

Abstract
The incidence of pediatric Inflammatory Bowel Disease, which includes Crohn's and ulcerative colitis, has risen alarmingly in the Western and developing world in recent decades. Epidemiological (including monozygotic twin and migrant) studies highlight the substantial role of environment and nutrition in IBD etiology. Here we review the literature supporting the developmental and environmental origins hypothesis of IBD. We also provide a detailed exploration of how the human microbiome and epigenome (primarily through DNA methylation) may be important elements in the developmental origins of IBD in both children and adults.
Introduction
Inflammatory Bowel Diseases (IBD), including Ulcerative Colitis (UC) and Crohn's Disease (CD), are disorders characterized by chronic inflammatory destruction of the gastrointestinal mucosa(1). The etiology of IBD is unknown, but the diseases are thought to arise secondary to an uncontrolled mucosal immune response in the background of host genetic susceptibility, environmentally induced predisposition, and gut microbial dysbiosis. The key feature of pathogenesis is believed to be a dysregulated immune response against the commensal microbiota(2).
A recent systematic review chronicles the relative contribution of genetics, nutrition, environment, and other factors on early-onset vs. late-onset IBD(3). For children with rare, very early-onset IBD, genetic predisposition appears to play a more important role, while environmental factors and gut microbiota are likely more involved in the disease etiology and natural history of patients who present with the diseases at a later age. The incidence of IBD, particularly in pediatric populations, has been rising in the Western world and in developing nations at an alarming rate(4–6). The prevailing theory behind this surge in light of the geographical distribution of IBD is that the environmental and nutritional factors associated with Westernization are at fault(7). The purpose of this review is to highlight the environmental and nutritional origins hypothesis of IBD. We will further explore how select environmental and nutritional factors may affect host epigenetics and commensal microbiota.
Epidemiological Evidence Supports The Environmental Origins of IBD
General Epidemiology of IBD
IBD prevalence is highest in Western Europe, North America, and Australia and steeply declines outside of the developed world(8,9). In recent decades, however, IBD incidence has increased in previously low prevalence areas, such as South America and Asia, and is thought to be correlated with industrialization and Westernization(8,10). Particularly, pediatric IBD incidence is increasing at an alarming rate in both developed and developing countries,(8,11) although some investigations indicate a relative stabilization of incidence in certain high prevalence areas(12). Generally, UC appears before CD where IBD is on the rise(11). Many of these observations may be a byproduct of increased physician access and improving healthcare systems in developing nations. The rise in IBD, however, that has been observed in Eastern Europe, a region of comparatively quality healthcare, over the last 25 years strongly associates with the steady appropriation of a Western lifestyle(13–15). Prevalence of IBD follows a North-South gradient in the United States and an East-West gradient in Canada. These gradients likely reflect differences in population density, urbanity, and environmental exposure within each nation rather than genetic differences or access to healthcare(16–18). A recent meta-analysis of previous epidemiological studies, mostly from North America and Western Europe, discovered a modest, though significant, increase of pediatric CD incidence associated with increasing latitude and low daily ultraviolet radiation levels (19). The authors suggest that diminished daily ultraviolet radiation levels at higher latitudes might affect vitamin D synthesis, immunologically predisposing children to the disease. Within-region observations, such as the above, dampen the theorized role of genetics and point to the greater contribution of environment and nutrition in IBD etiology.
The Environmental/Nutritional Origins of IBD
The rise in IBD incidence in developed and developing nations has coincided with a plethora of factors associated with Westernization including improved hygiene, increased access to and consumption of food that has changed in composition and processing, sedentary lifestyle, antibiotic use, refrigeration, urbanization, etc. (described, briefly, in Table 1). It should be emphasized that many of the environmental/nutritional factors contributing to IBD have been disputed or questioned in the literature. Furthermore, there are several considerations that are important to exercise when assessing environmental factor contribution to IBD etiology:
IBD is specific to humans (since similar disorders are still markedly different even in non-human primates(20)). Therefore, animal environmental/nutritional model studies of IBD etiology are inherently flawed.
Dependable results from human epidemiologic studies (even if prospective and well-controlled) are very difficult to attain for ethical and technical reasons(21).
Secondary to the limitations above, it is almost impossible to exclusively examine a single environmental factor in human IBD etiology.
Table 1.
