

Abstract
Indoleamine 2,3-dioxygenase-1 (IDO1) is a promising therapeutic target for the treatment of cancer, chronic viral infections, and other diseases characterized by pathological immune suppression. Recently important advances have been made in understanding IDO1’s catalytic mechanism. Although much remains to be discovered, there is strong evidence that the mechanism proceeds through a heme-iron bound alkylperoxy transition or intermediate state. Accordingly, we explored stable structural mimics of the alkylperoxy species and provide evidence that such structures do mimic the alkylperoxy transition or intermediate state. We discovered that O-benzylhydroxylamine, a commercially available compound, is a potent sub-micromolar inhibitor of IDO1. Structure-activity studies of over forty derivatives of O-benzylhydroxylamine led to further improvement in inhibitor potency, particularly with the addition of halogen atoms to the meta position of the aromatic ring. The most potent derivatives and the lead, O-benzylhydroxylamine, have high ligand efficiency values, which are considered an important criterion for successful drug development. Notably, two of the most potent compounds demonstrated nanomolar-level cell-based potency and limited toxicity. The combination of the simplicity of the structures of these compounds and their excellent cellular activity makes them quite attractive for biological exploration of IDO1 function and antitumor therapeutic applications.
Keywords: IDO1 inhibition, O-alkylhydroxylamines, antitumor therapy, rational drug design
Graphical Abstract
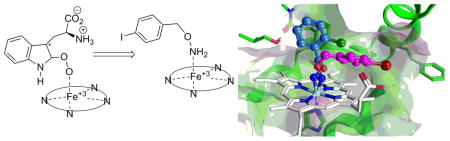
Introduction
Immune escape by tumors is a fundamental aspect of disease progression resulting from immunoediting of tumors as they interact with the host immune system1,2. Key to this process from a therapeutic standpoint is that tumors are selected to engender a tolerogenic microenvironment that actively suppresses the ability of the immune system to mount an effective response3. Ongoing progress in understanding the cellular and molecular mechanisms that govern the pathological state of tumor immune tolerance has revealed several protein targets which provide the potential for therapeutic intervention4. One central player is the immunomodulatory enzyme indoleamine 2,3-dioxygenase, IDO1, formerly known as IDO before the discovery of a second isoform5,6. IDO1 can contribute to immune escape when expressed directly in tumor cells or when expressed in immunosuppressive antigen presenting cells such as tolerogenic dendritic cells or tumor associated macrophages7,8. For both cases, experimental results suggest that IDO1 inhibition may restore an effective antitumor immune response and thus provide a method to treat malignant diseases in combination with chemotherapeutic agents and/or immunotherapy-based strategies9. In fact, there are currently three drugs in clinical trials testing IDO1 inhibition as a strategy for the treatment of cancer10. Clearly there is an interest and a need for further development of potent IDO1 inhibitors to adequately address this therapeutic opportunity11,12.
IDO1 is an extrahepatic, tryptophan (Trp) metabolizing enzyme, 13–15 which catalyzes the initial and rate-limiting step along the kynurenine pathway. The oxidative metabolism of Trp by IDO1 involves the coordination of molecular oxygen to a ferrous heme iron and its subsequent addition across the C-2/C-3 bond of the indole ring. Two alkylperoxy transition or intermediate states resulting from dioxygen insertion to C-2 or C-3 carbon of the indole ring (Figure 1, compound 1 and 2) have been proposed13,16–18. Here we sought to rationally design a new family of IDO1 inhibitors by mimicking the alkylperoxy species as illustrated in Figure 1, in which one oxygen atom of the peroxo moiety is substituted with an electronegative atom, N (compound 3). To simplify compound 3, the alkylperoxy moiety is further reduced to Ar-C-X-Y, where Ar is an aryl ring and C is a linker containing a carbon moiety (such as methylene or carbonyl). In related work, two previously reported IDO1 inhibitors in the scientific literature demonstrate a similar theme in their inhibitor design: The hydroxyamidines reported in 2009 by Incyte19 and the recent report of alkylhydrazine derivative inhibitory activity by Ching et al.20. Neither of these reports discussed the potential mimicry of the proposed alkylperoxy intermediate or related rational design idea. Contemporaneous with our studies, O-alkylhydroxylamine IDO1 inhibitors were reported in the patent literature by NewLink Genetics21. With the rise of chemical library screening for drug lead development, mechanism-based rational drug design ideas are less frequently used, but the current work illustrates there is still merit in their use as they proved a simple and efficient way to identify a new IDO1 inhibitor lead.
Figure 1.

Proposed Alkylperoxy Transition or Intermediate State of IDO1 (1 and 2) and Their Mimic (3).
Results and Discussion
Discovery of New Structural Class of IDO1 Inhibitors
To explore this rational drug design idea, we screened commercially available compounds with a general structure of Ar-X-Y or Ar-C-X-Y (Table 1). Gratifyingly, a screen of a few dozen commercial compounds yielded several micromolar or sub-micromolar potency molecules. Similar to the recent report from the Ching group20, phenylhydrazine was found to be a highly potent inhibitor of IDO1. This is not surprising since phenylhydrazine has been found to interact with other heme-containing proteins, such as hemoglobin,22 cytochrome P450,23 catalase24 and myoglobin25. In fact, phenylhydrazine can be oxidized by heme iron to generate phenyl radicals that can subsequently react to covalently modify the heme prosthetic group26. Although we did not see any signs of irreversible inhibition by phenylhydrazine, we chose to focus our efforts on O-benzylhydoxylamine, the second best inhibitor with an IC50 of 0.90 μM, as we were more interested in developing a reversible inhibitor of IDO1. Nonetheless, all the compounds in Table 1 represent a new class of IDO1 inhibitors: alkylperoxy mimics. The therapeutic potential of IDO1 inhibition, as well as the commercial availability and sub-micromolar level potency of these compounds, made this discovery an important opportunity. Our efforts at developing peroxy structural mimics, in particular, optimizing the O-alkylhydroxylamine structural class, are described below.
Table 1.
IC50 of Commercially Available Ar-X-Y or Ar-C-X-Y Compoundsa
| Compound Structure | IC50 (μM) |
|---|---|
| Ph-NH-NH2 | 0.23±0.25 |
| Ph-CH2-O-NH2 | 0.81±0.081 |
| Ph-CH2-NH-OH | 6.0 |
| Ph-NH-OH | 9.2 |
| Ph-C(=O)-NH-OH | 16 |
| Ph-CH2-NH-NH2 | 71 |
values are based on single point inhibition curves run once for each compound, but repeated 2x IC50 for the most potent compounds (initial IC50 values below 1.0 μM). These results are reported as the averages ± SD.
Chemistry
The O-alkylhydroxylamine derivatives were synthesized from the analogous alcohol through a one pot process involving the Mitsunobu reaction with N-hydroxyphthalimide27 and subsequent deprotection of the phthalimide group with hydrazine (Scheme 1)28. The O-alkyl hydroxylamines were purified and isolated as their hydrochloride salts. Alcohols that were not commercially available were synthesized via reduction of the appropriate aldehyde with NaBH4 (Scheme 2).
Scheme 1.

Mitsunobu Reaction to Convert Alcohols to O-Alkyl Hydroxylamines
Scheme 2.
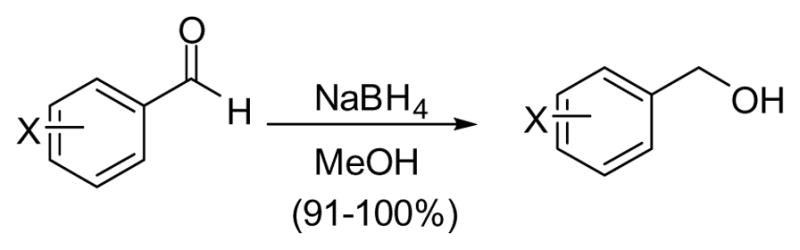
Synthesis of Monoaryl Alcohols
Results and Discussion
IDO1 Inhibition by O-Alkylhydroxylamines
In an effort to further optimize the O-benzylhydroxylamine lead, we explored two general modifications of the O-benzylhydroxylamine structure: (1) alteration of the carbon linker between the aryl ring and the hydroxylamine group; (2) substitution of the aryl ring (Figure 2).
Figure 2.
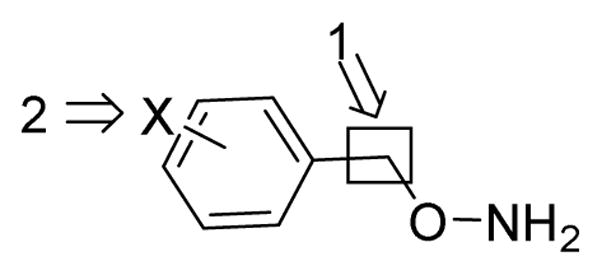
Two Modifications of O-Benzylhydroxylamine Lead Compound
Alternation of the carbon linker between the aryl ring and the peroxy mimic moiety
Linker length and flexibility was explored with the compounds shown in Table 2. It quickly became clear that any modifications made to the carbon linker in the lead compound resulted in a dramatic decrease in inhibitor potency. Attempts at rigidifying the hydroxylamine moiety in the benzylic position (compounds 3, 4 and 6) were unsuccessful as well.
Table 2.
Inhibition Data for Monoaryl Hydroxylamines with Various Linkers between the Aryl and the Hydroxylamine Groupa
| Compd | Structure | IC50 (μM) |
|---|---|---|
| Lead |
|
0.81±0.081 |
| 1 |
|
5.2 |
| 2 |
|
7.1 |
| 3 |
|
10 |
| 4 |
|
10 |
| 5 |
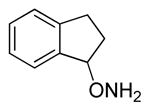
|
20 |
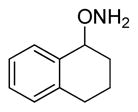
| 6 |
|
26 |
| 7 |
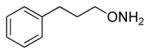
|
82 |
IC50 values are based on single point inhibition curves run once for each compound except for the lead compound which was repeated 2x. These results are reported as the average ± SD.
Substitution of the Aryl Ring
Derivatization of the O-benzylhydroxylamines via substitution on the aromatic ring initially explored potential interactions with the amino acid residues located in the active site (C129, S167, Y126) by adding hydrogen donating or accepting substitutions to the phenyl ring. Previous work from our laboratory had successfully exploited these interactions in the optimization of the phenyl-imidazole structural series29. However, addition of a hydroxyl group at the ortho (29) or para (31) position substantially reduced inhibition (Table 3). Likewise, ortho, meta, or para- substitution with a methoxy also proved deleterious (24, 26 and 30). Halogen substitution was the only successful modification that increased inhibitor potency, which contrasts with the results of the phenyl-imidazole study29. Addition of iodine, bromine, chlorine or trifluoromethyl substituents to either ortho, meta or para positions of O-benzylhydroxylamine (8–15) proved beneficial with the greatest gain in IDO1 inhibitor potency seen with halogens in the meta position. In contrast to other halogen substitutions, the more electronegative fluorine containing inhibitors (15, 16 and 18) were essentially equipotent or slightly less potent than the lead.
