

Abstract
Background
Although most prior cognitive studies of β-amyloidopathy in Parkinson disease (PD) focused on cortical plaque deposition, recent post-mortem studies point to an important role of striatal β-amyloid plaque deposition.
Objective
To investigate the relative contributions of striatal and cortical β-amyloidopathy to cognitive impairment in PD.
Methods
Patients with PD (n=62; age 68.9±6.4 years, Hoehn and Yahr stage 2.7±0.5, Montreal Cognitive Assessment score 25.2±3.0) underwent [11C]Pittsburgh compound B β-amyloid, [11C]dihydrotetrabenazine monoaminergic and [11C]methyl-4-piperidinyl propionate acetylcholinesterase brain positron emission tomography imaging and neuropsychological assessment. [11C]Pittsburgh compound B β-amyloid data from young to middle-aged healthy subjects were used to define elevated [11C]Pittsburgh compound B binding in the patients.
Results
Elevated cortical and striatal β-amyloid deposition were present in 38% and 16%, respectively, of this predominantly non-demented cohort of patients with PD. Increased striatal β-amyloid deposition occurred in half of all subjects with increased cortical β-amyloid deposition. In contrast, increased striatal β-amyloid deposition did not occur in the absence of increased cortical β-amyloid deposition. Analysis of covariance using global composite cognitive z-scores as the outcome parameter showed significant regressor effects for combined striatal and cortical β-amyloidopathy (F=4.18, P=0.02) after adjusting for covariate effects of cortical cholinergic activity (F=5.67, P=0.02), caudate nucleus monoaminergic binding, duration of disease and age (total model: F=3.55, P=0.0048). Post-hoc analysis showed significantly lower cognitive z-score for combined striatal and cortical β-amyloidopathy compared to cortical-only β-amyloidopathy and non-β-amyloidopathy subgroups.
Conclusions
The combined presence of striatal and cortical β-amyloidopathy is associated with greater cognitive impairment than cortical β-amyloidopathy alone in PD.
Keywords: Acetylcholinesterase, β-amyloid, cognitive impairment, cortex, dopamine, Parkinson disease, PET, striatum
Introduction
Proteinopathic degeneration of important subcortical projection systems, including nigrostriatal dopaminergic and corticopetal cholinergic projections, limbic and cortical pathology associated with α-synuclein deposits (Lewy bodies and Lewy neurites; α–synucleinopathy), and Alzheimer fibrillary β-amyloid plaque deposition (β-amyloidopathy) likely contribute to cognitive decline in Parkinson disease (PD) 1, 2. Information on the relative frequencies of different proteinopathies in PD comes mainly from post-mortem studies. One recent large post-mortem series of PD dementia (PDD) subjects, for example, reports "pure" or exclusive Lewy body pathology in 38%, combined Lewy body pathology and β-amyloid plaques in 59%, and combined Lewy bodies, β-amyloid plaques and neurofibrillary tangles in 3% 3.
Most in vivo imaging and post-mortem neuropathological studies of comorbid β-amyloidopathy in relationship to cognitive functions in PD focused on cortical plaque deposition. We previously reported, for example, a significant relationship between in vivo measures of elevated cortical β-amyloid deposition and greater cognitive impairment in PD 4. Recent neuropathological studies, however, point to an important role for striatal β-amyloidopathy in the development of cognitive impairment in PD 5-7, with evidence indicating that striatal β-amyloid deposition is significantly greater in demented than non-demented PD patients 8. These post-mortem findings suggest that both cortical and also striatal β-amyloidopathy in PD have pathophysiological impact in PD. Using in vivo β-amyloid PET imaging, we investigated the relative respective contributions of cortical and striatal β-amyloidopathy to cognitive impairment in PD while controlling for the magnitude of cortical cholinergic and nigrostriatal dopaminergic neurodegenerations.
Subjects and Methods
Subjects
This cross-sectional study involved 62 subjects with PD (46 males, 16 females), mean age 68.9±6.4 (range 55-84) and mean duration of motor disease of 6.5±4.7 (range 0.5-20) years. PD subjects met the UK Parkinson’s Disease Society Brain Bank clinical diagnostic criteria 9. The diagnosis of PD was consistent with the presence of typical nigrostriatal dopaminergic denervation as revealed by [11C]dihydrotetrabenazine (DTBZ) vesicular monoamine type 2 (VMAT2) PET imaging in all subjects. The majority of subjects were on dopaminergic drugs but none were on anti-cholinergic or cholinergic drugs. The mean Hoehn and Yahr (HY) stage was 2.7±0.5 (range 1.5-4.0) 10. The mean Movement Disorder Society-revised Unified Parkinson Disease Rating Scale (UPDRS) in the practically defined “off” state (i.e., the examination was performed in the morning after overnight withholding of dopaminergic medication) was 36.3±14.3 (range 15-70) 11. The subjects in this study were recruited based on at least one recognized risk factor for PDD, such as having cognitive complaints, older age, or greater motor symptom severity, but no existing clinical PDD diagnosis. The mean Montreal Cognitive Assessment (MOCA) test score was 25.2±3.0 (range 15-30) 12.