Non-exhaustive description of environmental/nutritional factors identified in epidemiological studies as being associated with IBD and the known effects of those factors on the microbiome and epigenome. Importantly, many of these factors have been disputed or questioned in the literature, repeatedly emphasizing the limitations of human epidemiologic observations.
| Factor | Observed IBD Associations | Effects of Factor on Microbiome | Effects of Factor on Epigenome | |
|---|---|---|---|---|
| Encironmental | Helminth Infection | Protective against IBD 145,146 | Reduces bacterial attachment, alters community structure, increases diversity 147,148 | Affects methylation signature in host T-cell response 149 |
| Antibiotic Use in Childhood | Strongly associated with CD incidence but generally not withUC150-153 Antibiotic use for treatment of gastroenteritis – strongly associated with both UC and CD 150,154,155 |
Antibiotic use has substantial effects on microbiome composition/function, decreases diversity over time, and predisposes hosts to infection 156 | Antibiotics have trans-generational effects on sperm viability in insect models 157 | |
| Urbanization | IBD incidence is high in urban environments 158-160 Rural, farming upbringing is protective against IBD161 Air pollutants may increase risk of early onset IBD 162,163 |
Rural and urban community structures and functions differ 164,165 | Insufficient Information | |
| Smoking | Protective (and therapeutic) in UC 166-168 Associated with increased risk and prognosis in CD 169-171 |
Cessation in results in increased microbial diversity; gut microbiome composition changes to one associated with improved energy harvest 172,173 CD patients that smoke have clinically relevant dysbiosis 174 |
Maternal smoking – persistent, altered methylation of development- and metabolism-associated fetal genes 175-179 In adults – global, persistent methylation changes 180-182 |
|
| Preterm Birth | Increases risk of IBD 183 | Reduced diversity, high risk of pathogenic colonization, decreased stability 184 | Extreme preterm birth transiently affects methylation profile; select regions show persistence into adulthood185 | |
| Gastroenteritis | Strongly associated with increased IBD risk 150,154 | Intestinal microbiota can promote infection with and replication of enteric viruses 186,187 | Insufficient Information | |
| Mycobacterial Infection | M. avium paratuberculosis (MAP) frequently identified in CD patients 188-190 | Insufficient Information | MAP inhibits chromatin remodeling in host macrophages 191 | |
| Appendectomy | Protective for U C 192-194 More frequent, though not a risk factor, for CD 193-196 |
Limited evidence suggests microbiota in normal and diseased appendixes differ 197 | Insufficient Information | |
| Nutritional | Breastfeeding | Generally found to protect against IBD 198 | Promotes microbial homeostasis and microbiome plasticity later in life 158,199,200 | Breast milk contains SCFAs, which have epigenome-modifying properties (including DNA methylation and histone modification) |
| Non-Western Dietary Practices | Fasting protected against (murine) colitis; lack of fasting predisposes colitis, presumably leptin-mediated 201-205 Monotonous diet protects against colitis in humans and murine models 30,206 |
Variable diet reduces microbial diversity in mice 30 Fasting alters microbiome composition and diversity in species – dependent manner, generally associated with reduced inflammation 207-209 |
Caloric restriction during the prenatal and adolescent periods has potent, persistent effects on DNA methylation 81,210-212 | |
| High Fat / High Carb Diet | High intake of trans-unsaturated fat associated with increased UC risk; High intake of long-chain n-3 fatty acid associated with reduced UC risk213 However, dietary reversal after n-6 fat consumption protects against murine colitis 25 |
Stimulate expansion of organisms within the microbiome that are efficient at energy harvest which then promote inflammation, hyperphagia, hyperlipidemia, gut permeability, etc. 68,214 | Associated with specific methylation differences affecting metabolic pathways in pediatric population 79 | |
| Dietary Fiber | Increased fiber consumption 215 Cellulose supplementation ameliorates murine colitis 64 |
Decreased fiber consumption dramatically alters composition, gene content, richness, and SCFA production 206 | Dietary fiber is fermented to release SCFAs, which have validated epigenome-modifying properties (including DNA methylation and histone modification) 216,217 | |
| Dietary Protein (meat and dairy) | Diet high in animal protein increases risk of IBD 218 | Reliably alter composition and function: Increase in bile-tolerant organisms, fewer (anti-inflammatory) butyrate producers60 | Insufficient Information | |
| Vitamin D | Latitude associates with IBD risk, presumably due to Vitamin D intake 17,19 | Regulates gut microbiota to protect against colitis in mice 219 Vitamin D deficiency increases C. difficile infection risk 220,221 |
Pre-natal vitamin D deficiency epigenetically reduces immune cell development in mice 222 | |
| Food Additives | Dietary emulsifiers promote murine colitis and metabolic syndrome in a microbiome mediated manner67 Micronutrient supplementation of maternal diet augments murine colitis in microbiome-mediated manner75,76 |
Emulsifiers alter microbiome composition and function, microbiome transplant of treated mice results in metabolic disease in recipient 67 Artificial sweeteners induce microbiome-mediated glucose intolerance65 Maternal methyl donor supplementation alters murine clonic mucosal microbiome, arguments colitis 76 |
Supplementation of murine maternal diet with methyl donors modified colonic mucosal epigenome and conferred colitis susceptibility80 | |
| Refrigeration | Associates with CD development27 | Insufficient Information | Insufficient Information |
In regard to the third consideration, we bring a few examples to demonstrate the complexity and interactive nature of environmental elements relevant for IBD pathogenesis based on epidemiologic and animal model studies.