Table 3.
Inhibition Data for Monoaryl Hydroxylamines with Ring Substitutiona
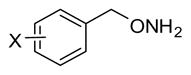
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd | X | IC50(μM) | Compd | X | IC50(μM) | Compd | X | IC50(μM) |
| 8 | 4-I | 0.22± 0.066 | 16 | 3-F | 1.0 | 24 | 2-OCH3 | 5.4 |
| 9 | 3-Cl | 0.30± 0.015 | 17 | 4-Cl | 1.3 | 25 | 4-CF3 | 8.9 |
| 10 | 3-Br | 0.32± 0.042 | 18 | 2-F | 1.4 | 26 | 3-OCH3 | 9.5 |
| 11 | 3-I | 0.34± 0.12 | 19 | 4-Br | 1.6 | 27 | 4-Ph | 12 |
| 12 | 3-CF3 | 0.41 | 20 | 2-Br | 1.6 | 28 | 4- CH(CH3)2 | 15 |
| 13 | 2-I | 0.57 | 21 | 2-CF3 | 2.5 | 29 | 2-OH | 19 |
| 14 | 2-Cl | 0.63 | 22 | 3-CH3 | 2.7 | 30 | 4-OCH3 | 24 |
| Lead | H | 0.81±0.081 | 23 | 4-NO2 | 2.7 | 31 | 4-OH | 61 |
| 15 | 4-F | 0.98 | ||||||
IC50 values are based on single point inhibition curves run once for each compound, but repeated at least 2x for the most potent compounds (initial IC50 values below 0.4 μM). These results are reported as the averages ± SD.
The benefits of halogen aromatic substitution have been recognized before in several previous reports of structure-activity relationship studies19,30–35. There are three potential explanations for the benefits of halogen substitution: Favorable pi stacking interactions between the halo-aromatics and aromatic amino acid side chains in the IDO1 active site; halogen bonding between the halogen atoms of the inhibitor and Lewis basic sites in the IDO1 active site36; or beneficial hydrophobic interactions between the halogen and a complementary pocket in the active site. Favorable pi stacking interactions between the three relatively more electron rich Phe residues (F163, 164 and 226) in the active site and the electron poor halo-aromatic pi systems in the inhibitors would benefit binding. Contradicting that assessment would be the fluorobenzene derivatives (15, 16, and 18) which afforded inhibition essentially equivalent to the lead, O-benzylhydroxylamine, or slightly less potent. The weaker activity of the fluorine substitutions versus the chlorine, bromine and iodine would be consistent with halogen bonding events since fluorine is too electronegative and has too small a sigma hole for effective halogen bonding37,38. A more traditional explanation of simple favorable hydrophobic interactions by the halogens seems less likely due to the weaker activity seen with 3-methyl (22) and 4-isopropyl (28), which are considered roughly isosteric with chlorine and iodine respectively39.
Disubstitution of the Aryl Ring
Synergistic effects of multiple substituents were also explored with a particular focus on maximizing the halogen benefits. However, as depicted in Table 4, minimal additive effects were observed when combining beneficial substitutions from the monoaryl system. None of the disubstituted derivatives was more potent than the most potent monosubstituted derivatives, i.e. the C-3 substituted compounds.
Table 4.
Inhibition Data for Monoaryl Hydroxylamines with Ring Disubstitutiona
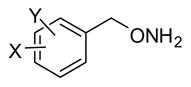
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd | R | IC50(μM) | Compd | R | IC50(μM) | Compd | R | IC50(μM) |
| 32 | 2,3-Cl2 | 0.45 | 37 | 3,5-Cl2 | 0.90 | 42 | 2-CF3, 4-F | 2.4 |
| 33 | 3-CF3,4-Cl | 0.49 | 38 | 2-F,4-CF3 | 1.1 | 43 | 3,5-F2 | 3.4 |
| 34 | 2-F, 5-CF3 | 0.57 | 39 | 2,4-F2 | 1.4 | 44 | 3,5-Br2 | 4.2 |
| 35 | 2,4-Cl2 | 0.63 | 40 | 2,5-(OCH3)2 | 1.7 | 45 | 3,5-(CF3)2 | 4.6 |
| 36 | 2-Cl, 4-I | 0.73 | 41 | 3,4-Cl2 | 2.1 | 46 | 2-OH, 3-OCH3 | 55 |
| Lead | H | 0.81±0.081 | ||||||
IC50 values are based on single point inhibition curves run once for each compound.
Spectroscopic Analysis of Monoaryl Hydroxylamine Binding
Given the perplexing results, especially the contradictions with the phenyl-imidazole work, we further explored the binding mechanism of the monoaryl hydroxylamines, including the lead compound, a monosubstituted derivative (8) and a disubstituted derivative (40), by UV-Vis absorption spectroscopic studies. As shown in Figure 3B, in the absence of inhibitors, the deoxy ferrous enzyme exhibits Soret and Q band at 427 and 556 nm, characteristic for a typical five-coordinate high-spin heme species. Binding of the lead compound, 8 and 40, led to shift of the Soret band to 419–421 nm, and the appearance of two new Q bands at 531/556 nm, characteristic of a six-coordinate low-spin species, indicating the coordination of the monoaryl hydroxylamines to the heme iron, possibly via the amine group. Although definitive proof of the coordination of the amine group to the heme iron requires further studies, the result strongly supports the proposed mimicry of the alkylperoxy transition or intermediate state by the monoaryl hydroxylamines.
Figure 3.

Absorption spectra of ferric (A), deoxy ferrous (B) and CO-bound (C) IDO1 in the presence of 500 μM inhibitors. Ref spectra were obtained in the absence of inhibitors. The 616 nm band labeled with an asterisk is of unknown origin.
In addition to the deoxy enzyme, we also studied inhibitor binding to the CO-bound ferrous enzyme, where CO was used as a surrogate for O2. The CO-bound IDO1 exhibits Soret and Q band at 421 and 538/568 nm, typical for a CO bound six-coordinate low-spin heme species (Figure 3C). The addition of the lead compound resulted in a spectrum similar to that of the inhibitor-bound deoxy species shown in (B), indicating that inhibitor binding prevents CO binding to the heme iron. In contrast, the addition of 8 or 40 led to a spectrum, consistent with a mixture of inhibitor-bound deoxy spectrum (B) and CO-bound spectrum (C), indicating that inhibitor binding partially prevents CO binding to the heme iron. Comparative studies suggested that the affinity of these inhibitors towards the ferric enzyme (Figure 3A) is much weaker than the ferrous enzyme.
Modeling of Monoaryl Hydroxylamine Binding to Guide Drug Development
With confirmation that the monoaryl hydroxylamine compounds inhibit IDO1 by coordinating to the heme iron, we then turned to docking studies using Gold (v5.1) to determine their potential binding modes (Figure 4). Unsubstituted monoaryl inhibitor, O-benzylhydroxylamine, was predicted to have two equally plausible binding modes: in the interior of the cavity near residues F163 and S167, or in the entrance of the cavity between residues F226 and the heme propionate group. Ortho-substituted inhibitors (C-2 substitution, Figure 5) were predicted to bind in the back of the cavity (blue structure in Figure 4), while the para-substituted inhibitors (C-4 substitution, Figure 5) appeared to preferentially bind in the cavity entrance (magenta structure in Figure 4). The two outliers to this trend were compound 29 (2-OH) and compound 31 (4-OH). For compound 31, docking predicted the formation of a hydrogen bond with the S167 in the back of the cavity, while for compound 29 docking suggested hydrogen bonding with the heme propionate in the front of the cavity. Nonetheless, assessment of the inhibition data does not support that either of these predicted hydrogen bonds enhance the binding affinity in comparison with the halogenated derivatives; this stands in contrast to earlier reported hydroxyl-substituted 4-phenyl-imidazole inhibitors29. Docking of the meta substituted analogs to IDO1, however, did not define a consistent binding mode as the aryl ring could be equally positioned either in the back or the entrance of the cavity (C-3 substitution, Figure 5).
Figure 4.

Compounds 8 and 14 Docked in the IDO1 Active Site. Compound 14 (blue; chlorine atom green) binds in the interior of the active site, while 8 (magenta; iodine atom red) prefers to bind in the active site entrance when docked. The heme is shown in white and the green represents active site structure of IDO1.
Figure 5.

Structure-Activity Relationships (SAR) of Substituted Monoaryl Hydroxylamines with Predicted Binding Mode. Plot of pIC50 values for the substituted hydroxylamine compounds. The x-axis lists the substitution number on the phenyl ring with disubstituted represented by the sum of the two numbers. The data points are colored based on docked binding mode in the back (blue), front (pink) or either location (gray) of the IDO1 active site.
Detailed Enzyme Inhibition Studies
As further confirmation of the inhibitory characteristics of the hydroxylamine structural class, inhibitory constants (Ki) were determined for two of the most potent hydroxylamine inhibitors. Analysis showed Ki values of 164 and 154 nM for two O-alkylhydroxylamines, 8 and 9, respectively These potency values yield ligand efficiencies of 0.93 for both compounds40,41,42. Given the strong correlation between successful drugs and high ligand efficiencies, the O-alkylhydroxylamines represent a promising class of IDO1 inhibitors43. Based on Lineweaver-Burk graphical analysis, both molecules demonstrated an uncompetitive mode of inhibition. Additionally, it was determined that inhibition of IDO1 activity by these two compounds was reversible and appeared to be the result of one-to-one interaction between the O-alkylhydroxylamine inhibitor and IDO1 based on dose-response studies. Details of the procedures and graphs for these studies can be found in the Supplementary Data section (see Figures S1–S3). An uncompetitive mode of inhibition would seem inconsistent with the demonstrated binding at the heme iron from the spectroscopic studies. However, 4-phenylimidazole has been crystallized in IDO1 bound to the heme iron44 and it demonstrates noncompetitive inhibition45. Furthermore, 4-phenylimidazole derivatives have also demonstrated an uncompetitive inhibition mode29 and presumably they are also binding at the IDO1 active site, in direct competition with the Trp binding location. Therefore, uncompetitive or non-competitive inhibition mode does not preclude binding in the active site or to the heme iron. One explanation for such behavior is that the inhibitors are actually in direct competition for binding at the heme iron with the other substrate in the reaction, oxygen. In an assay that modifies the concentration of tryptophan, an inhibitor that competes with oxygen would likely afford an uncompetitive inhibition mode.
Selectivity Studies
The simplicity of the O-alkylhydroxylamines makes concerns about their selectivity warranted, especially given their primary mechanism of attraction to IDO1, heme iron binding. To analyze the selectivity of two of the top compounds, 8 and 9, two additional heme iron containing enzymes, catalase and CYP3A4, were screened for inhibition. As noted in Table 5, neither 8 nor 9 showed inhibition of catalase; perhaps this is not surprising given catalase’s small natural substrate, hydrogen peroxide, however, this result does demonstrate that the inhibitory activity of these compounds is not attributable to an indirect effect on the catalase in the reaction mixture that is required to protect IDO1 from oxidative damage. CYP3A4 was inhibited at the low micromolar level by both 8 and 9. However, this is still 22- and 48-fold less potent than IDO1 based on the IC50 values for 8 and 9, respectively. This suggests that the O-alkylhydroxylamines are selective, but they do demonstrate some promiscuity and as studies advance, they will need to be closely monitored for off-target activity.