This study was approved by and study procedures were followed in accordance with the ethical standards of the Institutional Review Board of the University of Michigan. Written informed consent was obtained from all subjects.
Each subject underwent a detailed cognitive examination (while on dopaminergic medications) following the approach previously reported for cognitive impairment in PD 13. These tests included measures of verbal memory: California Verbal Learning Test 14; executive/reasoning functions: WAIS III Picture Arrangement test 15, and Delis-Kaplan Executive Function System Sort ing Test 16; attention/psychomotor speed as absolute time on the Stroop 1 test 17; and visuospatial function: Benton Judgment of Line Orientation test 18. Composite z-scores were calculated for these different cognitive domains based on normative data from a database of non-PD control subjects with similar age, education and gender distribution as the PD subjects in our center 19. A global composite z-score was calculated as the mean of the domain z-scores. After completion of detailed neuropsychological testing and assessment of functional abilities three subjects met criteria for mild dementia.
All subjects in this study completed three PET studies: [11C]Pittsburgh compound B (PIB) β-amyloid, [11C]DTBZ monoaminergic, and [11C]methyl-4-piperidinyl propionate (PMP) acetylcholinesterase brain PET. Cognitive and motor (axial motor impairment, including freezing of gait) correlates of cortical (but not striatal) β-amyloidopathy of sub-sets of the subjects from the present study have been reported previously 4, 20, 21. Furthermore, detailed analyses regarding relative cholinergic and dopaminergic determinants of cognitive impairment in PD from an expanded pooled data set from our center has also been previously reported 22.
Imaging techniques
MRI was performed on a 3 Tesla Philips Achieva system (Philips, Best, The Netherlands) and PET imaging was performed in 3D imaging mode with an ECAT Exact HR+ tomograph (Siemens Molecular Imaging, Inc., Knoxville, TN) as previously reported 19.
[11C]DTBZ, [11C]PMP, and [11C]PIB were prepared as described previously 23, 24. Dynamic PET scanning was performed for 70 minutes using a bolus dose of 15 mCi [11C]PMP dose. Bolus/infusion protocols were used for [11C]DTBZ (15 mCi) in 60 minutes and for [11C]PIB (18 mCi) in 70 minutes 25.
Image Analysis
Imaging registration and volume of interest definition were performed as previously reported 19. Cortical AChE [11C]PMP hydrolysis rates (k3) were estimated using the striatal volume as the tissue reference 26.
[11C]PIB and [11C]DTBZ distribution volume ratio (DVR) were estimated using the Logan plot graphical analysis method using cerebellar cortical and supratentorial cortical reference tissues, respectively 27. Caudate nucleus [11C]DTBZ and global cortical and striatal [11C]PIB DVRs were calculated.
Data Analysis
The major aim of the study was to investigate the relative contributions of cortical and striatal β-amyloidopathy to cognitive impairment in PD. We used [11C]PIB β-amyloid data from young to middle-aged healthy subjects (n=11, 5M/6F), mean age 46.7±3.4 (range 40-50 yr) to define elevated [11C]PIB binding in the patients because of age-related progressive β-amyloid deposition in a subset of cognitively normal elderly individuals 28. The maximum normal range cortical and striatal [11C]PIB DVR seen in our healthy middle-aged control subjects’ ratios were 1.17 (mean 1.12±0.04) and 1.50 (mean 1.38±0.06), respectively. PD subjects with values exceeding the maximum thresholds were defined as exhibiting increased regional β-amyloid binding.
Analysis of covariance (ANCOVA) was used to evaluate the effects of the β-amyloidopathy grouping factor on cognitive z-scores while adjusting for cortical AChE hydrolysis rate, caudate nucleus monoaminergic terminal DVR, duration of disease and age covariates. Duncan Multiple Range post-hoc analysis was used to evaluate differences between the β-amyloidopathy subgroups. All analyses were performed using SAS version 9.2, SAS institute (Cary, North Carolina).
Results
Relative frequency of striatal and cortical β-amyloidopathy
Increased striatal and cortical β-amyloid deposition were present in approximately 16% and 38%, respectively, of this predominantly non-demented PD cohort. Increased striatal β-amyloid deposition occurred in half of all subjects with increased cortical β-amyloid deposition. In contrast, increased striatal β-amyloid deposition did not occur in the absence of increased cortical β-amyloid deposition (Table 1).
Table 1.
