

Abstract
Glucose-stimulated insulin secretion (GSIS) involves interplay between metabolic and cationic events. Several lines of evidence suggests novel regulatory roles for small G proteins (Rac1, Cdc42, Rab27A) in cytoskeletal remodeling and docking of insulin granules on the plasma membrane for insulin secretion. Emerging evidence implicates novel roles for post-translational prenylation (farnesylation and geranylgeranylation) of G proteins for their targeting to appropriate membranous compartments. While several recent studies were focused on prenylating enzymes in the islet β-cell, a significant knowledge gap exists on regulatory roles and function of enzymes that mediate intracellular generation of prenyl pyrophosphate substrates (farnesyl and geranylgeranyl pyrophosphates) for prenyltransferases. Recent work published in the Journal of Pathology by Jiang and associates highlights requisite roles for geranylgeranyl pyrophosphate synthase (GGPPS) in islet β-cell function in health and diabetes. These studies are timely and will form the basis for a series of new investigations to further validate roles for G protein prenylation in GSIS under physiological conditions. They also pave the path toward the identification of potential defects in these signaling pathways in β-cell models of impaired insulin secretion including metabolic stress and diabetes.
Keywords: Glucose, G proteins, Geranylgeranylation, Rab27A, Islet β-cell, Insulin Secretion, Diabetes
Introduction
Insulin secretion from pancreatic β-cells is regulated principally by the ambient concentrations of glucose. However, the molecular and cellular mechanisms underlying the stimulus-secretion coupling of glucose-stimulated insulin secretion (GSIS) remain only partially understood. It is well established that the signaling steps involved in GSIS require well-regulated trafficking of insulin-laden secretory granules for their docking and fusion with the plasma membrane [1]. Emerging evidence also suggests that such cellular events are under the fine control of small GTP-binding proteins (G proteins), which have been implicated in cytoskeletal remodeling to facilitate granule movement toward the plasma membrane [2,3]. Published evidence from multiple laboratories demonstrated critical involvement of G proteins Rac1, Cdc42, ARF6 and Rabs in GSIS from normal rat islets, human islets, and clonal β-cells [2–5].
G proteins undergo post-translational prenylation, which “glues” them to the membrane
The majority of small G proteins and the γ-subunits of trimeric G proteins undergo post-translational modification steps (farnesylation and geranylgeranylation) at their C-terminal cysteine residues. Such modifications are requisite for targeting the modified G proteins to relevant membranous compartments (“gluing”) for optimal interaction with their effector proteins [3,6,7]. The prenylating enzymes farnesyltransferase (Ftase) and geranylgeranyltransferase (GGTase) catalyze the incorporation of either a 15-carbon (farnesyl) or a 20-carbon (geranylgeranyl) derivative of mevalonic acid into the C-terminal cysteine residues of the candidate proteins [3,5,7]. To date, three distinct protein prenyl transferases have been described in the literature. FTase and GGTase-I are also referred to as CAAX PPTases because they share the CAAX motif around the C-terminal cysteine region of their substrate proteins. GGTase-II (Rab GGTase) prenylates the Rab subfamily of proteins at a different motif, and hence this group of PPTases is often referred to as non-CAAX PPTases [3,6,7]. Recent studies have characterized FTase, GGTase-I and GGTase-II in a variety of pancreatic β-cells [3,7]. Pharmacological inhibition or siRNA-mediated knockdown of subunits of FTase, GGTase-I and GGTase-II culminates in suppression of GSIS suggesting critical roles for prenylation in islet function [3,5,7]. Interestingly, however, very little is known to date about the enzyme that mediates formation of geranylgeranyl-PP (geranylgeranyl-PP synthase; GGPPS; Figure 1).
Figure 1. Regulatory roles GGPPS in GSIS.