Table 5.
Inhibition of catalase and CYP3A4 by 8 and 9
| Compound | IDO1 | Catalase | CYP3A4 | ||
|---|---|---|---|---|---|
| IC50 (μM) | IC50 (μM) | Fold | IC50 (μM) | Fold | |
| 8 | 0.33 | > 100 | > 300 | 7.2 | 22 |
| 9 | 0.31 | > 100 | > 320 | 15 | 48 |
Cell-based Assays
To assess the therapeutic potential of the hydroxylamine structural class, two of the most potent compounds, 8and 9, were tested for cell-based activity in two cell systems: HeLa, expressing native human IDO1 induced with IFNγ, and Trex, expressing recombinant human IDO1 induced with doxycycline (Table 7; graphs found in Supplementary Data, Figure S4). Both compounds demonstrated nanomolar level activity in both the HeLa and Trex cell lines. Compounds 8 and 9 were more potent in the cell-based assay than in the isolated enzyme assay. Although such a result is generally considered inconsistent with normal drug activity, this irregularity has been seen repeatedly with IDO1 studies19,46 and has been attributed to deficiencies in the isolated enzyme assay, and specifically problems with controlling IDO1 redox activity.
Table 7.
IDO1 IC50 values in HeLa and Trex cell-based assays
HeLa assay results are the averages of three runs with standard deviations
Trex assay was a single run
Cytotoxicity studies with HeLa cells also demonstrated good cell viability up to concentrations of 100 μM of 8 and 9 (see Supplementary Data, Figure S4). Assessment of impact of human serum protein binding indicated a substantial reduction in inhibitory activity for one of the compounds, 8, while the inhibitory activity of 9 was relatively unaffected (see Supplementary Data, Figure S5). This analysis indicates that it should be possible to develop O-alkylhydroxylamine inhibitors that are not unduly compromised by serum protein binding.
Cumulatively, the cell-based potency and the cytotoxicity studies dramatically illustrate the therapeutic potential for the hydroxylamines. To the best of our knowledge, there are only two other structural classes reported to date with similar nanomolar-level cell-based potency19,32,33,46 and both of these have been developed for clinical evaluation.
Conclusion
A new structural class of IDO1 inhibitors, O-alkylhydroxylamines, has been discovered. The parent compound, O-benzylhydroxylamine, exhibits sub-micromolar inhibition and, unlike many similarly potent IDO1 inhibitors, is an inexpensive, commercially available compound. Structure-activity studies of the O-benzylhydroxylamine lead have explored the chemical space around this structure and led to some modest improvements in potency. Halogenation of the aromatic ring was particularly successful in improving potency. Spectroscopic studies have shown that the O-alkylhydroxylamine compounds coordinate to the heme iron, which provide preliminary support for these compounds as alkylperoxy transition or intermediate state mimics. More importantly, among the strongest enzymatic inhibitors in the O-alkylhydroxylamine class, two exhibited submicromolar cell-based potency with minimal toxicity. With ligand efficiencies of 0.9, these compounds hold significant promise as therapeutic tools. Translating enzymatic potency to cell-based potency has been a major roadblock to inhibitor development for other structural classes such as the naphthoquinones47. Furthermore, two O-alkylhydroxylamine derivatives, 8 and 9, demonstrated exceptional potency on par with the two compounds INCB02436048 and NLG91949 that are currently being evaluated in clinical trials. Given the exciting potential of these rather simple molecules to meet the burgeoning interest in IDO1 inhibition for both biological investigations of the kynurenine pathway and therapeutic applications, we believe that the O-alkyl hydroxylamines will be a useful structural class of inhibitors.
Experimental Section
Chemical synthesis
General Procedures
All reactants and reagents were commercially available and were used without further purification unless otherwise indicated. Anhydrous THF was freshly distilled from Na and benzophenone. All reactions were carried out under an inert atmosphere of argon or nitrogen unless otherwise indicated. Concentrated refers to the removal of solvent with a rotary evaporator at normal water aspirator pressure followed by further evacuation with a direct-drive rotary vane vacuum pump. Thin layer chromatography was performed using silica gel 60 Å precoated glass or aluminum backed plates (0.25 mm thickness) with fluorescent indicator, which were cut. Developed TLC plates were visualized with UV light (254 nm), iodine, p-anisaldehyde or ninhydrin. Flash column chromatography was conducted with the indicated solvent system using normal phase silica gel 60 Å, 230–400 mesh. Yields refer to chromatographically and spectroscopically pure (>95%) compounds except as otherwise indicated. Melting points were determined using an open capillary and are uncorrected. 1H NMR spectra were recorded at 400 MHz and 13C NMR spectra were recorded at 100 MHz. Chemical shifts are reported in δ values (ppm) relative to an internal reference (0.05% v/v) of tetramethylsilane (TMS, 0.0) for 1H NMR and the CD3OD solvent peak (49.0) for 13C NMR. Peak splitting patterns in the 1H NMR are reported as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad. The attached proton test (APT) experiment was conducted for 13C NMR analysis; methylene and quaternary carbons were identified as negative peaks, while methyl and methine had positive peaks. HPLC was conducted on an Agilent 1100 with an Ascentis Express C-18 column (100 × 4.6 mm, 2.7 μm) and a mobile phase of 80:20 MeCN:H2O. GC analyses were performed on the free hydroxylamine with an EI-MS detector fitted with a 30 m × 0.25 mm column filled with cross-linked 5% PH ME siloxane (0.25 μm film thickness); gas pressure 7.63 psi He. Analysis of samples involved heating from 70 to 250°C (10°C/min) and finally holding at 250°C for 7 min. The free hydroxylamine compounds were generated by treating the hydrochloride salts with saturated NaHCO3 and extracting with EtOAc. All compounds were found to be >95% purity by elemental analysis, GC, or HPLC as indicated.
General Synthesis of O-Alkyl Hydroxylamines
To a solution of alcohol (1 mmol) in freshly distilled THF (5 ml) was added triphenylphosphine (1.1 mmol) and N-hydroxylphthalimide (1.1 mmol). After the solution was cooled to 0°C diisopropylazodicarboxylate (1.1 mmol) was added dropwise. The solution was allowed to warm to room temperature over 3 hours. Reaction progress was monitored by TLC (1:1 heptanes:ethyl acetate). Hydrazine monohydrate (1.1 mmol) was then added and the solution was allowed to stir for 30 min. The resulting reaction mixture was filtered to remove the white precipitate. The filtrate was concentrated and subjected to flash chromatography (1:1 heptanes/ethyl acetate). The resulting product was dissolved in ether and treated with HCl (2.0 M solution in ether) to afford the HCl salt of the O-alkylhydroxylamine. Contaminating diisopropyl hydrazinodicarboxylate could be washed away from the HCl salt with dichloromethane.
O-(α-Cyclopropyl)benzylhydroxylamine hydrochloride (1)
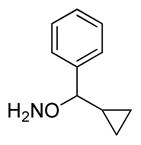
Synthesized from α-cyclopropyl benzyl alcohol according to the general procedure to afford 1 as white crystals in 96% yield. 1H NMR (400 MHz, CD3OD) δ 7.46-7.41 (br s, 5H, ArH), 4.86 (s, 1H, ArCH, J=8 Hz), 1.36-1.29 (m, 1H, ArCH(ONH2)CH), 0.87-0.81 (m, 1H, CH2), 0.65-0.57 (m, 2H, CH2), 0.36-0.31 (m, 1H, CH2). 13C NMR (100 MHz, CD3OD) δ 138.36, 130.66, 130.19, 128.63, 92.98, 16.05, 6.14, 2.64. Anal. calcd. for C10H14ClNO: C= 60.15; H=7.07; N=7.01. Found: C=60.05%, H = 7.35%, N =6.91%.
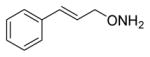
O-(3-Phenyl-2-propenyl)-1-hydroxylamine hydrochloride (2)
Synthesized from 3-phenyl-2-propen-1-ol according to the general procedure to afford 2 as white crystals in 90% yield. 1H NMR (400 MHz, CD3OD) δ 7.53-7.47 (m, 2H, ArH), 7.34-7.29 (m, 3H, ArH), 6.85 (d, 1H, ArCH, J=16 Hz) 6.40-6.33 (m, 1H, ArCHCH), 4.69 (dd, 2H, ArCHCHCH2, J=6.9, 1.1 Hz). 13C NMR (100 MHz, CD3OD) δ 139.57, 137.10, 129.93, 129.88, 128.13, 121.47, 77.00. Anal. calcd. for C9H12ClNO: C, 58.23; H, 6.52; N, 7.54Found: C=58.71, H = 6.95, N =7.49.
O-(1,2,3,4-Tetrahydro-1-naphthalene)-hydroxylamine hydrochloride (3)
Synthesized from 1,2,3,4-tetrahydro-1-naphthalenol according to the general procedure to afford 3 in 62% yield as white crystals. 1H NMR (400 MHz, CD3OD) δ 7.40 (d, 1H, ArH, J=8 Hz), 7.31 (t, 1H, ArH, J=4 Hz), 7.25-7.17 (m, 2H, ArH), 5.14 (t, 1H, ArCH, J=2 Hz), 2.90-2.75 (m, 2H, ArCH2), 2.32-2.27 (m, 1H, ArCH2CH2), 2.01-1.94 (m, 2H, ArCH2CH2CH2), 1.87-1.81 (m, 1H, ArCH2CH2). 13C NMR (100 MHz, CD3OD) δ 139.93, 132.27. 131.63, 130.44, 130.33, 127.16, 82.10, 29.55, 27.51, 18.67. Anal. calcd. for C10H14ClNO: C=60.15; H=7.07; N= 7.01Found: C=60.13, H = 7.34, N =6.91.
O-(2,3-Dihydro-1H-inden-2-yl)-hydroxylamine hydrochloride (4)
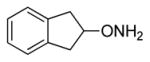
Synthesized from 2,3-dihydro-1H-inden-2-ol according to the general procedure to afford 4 as white crystals in 81% yield. Mp=132–133.5°C. 1H NMR (400 MHz, CD3OD) δ 7.29-7.25 (m, 2H, ArH), 7.22-7.18 (m, 2H, ArH), 5.07-5.02 (m, 1H, ArCH2CH), 3.37 (d, 1H, ArCHCH2, J=4 Hz), 3.33 (d, 1H, ArCHCH2, J=4 Hz), 3.19 (d, 1H, ArCHCH2, J=4 Hz), 3.15 (d, 1H, ArCHCH2, J=4 Hz. 13C NMR (100 MHz, CD3OD) δ 139.18, 126.73, 124.25, 86.05, 37.65. GC tR = 3.182 min.
