mice treated with piperonyl butoxide or decabromodiphenyl ether. Toxicol Pathol. 2013;41:1078–1092. doi: 10.1177/0192623313482055. [DOI] [PubMed] [Google Scholar]
- 11.Huang W, Zhang J, Washington M, Liu J, Parant JM, Lozano G, Moore DD. Xenobiotic stress induces hepatomegaly and liver tumors via the nuclear receptor constitutive androstane receptor. Mol Endocrinol. 2005;19:1646–1653. doi: 10.1210/me.2004-0520. [DOI] [PubMed] [Google Scholar]
- 12.Kanno Y, Suzuki M, Miyazaki Y, Matsuzaki M, Nakahama T, Kurose K, Sawada J, Inouye Y. Difference in nucleocytoplasmic shuttling sequences of rat and human constitutive active/androstane receptor. Biochim Biophys Acta. 2007;1773:934–944. doi: 10.1016/j.bbamcr.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 13.Kojetin DJ, Burris TP. Small molecule modulation of nuclear receptor conformational dynamics: implications for function and drug discovery. Mol Pharmacol. 2013;83:1–8. doi: 10.1124/mol.112.079285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Timsit YE, Negishi M. CAR and PXR: the xenobiotic-sensing receptors. Steroids. 2007;72:231–246. doi: 10.1016/j.steroids.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jinno H, Tanaka-Kagawa T, Hanioka N, Ishida S, Saeki M, Soyama A, Itoda M, Nishimura T, Saito Y, Ozawa S, Ando M, Sawada J. Identification of novel alternative splice variants of human constitutive androstane receptor and characterization of their expression in the liver. Mol Pharmacol. 2004;65:496–502. doi: 10.1124/mol.65.3.496. [DOI] [PubMed] [Google Scholar]
- 16.Carazo FA, Smutny T, Hyrsova L, Berka K, Pavek P. Chrysin, baicalein and galangin are indirect activators of the human constitutive androstane receptor (CAR) Toxicol Lett. 2015;233:68–77. doi: 10.1016/j.toxlet.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 17.Moore LB, Parks DJ, Jones SA, Bledsoe RK, Consler TG, Stimmel JB, Goodwin B, Liddle C, Blanchard SG, Willson TM, Collins JL, Kliewer SA. Orphan nuclear receptors constitutive androstane receptor and pregnane X receptor share xenobiotic and steroid ligands. J Biol Chem. 2000;275:15122–15127. doi: 10.1074/jbc.M001215200. [DOI] [PubMed] [Google Scholar]
- 18.di MA, De ME, Ascenzi P, Marino M. Nuclear receptors CAR and PXR: Molecular, functional, and biomedical aspects. Mol Aspects Med. 2009;30:297–343. doi: 10.1016/j.mam.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 19.Moore LB, Maglich JM, McKee DD, Wisely B, Willson TM, Kliewer SA, Lambert MH, Moore JT. Pregnane X receptor (PXR), constitutive androstane receptor (CAR), and benzoate X receptor (BXR) define three pharmacologically distinct classes of nuclear receptors. Mol Endocrinol. 2002;16:977–986. doi: 10.1210/mend.16.5.0828. [DOI] [PubMed] [Google Scholar]
- 20.Jyrkkarinne J, Windshugel B, Makinen J, Ylisirnio M, Perakyla M, Poso A, Sippl W, Honkakoski P. Amino acids important for ligand specificity of the human constitutive androstane receptor. J Biol Chem. 2005;280:5960–5971. doi: 10.1074/jbc.M411241200. [DOI] [PubMed] [Google Scholar]
- 21.Dring AM, Anderson LE, Qamar S, Stoner MA. Rational quantitative structure-activity relationship (RQSAR) screen for PXR and CAR isoform-specific nuclear receptor ligands. Chem Biol Interact. 2010;188:512–525. doi: 10.1016/j.cbi.2010.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Molnar F, Kublbeck J, Jyrkkarinne J, Prantner V, Honkakoski P. An update on the constitutive androstane receptor (CAR) Drug Metabol Drug Interact. 2013;28:79–93. doi: 10.1515/dmdi-2013-0009. [DOI] [PubMed] [Google Scholar]
- 23.Huang W, Zhang J, Wei P, Schrader WT, Moore DD. Meclizine is an agonist ligand for mouse constitutive androstane receptor (CAR) and an inverse agonist for human CAR. Mol Endocrinol. 2004;18:2402–2408. doi: 10.1210/me.2004-0046. [DOI] [PubMed] [Google Scholar]
- 24.Lau AJ, Yang G, Rajaraman G, Baucom CC, Chang TK. Differential effect of meclizine on the activity of human pregnane X receptor and constitutive androstane receptor. J Pharmacol Exp Ther. 2011;336:816–826. doi: 10.1124/jpet.110.175927. [DOI] [PubMed] [Google Scholar]
