

Abstract
Arthrogryposis, renal dysfunction and cholestasis (ARC) syndrome is a rare disorder associated with platelet abnormalities resembling Gray Platelet Syndrome. Affected patients have normal platelet numbers but abnormal morphology and function. Bleeding symptomatology ranges from post-procedural to spontaneous life-threatening hemorrhage. We report a patient with ARC syndrome and compound heterozygous mutations in VPS33B who presented with significant bleeding requiring numerous admissions and transfusions. She was treated with prophylactic platelet transfusions and ε-aminocaproic acid. This was well tolerated and significantly decreased transfusion requirements and admissions for bleeding. Our experience provides support for consideration of prophylactic measures in these patients as well as the possibility of using prophylaxis in related disorders.
Keywords: platelet function disorder, ARC syndrome, epsilon aminocaproic acid, platelets, hemorrhage, gray platelet syndrome
Clinical course
A two-month-old infant presented with failure to thrive and glycosuria. She was born to two healthy non-consanguineous parents of Mexican descent. Pregnancy, delivery and postnatal course were uncomplicated except for the presence of rocker bottom feet. Her older brother had been diagnosed at three months of age with Fanconi-Bickel syndrome (a glycogen storage disease characterized by accumulation in the liver and kidneys) and died two years later after massive hemorrhage post-liver biopsy. On presentation, she had findings consistent with generalized proximal tubular dysfunction including glucosuria, tubular proteinuria, and hypophosphatemia. This was felt to be consistent with Fanconi syndrome. For her failure to thrive, a nasogastric tube was placed for supplemental nutrition. One week later, she developed significant epistaxis requiring packed red blood cell (pRBC) transfusion and ε-aminocaproic acid. Laboratory studies at that time revealed normal complete blood counts, prothrombin time, activated partial thromboplastin time, von Willebrand factor panel, and factor XIII.
Over the following two years, she did not have any significant bleeding despite multiple surgeries, including laparoscopic Nissen fundoplication and gastrostomy tube placement. She later underwent tonsillectomy and adenoidectomy for obstructive sleep apnea and subsequently developed hemorrhage requiring admission to the pediatric intensive care unit (PICU). Hemostasis was achieved after several cauterization attempts and treatment with ε-aminocaproic acid, oxymetazoline, and pRBC transfusion. Platelet function testing at that time was performed using a Chronolog lumiaggregometer with ATP chemiluminescence (see Supplementary Table I for details). The results demonstrated reduced platelet aggregation and secretion to all agonists except ristocetin, consistent with a secretion defect. Evaluation of her peripheral smear revealed significantly hypogranulated platelets (Figure 1), and hepatomegaly and transaminitis were noted.
Figure 1.
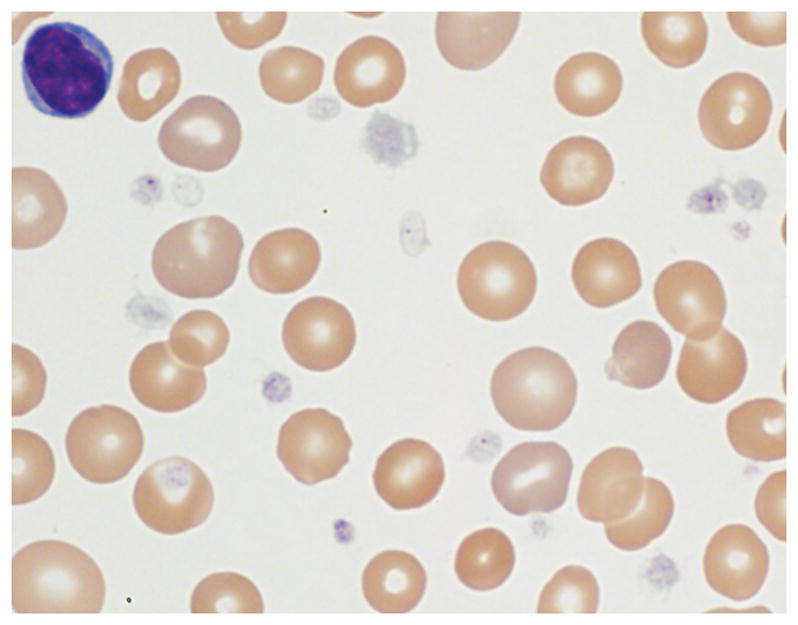
Peripheral blood smear demonstrating varying degrees of platelet hypogranulation. Magnification, 1000x.