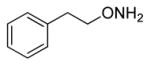
O-Phenethylhydroxylamine hydrochloride (5)
Synthesized from phenethyl alcohol according to the general procedure to afford 5 as white crystals in 56% yield. Mp = 110–111°C. 1H NMR (400 MHz, CD3OD) δ 7.28-7.21 (m, 5H, ArH), 4.25 (t, 2H, ArCH2, J=8 Hz), 2.98 (t, 2H, ArCH2CH2, J=8 Hz). 13C NMR (100 MHz, CD3OD) δ 139.28, 129.97, 129.61, 127.83, 76.86, 35.08. Anal. calcd. for C8H11NO-0.9375 HCl: C=56.07, H=7.03, N=8.06. Found: C=55.88%, H = 6.56%, N =8.08%.
O-(2–3-Dihydro-1H-inden-1-yl)hydroxylamine (6)
Synthesized from 2,3-dihydro-1H-inden-ol according to the general procedure to afford 6 as white crystals in 78% yield. Mp=150.5–152°C. 1H NMR (CD3OD) δ 7.49 (d, 1H, ArH, J=8 Hz), 7.40-7.33 (m, 2H, ArH), 7.30-7.26 (m, 1H, ArH), 5.59 (d, 2H, CHONH2, J=4 Hz), 3.18-3.10 (m, 1H, ArCH2), 2.96-2.89 (m, 1H, ArCH2), 2.48-2.39 (m, 1H, ArCH2CH2), 2.36-2.31 (m, 1H, ArCH2CH2). 13C NMR (100 MHz, CD3OD) δ 146.69, 139.10, 131.42, 127.95, 127.02, 126.28, 90.29, 31.90, 30.83. GC tR = 6.781 min.
O-(3-Phenylpropyl)-hydroxylamine hydrochloride (7)
Synthesized from 3-phenylpropanol according to the general procedure to afford 7 as white crystals in 95%yield. Mp=168–169°C.1H NMR (400 MHz, CD3OD) δ 7.33-7.25 (m, 2H, ArH), 7.21-7.13 (m, 3H, ArH), 4.02 (t, 2H, CH2ONH2, J=8 Hz), 2.72 (t, 2H, ArCH2, J=8 Hz), 2.03-1.96 (m, 2H, ArCH2CH2,). 13C NMR (100 MHz, CD3OD) δ 142.15, 129.66, 129.51, 127.33, 75.63, 32.63, 30.57. GC tR = 6.425 min.
4-(Iodobenzyl)hydroxylamine hydrochloride (8)
Synthesized from 4-iodobenzyl alcohol according to the general procedure to afford 8 as white crystals in 70% yield. Mp =210–212°C 1H NMR (400 MHz, CD3OD) δ 7.80 (d, 2H, ArH, J=4 Hz), 7.22 (d, 2H, ArH J=4 Hz), 4.99 (s, 1H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 139.03, 133.86, 132.02, 96.09, 77.16. GC tR = 8.828 min.
O-(3-Chlorobenzyl)hydroxylamine hydrochloride (9)
Synthesized from the respective alcohol according to the general procedure to afford 9 as white crystals in 50% yield. Mp =144–148°C. 1H NMR (400 MHz, CD3OD) δ 7.49 (s, 1H, ArH), 7.45-7.35 (m, 1H, ArH), 5.04 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 139.49, 135.59, 131.42, 130.55, 130.12, 128.56, 77.02. GC tR = 6.251 min.
O-(3-Bromobenzyl)hydroxylamine hydrochloride (10)
Synthesized from 3-bromobenzenyl alcohol according to the general procedure to afford 10 as white crystals in 98%yield. Mp=210–211°C. 1H NMR (400 MHz, CD3OD) δ 7.66-7.56 (m, 2H, ArH), 7.43 (d, 1H, ArH, J=8 Hz),7.37 (t, 1H, ArCH, J=8 Hz), 5.03 (s, 2H). 13C NMR (100 MHz, CD3OD) δ 136.78, 133.62, 133.14, 131.71, 129.02, 123.59, 77.03. Anal. calcd. for C7H9ClBrNO:C=35.25; H=3.80; N=5.87. Found: C=35.37%, H = 3.52%, N =5.76%.
O-(3-Iodobenzyl)hydroxylamine hydrochloride (11)
Synthesized from the respective alcohol according to the general procedure to afford 11 as white crystals in 41% yield. Mp=209–214°C. 1H NMR (400 MHz, CD3OD) δ 7.82-7.77 (m, 2H, ArH ), δ 7.44 (d, 1H, ArH, J=8 Hz), 7.21 (t, 1H, ArH, J=8 Hz), 4.98 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 139.79, 139.27, 136.74, 131.77, 129.62, 95.02, 77.04. GC tR =8.732 min.
O-(3-Trifluoromethylbenzyl)hydroxylamine hydrochloride (12)
Synthesized from 3-trifluoromethylbenzyl alcohol according to the general procedure to afford 12 as white crystals in 88% yield. Mp=164–165°C 1H NMR (400 MHz, CD3OD) δ 7.77-7.72 (m, 3H, ArH), 7.65 (t, 1H, ArH, J=8 Hz), 5.14 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 134.40, 132.62, 130.7 (q, JC–F=40 Hz), 129.50, 125.92 (q, JC–F=4 Hz), 125.51 (q, JC–F=4 Hz), 123.99 (d, JC–F =270 Hz), 75.71. GC tR = 4.170 min.
O-(2-Iodobenzyl)hydroxylamine hydrochloride (13)
Synthesized from the respective alcohol according to the general procedure to afford 13 as white crystals in 43% yield. Mp=119–120°C. 1H NMR (400 MHz, CD3OD) δ 7.95 (d, 1, ArH, J=8 Hz), 7.51-7.44 (m, 2H, ArH), 7.16 (t, 1H, ArH, J=8 Hz), 5.14 (s, 1H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 141.12, 137.07, 132.35, 132.01, 129.83, 100.05, 81.35. Anal. calcd. for C7H9ClINO-H2O: C, 27.70; H, 3.65; N, 4.61. Found: C=27.84, H =3.66, N =4.48.
O-(2-Chlorobenzyl)hydroxylamine hydrochloride (14)
Synthesized from the respective alcohol according to the general procedure to afford 14 as white crystals in 75% yield. Mp=148–150°C. 1H NMR (400 MHz, CD3OD) δ 7.57 (d, 1H, ArH, J= 8 Hz), 7.48-7.35 (m, 3H, ArH), 5.22 (s, 2H, ArH). 13C NMR (100 MHz, CD3OD) δ 135.63, 132.68, 132.21, 131.89, 130.74, 128.37, 74.92. Anal. calcd. for C7H9Cl2NO: C, 43.33; H, 4.67; N, 7.22. Found: C=43.01, H =4.72, N =7.03.
O-(4-Fluorobenzyl)hydroxylamine hydrochloride (15)
Synthesized from 4-fluorobenzyl alcohol according to the general procedure to afford 15 as white crystals in 85% yield. Mp=219–220°C. 1H NMR (400 MHz, CD3OD) δ 7.49 (t, 2H, ArH, J=4 Hz) 7.16 (t, 2H, ArH, J=4 Hz), 5.02 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 164.90 (d, JC–F=240 Hz), 132,87 (d, JC–F=9 Hz), 130.53 (d, JC–F=3 Hz), 116.78 (d, JC–F=22 Hz), 77.31. Anal. calcd. for C7H9ClFNO: C, 47.34; H, 5.11; N, 7.89. Found: C=47.43, H = 4.94, N =7.81.
O-(3-Fluorobenzyl)hydroxylamine hydrochloride (16)
Synthesized from 3-fluorobenzyl alcohol according to the general procedure to afford 16 as white crystals in 81% yield. Mp=245°C (decomp).1H NMR (400 MHz, CD3OD) δ 7.45 (t, 1H, ArH, J=8 Hz) 7.28-7.15 (m, 3H, ArH), 5.07 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 164.22 (d, JC–F=240 Hz), 136.91 (d, JC–F=8 Hz), 131.80 (d, JC–F=8 Hz), 126.04 (d, JC–F=3 Hz), 117.31 (d, JC–F=21 Hz), 116.85 (d, JC–F=22 Hz), 77.12 (d, JC–F=2 Hz). GC tR = 3.981 min.
O-(4-Chlorobenzyl)hydroxylamine hydrochloride (17)
Synthesized from 4-chlorobenzyl alcohol according to the general procedure to afford 17 as white crystals in 70% yield. Mp=226–228.5°C. 1H NMR (400 MHz, CD3OD) δ 7.44 (br s, 4H, ArH), 5.05 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 136.54, 133.11, 132.05, 130.01, 77.13. Anal. calcd. for C7H9Cl2NO: C=43.44; H=4.67; N=7.22. Found: C=43.32%, H =4.59%, N =7.15%.
O-(2-Fluorobenzyl)hydroxylamine hydrochloride (18)
Synthesized from the respective alcohol according to the general procedure to afford 18 as white crystals in 90% yield. Mp=182–184°C. 1H NMR (400 MHz, CD3OD) δ 7.58-7.45 (m, 2H, ArH), 7.27-7.19 (m, 2H, ArH), 5.17 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 162.93 (d, JC–F=247 Hz), 133.26 (d, JC–F=8 Hz), 133.19 (d, JC–F=4 Hz), 125.82 (d, JC–F=3 Hz), 121.48 (d, JC–F=14 Hz ), 116.71 Hz (d, JC–F=21 Hz), 71.73 (d, JC–F=4 Hz). GC tR = 4.067 min.
O-(4-Bromobenzyl)hydroxylamine hydrochloride (19)
Synthesized from 4-bromobenzyl alcohol according to the general procedure to afford 19 as white crystals in 92% yield. Mp=232–236°C. 1H NMR (400 MHz, CD3OD) δ 7.58 (d, 2H, ArH, J=4 Hz), 7.37 (d, 2H, ArH, J=4 Hz), 5.02 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 133.60, 133.11, 132.29, 124.70, 77.23. GC tR =7.383 min.
O-(2-Bromobenzyl)hydroxylamine hydrochloride (20)
Synthesized from 2-bromobenzyl alcohol according to the general procedure to afford 20 as white crystals in 87% yield. Mp=95–98°C. 1H NMR (400 MHz, CD3OD) δ 7.70 (dd, 1H, ArH, J=4, 1 Hz), 7.54 (dd, 1H, ArH, J=4, 1 Hz), 7.46 (dt, 1H, ArH, J=7, 1 Hz), 7.37 (dt, 1H, ArH, J=7, 1 Hz), 5.20 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 134.38, 133.83, 132.71, 132.56, 129.19, 125.47, 77.32. Anal. calcd. for C7H9ClBrNO C=35.25; H=3.80; N=5.87. Found: C=35.14%, H = 3.57%, N =5.86%.