First, increased n-6 polyunsaturated fat (PUFA) consumption has been associated with UC development in a large scale human prospective trial(22). Concomitantly, one major dietary source of n-6 PUFA is fried foods, which have been observed as significant component of a pre-illness diet in Crohn's disease patients(23). Additionally, consumption of potato skin derived glycoalkaloids (through French fries, for example) has been shown to worsen colitis in 2 different mouse models of IBD(24). These studies underscore the complex dietary attributes of one single nutritional component (n-6 PUFA linked to fried foods in this example) that can be relevant for IBD development. In a recent work, we have not found any significant effect of isolated n-6 PUFA supplementation on acute colitis susceptibility in mice. Rather, the transient pediatric supplementation of n-6 PUFA resulted in acute colitis protection in young adult animals(25).
For our second example illustrating the difficulty of associating any single environmental factor to IBD etiology, we examine the proposed contribution of refrigeration, as a consequence of industrialization, to the development of CD(26). It is thought to do so by inducing increased exposure to psychotropic bacteria such as Yersinia spp and Listeria spp (i.e. the cold-chain hypothesis). This hypothesis was further substantiated by a record of significantly earlier pediatric exposure to a home fridge in CD patients than in controls(27). The consumption of refrigerated foods, however, has many secondary effects that could be relevant for IBD pathogenesis. Refrigeration results in the consumption of less pickled food and increases dietary diversity, both of which are highlighted as major characteristics of the Westernization of global dietary habits.(28). The consumption of acetate (a major component of pickling), for instance, has been shown to be protective against murine colitis(29). Additionally, we have highlighted the potential for increased dietary diversity to possibly contribute to IBD pathogenesis based on a murine colitis model and human epidemiology(30). Consequently, decreased consumption of pickled foods and increased dietary diversity resulting from the expansion of refrigeration may be important contributors to its association with IBD development (in addition to the hypothesized cold-chain effect). Importantly, both acetate and dietary diversity have functional and compositional relevance to the intestinal microbiota, emphasizing its physiologic role in communicating environmental/nutritional exposures to host physiology and pathology.
In spite of the difficulties in interpreting environmental influences on IBD susceptibility, an emerging common theme from animal model(31,32) and human epidemiological observations is that pediatric (especially infantile) disruption of the gut microbiome maturation process can significantly contribute to colitis susceptibility in animals and to IBD in humans. Prematurity, infantile exposure to antibiotics, pediatric exposure to refrigeration (see above), and plausibly all environmental factors relevant in IBD etiology can have significant effects on gut microbiota composition during critical periods of development. Microbiome composition changes during such developmental periods may lead to persistent modifications in host physiology (and vice-versa) that could alter predisposition to IBD later in life. This will be discussed further in a later section.