The proportion of patients with negative versus positive cortical and striatal β-amyloid deposition (n=62).
| Negative cortical β-amyloid deposition |
Positive cortical β- amyloid deposition |
||
|---|---|---|---|
| Negative striatal β-amyloid deposition |
n=39 (62.9%) | n=13 (21.0%) | n=52 (83.9%) |
| Positive striatal β-amyloid deposition |
n=0 (0%) | n=10 (16.1%) | n=10 (16.1%) |
| n=39 (62.9%) | n=23 (37.1%) | Total: n=62 (100%) |
The mean cortical β-amyloid PIB DVR in the different amyloidopathy subgroups were as following: Combined cortical and striatal β-amyloidopathy subgroup (n=10; 1.55±0.19), cortical-only β-amyloidopathy subgroup (n=13, 1.22±0.04) and non-β-amyloidopathy subgroup (n=39, 1.09±0.06). Corresponding mean striatal β-amyloid PIB DVR in the different subgroups were as following: Combined cortical and striatal β-amyloidopathy subgroup (1.88±0.20), cortical-only β-amyloidopathy subgroup (1.40±0.06) and non-β-amyloidopathy subgroup (1.29±0.07). Three subjects with mild dementia had cortical and striatal PIB DVR values in the average range of the combined cortical and striatal β-amyloidopathy subgroup.
Table 2 lists the mean clinical variables for each of the subgroups. None of these variables remained significant after correction for type I statistical error inflation. There was a non-significant trend for older age in the patients in the combined striatal and cortical β-amyloidopathy subgroup compared to the non-β-amyloidopathy and the cortical-only β-amyloidopathy subgroups. There was also a non-significant trend toward more severe motor parkinsonism in the combined striatal and cortical β-amyloidopathy subgroup.
Table 2.
Mean (±SD) values of demographic and clinical variables in the different β-amyloidopathy subgroups. ANOVA F-values with level of significance are listed in the last column. None of the variables were significant after correction for multiple testing.
| Non-β- amyloidopathy subgroup (n=39) |
Cortical-only β- amyloidopathy subgroup (n=13) |
Combined cortical and striatal β- amyloidopathy subgroup (n=10) |
ANOVA F-value, P level |
|
|---|---|---|---|---|
| Age | 67.3+6.3 | 70.2±5.4* | 73.3±6.3 | F=4.13, P=0.02 |
| Duration of disease |
6.4+4.4 | 5.0±4.1 | 8.7±6.1 | F=1.84, P=0.17 |
| Hoehn & Yahr stage |
2.6±0.4 | 2.8±0.5 | 2.9±0.5 | F=2.72, P=0.07 |
| MDS UPDRS motor scores |
33.6±13.5 | 38.6±15.3 | 43.7±14.2 | F=2.30, P=0.11 |
Cognitive performance in the different β-amyloidopathy subgroups: combined striatal and cortical versus the cortical-only β-amyloidopathy versus the non-β-amyloidopathy subgroups
ANCOVA using global composite cognitive z-scores as the outcome parameter showed a significant regressor effect for the β-amyloidopathy grouping factor (F=4.18, P=0.02) while adjusting for covariate effects of cortical AChE activity (F=5.67, P=0.02), caudate nucleus monoaminergic terminal binding, duration of disease and age (total model: F=3.55, P=0.0048; Table 3). The model showed that the β-amyloidopathy grouping factor and cortical AChE activity were both independent significant predictors of cognitive impairment in the patients. Duncan Multiple Range post-hoc analysis showed significantly lower global composite cognitive z-scores for the combined striatal and cortical β-amyloidopathy (mean z-score: −1.57, adjusted: −1.52, SD: 0.88, confidence interval, CI: −2.09 to −0.95) compared to cortical-only β-amyloidopathy (mean z-score: −0.53, adjusted: −0.45, SD: 0.79, confidence interval, CI: −0.93 to 0.03) and non-β-amyloidopathy subgroups (mean z-score: −0.81, adjusted: −0.85, SD: 0.95, confidence interval, CI: −1.13 to −0.57); the latter two subgroups were not significantly different from each other (Figure 1).
Table 3.
Analysis of covariance using global composite cognitive z-scores as outcome parameter showed significant regressor effects for amyloidopathy grouping factor while adjusting for covariate effects of cortical AChE activity, caudate nucleus monoaminergic terminal binding, duration of disease and age. Duncan Multiple Range post hoc analysis showed significantly lower global composite cognitive z-scores for the combined striatal and cortical amyloidopathy sub-group (mean z-score: -1.57+0.88) compared to cortical-only amyloidopathy (mean z-score: -0.53+0.79) and non-amyloidopathy subgroups (mean z-score: −0.81±0.95); the latter two were not significantly different from each other.
| Amyloidopathy grouping factor effect |
Cortical AChE activity covariate effects |
Caudate nucleus DTBZ binding covariate effect |
Duration of disease covariate effect |
Age covariate effect |
Overall model | |
|---|---|---|---|---|---|---|
| Global cognitive z-score |
F=4.18, P=0.02 |
F=5.67, P=0.02 |
F=1.17, P=0.38 |
F=0.70, P=0.38 |
F=0.55, P=0.46 |
F=3.55, P=0.0048 |
Figure 1.