- 25.Kublbeck J, Jyrkkarinne J, Molnar F, Kuningas T, Patel J, Windshugel B, Nevalainen T, Laitinen T, Sippl W, Poso A, Honkakoski P. New in vitro tools to study human constitutive androstane receptor (CAR) biology: discovery and comparison of human CAR inverse agonists. Mol Pharm. 2011;8:2424–2433. doi: 10.1021/mp2003658. [DOI] [PubMed] [Google Scholar]
- 26.Kanno Y, Tanuma N, Takahashi A, Inouye Y. TO901317, a potent LXR agonist, is an inverse agonist of CAR. J Toxicol Sci. 2013;38:309–315. doi: 10.2131/jts.38.309. [DOI] [PubMed] [Google Scholar]
- 27.Kanno Y, Tanuma N, Yatsu T, Li W, Koike K, Inouye Y. Nigramide J is a novel potent inverse agonist of the human constitutive androstane receptor. Pharmacol Res Perspect. 2014;2:2. doi: 10.1002/prp2.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zamek-Gliszczynski MJ, Mohutsky MA, Rehmel JL, Ke AB. Investigational small-molecule drug selectively suppresses constitutive CYP2B6 activity at the gene transcription level: physiologically based pharmacokinetic model assessment of clinical drug interaction risk. Drug Metab Dispos. 2014;42:1008–1015. doi: 10.1124/dmd.114.057018. [DOI] [PubMed] [Google Scholar]
- 29.Lim YP, Cheng CH, Chen WC, Chang SY, Hung DZ, Chen JJ, Wan L, Ma WC, Lin YH, Chen CY, Yokoi T, Nakajima M, Chen CJ. Allyl isothiocyanate (AITC) inhibits pregnane X receptor (PXR) and constitutive androstane receptor (CAR) activation and protects against acetaminophen- and amiodarone-induced cytotoxicity. Arch Toxicol. 2015;89:57–72. doi: 10.1007/s00204-014-1230-x. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Wang L, Hu M, Li Y, Wang S, Ding Q, Hu D, Yang H, Zhao J, Zhang L, Li Y. Method for synthesis of 5-acetyl-3-chloroiminodibenzyl. 102010349A. CN Patent. 2011 Apr 13;
- 31.Stark A, Wunderlich H, Ehben H, Mueller R, Oestreich E, Kuehne W. Preparation of 3-carbethoxyamino-5-dimethylaminoacetyl-10,11-dihydro-5H-dibenzo[b,f]azepine and its hydrochloride. 293346A5. DD Patent. 1991 Aug 29;
- 32.Wunderlich H, Stark A, Carstens E, Lohmann D, Gritsenko AN, Skoldinov AP. New derivatives of 10,11-dihydro-5H-dibenz[b,f]azepine with antiarrhythmic efficacy. Pharmazie. 1985;40:827–830. [PubMed] [Google Scholar]
- 33.Honda E, Ishichi Y, Kimura E, Yoshikawa M, Kanzaki N, Nakagawa H, Terao Y, Suzuki A, Kawai T, Arakawa Y, Ohta H, Terauchi J. Design, synthesis, and biological activities of 1-aryl-1,4-diazepan-2-one derivatives as novel triple reuptake inhibitors. Bioorg Med Chem Lett. 2014;24:3898–3902. doi: 10.1016/j.bmcl.2014.06.046. [DOI] [PubMed] [Google Scholar]
- 34.Ikegashira K, Oka T, Hirashima S, Noji S, Yamanaka H, Hara Y, Adachi T, Tsuruha J, Doi S, Hase Y, Noguchi T, Ando I, Ogura N, Ikeda S, Hashimoto H. Discovery of conformationally constrained tetracyclic compounds as potent hepatitis C virus NS5B RNA polymerase inhibitors. J Med Chem. 2006;49:6950–6953. doi: 10.1021/jm0610245. [DOI] [PubMed] [Google Scholar]
- 35.Guthrie DB, Geib SJ, Curran DP. Synthesis of highly enantioenriched 3,4-dihydroquinolin-2-ones by 6-exo-trig radical cyclizations of axially chiral alpha-halo-ortho-alkenyl anilides. J Am Chem Soc. 2009;131:15492–15500. doi: 10.1021/ja9066282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hromatka O, Knollmueller M, Maior KA, Gotschy F. 1-Acetylaminoanthraquinones with basic substituents. Monatsh Chem. 1969:1393–1399. [Google Scholar]
- 37.Flor PJ, Marzinzik A, Nozulak J, Ofner S, Roy B, Spanka C. Preparation of substituted diamines as mGluR7 regulators. 2007025709A2. WO Patent. 2007 Mar 8;
- 38.Song HY, Ngai MH, Song ZY, MacAry PA, Hobley J, Lear MJ. Practical synthesis of maleimides and coumarin-linked probes for protein and antibody labelling via reduction of native disulfides. Org Biomol Chem. 2009;7:3400–3406. doi: 10.1039/b904060a. [DOI] [PubMed] [Google Scholar]