O-(2-Trifluoromethylbenzyl)hydroxylamine hydrochloride (21)
Synthesized from 2-trifluoromethylbenzyl alcohol according to the general procedure to afford 21 as white crystals in 78% yield. Mp=115–119°C.1H NMR (400 MHz, CD3OD) δ 7.81 (d, 1H, ArH, J=8 Hz), 7.76-7.66 (m, 2H, ArH), 7.63 (t, 1H, ArH, J=8 Hz), 5.26 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 133.89, 132.94, 132.41, 131.13, 130.10 (q, JC–F 40 Hz), 127.50 (q, JC–F=6 Hz), 125.53 (d, JC–F=271 Hz), 74.25. Anal. calcd. for C8H9ClF3NOC=42.22; H=3.99; N=6.15. Found: C=42.10%, H 4.15%, N =6.05%.
O-[(3-Methylphenyl)-methyl]-hydroxylamine hydrochloride (22)
Synthesized from 3-methyl benzenemethanol according to the general procedure to afford 22 as white crystals in 94% yield. Mp =187–188.5°C. 1H NMR (400 MHz, CD3OD) δ 7.33-7.22 (m, 4H, ArH), 5.01 (s, 2H, ArCH2), 2.37 (s, 3H, ArCH3). 13C NMR (100 MHz, CD3OD) δ 139.84, 134.12, 131.30, 131.02, 129.79, 127.49, 78.17, 21.23. Anal. calcd. for C8H12ClFNO C=55.34; H=6.97; N=8.07. Found: C=55.47%, H = 7.22%, N =8.06%.
O-(4-Nitrobenzyl)hydroxylamine hydrochloride (23)
Synthesized from the respective alcohol according to the general procedure to afford 23 as off-white crystals in 54% yield. Mp =199–199.5°C. 1H NMR (400 MHz, CD3OD) δ 8.29 (d, 2H, ArH, J=12 Hz), 7.70 (d, 2H, ArH, J=12 Hz), 5.19 (s, ArCH2, 2H). 13C NMR (100 MHz, CD3OD) δ 149.92, 141.55, 130.98, 124.90, 76.56. Anal. calcd. for C7H9ClN2O3:C=41.09; H=4.43; N=13.69. Found: C=41.20%, H =4.42%, N =13.42%.
O-(2-Methoxybenzyl)hydroxylamine hydrochloride (24)
Synthesized from respective alcohol according to the general procedure to afford 24 as white crystals in 64% yield. Mp=102–104°C. 1H NMR (400 MHz, CD3OD) δ 7.42 (t, 1H, ArH, J=8 Hz), 7.37 (d, 1H, ArH,, J=4 Hz), 7.06 (d, 1H, ArH, J=8 Hz), 6.99 (t, 1H, ArH, J=8 Hz), 5.09 (s, 2H, ArCH2), 3.88 (s, 3H, CH3). 13C NMR (100 MHz, CD3OD) δ 159.72, 132.67, 122.43, 121.75, 112.14, 73.34, 56.09. GC tR = 6.953 min.
O-(4-Trifluoromethylbenzyl)hydroxylamine hydrochloride (25)
Synthesized from the respective alcohol according to the general procedure to afford 25 as white crystals in 99% yield. Mp= 179–181°C. 1H NMR (400 MHz, CD3OD) δ 7.77 (d, 2H, ArH, J=8 Hz), 7.67 (d, 2H, ArH, J=8 Hz),5.16 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 138.81, 132.52 (d, JC–F=32 Hz), 130.73, 126.84 (q, JC–F=4 Hz), 124.09, 77.07. Anal. calcd. for C8H9ClF3NO: C=42.22; H=3.99; N=6.15 Found: C=41.98%, H = 3.89%, N =6.34%.
O-(3-Methoxybenzyl)hydroxylamine hydrochloride (26)
Synthesized from 3-methoxybenzenyl alcohol according to the general procedure to afford 26 as white crystals in 98% yield. Mp=126–127.5°C. 1H NMR (400 MHz, CD3OD) δ 7.34 (t, 1H, ArH, J=8 Hz), 7.03-6.97 (m, 3H, ArH), 5.04 (s, 2H, ArCH2), 3.81 (s, 3H, CH3OAr). 13C NMR (100 MHz, CD3OD) δ 161.44, 135.64, 130.99, 122.46, 116.15, 116.74, 78.00, 55/76. Anal. calcd. for C8H12ClNO2. C=50.67; H=6.38; N=7.39. Found: C=50.52%, H =6.07%, N =7.36%.
O-(4-Phenylbenzyl)hydroxylamine hydrochloride (27)
Synthesized from the respective alcohol according to the general procedure to afford 27 as white crystals in 75% yield. Mp=187–188°C. 1H NMR (400 MHz, CD3OD) δ 7.71 (d, 2H, ArH, J=8 Hz), 7.62 (d, 2H, ArH, J=8 Hz), 7.52 (d, 2H, ArH, J=8 Hz), 7.44 (t, 2H, ArH, J=4 Hz), 7.37 (t, 1H, ArH, , J=4 Hz), 5.07 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 143.93, 141.55, 133.21, 131.05, 129.98, 128.83, 128.03, 127.94, 77.98. GC tR = 12.779 min.
O-(4-Isopropylbenzyl)hydroxylamine hydrochloride (28)
Synthesized from the respective alcohol according to the general procedure to afford 28 as white crystals in 89%yield. Mp=159–160°C. 1H NMR (400 MHz, CD3OD) δ 7.39 (d, 2H, ArH, J=8 Hz), 7.33 (d, 2H, ArH, J=8 Hz), 5.03 (s, 2H, ArCH2),2.99-2.92 (m, 1H, (CH3)2CHAr), 1.27 (d, 6H, J=8 Hz). 13C NMR (100 MHz, CD3OD) δ 150.55, 130.30, 129.36, 126.60, 76.69, 33.86, 22.89. Anal. calcd. for C10H16ClNO C=59.55; H=8.00; N=6.94. Found: C=59.46%, H = 7.77%, N =7.02%.
O-(2-Hydroxybenzyl)hydroxylamine hydrochloride (29)
Synthesized from the respective alcohol according to the general procedure to afford 29 as white crystals in 79% yield. Mp=134–135°C. 1H NMR (400 MHz, CD3OD) 7.35-7.25, (m, 2H, ArH) 6.93-6.85 (m, 2H, ArH), 5.10 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 157.79, 132.82, 132.37, 120.86, 120.78, 116.46, 73.63. GC tR = 7.582 min.
O-(4-Methoxybenzyl)hydroxylamine hydrochloride (30)
Synthesized from 4-methoxy benzyl alcohol according to the general procedure to afford 30 as white crystals in 87%yield. Mp=125–127°C. 1H NMR (400 MHz, CD3OD) δ 7.39 (d, 2H, ArH, J=8 Hz), 6.99 (d, 2H, ArH, J=8 Hz), 4.98 (s, 2H, ArCH2), 3.83 (s, 3H, ArOCH3). 13C NMR (100 MHz, CD3OD) δ 162.17, 135.55, 132.21, 126.00, 124.20, 115.13, 77.75, 55.64. Anal. calcd. for C8H12ClNO2: C=50.67; H=6.38; N=7.39. Found: C=50.84%, H = 6.53%, N =7.39%.
O-(4-Hydroxylbenzyl)hydroxylamine hydrochloride (31)
Synthesized from 4-hydroxybenzyl alcohol according to the general procedure to afford 31 as white crystals in 44% yield. Mp=189–191°C. 1H NMR (400 MHz, CD3OD) δ 7.29 (d, 2H, ArH, J=8 Hz), 6.85 (d, 2H, ArH, J=8 Hz), 4.93 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 160.14, 132.50, 124.86, 116.63, 78.11. HPLC tR = 0.475 min.
O-(2,3-Dichlorobenzyl)hydroxylamine hydrochloride (32)
Synthesized from 2,3-dichlorobenzyl alcohol according to the general procedure to afford 32 as white crystals in 48% yield. Mp=184.5–185.5°C. 1H NMR (400 MHz, CD3OD) δ 7.64 (dd, 1H, ArH, J=8, 1 Hz), 7.51 (dd, 2H, ArH, J=8, 1 Hz), 7.39 (t, 1H, ArH, J=8 Hz), 5.24 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 134.63, 134.61, 133.84, 132.88, 130.96, 129.32, 75.38. GC tR = 8.752 min.
O-(4-Chloro-3-trifluoromethyl)benzyl)hydroxylamine hydrochloride (33)
Synthesized from the respective alcohol according to the general procedure to afford 33 (12% yield) as white crystals.). Mp=120–122°C. 1H NMR (400 MHz, CD3OD) δ 7.86 (s, 1H, ArH), 7.69 (2, 2H, ArH), 5.10 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 135.33, 134.30 (d, JC–F=3 Hz), 133.35, 129.54 (q, JC–F=30 Hz), 129.49, 124.13 (d, JC–F=271 Hz), 76.39. Anal. calcd. for C8H8Cl2F3NO-0.25 HCl: C=35.43; H=3.08; N=5.17. Found: C=35.11%, H =2.81%, N =5.11%.
O-(2-Fluoro-5-Trifluoromethylbenzyl)hydroxylamine hydrochloride (34)
Synthesized from the respective alcohol according to the general procedure to afford 34 as white crystals in 40% yield. 174.5–175°C. 1H NMR (400 MHz, CD3OD) δ 7.89-7.83 (m, 2H, ArH), 7.42 (t, 1H, ArH, J=8 Hz), 5.22 (d, 2H, ArCH2, 4JC–F=12 Hz). 13C NMR (100 MHz, CD3OD) δ 164.70 (d, JC–F=254 Hz), 130.56, 130.50 (d, JC–F=15 Hz), 128.42, 125.02 (d, JC–F=269 Hz), 123.03 (d, JC–F=16 Hz), 117.95 (d, JC–F=22 Hz), 70.95 (d, JC–F=4 Hz). Anal. calcd. for C8H8ClF4NO-0.25 HCl: C=37.72, H=3.27, N=5.50. Found: C=37.61%, H =3.05%, N =5.36%.
O-(2,4-Dichlorobenzyl)hydroxylamine hydrochloride (35)
Synthesized from 2,4-dichloro benzyl alcohol according to the general procedure to afford 35 as white crystals in 69% yield. Mp=184.5–185.5°C. 1H NMR (400 MHz, CD3OD) δ 7.57-7.54 (m, 2H, ArH), 7.44-7.41 (m, 1H, ArH), 7.40 (dd, 1H, ArH, J=2.0 Hz, 8.2 Hz), 5.19 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 137.50, 136.74, 133.94, 131.07, 130.71, 128.88, 74.40. Anal. calcd. for C7H8Cl3NO C=36.80; H=3.53; N=6.13. Found: C=36.89%, H =3.50%, N =5.88%.