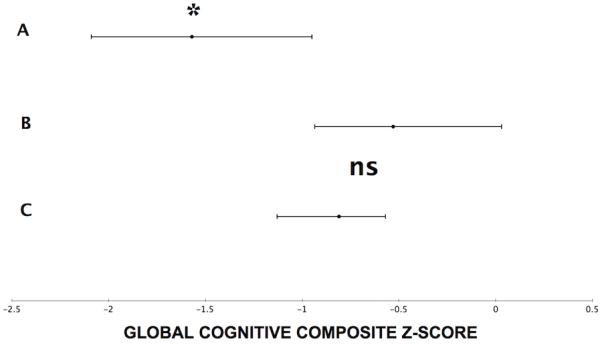
Line plots showing the mean value and confidence interval ranges for the global cognitive composite z-score values for the three amyloidopathy subgroups: A= combined striatal and cortical amyloidopathy subgroup, B= cortical-only amyloidopathy subgroup, and C= non-amyloidopathy subgroup. *Duncan Multiple Range post hoc analysis showed significantly lower global composite cognitive z-scores for the combined striatal and cortical amyloidopathy sub-group (A) compared to cortical-only amyloidopathy (B) and non-amyloidopathy subgroups (C); the latter two were not significantly different from each other (ns).
Post hoc analysis: Cognitive domains
Post hoc analyses were performed to investigate the relationship between the β-amyloidopathy grouping factor and the four individual cognitive domain z-scores (verbal learning, attention, visuospatial and executive functions) while controlling for covariate effects of cortical cholinergic and caudate nucleus monoaminergic terminal densities, duration of disease and age. Significant regressor effects for β-amyloidopathy grouping factor were seen for the verbal learning (F=4.18, P=0.021) and attention (F=3.90, P=0.026) cognitive domain z-scores. Duncan Multiple Range post-hoc analysis showed significantly lower verbal learning and attention cognitive domain z-scores for the combined striatal and cortical β-amyloidopathy compared to cortical-only β-amyloidopathy and non-β-amyloidopathy subgroups; the latter two were not significantly different from each other. No significant regressor effects for β-amyloidopathy grouping factor were seen for visuospatial or executive function cognitive domain z-scores.
We performed additional linear post hoc analysis using β-amyloid PIB DVR binding to complement our β-amyloidopathy subgroup findings. As cortical β-amyloid PIB DVR were higher in the combined cortical and striatal β-amyloidopathy subgroup compared to the other amyloidopathy sub-groups, we evaluated relative cognitive effects of global (i.e., cortical and striatal) versus cortical or striatal β-amyloid plaques using absolute β-amyloid PIB DVR binding values. For this purpose, we computed percentage scores of cortical and striatal PIB DVR based on the mean PIB DVR in the healthy middle-aged control subjects and summed the cortical and striatal PIB DVR percentage scores in order to derive a global PIB DVR measure. Stepwise regression was then used to predict global and cognitive domain scores using the global, cortical and striatal PIB DVR percentage scores together with the cholinergic and dopaminergic PET measures. Results confirmed our subgroup findings and showed that global composite cognitive z-scores were best predicted by cortical AChE (F=9.42, P=0.0032) and global PIB DVR (F=4.24, P=0.04; total model F=7.09, P=0.0018). Verbal learning cognitive domain z-scores were predicted by cortical PIB DVR (F=9.55, P=0.003) and cortical AChE activity (F=7.42, P=0.008; total model F= 9.00, P=0.0004). Attention cognitive domain z-scores were predicted by striatal PIB DVR (F=4.86, P=0.03) and caudate nucleus dopaminergic activity (F=4.64, P=0.035; total model F=5.53, P=0.006). There were no significant predictors for the visuospatial cognitive domain subscores (total model F=2.34, P=0.13). Executive functions cognitive domain scores were predicted by cortical AChE activity (F=9.05, P=0.0038; total model F=6.74, P=0.0023).