At five years of age, she developed nephrotic syndrome with significant glomerular proteinuria and hypoalbuminemia and was treated with steroids. She had rapid decline of renal function and required initiation of dialysis three months later. That year, she was seen by genetics who concluded that her brother’s liver biopsy results and clinical picture were not consistent with Fanconi-Bickel syndrome. Molecular testing revealed compound heterozygous mutations in VPS33B (vacuolar protein sorting 33B), consistent with a diagnosis of arthrogryposis, renal dysfunction and cholestasis (ARC) syndrome(1). The first mutation, defined as c.1609_1657+9del, is a deletion that encompasses the forty-nine terminal coding nucleotides of exon 21, including the splice donor site for intron 21 and nine additional adjacent intronic nucleotides. The second mutation, c.1225+5G>C, is at the junction of exon 16 and intron 16, altering the universal GT splice donor sequence.
She continued to have recurrent and life-threatening episodes of hemorrhage, primarily epistaxis, requiring frequent hospitalizations and PICU admissions. Due to the frequency and severity of these episodes, the decision was made to initiate short term prophylactic treatment with pooled leukocyte-reduced irradiated platelets, in order to facilitate adequate mucosal healing. Initially she received once weekly transfusions but continued to have frequent bleeding requiring admission. There was a significant improvement in symptoms as well as decreased admissions with twice weekly transfusions (Figure 2). After 6 months, she was trialed off prophylactic transfusions but quickly developed hemorrhage requiring admission, as well as platelet and pRBC transfusions. Therefore, prophylactic platelet transfusions were resumed on a weekly basis, along with indefinite administration of oral ε-aminocaproic acid adjusted for her renal failure. She had three minor admissions for bleeding from her hemodialysis catheter and gastrostomy tube, two of which required additional platelet transfusions. She tolerated platelet prophylaxis well with appropriate platelet count increment demonstrated throughout her treatment course. Approximately 16 months after initiation of prophylaxis, she had a prolonged hospitalization for line-associated fungal and bacterial sepsis and passed away shortly thereafter. During the period from resumption of transfusions and initiation of ε-aminocaproic acid until her death, there were only three episodes of bleeding requiring admission, two of which were associated with manipulation of her dialysis catheter.
Figure 2. Number of admissions, pRBC and platelet transfusions related to hemorrhagic events over the course of the prophylactic regimen.

The patient’s medical record was reviewed over the 30 month period before her death. Prophylactic platelet infusions were not included. Admissions and transfusions clearly unrelated to bleeding were not included, e.g. admissions for infection or anemia secondary to renal failure.
Case discussion
ARC syndrome is a rare autosomal recessive multisystem disorder of childhood which has been found to be caused by mutations in either the VPS33B or VIPAR (VIPAR, VPS33B-interacting protein involved in polarity and apical protein restriction, also known as VPS16B, current gene nomenclature is VIPAS39) proteins(1). Patients typically present with renal tubular dysfunction, cholestasis, severe failure to thrive and joint contractures. Other common features include ichthyosis, defects in platelet alpha granule biogenesis, and central nervous system abnormalities. In the largest published cohort of patients, the majority of patients did not survive past one year of life(2). Our patient lived to 8.5 years which, to our knowledge, is the longest survival reported in a patient with ARC syndrome. One of the mutations found in our patient, c.1225+5G>C, has been reported to be associated with an attenuated phenotype with two reported cases at ages 3 and 5.5 years at time of publication(3). These patients were also of Latino ancestry, similar to our patient. This mutation has been shown to result in a truncated protein that may maintain some residual wild type function due to the formation of VPS33B-VIPAR complex-containing clusters(3).
Morbidity and mortality in patients with ARC syndrome is affected by a range of clinical manifestations including bleeding, infection, and dehydration. A review of cases in the literature reveals a significant number of patients with significant morbidity and mortality secondary to a bleeding diathesis (Supplementary Table II)(2–14). There is a wide range of platelet abnormalities in patients with ARC syndrome. One cohort found 7 out of 62 patients with either thrombocytopenia or abnormal platelet morphology resembling Gray Platelet syndrome due to loss of alpha granules. Eleven of those same 62 patients experienced severe bleeding (either spontaneous or secondary to organ biopsy). However, 9 out of these 11 patients with severe bleeding had a normal platelet count and morphology(2). Platelet function testing in these patients may be informative as those in whom aggregation or PFA-100 testing was performed had demonstrated abnormal responses(2, 9, 11, 15). Findings on platelet aggregation in these patients has been variable. Our patient demonstrated decreased aggregation to all agonists except ristocetin, while others have found abnormal responses to ADP and collagen(9, 15), and decreased aggregation to arachidonic acid, epinephrine and ADP(11). One note of caution is that ARC syndrome diagnoses in some studies were solely by the evaluation of peripheral blood smears. These are less reliable in proving the absence of alpha granules when compared with transmission electron microscopy as demonstrated in patient studies and mouse models(15–17).