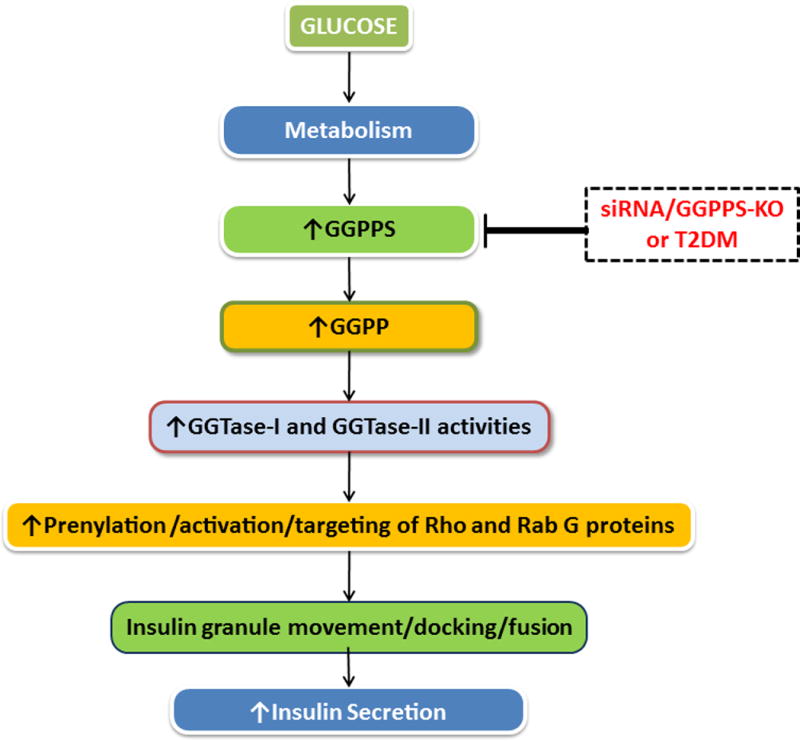
Glucose metabolism in pancreatic islet β-cells leads to catalytic activation of GGPPS resulting in increased intracellular GGPP, which, in turn, is utilized by GGTases to geranylgeranylate candidate G proteins. GGTase-I geranylgeranylates small G proteins including Rac1, Cdc42, Rho and Rap1, whereas GGTase-II catalyzes geranylgeranylation of Rab subfamily of G proteins (Rab27A; ref. [8]). Numerous studies involving pharmacological, biochemical, molecular biological and physiological approaches have demonstrated that geranylgeranylation of these G proteins is requisite for actin cytoskeletal remodeling, insulin granule transport and docking on to the plasma membrane for fusion and exocytotic secretion of insulin under physiological insulin secretion. In support of this original hypothesis [3,5,7] using a variety of experimental approaches (siRNA and conditional deletion in pancreatic β-cells) Jiang et al [8] have demonstrated that functional inactivation of GGPPS leads to reduced intracellular GGPP, altered geranylgeranylation and membrane association of Rab27A and impaired insulin secretion in in vitro and in vivo models of metabolic stress and T2DM.
Evidence for potential alterations in G-protein prenylation in the diabetic β-cell
Despite considerable evidence in support of the overarching hypothesis that G protein activation is essential for GSIS to occur, very little is known about potential alterations in G protein function in the islet β-cell under the duress of metabolic stress and diabetes. In this context, Jiang et al have recently published a paper in this journal [8] demonstrating critical roles for GGPPS in islet function in health and diabetes. Using a variety of experimental approaches these investigators demonstrated novel regulatory roles for GGPPS in islet β-cell function during the onset of T2DM. Catalytic activation of GGPPS in islets from db/db mice is increased during the initial compensatory period followed by a sharp decline during the onset of insulin secretory abnormality. Conditional deletion of GGPPS in the islet β-cell resulted in depletion of intracellular GGPP, geranylgeranylation and membrane targeting of Rab27A, and reduced the number of insulin granules beneath the plasma membrane suggesting significant defects in granule docking in GGPPS-null mice; these defects culminated in attenuated GSIS. Over-expression of GGPPS or provision of exogenous GGPP significantly restored GSIS in islets from GGPPS-null mice, affirming the postulation that defects in GGPPS pathway (generation of GGPP, geranylgeranylation of Rab27A and granule docking) cause islet β-cell dysfunction in diabetes [8].
Perspective and future directions
The observations by Jiang et al [8] will undoubtedly form the basis for immediate investigations in the field, which might fill the knowledge gaps in our current understanding of regulatory roles of G proteins in islet biology. Some of these are highlighted below.
First, it is likely that decreased levels of GGPP due to altered expression and defective catalytic function of GGPPS under diabetic conditions could result in impaired prenylation of other key signaling proteins mediated by GGTase-I (e.g., Cdc42, Rac1, Rho and Rap1; Figure 1), which have been implicated in islet function including GSIS [2,3,7]. Thus, functional inactivation of these proteins consequential to attenuated GGPPS could contribute to islet dysregulation.
Second, geranylgeranylation of other Rab G proteins, which are substrates for GGTase-II could also be affected resulting in the dysregulation of the islet β-cell in diabetes [3–5]. More importantly, in addition to defects in geranylgeranylation and membrane association of specific G proteins (Rab27A) in GGPPS-null mouse islets, it is necessary to quantify their functional activation (i.e., their GTP-bound conformation). It is noteworthy that recent studies from our laboratory have demonstrated significant activation of Rac1, a geranylgeranylated protein, in in vitro and in vivo models of glucolipotoxicity, exposure to pro-inflammtory cytokines and T2DM [9, 10]. These observations alongside the findings of Jiang and coworkers [8] emphasize the need for quantification of G protein activation in model systems in which the catalytic functions of GGPPS, and potentially FPS, are compromised consequential to metabolic stress and diabetes.