Discussion
There are probably at least two major sources of pathogenic proteinopathy in PD; variations within the spectrum of α–synucleinopathy and the effects of additional, probably independent, pathologic processes involving β-amyloid, and less commonly tau deposition. In this study, we assessed the regional effects of β-amyloidopathy in PD. Our data indicate that cortical β-amyloidopathy is present in approximately one third of subjects in this predominantly non-demented cohort. The one-third relative frequency of β-amyloidopathy in our subjects is consistent with the expected occurrence of cerebral β-amyloid deposition in otherwise cognitively normal elders 28, and may be a function of older age in PD 29, or longer duration of disease 29. As expected, our in vivo estimate of the relative frequency of β-amyloidopathy in PD is lower than post-mortem estimates 3. Post-mortem studies, typically, reflect changes found in end-stage disease and may not represent pathological changes at earlier disease stages with less severe cognitive impairments. The subjects in this study were at risk of development of dementia based on older age, presence of cognitive complaints, or more severe motor disease. Although these factors may put them at higher risk of β-amyloid deposition, the prevalence of β-amyloidopathy of about one in three is still relatively low compared to (approximately) half of post-mortem estimates 3.
Striatal β-amyloidopathy in our cohort, however, was less frequent, approximately one in six PD subjects. Increased striatal β-amyloid deposition occurred in half of all subjects with increased cortical β-amyloid deposition. In contrast, increased striatal β-amyloid deposition did not occur in the absence of increased cortical β-amyloid deposition. These findings imply a sequential or two-step process of β-amyloidopathy in PD where cortical changes antecede striatal plaque deposition. This is in keeping with post-mortem findings from patients with AD where a distinct sequence in which the regions are hierarchically involved has been observed 30. This sequential nature of plaque deposition in PD needs to be confirmed in a prospective cohort study.
Accumulating post-mortem evidence indicates that comorbid cerebral β-amyloidopathy in PDD is associated with worse clinical outcome, shorter time to dementia conversion, and reduced survival rates compared to PDD patients with "pure" Lewy body pathology 3, 31. These findings agree with in vivo CSF studies where abnormally low levels of β-amyloid are associated with more rapid cognitive decline in PD 32. The comorbid presence of two neurodegenerations (α-synucleinopathy and β-amyloidopathy) in some patients with PDD suggests additive or even synergistic detrimental interactions of these proteinopathies 33.
Interestingly, we found that the presence of combined striatal and cortical β-amyloidopathy was a more robust determinant of cognitive impairment in PD compared to cortical-only β-amyloidopathy. A post-mortem study found greater frequency of striatal β-amyloid deposition in PDD compared to PD and found that β-amyloid deposition in the striatum strongly correlated with dementia in PD 8. Another post-mortem study indicated that higher striatal β-amyloid plaque density predicted the presence of dementia in a mixed cohort of patients with AD and non-demented control subjects 34. The majority of prior in vivo β-amyloid PET imaging studies describe associations between cortical β-amyloid plaque deposition and cognitive changes in Lewy body disorders, including PD, but do not report on the relationship between striatal β-amyloid deposition and cognitive functions 35-38. Furthermore, most of these studies report on global measures of cognition rather than a detailed neuropsychological testing battery. Although Foster and colleagues reported also on correlations between cognitive measures and both cortical and striatal β-amyloid binding in patients with Lewy body disorders, including PD, they did not directly compare cortical to striatal β-amyloid measures 35. The presence of β-amyloid plaques in the striatum, a key node in several cognitively important circuits, is likely to further aggravate cognitive deficits in PD.
We reported previously that even relatively low levels of cortical β-amyloidopathy in PD correlate with cognitive dysfunction 4. These findings suggest that amyloidopathy, even at mildly elevated levels, may exert pathologic effects in PD subjects. A fraction of cognitively normal elderly individuals, however, exhibit higher β-amyloid burdens than documented in our PD subjects, and Alzheimer disease patients appear to become symptomatic at considerably higher levels of amyloid burden 39. In PD, however, β-amyloidopathy occurs in the context of a-synucleinopathy and other degenerations, which likely reduce the ability of the brain to adapt to increased β-amyloid burden.
We found that 'global' (defined as summed effects of cortical and striatal) β-amyloid plaque burden best predicted global composite cognitive z-scores compared to either cortical or striatal measures alone. We also found regional effects on specific cognitive domains. Cortical β-amyloid PIB DVR best predicted verbal learning deficits, while striatal β-amyloid PIB DVR best predicted attention cognitive domain z-scores. These findings underscore the heterogeneity of cognitive impairment in PD related to β-amyloid plaque deposition in PD. We found also that the effects of cerebral β-amyloidopathy were independent from effects of cortical cholinergic and caudate nucleus dopaminergic denervations that are key contributors to the neurochemical processes underlying the cognitive decline in PD 22. These data provide further support for a multifactorial basis of cognitive deficits in PD extending beyond the effects of α-synuclein deposition. These findings also emphasize the multisystem subcortical-cortical substrates of the cognitive decline in PD.
A limitation of our study was that because of the cross-sectional design no inference about the risk of conversion to dementia could be made. Our study subjects were clinically recruited based on known risk factors for dementia in PD with only three subjects meeting criteria for mild dementia after detailed neuropsychological evaluation. Future studies in PD subjects across the full spectrum of cognitive impairment are needed to investigate the relative impact of striatal β-amyloid plaques as the frequency of β-amyloidopathy appears higher in demented PD subjects 40.