We believe that the use of continuous ε-aminocaproic acid prophylaxis, in addition to prophylactic platelet transfusions, increased our patient’s overall survival as well as quality of life. In our patient, the use of prophylactic platelet transfusions alone did not significantly decrease the number of admissions for bleeding. The addition of ε-aminocaproic acid resulted in significantly decreased admissions for bleeding, reduced unscheduled platelet transfusions, and decreased pRBC transfusions (Figure 2). Review and extrapolation of transfusion incidence prior to prophylaxis suggests that this plan was at least equivalent and may have decreased the total number of platelet transfusions. The decision to proceed with prophylactic transfusions was not made lightly given the risk for the development of antibodies. This never became a problem but should be carefully weighed in the consideration of any prophylactic transfusion program. It is unclear whether the use of ε-aminocaproic acid alone would have resulted in a similar reduction in admissions for bleeding and transfusions. Multiple studies suggest a role for prophylactic ε-aminocaproic acid in addition to prophylactic platelet transfusions in patients with hemostatic abnormalities(18), and this is supported by our case.
Conclusion
This case highlights the importance of a thorough evaluation of platelets, including morphology and functional studies, in patients with ARC syndrome. The use of prophylactic measures, such as continuous ε-aminocaproic acid with or without prophylactic platelet transfusions should be considered in these patients in whom the risk of bleeding complications justifies the benefit of this approach.
Supplementary Material
Platelet aggregation results
Literature review of previously reported ARC syndrome patients and bleeding symptoms.
Abbreviations Key
- ARC
Arthrogryposis, renal dysfunction, and cholestasis
- pRBC
Packed red blood cell
- PICU
Pediatric intensive care unit
- VPS33B
Vacuolar protein sorting 33B
- VIPAR
VPS33B-interacting protein involved in polarity and apical protein restriction
- PFA
Platelet function analyzer
- ADP
Adenosine diphosphate
Footnotes
Conflict of Interest Statement
Jordan Shavit has the following conflicts of interest: Advancing Science through Pfizer-Investigator Research Exchange, Bayer Hemophilia Awards Program, National Hemophilia Foundation/Novo Nordisk (grants) and Bayer, Baxter, CSL Behring, and Octapharma (consulting). No other authors had any conflicts of interest to report.
References
- 1.Gissen P, Johnson CA, Morgan NV, Stapelbroek JM, Forshew T, Cooper WN, McKiernan PJ, Klomp LW, Morris AA, Wraith JE, McClean P, Lynch SA, Thompson RJ, Lo B, Quarrell OW, DiRocco M, Trembath RC, Mandel H, Wali S, Karet FE, Knisely AS, Houwen RH, Kelly DA, Maher ER. Mutations in VPS33B, encoding a regulator of SNARE-dependent membrane fusion, cause arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome. Nat Genet. 2004;36:400–4. doi: 10.1038/ng1325. [DOI] [PubMed] [Google Scholar]
- 2.Gissen P, Tee L, Johnson CA, Genin E, Caliebe A, Chitayat D, Clericuzio C, Denecke J, DiRocco M, Fischler B, FitzPatrick D, Garcia-Cazorla A, Guyot D, Jacquemont S, Koletzko S, Leheup B, Mandel H, Sanseverino MT, Houwen RH, McKiernan PJ, Kelly DA, Maher ER. Clinical and molecular genetic features of ARC syndrome. Hum Genet. 2006;120:396–409. doi: 10.1007/s00439-006-0232-z. [DOI] [PubMed] [Google Scholar]
- 3.Smith H, Galmes R, Gogolina E, Straatman-Iwanowska A, Reay K, Banushi B, Bruce CK, Cullinane AR, Romero R, Chang R, Ackermann O, Baumann C, Cangul H, Cakmak Celik F, Aygun C, Coward R, Dionisi-Vici C, Sibbles B, Inward C, Kim CA, Klumperman J, Knisely AS, Watson SP, Gissen P. Associations among genotype, clinical phenotype, and intracellular localization of trafficking proteins in ARC syndrome. Hum Mutat. 2012;33:1656–64. doi: 10.1002/humu.22155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eastham KM, McKiernan PJ, Milford DV, Ramani P, Wyllie J, van’t Hoff W, Lynch SA, Morris AA. ARC syndrome: an expanding range of phenotypes. Arch Dis Child. 2001;85:415–20. doi: 10.1136/adc.85.5.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Di Rocco M, Callea F, Pollice B, Faraci M, Campiani F, Borrone C. Arthrogryposis, renal dysfunction and cholestasis syndrome: report of five patients from three Italian families. Eur J Pediatr. 1995;154:835–9. doi: 10.1007/BF01959793. [DOI] [PubMed] [Google Scholar]