O-(2-Chloro,4-iodobenzyl)hydroxylamine hydrochloride (36)
Synthesized from the respective alcohol according to the general procedure to afford 36 as white crystals in 80% yield. Mp=117–119°C. 1H NMR (400 MHz, CD3OD) δ 7.91 (s, 1H, ArH), 7.78 (d, 1H, ArH, J=8 Hz ), 7.31 (d, 1H, ArH, J= 8 Hz), 5.17 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 139.26, 137.91, 136.43, 133.89, 131.89, 96.49, 74.51. Anal. cald. for C7H8Cl2INO: C=26.28; H=2.52; N=4.38. Found: C=26.62%, H =2.15%, N =4.34%.
O-(3,5-Dichlorobenzyl)hydroxylamine hydrochloride (37)
Synthesized from respective alcohol according to the general procedure to afford 37 as white crystals in 43% yield. 1H NMR (400 MHz, CD3OD) δ 7.52 (s, 1H, ArH), 7.45 (d, 2H, ArH), 5.05 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 138.30, 136.58, 130.41, 128.66, 76.35. GC tR = 8.248 min.
O-(2-Fluoro, 4-trifluoromethylbenzyl)hydroxylamine hydrochloride (38)
Synthesized from 2-fluoro-4-trifluoromethyl benzyl alcohol according to the general procedure to afford 38 as white crystals in 67% yield. Mp=228–231°C.1H NMR (CD3OD) δ 7.75 (t, 1H, ArH, J=8 Hz), 7.61 (t, 2H, ArH, J=8 Hz), 5.23 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 162.41 (d, JC–F=250 Hz), 134.84 (dd, JC–F=42, 8 Hz), 133.79 (d, JC–F=4 Hz), 126.00 (d, JC–F=14 Hz), 124.46 (d, JC–F=270 Hz), 122.67 (t, JC–F=4 Hz), 114.20 (dq, JC–F=25, 4 Hz), 70.94 (d, JC–F=3 Hz). Anal. calcd. for C8H8ClF4NO: C=39.12; H=3.28; N=5.70. Found: C=39.06%, H =3.02%, N =5.66%.
O-2,4-(Difluorobenzyl)hydroxylamine hydrochloride (39)
Synthesized from the respective alcohol according to the general procedure to afford 39 as white crystals in 30% yield. Mp=155–157°C. 1H NMR (400 MHz, CD3OD) δ 7.54 (q, 1H, ArH, J=4 Hz), 7.10-7.03 (m, 2H, ArH), 5.12 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 165.63 (d, JC–F=249 Hz), 163.23 (d, JC–F=231 Hz), 134.70 (d, JC–F=5 Hz), 117.83, 112.92 (d, JC–F=4 Hz), 105.16 (t, JC–F=26 Hz), 71.12 (d, JC–F=4 Hz). Anal. calcd. for C7H8ClF2NO: C=42.99; H=4.12; N=7.16. Found: C=43.15%, H = 3.74%, N =6.89%.
O-(2,5-Dimethoxybenzyl)hydroxylamine hydrochloride (40)
Synthesized from 2,5-dimethoxybenzyl alcohol according to the general procedure to afford 40 as white crystals in 90% yield. Mp=115–120°C. 1H NMR (400 MHz, CD3OD) δ 6.99-6.95 (m, 3H, ArH), 5.05 (s, 2H), 3.83 (s, 3H), 3.76 (s, 3H). 13C NMR (100 MHz, CD3OD) δ 155.11, 153.71, 123.23, 118.26, 116.95, 113.27, 73.22, 56.56, 56.20. Anal. calcd. for C9H14Cl3NO3: C=49.21; H=6.42; N=6.38. Found: C=49.14%, H =6.76%, N =5.94%.
O-(3,4-Dichlorobenzy)lhydroxylamine hydrochloride (41)
Synthesized from 3,4-dichlorobenzyl alcohol according to the general procedure to afford 41 as white crystals in 48%yield. Mp=193.5–195.5°C. 1H NMR (400 MHz, CD3OD) δ 7.64 (s, 1H, ArH), 7.60 (d, 1H, ArH, J=8 Hz), 7.39 (dd, 1H, ArH, J=8, 1 Hz), 5.06 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 135.13, 134.57, 133.78, 132.28, 132.11, 130.06, 76.42. Anal. calcd. for C7H8Cl3NO C=36.80; H=3.53; N=6.13. Found: C=36.40%, H =3.08%, N =6.10%.
O-(2-Trifluoromethyl,4-fluorobenzyl)hydroxylamine hydrochloride (42)
Synthesized from respective alcohol according to the general procedure to afford 42 as white crystals in 61% yield. Mp=118–119°C. 1H NMR (400 MHz, CD3OD) δ 7.59 (dd, 1H, ArH, J=8, 1 Hz), δ 7.51 (dd, 1H, ArH, J=8, 1 Hz), 7.47 (dt, 1H, ArH, J=4, 1 Hz), 5.22 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 164.21 (d, JC–F=249 Hz), 135.99 (d, JC–F=8 Hz), 132.41 (q, JC–F=32 Hz), 128.62, 124.57 (d, JC–F=272 Hz), 120.65 (d, JC–F=21 Hz), 115.35 (m), 73.55 (d, JC–F=2 Hz). GC tR =3.903 min.
O-(3,5-Diflourobenzyl)hydroxylamine hydrochloride (43)
Synthesized from 3,5-diflourobenzyl alcohol according to the general procedure to afford 43 as white crystals in 92% yield. Mp=200.5–201°C.1H NMR (400 MHz, CD3OD) δ 7.10 (d, 2H, ArH,, J=4 Hz), 7.03 (t, ArH, J=8 Hz), 5.09 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 164.66 (dd, JC–F=247, 13 Hz), 138.65 (t, JC–F=9 Hz ), 112.89 (dd, JC–F=12, 7 Hz ), 105.57 (t, JC–F=25 Hz ), 76.45. GC tR =3.717 min.
O-(3,5-Dibromobenzyl)hydroxylamine hydrochloride (44)
Synthesized from 3,5-dibromo benzyl alcohol according to the general procedure to afford 44 as white crystals in 79%yield. Mp=197–200°C. 1H NMR (400 MHz, CD3OD) δ 7.79 (t, 1H, ArH, J=1 Hz), 7.62 (d, 2H, ArH, J=1 Hz), 5.03 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 138.71, 135.95, 132.02, 124.22, 76.16. GC tR = 10.513 min.
O-(3,5-Bis(triflouromethyl-benzyl)hydroxylamine hydrochloride (45)
Synthesized from 3,5-(bis)triflouromethyl benzyl aclohol according to the general procedure to afford 45 as white crystals in 93% yield. Mp-195–197°C.1H NMR (400 MHz, CD3OD) δ 8.08 (s, 2H, ArH), 8.04 (s, 1H, ArH), 5.26 (s, 2H, ArCH2). 13C NMR (100 MHz, CD3OD) δ 137.90, 133.22 (q, JC–F=33 Hz), 130.60 (d, JC–F=3 Hz), 124.59 (d, JC–F=271 Hz), 124.13 (t, JC–F=3 Hz), 76.14. Anal. calcd. for C9H8ClF6NO C=36.57; H=2.73; N=4.74. Found: C=36.55%, H = 2.49%, N =4.64%.
O-(2-Hydroxy-3-methoxybenzyl)hydroxylamine hydrochloride (46)
Synthesized from 2-hydroxy-3-methoxybenzyl alcohol according to the general procedure to afford 46 as white crystals in 45%yield. Mp=176–178°C.1H NMR (400 MHz, CD3OD) δ 7.03 (d, 1H, ArH, J=8 Hz), 6.91-6.82 (m, 2H, ArH), 5.08 (s, 2H, ArCH2), 3.87 (s, 3H, ArOCH3). 13C NMR (100 MHz, CD3OD) δ 149.13, 147.21, 124.15, 120.61, 120.56, 113.91, 73.28, 56.56. Anal. Calcd. for C8H12ClNO3 C=46.73; H=5.88; N=6.81. Found: C=46.38%, H = 5.71%, N =6.72%.
Biochemical assays
Recombinant human IDO1 was expressed and purified as described.50 Inhibition assays to determine IC50 and Ki values were performed in a 96-well microtiter plate as described by Littlejohn et.al.50 with some modification. Under these assay conditions, we have previously determined that the recombinant hIDO1 follows Michaelis-Menten kinetics51. Briefly, the reaction mixture contained 50 mM potassium phosphate buffer (pH 6.5), 40 mM ascorbic acid, 400 μg/ml catalase, 20 μM methylene blue and ~27 nM purified recombinant IDO1 per reaction. The reaction mixture was added to the substrate, L-tryptophan (L-Trp), and the inhibitor. For IC50 determinations, inhibitor solutions were made fresh and serially diluted and the L-Trp substrate was tested at 100 μM (Km=80 μM). For Ki determinations, the assay was set up similarly except that serial dilutions of inhibitors were matrixed with serial dilutions of L-Trp substrate. The reaction was carried out at 37°C for 60 min and stopped by the addition of 30% (w/v) trichloroacetic acid. The plate was then incubated at 65 °C for 15 min to convert N-formylkynurenine to kynurenine and centrifuged at 1250 g for 10 min. Lastly, 100 μl supernatant from each well was transferred to a new 96 well plate and mixed at equal volume with 2% (w/v) p-dimethylamino-benzaldehyde (Ehrlich’s reagent) in acetic acid. The yellow color generated from the reaction with kynurenine was measured at 490 nm using a Synergy HT microtiter plate reader (Bio-Tek, Winooski, VT). As an initial compound screen, assessments of IC50 values were performed as single point dilution series. The most potent compounds were subsequently retested two or more times with the results reported as averages. The data were analyzed using Graph Pad Prism 4 software (Graph Pad Software Inc., San Diego, CA) and the Enzyme Kinetics module in SigmaPlot version 10 (Systat Software Inc, San Jose, CA). (Note that a separate experiment analyzing the possible competition between kynurenine and four different O-alkylhydroxylamine inhibitors for p-dimethylaminobenzaldehyde showed no impact on the visible absorbance reading from the presence of any one of the four inhibitors.)
Spectroscopic measurements
The UV-Vis spectra were obtained at room temperature using a UV2401 spectrophotometer (Shimadzu Scientific Instruments, Inc., Columbia, MD) with a slit width of 1 nm in a 1 cm path-length quartz cuvette. The proteins (5 μM) were buffered with a 100 mM pH of 7.4 Tris buffer. The deoxy samples were prepared by injecting ~10-fold excess sodium dithionite into the protein samples pre-purged with nitrogen gas with a gas-tight syringe. The CO adducts were prepared by adding 200 μl CO gas (1 atmosphere) into the deoxy samples.