Migrant Population Studies
Interestingly, studies focusing on immigrants further point to a substantial contribution of environment and diet to IBD incidence. Migrants represent a unique study cohort – clearly, the genetic susceptibility of an immigrant to IBD remains static, yet they experience a dramatic change in environment and diet upon arrival to a new country. Epidemiological evidence suggests that exposure to environmental factors associated with IBD has greater influence on IBD incidence during early life, although transient exposures to higher levels of industrialization even in young adulthood may increase one's chances for developing UC(33). Recent studies, conducted in Canada and Sweden, show that immigrants have lower IBD incidence, with decreasing risk for each additional year of age at immigration(6,34). Furthermore, children of some, but not all, migrant groups are less likely to be diagnosed with IBD(6). The heterogeneity of IBD incidence appropriation in children of migrants indicates that early life exposure to a Western environment cannot exclusively predict likelihood of disease. Variation in different ethnic groups towards preserving dietary habits within the family following migration, however, could explain the heterogeneous IBD incidence in first generation immigrant children. This latter hypothesis would imply that nutritional influences are more important in IBD pathogenesis during postnatal pediatric development than other environmental factors. Second generation immigrants, however, have a similar IBD incidence rate as non-immigrants(35). These studies indicate that environmental exposures (including maternal diet) in prenatal life and during pediatric development (perhaps diet being the most important postnatally) are more critical than genetic predisposition towards IBD pathogenesis.
Genetics and Monozygotic Twin Studies
Over 160 genetic susceptibility loci have been linked to IBD(36). The vast majority of these loci contribute to disease development with low odds ratios (1-1.5) revealing the complexity of the limited genetic attribution (13.6% for CD and 7.5% for UC) to these disorders. There is also diverse genetic susceptibility to IBD in different ethnic backgrounds in spite of similar disease phenotypes(37–41). This genetic diversity suggests that changes common to industrialization induce non-genetic modifications in genetically vulnerable hosts that may be more important than the genetic predisposition itself towards inducing disease.
Observations in monozygotic (MZ) twins revealed low concordance for IBD within pairs. Concordance rates are overall less than 50% for IBD with CD higher (27-56%) than UC (15-19%)(42). This suggests that genetic predisposition is insufficient for IBD development – and that environmental factors are playing a bigger role in disease pathogenesis, especially in UC. In the meantime, it is MZ twins who, unless they are raised apart, share the same environmental and nutritional influences. Therefore, based on MZ twins, genetic and epidemiologic observations, it appears that genetic predisposition and shared environment/nutrition only heighten the probability of developing IBD, but are insufficient to instigate the onset of disease. Stochastic biological factors influenced by genetics and the environment appear to be key elements in the pathogenesis of the disorders (Figure 1.). Although frequently overlooked, physiologic noise (or stochasticity), defined as an unpredictable disturbance to a biological system, is an inherent part of human biology(43,44). IBD are prime examples where physiologic stochasticity is likely to play a significant etiologic role(45). It is, of course, mindboggling to consider that unique instigators may offset each case of IBD stochastically within the complex and interactive biologic systems involved in the pathology of the diseases.
Figure 1.
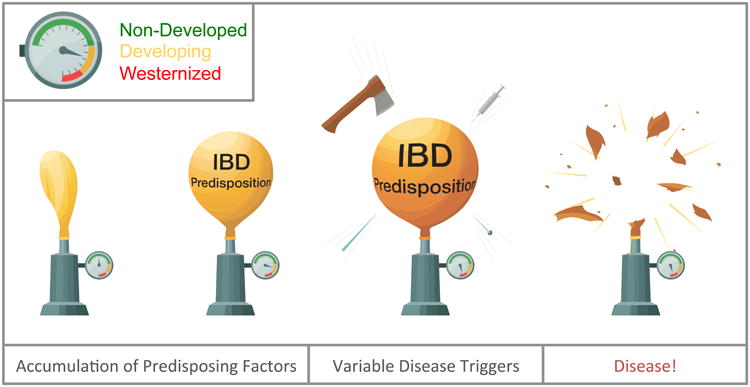
Schematic demonstration of the role of stochastic biological factors in the initiation of IBD. In genetically predisposed individuals, environmental and nutritional factors contribute to IBD predisposition (balloon inflation), where degree of predisposition positively correlates with Westernization/urbanization (pressure gauge). Stochastic biological factors (sharp objects), however, give impetus for the onset of the disease. Such factors may exert their critical effect in any of the intercalating biological systems that participate in IBD pathogenesis. Disease presentation is similar, however, irrespective of the critical trigger secondary to the intimately interactive systems (microbiome-mucosa-immune system).
Criteria for Interactive Systems
Out of these interactive systems, the microbiome and the host epigenome fulfill certain criteria, which make them prime candidates for communicating the environmental, nutritional, and developmental origins of IBD. Such criteria are:
Developmental maturation/modification - Biologic systems that are in flux during critical developmental periods are most likely to respond to environmental stimuli
Environmental/nutritional responsiveness
Stability once adjusted to environmental/nutritional influences
Penetrance (i.e. able to convey phenotypically relevant effects)
In the next section, we will highlight how the epigenome and the commensal microbiome fulfill the above criteria.