Our findings suggest also that anti-amyloid therapies deserve further study as potential therapeutic interventions to mitigate cognitive decline in PD. The selection of eligible subjects for anti-amyloid treatment trials may be challenging as only a subset of PD subjects may develop β-amyloidopathy. The identification of at-risk candidates may be particularly impactful in PD, however, since the relatively low level of amyloidopathy may provide a unique window of opportunity to intervene earlier in the β-amyloid cascade and might attenuate synergistic interactions with α–synuclein pathology.
We conclude that the combined presence of striatal and cortical amyloidopathy is associated with greater cognitive impairment than cortical amyloidopathy alone in PD.
Acknowledgements
The authors thank Christine Minderovic, Virginia Rogers, the PET technologists, cyclotron operators, and chemists, for their assistance. This work was supported by the Department of Veterans Affairs; the Michael J. Fox Foundation; and the NIH [grant numbers P01 NS015655, P50 NS091856 and RO1 NS070856].
Dr. Frey has research support from the NIH, GE Healthcare and AVID Radiopharmaceuticals (Eli Lilly subsidiary). Dr. Frey also serves as a consultant to AVID Radiopharmaceuticals, MIMVista, Inc, Bayer-Schering and GE healthcare. He also holds equity (common stock) in GE, Bristol-Myers, Merck and Novo-Nordisk.
Dr. Muller has research support from the NIH, Michael J. Fox Foundation and the Department of Veteran Affairs.
Dr. Petrou has research support from the Radiological Society of North America, NIH, Pfizer Inc., Greatergood Foundation, Department of Veterans Affairs. Dr. Petrou serves as a consultant to Neuralstem.
Dr. Kotagal has research support from the American Academy of Neurology Clinical Research Training Fellowship and the Blue Cross Blue Shield Foundation of Michigan.
Dr. Koeppe receives research support from NIH (NINDS, NIA).
Dr. Scott receives Editorial Royalties from Wiley, is an owner of SynFast Consulting, LLC, and has received research funding from the University of Michigan, GE Healthcare, Bristol-Myers Squibb, Bayer Pharma AG, Eli Lilly, and Molecular Imaging Research.
Dr. Albin serves on the editorial boards of Neurology, Experimental Neurology, and Neurobiology of Disease. He receives grant support from the National Institutes of Health, CHDI, Michael J. Fox Foundation, and the Dept. of Veterans Affairs. Dr. Albin serves on the Data Safety and Monitoring Boards of the PRIDE-HD and LEGATO trials.
Dr. Bohnen has research support from the NIH, Department of Veteran Affairs, and the Michael J. Fox Foundation.
Funding agencies: This work was supported by the Department of Veterans Affairs; the Michael J. Fox Foundation; and the NIH [grant numbers P01 NS015655, P50 NS091856 and RO1 NS070856].
Abbreviations
- AChE
Acetylcholinesterase
- DVR
Distribution volume ratio
- PD
Parkinson disease
- PDD
Parkinson disease with dementia
- PIB
Pittsburgh compound B
- PET
positron emission tomography
- PMP
methyl-4-piperidinyl propionate
- VMAT2
vesicular monoamine transporter type 2
Footnotes
Relevant conflicts of interest/financial disclosures: The authors have no relevant financial or conflict of interest to disclose.
Authors' roles:
1. Research Project: A. Conception, B. Organization, C. Execution
2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique
3. Manuscript: A. Writing of the First Draft, B. Review and Critique
Neha Shah: 1C, 2C, 3A
Dr. Frey: 1A, 1B, 1C, 2A, 2C, 3B.
Dr. Muller: 1A, 1B, 1C, 2A, 2C, 3B
Dr. Petrou: 2C, 3B
Dr. Kotagal: 2C, 3B
Dr. Koeppe: 1A, 1B, 1C, 2C, 3B.
Dr. Scott: 1A, 1B, 1C, 2C, 3B.
Dr. Albin: 1A, 1B, 1C, 2C, 3B.
Dr. Bohnen: 1A, 1B, 1C, 2A, 2B, 3A.
Financial Disclosures
Neha Shah has nothing to disclose.