- 6.Coleman RA, Van Hove JL, Morris CR, Rhoads JM, Summar ML. Cerebral defects and nephrogenic diabetes insipidus with the ARC syndrome: additional findings or a new syndrome (ARCC-NDI)? Am J Med Genet. 1997;72:335–8. [PubMed] [Google Scholar]
- 7.Deal JE, Barratt TM, Dillon MJ. Fanconi syndrome, ichthyosis, dysmorphism, jaundice and diarrhoea--a new syndrome. Pediatr Nephrol. 1990;4:308–13. doi: 10.1007/BF00862505. [DOI] [PubMed] [Google Scholar]
- 8.Hayes JA, Kahr WH, Lo B, Macpherson BA. Liver biopsy complicated by hemorrhage in a patient with ARC syndrome. Paediatr Anaesth. 2004;14:960–3. doi: 10.1111/j.1460-9592.2004.01301.x. [DOI] [PubMed] [Google Scholar]
- 9.Kim SM, Chang HK, Song JW, Koh H, Han SJ. Agranular platelets as a cardinal feature of ARC syndrome. J Pediatr Hematol Oncol. 2010;32:253–8. doi: 10.1097/MPH.0b013e3181c3a8d0. [DOI] [PubMed] [Google Scholar]
- 10.Nezelof C, Dupart MC, Jaubert F, Eliachar E. A lethal familial syndrome associating arthrogryposis multiplex congenita, renal dysfunction, and a cholestatic and pigmentary liver disease. J Pediatr. 1979;94:258–60. doi: 10.1016/s0022-3476(79)80839-2. [DOI] [PubMed] [Google Scholar]
- 11.Saadah OI, Bokhari BE, Alshaeri TM, Jastaniah W. Haematological manifestations of arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome: a case report. Arab J Gastroenterol. 2013;14:26–8. doi: 10.1016/j.ajg.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 12.Tekin N, Durmus-Aydogdu S, Dinleyici EC, Bor O, Bildirici K, Aksit A. Clinical and pathological aspects of ARC (arthrogryposis, renal dysfunction and cholestasis) syndrome in two siblings. Turk J Pediatr. 2005;47:67–70. [PubMed] [Google Scholar]
- 13.Jang JY, Kim KM, Kim GH, Yu E, Lee JJ, Park YS, Yoo HW. Clinical characteristics and VPS33B mutations in patients with ARC syndrome. J Pediatr Gastroenterol Nutr. 2009;48:348–54. doi: 10.1097/mpg.0b013e31817fcb3f. [DOI] [PubMed] [Google Scholar]
- 14.Elmeery A, Lanka K, Cummings J. ARC syndrome in preterm baby. J Perinatol. 2013;33:821–2. doi: 10.1038/jp.2013.62. [DOI] [PubMed] [Google Scholar]
- 15.Lo B, Li L, Gissen P, Christensen H, McKiernan PJ, Ye C, Abdelhaleem M, Hayes JA, Williams MD, Chitayat D, Kahr WH. Requirement of VPS33B, a member of the Sec1/Munc18 protein family, in megakaryocyte and platelet alpha-granule biogenesis. Blood. 2005;106:4159–66. doi: 10.1182/blood-2005-04-1356. [DOI] [PubMed] [Google Scholar]
- 16.Bem D, Smith H, Banushi B, Burden JJ, White IJ, Hanley J, Jeremiah N, Rieux Laucat F, Bettels R, Ariceta G, Mumford AD, Thomas SG, Watson SP, Gissen P. VPS33B regulates protein sorting into and maturation of alpha-granule progenitor organelles in mouse megakaryocytes. Blood. 2015 Jul 9;126:133–43. doi: 10.1182/blood-2014-12-614677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Urban D, Li L, Christensen H, Pluthero FG, Chen SZ, Puhacz M, Garg PM, Lanka KK, Cummings JJ, Kramer H, Wasmuth JD, Parkinson J, Kahr WH. The VPS33B-binding protein VPS16B is required in megakaryocyte and platelet alpha-granule biogenesis. Blood. 2012 Dec 13;120:5032–40. doi: 10.1182/blood-2012-05-431205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wardrop D, Estcourt LJ, Brunskill SJ, Doree C, Trivella M, Stanworth S, Murphy MF. Antifibrinolytics (lysine analogues) for the prevention of bleeding in patients with haematological disorders. Cochrane Database Syst Rev. 2013;7:Cd009733. doi: 10.1002/14651858.CD009733.pub2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Platelet aggregation results
Literature review of previously reported ARC syndrome patients and bleeding symptoms.