The Microbiome and The Host Epigenome are Potential Elements in The Environmental Origins of IBD
The Microbiome in the Environmental Origins of IBD
The human microbiome is defined as the complete population of bacteria, fungi, and viruses that live in and on the human body (all their molecular components as well, dead or alive)(46). In humans, the gastrointestinal system houses the largest quantity and diversity of microbiota (i.e. the live microbiome)(47). Generally, the enteric microbiota exists in symbiosis with the host, and plays important roles in digestion, immune system maturation, and epithelial barrier integrity, in addition to acting as a competitive “barrier” to pathogenic invasion(48). The microbiota can act as an environmental sentinel, able to quickly respond to external stimuli such as dietary/environmental change, with protein-coding bacterial genes outnumbering human host genes approximately 360:1(49).
Gastrointestinal microbiome composition, microbial function, and metabolic activity of gut microbiota are perturbed in patients suffering from IBD, though whether this is an initiator or consequence of the disease is unclear(50,51). Abnormal secretion of antibodies against common commensal microbiota, including common enteric fungi, in IBD-affected tissues suggests that the mechanisms responsible for tolerance induction are obstructed(52,53). A dysregulated immune response toward enteric microbes results in the down-regulation of tight junction proteins, allowing bacteria to infiltrate the epithelial barrier(52,54–56). This results in further inflammation and exaggerates dysbiosis (i.e. abnormal microbiome composition)(54,57). The current paradigm states that IBD pathology is characterized by an aberrant inflammatory response to microbiome, resulting in the subsequent destruction of the gastrointestinal mucosa and further bacterial/microbial encroachment.
In relation to the nutritional and environmental origins of IBD, the microbiome appears to be an important element in postnatal life by fulfilling the above outlined “systems criteria”:
The human microbiome goes through an intense evolution from birth that stabilizes following 3 years of age, but proceeds at lower velocity to young adulthood(58,59). Therefore, environmental influences such as infantile exposure to antibiotics, pediatric exposure to refrigerated foods, and even young adulthood exposure to high levels of industrialization could modulate its composition to promote IBD.
The human microbiome is highly responsive to nutritional changes. Rapid microbiome shifts can occur upon drastic dietary modification even in adults,(60) where microbiome composition is otherwise relatively stable over several months to years(61). The microbial dietary shifts appear to depend mostly on the nutritional impact, rather than the host genotype based on murine studies(62). Therefore, it is conceivable that environmental/nutritional changes of industrialization could induce similar IBD prone microbiome modifications in different ethnic backgrounds.
As previously stated, the human microbiome possesses stability(61). Individual microbiomes remain separated in spite of significant nutritional changes over short periods of time in adults, and long-term dietary influences link to distinct enterotypes(63). Therefore, once a nutritional change modifies the microbiome during a critical developmental period (i.e. childhood), IBD promoting characteristics could be carried on to young adulthood, when the onset of disease peaks. We have observed both transient(64) and persistent(25) microbiome-associated pediatric dietary influences to modulate IBD predisposition in young adult mouse models.
The mammalian, including human, microbiome can modulate the host phenotype. A large number of emerging studies utilizing broad-spectrum antibiotics and fecal microbiota transplantation into germ free animals(65) or even humans with disease(66) show the significant phenotype-modulating capacity of the microbiome. Our work demonstrates that mice fed a high n-6 fat diet in early life show prolonged protection against DSS-induced colitis, but only when the diet is reversed prior to insult. Critically, this protection appeared to be mediated by the microbiota as transplantation of the n-6 microbiome into germ free mice transmitted significant protection against colitis(25). Similar studies demonstrate that capacity of the microbiome to transmit the metabolic phenotypes of the donor(67,68). The most clinically relevant application of the transmissive properties of the microbiome is in the treatment of C. difficile infection. Fecal microbiome transplantation not only restores microbiome composition/function, it also reinstates normal bile acid composition and re-establishes gut homeostasis(69–71).