Third, while the investigations of Jiang and associates have focused on GGPPS, it would be interesting to critically evaluate the kinetics of FPPS activity and the intracellular concentrations of FPP in normal and diabetic islet β-cells to determine contributory roles of these signaling proteins in islet function in health and diabetes. It should be also kept in mind that decreased GGPPS activity, as shown in the diabetic islet β-cell, could result in increased levels (accumulation) of FPP, which, in turn could lead to farnesylation of proteins (Ras; [3, 7]). It is notable that the studies of Jiang and colleagues have identified no significant defects in farnesylation and membrane association of H-Ras, a farnesylated protein, in GGPPS-depleted islet β-cells [8]. Interestingly, recent studies from this group [11] have demonstrated a significant increase in farnesylation of H-Ras in GGPPS −/− Sertolli cells, thus suggesting clear differences in the two model systems. Again, as stated above, it is critical to quantify the degree of activation of these G proteins (farnesylated and geranylgeranylated) in GGPPS- and FPPS- depleted pancreatic β-cells to further validate regulatory roles of these signaling cascades in the induction of islet dysfunction under conditions of metabolic stress, pre-diabetic and diabetic states.
In conclusion, data accrued from the investigations of Jiang et al [8] provide compelling evidence to implicate defects in roles for G protein geranylgeranylation in islet function, including translocation and docking of insulin-laden secretory granules to the plasma membrane for their fusion and secretion of insulin. They further affirm roles of geranylgeranylated Rab27A in these cellular events. More importantly, these findings also suggest significant defects in these signaling mechanisms in islets derived from an animal model of T2DM. Additional investigations along these lines should provide valuable insights in not only identifying novel targets that regulate islet function under normal physiological conditions, but also potential abnormalities in the functions of those proteins that could lead to dysregulation of insulin secretion and islet function under the duress of metabolic stress and diabetes.
Acknowledgments
The author thanks his former and current laboratory associates and collaborators for their contributions to the field. This research was supported in part by a Merit Review award from the Department of Veterans Affairs and the National Institutes of Health. AK is also the recipient of a Senior Research Career Scientist Award from the Department of Veterans Affairs.
Footnotes
Author contribution statement: AK wrote this commentary
Conflicts of interest: The author declares no conflict of interest.
References
- 1.Prentki M, Matschinsky FM, Madiraju SR. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 2013;18:162–185. doi: 10.1016/j.cmet.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 2.Wang Z, Thurmond DC. Mechanisms of biphasic insulin-granule exocytosis-roles of the cytoskeleton, small GTPases and SNARE proteins. J Cell Sci. 2009;122:893–903. doi: 10.1242/jcs.034355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kowluru A. Small G proteins in islet beta-cell function. Endocrine Rev. 2010;31:52–78. doi: 10.1210/er.2009-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cazares VA, Subramani A, Saldate JJ, et al. Distinct actions of Rab3 and Rab27 GTPases on late stages of exocytosis of insulin. Traffic. 2014;15:997–1015. doi: 10.1111/tra.12182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arora DK, Syed I, Machhadieh B, et al. Rab-geranylgeranyl transferase regulates glucose-stimulated insulin secretion from pancreatic beta-cells. Islets. 2012;4:354–358. doi: 10.4161/isl.22538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palsuledesai CC, Distefano MD. Protein prenylation: Enzymes, therapeutics, and biotechnology applications. ACS Chem Biol. 2015;10:51–62. doi: 10.1021/cb500791f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kowluru A, Kowluru RA. Protein prenylation in islet beta-cell function in health and diabetes: Putting the pieces of the puzzle together. Biochem Pharmacol. 2015 doi: 10.1016/j.bcp.2015.07.004. [in press] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang S, Shen D, Jia W-J, et al. GGPPS mediated Rab27A geranylgeranylation regulates beta-cell dysfunction during type 2 diabetes development via affecting insulin granule docked pool formation. J Pathol. 2015 doi: 10.1002/path.4652. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 9.Kowluru A. Friendly, and not so friendly, roles of Rac1 in islet beta-cell function: Lessons learnt from pharmacological and molecular biological approaches. Biochem Pharmacol. 2011;81:965–975. doi: 10.1016/j.bcp.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Syed I, Kyathanahalli CN, Jayaram B, et al. Increased phagocyte-like NADPH oxidase and ROS generation in type 2 diabetic ZDF rat and human islets: role of Rac1-JNK1/2 signaling pathway in mitochondrial dysregulation in the diabetic islet. Diabetes. 2011;60:2843–2852. doi: 10.2337/db11-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang X-X, Ying P, Diao F, et al. Altered protein prenylation in Sertolli cells is associated with adult infertility resulting from childhood mumps infection. J Exp Med. 2013;210:1559–1574. doi: 10.1084/jem.20121806. [DOI] [PMC free article] [PubMed] [Google Scholar]