Cell-based IDO1 inhibition and cytotoxicity assays
Compounds were evaluated for inhibitory activity against human IDO1 expressed endogenously in HeLa cells and exogenously in T-Rex cells. HeLa cells were seeded in a 96 well plate at a density of 10,000 cells per well in 100 μl DMEM + 10% fetal bovine serum + 1% Penicillin-Streptomycin and IDO1 expression was induced by the addition of IFNγ to a final concentration of 100 ng/mL. T-Rex cells containing a tet-regulated human IDO1 cDNA were seeded in a 96-well plate at a density of 10,000 cells per well in 100 μL of DMEM + 10% FBS + 1% Penicillin-Streptomycin and IDO1 expression was induced by the addition of 100 μL of media containing 20 ng/mL doxycycline. With both assays, IDO1 induction was allowed to proceed for 24 hours after which the media was discarded, the wells rinsed once, and serial dilutions of compound in 200 μL of DMEM +10% FBS with the final concentration of tryptophan adjusted to 100 μM. Following incubation at 37 °C for an additional 24 hrs, the assay was stopped by the addition of 50 μL of 50% (w/v) TCA to each well, and the cells were fixed by incubating for 1 hr at 4 °C.
Assessment of IDO1 activity
Compound IC50 values were assessed from single point dilution series with the most potent compounds subsequently retested two or more times and the results reported as averages. Following the TCA fixation step, the supernatants were transferred to a round-bottomed 96-well plate and incubated at 65 °C for 15 min. The plates were then centrifuged at 1250 × g for 10 min, and 100 μL of clarified supernatant was transferred to a new flat-bottomed 96-well plate and mixed at equal volume with 2% (w/v) p-dimethylaminobenzaldehyde in acetic acid. The yellow reaction was measured at 490 nm using a Synergy HT microtiter plate reader (Bio-Tek, Winooski, VT). Graphs of inhibition curves with IC50 values were generated using Prism v.5.0 (GraphPad Software, Inc.).
Assessment of cell viability
The single point dilution series used to determine IC50 values were also evaluated for cell viability. The TCA-fixed cells remaining in the 96-well plate following transfer of the media were processed essentially as described52. Fixed cells were washed four times in tap water, blotted, air-dried, and treated for 15 min at room temperature with 100 μL of 0.4% (w/v) sulfarhodamine B (SRB) (Sigma-Aldrich, St. Louis, MO) prepared in 1% acetic acid. Wells were then rinsed four times in 1% acetic acid, air-dried, and developed by adding 200 μL of 10 mM unbuffered Tris-HCl and incubating for 15 min at room temperature with gentle shaking. Staining intensity, proportional to cell number, was determined by reading the absorbance at 570 nm on a plate reader. Graphs of cell viability curves were generated using Prism v.5.0 (GraphPad Software, Inc.).
Docking calculations
Small molecules were constructed in MOE V2011.1 (Chemical Computing Group, Inc.) and ionized and hydrogens added using MOE’s WashMDB function. The small molecule conformation was minimized to a gradient of 0.01 with the MMFF94x53,54 using a distance-dependent dielectric constant of 1. The crystal structure of IDO1 bound with 4-phenylimidizole was used for docking calculations44. The 2-[N-cyclohexylamino]ethanesulfonic acid and 4-phenyl-1-imidazole ligands were removed from the active site docking, hydrogen atoms were added and tautomeric states and orientations of Asn, Gln, His residues were determined with Molprobity55,56. Hydrogens were then added to crystallographic waters using MOE. The Amber9957 force field in MOE was used, and iron was parameterized in the Fe+3 state. Dioxygen was not added to the iron. All hydrogens were minimized to an rms gradient of 0.01, holding the remaining heavy atoms fixed. A stepwise minimization followed for all atoms using a quadratic force constant (100 kcal-mole/Å) to tether the atoms to their starting geometries; for each subsequent minimization, the force constant was reduced by a half until reached 0.01 and was then followed by a cycle of minimization without applying any constraints. GOLD version 5.1 (Cambridge Crystallographic Data Centre) was used with coordination binding of the hydroxylamine nitrogen to the Fe+3 atom of the heme. Chemscore parameters, adapted for metal–ligand interactions, were used for scoring58. Fifty genetic algorithm (GA) docking runs were performed with the initial_virtual_pt_match_max = 2.5, all other parameters were set as defaults.
Supplementary Material
HIGHLIGHTS.
Rational drug design used to discover O-alkylhydroxylamines as IDO1 inhibitors.
Optimum inhibition with meta-halide substituted benzyl derivatives.
Heme iron binding of O-alkylhydroxylamines demonstrated.
Selective inhibition among several heme iron enzymes shown.
Nanomolar-level potency and limited toxicity in cells.
Acknowledgments
Financial support for this work was provided by the National Institutes of Health, NCI R01 CA109542-04A2 to J. M. L., G. C. P., A. J. M. and W. P. M., and GM086482 to S. R. Y. A.J.M. is also the recipient of grants from the Susan G. Komen for the Cure and receives additional funding support through NIH grant CA159337. G.C.P. is also the recipient of NIH grants CA159337 and CA159315. Additional support for this project was provided by grants to G.C.P. from the Charlotte Geyer Foundation and the Lankenau Hospital Foundation. W.P.M. receives additional financial support through NIH grant GM087291. Shreekari Tadepalli is recognized for contributions to the characterization of several compounds. A generous award (CHE-0958996) from the National Science Foundation enabling acquisition of the 400 MHz NMR spectrometer used in these studies is gratefully acknowledged. The authors would also like to thank Bryn Mawr College for financial support of this work. The authors are grateful to Richard Metz for providing purified recombinant IDO1 enzyme.
Abbreviations
- IDO1
indoleamine 2,3-dioxygenase isoform-1
- Trp
tryptophan
- UV-vis
ultraviolet-visible
- CYP3A4
cytochrome P-450 isoform 3A4
Footnotes
Full experimental details for the synthesis of alcohols that were not commercially available are available. Additional cell-based inhibition and viability assays are also presented. This material is available free of charge via the Internet at …
Conflict of interest: W.P.M., J.B.D., A.J.M. and G.C.P. declare a conflict of interest due to their various relationships with New Link Genetics Corporation, which has licensed IDO1-related intellectual property from the Lankenau Institute for Medical Research and Georgia Regents University. W.P.M., J.B.D. and A.J.M. are inventors and shareholders in the company. G.C.P. is an inventor and shareholder who has received grant support and compensation from the company in his role as a scientific advisor.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
William P. Malachowski, Email: wmalacho@brynmawr.edu.
Alexander J. Muller, Email: mullera@mlhs.org.
References
- 1.Schreiber RD, Old LJ, Smyth MJ. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 2.Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137–48. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 3.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263–74. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 4.Muller AJ, Scherle PA. Targeting the mechanisms of tumoral immune tolerance with small-molecule inhibitors. Nat Rev Cancer. 2006;6:613–625. doi: 10.1038/nrc1929. [DOI] [PubMed] [Google Scholar]
- 5.Metz R, Duhadaway JB, Kamasani U, Laury-Kleintop L, Muller AJ, Prendergast GC. Novel tryptophan catabolic enzyme IDO2 is the preferred biochemical target of the antitumor indoleamine 2,3-dioxygenase inhibitory compound D-1-methyl-tryptophan. Cancer Res. 2007;67:7082–7. doi: 10.1158/0008-5472.CAN-07-1872. [DOI] [PubMed] [Google Scholar]
- 6.Ball HJ, Yuasa HJ, Austin CJD, Weiser S, Hunt NH. Indoleamine 2,3-dioxygenase-2; a new enzyme in the kynurenine pathway. The International Journal of Biochemistry & Cell Biology. 2009;41:467–471. doi: 10.1016/j.biocel.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Muller AJ, Prendergast GC. Marrying immunotherapy with chemotherapy: why say IDO? Cancer Res. 2005;65:8065–8. doi: 10.1158/0008-5472.CAN-05-2213. [DOI] [PubMed] [Google Scholar]
- 8.Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117:1147–54. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med. 2005;11:312–9. doi: 10.1038/nm1196. [DOI] [PubMed] [Google Scholar]
- 10.ClinicalTrials.gov. National Institutes of Health. 2013;2013 a list of clinical trials. [Google Scholar]
- 11.Dolusic EF. Raphael Indoleamine 2, 3-dioxygenase inhibitors: a patent review (2008 – 2012) Expert Opinion on Therapeutic Patents. 2013;23:1367–1381. doi: 10.1517/13543776.2013.827662. [DOI] [PubMed] [Google Scholar]
- 12.Röhrig UF, Majjigapu SR, Vogel P, Zoete V, Michielin O. Challenges in the Discovery of Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitors. Journal of Medicinal Chemistry. 2015 doi: 10.1021/acs.jmedchem.5b00326. [DOI] [PubMed] [Google Scholar]
- 13.Sono M, Roach MP, Coulter ED, Dawson JH. Heme-Containing Oxygenases. Chem Rev. 1996;96:2841–2888. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 14.Botting NP. Chemistry and Neurochemistry of the Kynurenine Pathway of Tryptophan Metabolism. Chemical Society Reviews. 1995;24:401–12. [Google Scholar]
- 15.Sono M, Hayaishi O. The Reaction Mechanism of Indoleamine 2,3-Dioxygenase. Biochemical Reviews. 1980;50:173–81. [Google Scholar]
- 16.Chung LW, Li X, Sugimoto H, Shiro Y, Morokuma K. Density Functional Theory Study on a Missing Piece in Understanding of Heme Chemistry: The Reaction Mechanism for Indoleamine 2,3-Dioxygenase and Tryptophan 2,3-Dioxygenase. Journal of the American Chemical Society. 2008;130:12299–12309. doi: 10.1021/ja803107w. [DOI] [PubMed] [Google Scholar]