The Epigenome in the Environmental Origins of IBD
Epigenetic changes define molecular modifications that alter gene expression independently from genetic alterations in the DNA. Such functionally relevant genome-wide molecular modifications are frequently referred to as the “epigenome”. The epigenome may also be an important etiologic element in the environmental origins of IBD by fulfilling the above outlined “systems criteria”:
Epigenetic programming occurs during the prenatal and post-natal stages of life. A large, longitudinal (from birth to 18 months of age) study in twins has demonstrated that the epigenome changes rapidly in early life(72). Increasing epigenetic discordance between twins over time further underscores the role of non-shared environmental factors and, in part, stochasticity in the development of the epigenome during early life(72). Mouse studies highlight the post-infantile epigenetic maturation of the colonic mucosa as well(73). These findings indicate that the epigenetically responsive/vulnerable period in respect to colitis predisposition extends beyond infancy in mammals.
The human epigenome is responsive to environmental and nutritional changes(74). In mammals, supplementing the murine maternal diet with methyl-donor micronutrients can induce pronounced changes in colonic mucosal gene methylation and this associates with augmented colitis predisposition in the offspring(73,75,76). In Wistar rat offspring, a gestational diet rich in multivitamins inflicted specific epigenetic alterations, particularly affecting metabolic pathways(77). However, epigenome responsiveness is likely not limited to the prenatal period as our murine studies suggest that epigenetic maturation in the colonic mucosa continues post-natally through pediatric development (73). Similar findings in neonatal pigs have been made(78). In a study of preadolescent humans, quality of dietary fat consumed influenced DNA methylation of genes specifically involved in metabolic syndrome(79) underscoring the nutritional responsiveness of the human epigenome during childhood.
Epigenetic modifications, particularly DNA methylation, are stable over time. As mentioned above, supplementing the murine maternal diet with methyl-donors induced methylation changes still identifiable 3 months into the post-natal period(80). Perhaps the most famous example regarding environmental responsiveness of the epigenome in humans is the infamous Dutch Hunger Winter, throughout which pregnant women were deprived of food and nutrients during wartime – the effects of this prenatal programming were discernable in their children nearly 6 decades later (81).
Epigenetic changes in response to stimuli may exert phenotypically relevant effects in health and disease. The concept of epigenetic disease origins is erupting in all areas of medicine from neurology(82) to cardiology(83) and anesthesiology.(84) The potential importance for epigenetics in IBD was raised in 2000,(85) but is only recently receiving more attention(2,7,86,87). Although other epigenetic mechanisms besides DNA methylation may contribute to the development, progression and/or maintenance of IBD (e.g., histone modifications(86,88,89) and microRNAs(90–93)), only DNA methylation has been shown to be stably transmitted through repetitive cell divisions,(94,95) thereby having the capacity to permanently convey epigenetic information during the lifetime of an individual. Additionally, only DNA methylation has been described to directly communicate environmental exposures to phenotypic outcome in mammals(96). Recent work from Scotland has identified differentially methylated regions (DMRs) in peripheral blood leukocyte (PBL) DNA derived from treatment naïve pediatric CD patients(97). However, these had significant co-localization with IBD susceptibility single nucleotide polymorphisms (SNPs) indicating their genetic origins. As opposed to the Scottish study, we, in untreated pediatric IBD patients from the US, could not identify significant PBL DMRs following SNP exclusion despite using similar methodology(98). Significant cell-subset, variation-induced epigenetic noise in PBL may also interfere with consistent findings(99). Colonic mucosa, as the end-organ of IBD, has naturally been a tissue where DNA methylation associations of the disease group were extensively studied. Targeted(100) and non-biased(101) assessments of DNA methylation detected numerous colonic mucosal associates of inflammation in IBD. IBD treatments can modulate DNA methylation,(102) and most previous GI mucosal studies examined patients following treatments. We recently studied treatment naïve pediatric IBD cases and discovered a remarkable colonic mucosal epigenetic separation of UC compared to CD(103). Functional methylome studies indicate that such modifications may impact gene transcription relevant to UC(104). Our findings however, implicated that the majority of UC-specific DNA methylation variation resulted from mucosal inflammation and did not persist in patients achieving treatment-induced remission.(103) Therefore, much work needs to be done in the future to overcome the difficulties of epigenetic etiology research in humans. Causation-centered mouse studies, however, suggest that the epigenome's response to a nutritional stimulus can modulate colitis predisposition in mammals(80).