References
- 1.Churchyard A, Lees A. The relationship between dementia and direct involvement of the hippocampus and amygdala in Parkinson's disease. Neurology. 1997;49:1570–1576. doi: 10.1212/wnl.49.6.1570. [DOI] [PubMed] [Google Scholar]
- 2.Jellinger KA. Morphological substrates of mental dysfunction in Lewy body disease: an update. J Neural Transm. 2000;59(Suppl):185–212. doi: 10.1007/978-3-7091-6781-6_21. [DOI] [PubMed] [Google Scholar]
- 3.Kotzbauer PT, Cairns NJ, Campbell MC, et al. Pathologic Accumulation of alpha-Synuclein and Abeta in Parkinson Disease Patients With Dementia. Arch Neurol. 2012;69:1–6. doi: 10.1001/archneurol.2012.1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petrou M, Bohnen NI, Muller ML, Koeppe RA, Albin RL, Frey KA. Abeta-amyloid deposition in patients with Parkinson disease at risk for development of dementia. Neurology. 2012;79:1161–1167. doi: 10.1212/WNL.0b013e3182698d4a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalaitzakis ME, Graeber MB, Gentleman SM, Pearce RK. Striatal beta-amyloid deposition in Parkinson disease with dementia. J Neuropathol Exp Neurol. 2008;67:155–161. doi: 10.1097/NEN.0b013e31816362aa. [DOI] [PubMed] [Google Scholar]
- 6.Halliday GM, Song YJ, Harding AJ. Striatal beta-amyloid in dementia with Lewy bodies but not Parkinson's disease. J Neural Transm. 2011;118:713–719. doi: 10.1007/s00702-011-0641-6. [DOI] [PubMed] [Google Scholar]
- 7.Dugger BN, Serrano GE, Sue LI, et al. Presence of Striatal Amyloid Plaques in Parkinson's Disease Dementia Predicts Concomitant Alzheimer's Disease: Usefulness for Amyloid Imaging. Journal of Parkinson's disease. 2012;2:57–65. doi: 10.3233/JPD-2012-11073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalaitzakis ME, Walls AJ, Pearce RK, Gentleman SM. Striatal Abeta peptide deposition mirrors dementia and differentiates DLB and PDD from other Parkinsonian syndromes. Neurobiol Dis. 2011;41:377–384. doi: 10.1016/j.nbd.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 9.Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinicopathologic study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55:181–184. doi: 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goetz CG, Poewe W, Rascol O, et al. Movement Disorder Society Task Force report on the Hoehn and Yahr staging scale: status and recommendations. Mov Disord. 2004;19:1020–1028. doi: 10.1002/mds.20213. [DOI] [PubMed] [Google Scholar]
- 11.Goetz CG, Fahn S, Martinez-Martin P, et al. Movement Disorder Society-sponsored revision of the Unified Parkinson's Disease Rating Scale (MDS-UPDRS): Process, format, and clinimetric testing plan. Mov Disord. 2007;22:41–47. doi: 10.1002/mds.21198. [DOI] [PubMed] [Google Scholar]
- 12.Nasreddine ZS, Phillips NA, Bedirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc. 2005;53:695–699. doi: 10.1111/j.1532-5415.2005.53221.x. [DOI] [PubMed] [Google Scholar]
- 13.Aarsland D, Bronnick K, Larsen JP, Tysnes OB, Alves G. Cognitive impairment in incident, untreated Parkinson disease: the Norwegian ParkWest study. Neurology. 2009;72:1121–1126. doi: 10.1212/01.wnl.0000338632.00552.cb. [DOI] [PubMed] [Google Scholar]
- 14.Delis DC, Kramer JH, Kaplan E, Ober BA. California Verbal Learning Test Manual, Adult Version. Second The Psychological Corporation; San Antonio: 2000. [Google Scholar]
- 15.Wechsler D. WAIS III Technical Manual. The Psychological Corporation; San Antonio, TX: 1997. [Google Scholar]
- 16.Delis DC, Kaplan E, Kramer JH. Delis-Kaplan Executive Function System (D-KEFS): Examiner's manual. The Psychological Corporation; San Antonio, TX: 2001. [Google Scholar]
- 17.Stroop JR. Studies of interference in serial verbal reactions. J Exp Psychol. 1935;18:643–662. [Google Scholar]
- 18.Benton AL, Varney NR, Hamsher K. Judgment of Line Orientation, Form V. University of Iowa Hopsitals; Iowa City: 1975. [Google Scholar]
- 19.Bohnen NI, Mueller MLTM, Kotagal V, et al. Heterogeneity of cholinergic denervation in Parkinson disease. J Cereb Blood Flow Metab. 2012;32:1609–1617. doi: 10.1038/jcbfm.2012.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muller ML, Frey KA, Petrou M, et al. β-amyloid and postural instability and gait difficulty in Parkinson's disease at risk for dementia. Mov Disord. 2013;28:296–301. doi: 10.1002/mds.25213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bohnen NI, Frey KA, Studenski S, et al. Extra-nigral pathological conditions are common in Parkinson's disease with freezing of gait: An in vivo positron emission tomography study. Mov Disord. 