- 39.Muto T, Tanaka T, Maruoka H, Imajo S, Tomimori Y, Sato K, Yagi T. Preparation of 1,4-diazepane-3,5-dione derivatives as chymase inhibitors and pharmaceutical use thereof. 2007139230A1. WO Patent. 2007 Dec 6;
- 40.Kathuria A, Priya N, Chand K, Singh P, Gupta A, Jalal S, Gupta S, Raj HG, Sharma SK. Substrate specificity of acetoxy derivatives of coumarins and quinolones towards Calreticulin mediated transacetylation: investigations on antiplatelet function. Bioorg Med Chem. 2012;20:1624–1638. doi: 10.1016/j.bmc.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 41.Sams AG, Mikkelsen GK, Larsen M, Langgard M, Howells ME, Schroder TJ, Brennum LT, Torup L, Jorgensen EB, Bundgaard C, Kreilgard M, Bang-Andersen B. Discovery of phosphoric acid mono-{2-[(E/Z)-4-(3,3-dimethyl-butyrylamino)-3,5-difluoro-benzoylimino]-thiazol-3 -ylmethyl} ester (Lu AA47070): a phosphonooxymethylene prodrug of a potent and selective hA(2A) receptor antagonist. J Med Chem. 2011;54:751–764. doi: 10.1021/jm1008659. [DOI] [PubMed] [Google Scholar]
- 42.Zheng X, Bauer P, Baumeister T, Buckmelter AJ, Caligiuri M, Clodfelter KH, Han B, Ho YC, Kley N, Lin J, Reynolds DJ, Sharma G, Smith CC, Wang Z, Dragovich PS, Oh A, Wang W, Zak M, Gunzner-Toste J, Zhao G, Yuen PW, Bair KW. Structure-based identification of ureas as novel nicotinamide phosphoribosyltransferase (Nampt) inhibitors. J Med Chem. 2013;56:4921–4937. doi: 10.1021/jm400186h. [DOI] [PubMed] [Google Scholar]
- 43.Gale PA, Hiscock JR, Lalaoui N, Light ME, Wells NJ, Wenzel M. Benzimidazole-based anion receptors: tautomeric switching and selectivity. Org Biomol Chem. 2012;10:5909–5915. doi: 10.1039/c1ob06800h. [DOI] [PubMed] [Google Scholar]
- 44.Dietrich J, Hulme C, Hurley LH. The design, synthesis, and evaluation of 8 hybrid DFG-out allosteric kinase inhibitors: a structural analysis of the binding interactions of Gleevec, Nexavar, and BIRB-796. Bioorg Med Chem. 2010;18:5738–5748. doi: 10.1016/j.bmc.2010.05.063. [DOI] [PubMed] [Google Scholar]
- 45.Kim SH, Bok JH, Lee JH, Kim IH, Kwon SW, Lee GB, Kang SK, Park JS, Jung WH, Kim HY, Rhee SD, Ahn SH, Bae MA, Ha DC, Kim KY, Ahn JH. Synthesis and biological evaluation of cyclic sulfamide derivatives as 11beta-hydroxysteroid dehydrogenase 1 inhibitors. ACS Med Chem Lett. 2012;3:88–93. doi: 10.1021/ml200226x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen X, Duvadie R, Harrison T, Liu Q, Mao JYC, Patel S, Zecri F. Preparation of tetrahydropyran compounds as DGAT1 inhibitors. 2013163508A1. WO Patent. 2013 Oct 31;
- 47.Prakash TP, Swayze EE, Lima WF, Kinberger GA. Antisense oligonucleotide-conjugate complexes for diagnostic and therapeutic use. 2013033230A1. WO Patent. 2013 Mar 7;
- 48.Seganish WM, Mowery ME, Riggleman S, DeShong P. Palladium-catalyzed homocoupling of aryl halides in the presence of fluoride. Tetrahedron. 2005;61:2117–2121. [Google Scholar]
- 49.Foks H, Czarnocka-Janowicz A, Rudnicka W, Trzeciak H. Synthesis of new 5-substituted 1,2,4-triazole-3-thione derivatives, Phosphorus. Sulfur and Silicon and the Related Elements. 2000;164:67–81. [Google Scholar]
- 50.Guo X, Yang Q, Xu J, Zhang L, Chu H, Yu P, Zhu Y, Wei J, Chen W, Zhang Y, Zhang X, Sun H, Tang Y, You Q. Design and bio-evaluation of indole derivatives as potent Kv1.5 inhibitors. Bioorg Med Chem. 2013;21:6466–6476. doi: 10.1016/j.bmc.2013.08.041. [DOI] [PubMed] [Google Scholar]
- 51.Zhu J, Lin JB, Xu YX, Shao XB, Jiang XK, Li ZT. Hydrogen-bonding-mediated anthranilamide homoduplexes. Increasing stability through preorganization and iterative arrangement of a simple amide binding site. J Am Chem Soc. 2006;128:12307–12313. doi: 10.1021/ja064218i. [DOI] [PubMed] [Google Scholar]
- 52.Xu RX, Lambert MH, Wisely BB, Warren EN, Weinert EE, Waitt GM, Williams JD, Collins JL, Moore LB, Willson TM, Moore JT. A structural basis for constitutive activity in the human CAR/RXRalpha heterodimer. Mol Cell. 2004;16:919–928. doi: 10.1016/j.molcel.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 53.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.