Epigenome-Microbiome Interactions
We have already indicated that the host epigenome and the commensal microbiota are likely to act in concert to modulate IBD predisposition secondary to environmental/nutritional exposure during critical periods of life. This potential has been highlighted by others as well in recent reviews(105,106). Briefly, pathogenic and commensal microbiota (and their metabolites) may contribute to epigenetic changes in their human hosts through a variety of mechanisms. Pathogenic bacteria may initiate host epigenetic modifications to inhibit immune response to invasion and to encourage proliferation of infected tissues(107,108). Interestingly, a recent investigation by Chernov, et al. demonstrated that Mycoplasma, a common intracellular pathogen in colonic epithelia, produces DNA methyltransferases (DNMTs: enzymes catalyzing DNA methylation). As illustrated in Figure 2B, the mycoplasmal DNMTs had the capacity to localize into the host nucleus and alter host DNA methylation at genomic regions not methylated by host-derived DNMTs (109). These findings suggest that microbes could directly induce long-lasting unique epigenetic changes in the host.
Figure 2.

Direct and indirect modulation of the human epigenome by pathogenic and commensal microbiota: (A) Human colonic physiology and its spatial relationship with the microbiome and the short chain fatty acids (SCFAs) and metabolites it produces. (B) Mycoplasma, an intracellular microbial pathogen, synthesizes DNA methyltransferases (DNMT) complete with nuclear localization signals (NLS) that penetrate the host nucleus and result in de novo methylation. (C) Microbe-derived metabolites (folate, butyrate, and biotin) indirectly modulate the host epigenome: Folate enters the one carbon metabolism cycle (OCM Cycle) to affect the bioavailability of methyl groups, butyrate acts as a potent histone deacetylase (HDAC) inhibitor, biotin availability for biotinylation promotes chromosomal stability. (D) Commensal interactions with pattern recognition receptors (PRRs) cause downstream transcriptional changes, mediated primarily through methylation and acetylation, resulting in increased/decreased production of PRRs as well as extra-epithelial responses.
Commensal microbiota may also epigenetically modify the host genome through the production of Short Chain Fatty Acids (SCFAs) such as butyrate, through generating biotin and folate, and though their interaction with Pattern Recognition Receptors (PRRs) on cell surfaces (summarized in Figure 2C).
Butyrate is produced by bacteria as a consequence of dietary fiber digestion and is important for intestinal health(107,110). Well-studied groups of butyrate-producing bacteria include Faecalibacterium prausnitzii and many of the Roseburia species(111). The reduced representation, and sometimes complete absence, of these butyrate-producing bacteria is repeatedly observed in the gastrointestinal microbiome of both UC and CD patients(112–116). A number of in vivo and in vitro studies suggest that butyrate acts as a major nutrient for colonocytes, and can dampen intestinal inflammation (in part) by suppressing nuclear factor-B (Nf-kB) activation. Butyrate can also promote epigenetic remodeling in intestinal stem cells by acting as a histone deacetylase inhibitor(117–120). In UC patients, butyrate uptake is reduced in colonocytes and elements of the butyrate oxidation pathway are impaired(121–124). Concordantly, a double-blind, placebo-controlled clinical trial in UC patients demonstrated that supplementation of rectal 5-ASA enemas with butyrate resulted in a significant amelioration of disease activity(125). Biotinylation, the epigenetic process in which biotin is attached to histone groups, is important for suppressing retrotransposon activity and maintaining chromosomal stability. The human body uses diet- and bacteria-derived biotin to achieve this, as it is incapable of synthesizing its own (110). Some commensal genera (namely, Lactobacillus and Bifidobacteria) affect the bioavailability of methyl groups through their production of folate(126). Folate feeds into the one-carbon metabolism cycle, regulating methyl-donor availability and eventually can affect DNA methylation. Bifidobacteria and Lactobacillus populations are reported to be significantly reduced in IBD patients, and are associated with disease severity, though the mechanism of action is still unclear(127–129). Reports of blood folate concentrations in IBD patients are inconsistent: some groups report folate deficiency, while others present evidence for significantly higher folate concentrations in affected individuals(130–133). A potential, unexplored explanation for these inconsistent reports is that variability in microbiome composition between patients may be linked to folate bioavailability and resulting systemic concentrations of the molecule.