2014;29:1118–1124. doi: 10.1002/mds.25929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bohnen NI, Albin RL, Muller ML, et al. Frequency of cholinergic and caudate nucleus dopaminergic deficits across the predemented cognitive spectrum of Parkinson disease and evidence of interaction effects. JAMA Neurol. 2015;72:194–200. doi: 10.1001/jamaneurol.2014.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shao X, Hoareau R, Hockley BG, et al. Highlighting the Versatility of the Tracerlab Synthesis Modules. Part 1: Fully Automated Production of [F]Labelled Radiopharmaceuticals using a Tracerlab FX(FN) J Labelled Comp Radiopharm. 2011;54:292–307. doi: 10.1002/jlcr.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klunk WE, Engler E, Nordberg A, et al. Imaging brain amyloid in Alzheimer's disease using the novel positron emission tomography tracer, Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 25.Koeppe RA, Frey KA, Kume A, Albin R, Kilbourn MR, Kuhl DE. Equilibrium versus compartmental analysis for assessment of the vesicular monoamine transporter using (+)-alpha-[11C]dihydrotetrabenazine (DTBZ) and positron emission tomography. J Cereb Blood Flow Metab. 1997;17:919–931. doi: 10.1097/00004647-199709000-00001. [DOI] [PubMed] [Google Scholar]
- 26.Nagatsuka S, Fukushi K, Shinotoh H, et al. Kinetic analysis of [(11)C]MP4A using a high-radioactivity brain region that represents an integrated input function for measurement of cerebral acetylcholinesterase activity without arterial blood sampling. J Cereb Blood Flow Metab. 2001;21:1354–1366. doi: 10.1097/00004647-200111000-00011. [DOI] [PubMed] [Google Scholar]
- 27.Logan J, Fowler JS, Volkow ND, Ding YS, Wang GJ, Alexoff DL. A strategy for removing the bias in the graphical analysis method. J Cereb Blood Flow Metab. 2001;21:307–320. doi: 10.1097/00004647-200103000-00014. [DOI] [PubMed] [Google Scholar]
- 28.Chetelat G, La Joie R, Villain N, et al. Amyloid imaging in cognitively normal individuals, at-risk populations and preclinical Alzheimer's disease. Neuroimage Clin. 2013;2:356–365. doi: 10.1016/j.nicl.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Halliday G, Hely M, Reid W, Morris J. The progression of pathology in longitudinally followed patients with Parkinson's disease. Acta Neuropathol. 2008;115:409–415. doi: 10.1007/s00401-008-0344-8. [DOI] [PubMed] [Google Scholar]
- 30.Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 31.Compta Y, Parkkinen L, O'Sullivan SS, et al. Lewy- and Alzheimer-type pathologies in Parkinson's disease dementia: which is more important? Brain. 2011;134:1493–1505. doi: 10.1093/brain/awr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siderowf A, Xie SX, Hurtig H, et al. CSF amyloid {beta} 1-42 predicts cognitive decline in Parkinson disease. Neurology. 2010;75:1055–1061. doi: 10.1212/WNL.0b013e3181f39a78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsigelny IF, Crews L, Desplats P, et al. Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer's and Parkinson's diseases. PLoS One. 2008;3:e3135. doi: 10.1371/journal.pone.0003135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beach TG, Sue LI, Walker DG, et al. Striatal amyloid plaque density predicts Braak neurofibrillary stage and clinicopathological Alzheimer's disease: implications for amyloid imaging. J Alzheimers Dis. 2012;28:869–876. doi: 10.3233/JAD-2011-111340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Foster ER, Campbell MC, Burack MA, et al. Amyloid imaging of Lewy body-associated disorders. Mov Disord. 2010;25:2516–2523. doi: 10.1002/mds.23393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gomperts SN, Locascio JJ, Marquie M, et al. Brain amyloid and cognition in Lewy body diseases. Mov Disord. 2012;27:965–973. doi: 10.1002/mds.25048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gomperts SN, Locascio JJ, Rentz D, et al. Amyloid is linked to cognitive decline in patients with Parkinson disease without dementia. Neurology. 2013;80:85–91. doi: 10.1212/WNL.0b013e31827b1a07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lucero C, Campbell MC, Flores H, Maiti B, Perlmutter JS, Foster ER. Cognitive reserve and beta-amyloid pathology in Parkinson disease. Parkinsonism Relat Disord. 2015 doi: 10.1016/j.parkreldis.2015.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frey KA, Petrou M. Imaging Amyloidopathy in Parkinson Disease and Parkinsonian Dementia Syndromes. Clinical and translational imaging. 2015;3:57–64. doi: 10.1007/s40336-015-0104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petrou M, Dwamena BA, Foerster BR, et al. Amyloid deposition in Parkinson's disease and cognitive impairment: A systematic review. Mov Disord. 2015 doi: 10.1002/mds.26191. [DOI] [PMC free article] [PubMed] [Google Scholar]