- 17.Lewis-Ballester A, Batabyal D, Egawa T, Lu C, Lin Y, Marti MA, Capece L, Estrin DA, Yeh SR. Evidence for a ferryl intermediate in a heme-based dioxygenase. Proceedings of the National Academy of Sciences. 2009;106:17371–17376. doi: 10.1073/pnas.0906655106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Capece L, Lewis-Ballester A, Yeh SR, Estrin DA, Marti MA. Complete Reaction Mechanism of Indoleamine 2,3-Dioxygenase as Revealed by QM/MM Simulations. The Journal of Physical Chemistry B. 2012;116:1401–1413. doi: 10.1021/jp2082825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yue EW, Douty B, Wayland B, Bower M, Liu X, Leffet L, Wang Q, Bowman KJ, Hansbury MJ, Liu C, Wei M, Li Y, Wynn R, Burn TC, Koblish HK, Fridman JS, Metcalf B, Scherle PA, Combs AP. Discovery of Potent Competitive Inhibitors of Indoleamine 2,3-Dioxygenase with in Vivo Pharmacodynamic Activity and Efficacy in a Mouse Melanoma Model. Journal of Medicinal Chemistry. 2009;52:7364–7. doi: 10.1021/jm900518f. [DOI] [PubMed] [Google Scholar]
- 20.Fung SPS, Wang H, Tomek P, Squire CJ, Flanagan JU, Palmer BD, Bridewell DJA, Tijono SM, Jamie JF, Ching LM. Discovery and characterisation of hydrazines as inhibitors of the immune suppressive enzyme, indoleamine 2,3-dioxygenase 1 (IDO1) Bioorganic & Medicinal Chemistry. 2013;21:7595–7603. doi: 10.1016/j.bmc.2013.10.037. [DOI] [PubMed] [Google Scholar]
- 21.Mautino MJF, Marcinowicz-Flick A, Kesharwani T, Waldo J. Organization, W. I. P, editor. WO2009/073620. IDO inhibitors. 2009
- 22.Augusto OK, KL, Ortiz de Montellano PR. N-phenylprotoporphyrin IX formation in the hemoglobin-phenylhydrazin reaction. J Biol Chem. 1982;257:6231. [PubMed] [Google Scholar]
- 23.Jonen HGWJ, Prough RA, Estabrook RW. The reaction of phenylhydrazine with microsomal cytochrome P-450. Catalysis of heme modification. J Biol Chem. 1982;257:4404. [PubMed] [Google Scholar]
- 24.Ortiz De Montellano PRK, DE Inactivation of catalase by phenylhydrazine. Formation of a stable aryl-iron heme complex. J Biol Chem. 1983;258:10558–10563. [PubMed] [Google Scholar]
- 25.Ringe DP, GA, Kerr DE, Ortiz De Montellano PR, Kerr DE. Reaction of myoglobin with phenylhydrazine: A molecular doorstop. Biochemistry. 1984;23:2–4. doi: 10.1021/bi00296a001. [DOI] [PubMed] [Google Scholar]
- 26.Ortiz de Montellano PR, Kunze KL. Formation of N-phenylheme in the hemolytic reaction of phenylhydrazine with hemoglobin. Journal of the American Chemical Society. 1981;103:6534–6536. [Google Scholar]
- 27.Castro BR. Organic Reactions. John Wiley & Sons, Inc; 2004. Replacement of Alcoholic Hydroxyl Groups by Halogens and Other Nucleophiles via Oxyphosphonium Intermediates. [Google Scholar]
- 28.Macchia M, Jannitti N, Gervasi G, Danesi R. Geranylgeranyl Diphosphate-Based Inhibitors of Post-Translational Geranylgeranylation of Cellular Proteins. Journal of Medicinal Chemistry. 1996;39:1352–1356. doi: 10.1021/jm960127s. [DOI] [PubMed] [Google Scholar]
- 29.Kumar S, Jaller D, Patel B, LaLonde JM, DuHadaway JB, Malachowski WP, Prendergast GC, Muller AJ. Structure Based Development of Phenylimidazole-Derived Inhibitors of Indoleamine 2,3-Dioxygenase. J Med Chem. 2008;51:4968–4977. doi: 10.1021/jm800512z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsuno K, Takai K, Isaka Y, Unno Y, Sato M, Takikawa O, Asai A. S-Benzylisothiourea derivatives as small-molecule inhibitors of indoleamine-2,3-dioxygenase. Bioorganic & Medicinal Chemistry Letters. 2010;20:5126–5129. doi: 10.1016/j.bmcl.2010.07.025. [DOI] [PubMed] [Google Scholar]
- 31.Huang Q, Zheng M, Yang S, Kuang C, Yu C, Yang Q. Structure–activity relationship and enzyme kinetic studies on 4-aryl-1H-1,2,3-triazoles as indoleamine 2,3-dioxygenase (IDO) inhibitors. European Journal of Medicinal Chemistry. 2011;46:5680–5687. doi: 10.1016/j.ejmech.2011.08.044. [DOI] [PubMed] [Google Scholar]
- 32.Röhrig UF, Majjigapu SR, Grosdidier A, Bron S, Stroobant V, Pilotte L, Colau D, Vogel P, Van den Eynde BJ, Zoete V, Michielin O. Rational Design of 4-Aryl-1,2,3-Triazoles for Indoleamine 2,3-Dioxygenase 1 Inhibition. Journal of Medicinal Chemistry. 2012;55:5270–5290. doi: 10.1021/jm300260v. [DOI] [PubMed] [Google Scholar]
- 33.Cheng MF, Hung MS, Song JS, Lin SY, Liao FY, Wu MH, Hsiao W, Hsieh CL, Wu JS, Chao YS, Shih C, Wu SY, Ueng SH. Discovery and structure–activity relationships of phenyl benzenesulfonylhydrazides as novel indoleamine 2,3-dioxygenase inhibitors. Bioorganic & Medicinal Chemistry Letters. 2014;24:3403–3406. doi: 10.1016/j.bmcl.2014.05.084. [DOI] [PubMed] [Google Scholar]
- 34.Tojo S, Kohno T, Tanaka T, Kamioka S, Ota Y, Ishii T, Kamimoto K, Asano S, Isobe Y. Crystal Structures and Structure–Activity Relationships of Imidazothiazole Derivatives as IDO1 Inhibitors. ACS Medicinal Chemistry Letters. 2014 doi: 10.1021/ml500247w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Serra S, Moineaux L, Vancraeynest C, Masereel B, Wouters J, Pochet L, Frédérick R. Thiosemicarbazide, a fragment with promising indolamine-2,3-dioxygenase (IDO) inhibition properties. European Journal of Medicinal Chemistry. 2014;82:96–105. doi: 10.1016/j.ejmech.2014.05.044. [DOI] [PubMed] [Google Scholar]
- 36.Bissantz C, Kuhn B, Stahl M. A Medicinal Chemist’s Guide to Molecular Interactions. Journal of Medicinal Chemistry. 2010;53:5061–5084. doi: 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clark T, Hennemann M, Murray J, Politzer P. Halogen bonding: the σ-hole. J Mol Model. 2007;13:291–296. doi: 10.1007/s00894-006-0130-2. [DOI] [PubMed] [Google Scholar]
- 38.Lommerse JPM, Stone AJ, Taylor R, Allen FH. The Nature and Geometry of Intermolecular Interactions between Halogens and Oxygen or Nitrogen. Journal of the American Chemical Society. 1996;118:3108–3116. [Google Scholar]
- 39.Silverman RB. The Organic Chemistry of Drug Design and Drug Action. Elsevier Academic Press; 2004. [Google Scholar]
- 40.Ligand efficiency was calculated as follows: LE=1.37*pKi/number of heavy atoms.
- 41.Kuntz ID, Chen K, Sharp KA, Kollman PA. The maximal affinity of ligands. Proceedings of the National Academy of Sciences. 1999;96:9997–10002. doi: 10.1073/pnas.96.18.9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hopkins AL, Groom CR, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 43.Hopkins AL, Keseru GM, Leeson PD, Rees DC, Reynolds CH. The role of ligand efficiency metrics in drug discovery. Nat Rev Drug Discov. 2014;13:105–121. doi: 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- 44.Sugimoto H, Oda S, Otsuki T, Hino T, Yoshida T, Shiro Y. Crystal structure of human indoleamine 2,3-dioxygenase: catalytic mechanism of O2 incorporation by a heme-containing dioxygenase. Proc Natl Acad Sci U S A. 2006;103:2611–6. doi: 10.1073/pnas.0508996103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sono M, Cady SG. Enzyme kinetic and spectroscopic studies of inhibitor and effector interactions with indoleamine 2,3-dioxygenase. 1.Norharman and 4-phenylimidazole binding to the enzyme as inhibitors and heme ligands. Biochemistry. 1989;28:5392–5399. doi: 10.1021/bi00439a012. [DOI] [PubMed] [Google Scholar]
- 46.Yang S, Li X, Hu F, Li Y, Yang Y, Yan J, Kuang C, Yang Q. Discovery of Tryptanthrin Derivatives as Potent Inhibitors of Indoleamine 2,3-Dioxygenase with Therapeutic Activity in Lewis Lung Cancer (LLC) Tumor-Bearing Mice. Journal of Medicinal Chemistry. 2013;56:8321–8331. doi: 10.1021/jm401195n. [DOI] [PubMed] [Google Scholar]
- 47.Kumar S, Malachowski WP, DuHadaway JB, LaLonde JM, Carroll PJ, Jaller D, Metz R, Prendergast GC, Muller AJ. Indoleamine 2,3-Dioxygenase Is the Anticancer Target for a Novel Series of Potent Naphthoquinone-Based Inhibitors. J Med Chem. 2008;51:1706–1718. doi: 10.1021/jm7014155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Incyte Corporation. ClinicalTirals.gov [Internet] National Library of Medicine (US); Bethesda (MD): 2012. A Phase 1/2 Randomized, Blinded, Placebo Controlled Study of Ipilimumab in Combination With INCB024360 or Placebo in Subjects With Unresectable or Metastatic Melanoma. Available from http://clinicaltrials.gov/show/NCT01604889 NLM Identifier: NCT01604889. [Google Scholar]
- 49.NewLink Genetics Corporation. ClinicalTrials.gov [Internet] National Library of Medicine (US); Bethesda (MD): 2014. IDO Inhibitor Study for Advanced Solid Tumors. cited 2014 May 30 Available from http://clinicaltrials.gov/show/NCT02048709 NLM Identifier: NCT02048709. [Google Scholar]
- 50.Littlejohn TK, Takikawa O, Skylas D, Jamie JF, Walker MJ, Truscott RJ. Expression and purification of recombinant human indoleamine 2, 3-dioxygenase. Protein Expr Purif. 2000;19:22–9. doi: 10.1006/prep.2000.1214. [DOI] [PubMed] [Google Scholar]
- 51.Flick HEL, Judith M, Malachowski William P, Muller Alexander J. The Tumor-Selective Cytotoxic Agent β-Lapachone is a Potent Inhibitor of IDO1. International Journal of Tryptophan Research. 2013;6:35–45. doi: 10.4137/IJTR.S12094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82:1107–12. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 53.Halgren TA. MMFF VI. MMFF94s option for energy minimization studies. J Comput Chem. 1999;20:720–729. doi: 10.1002/(SICI)1096-987X(199905)20:7<720::AID-JCC7>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 54.Halgren TA. MMFF MMFF VII. Characterization of MMFF94, MMFF94s, and other widely available force fields for conformational energies and for intermolecular-interaction energies and geometries. J Comput Chem. 1999;20:740–774. doi: 10.1002/(SICI)1096-987X(199905)20:7<730::AID-JCC8>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 55.Word JM, Lovell SC, Richardson JS, Richardson DC. Asparagine and glutamine: Using hydrogen atom contacts in the choice of side-chain amide orientation. J Mol Biol. 1999;285:1735–1747. doi: 10.1006/jmbi.1998.2401. [DOI] [PubMed] [Google Scholar]
- 56.Lovell SC, Davis IW, Arendall WB, III, de Bakker PIW, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure Validation by Cα Geometry: φ,ψ and Cβ Deviation. Proteins: Structure, Function and Genetics. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 57.Ponder JW, Case DA. Force fields for protein simulations. Adv Protein Chem. 2003;66:27–85. doi: 10.1016/s0065-3233(03)66002-x. [DOI] [PubMed] [Google Scholar]
- 58.Kirton SB, Murray CW, Verdonk ML, Taylor RD. Prediction of binding modes for ligands in the cytochromes P450 and other heme-containing proteins. Proteins. 2005;58:836–44. doi: 10.1002/prot.20389. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.