Microbiota interact with intracellular and extracellular PRRs, resulting in transcriptional responses critical to tissue development, immunological maturation/surveillance, and normal physiological function (summarized in Figure 2D). Toll-like receptors (TLRs) are critical microbiome surveillance receptors as they bind microbe-associated molecular pattern (MAMP) molecules. These molecular interactions can stimulate the emergence of appropriate crypt-villus architecture in the intestinal mucosa. Germ free mice have an overall abnormal architecture of the gastrointestinal epithelium which can be rescued by microbial colonization(134,135). Immunologically, microbiota-mediated signaling through PRRs is essential for the post-natal maturation of gut-associated lymphoid tissue (GALT)(136), direct the conversion of CD4(+) T cells into Foxp3(+) T-regulatory cells(31,137), and are required for the establishment of a proper TH1/TH2 balance after birth(138,139). Furthermore, colonic region-specific TLR2 and TLR4 expression has been reported in SPF, but not germ-free mice, and microbiome transplantation reverses this region-specific expression pattern(140). It was later discovered that this microbiota-induced, region-specific expression associated with epigenetic alterations in the involved genes(141,142). In this way, commensal microbiota may modulate TLR expression, priming the host to respond to pathogenic threats later on. Later in life, microbe-mediated signaling via PRRs primarily affects gut homeostasis. Signaling through the TLR receptors, for example, regulates tight-junctions to maintain epithelial barrier integrity and stimulates the secretion of anti-microbial peptides to control commensal microbiota composition(143). Importantly, commensal microbiota are lifelong immunomodulators, promoting the intestinal expression of cytoprotective genes while suppressing pro-inflammatory genes(143).
The conceptualization of the microbiome as an epigenetic modulator is steadily gaining attention as researchers have begun to associate overall commensal microbiome composition, rather than select species, with epigenetic profiles in mice and in humans(141,142,144). Successive work has recently indicated strong associations between gut microbiome composition and promoter methylation of genes relevant for lipid metabolism, obesity and inflammation in PBL DNA(144).
Conclusion
Though a great many environmental and nutritional factors have been implicated in IBD, none have proven to be causal. This review presents a unique intersection of several compelling lines of evidence suggesting that environment and diet, through their profound effects on gut microbiome function and host epigenome modification, may play a primary role in IBD pathogenesis. This realization will hopefully fuel the near-future development of novel nutritionally- and epigenetically-focused preventative and therapeutic interventions for this highly morbid disease group.
Acknowledgments
Institutional Support provided by Baylor College of Medicine and by the National Institute of General Medical Science (Award Number T32 GM088129) to TYF; the content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health. RK was supported by the Gutsy Kids Fund including philanthropic donation from the Karen and Brock Wagner family, and was also supported by the Houston Men of Distinction.
Footnotes
No conflict of interest
References
- 1.Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. 2014;14:329–42. doi: 10.1038/nri3661. [DOI] [PubMed] [Google Scholar]
- 2.Ventham NT, Kennedy Na, Nimmo ER, et al. Beyond gene discovery in inflammatory bowel disease: the emerging role of epigenetics. Gastroenterology. 2013;145:293–308. doi: 10.1053/j.gastro.2013.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ruel J, Ruane D, Mehandru S, et al. IBD across the age spectrum-is it the same disease? Nat Rev Gastroenterol Hepatol. 2014;11:88–98. doi: 10.1038/nrgastro.2013.240. [DOI] [PubMed] [Google Scholar]
- 4.El-Matary W, SP M, CN B. Inflammatory bowel disease in children of Manitoba: 30 years' experience of a tertiary center. J Pediatr Gastroenterol Nutr. 2014;59:763–6. doi: 10.1097/MPG.0000000000000525. [DOI] [PubMed] [Google Scholar]
- 5.Benchimol EI, Fortinsky KJ, Gozdyra P, et al. Epidemiology of pediatric inflammatory bowel disease: A systematic review of international trends. Inflamm Bowel Dis. 2011;17:423–39. doi: 10.1002/ibd.21349. [DOI] [PubMed] [Google Scholar]
- 6.Benchimol EI, Mack DR, Guttmann A, et al. Inflammatory Bowel Disease in Immigrants to Canada And Their Children: A Population-Based Cohort Study. Am J Gastroenterol. 2015:1–11. doi: 10.1038/ajg.2015.52. [DOI] [PubMed] [Google Scholar]
- 7.Kellermayer R. Epigenetics and the developmental origins of inflammatory bowel diseases. Can J Gastroenterol. 2012;26:909–15. doi: 10.1155/2012/526408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ponder A, Long MD. A clinical review of recent findings in the epidemiology of inflammatory bowel disease. Clin Epidemiol. 2013;5:237–47. doi: 10.2147/CLEP.S33961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aujnarain A, Mack DR, Benchimol EI. The role of the environment in the development of pediatric inflammatory bowel disease. Curr Gastroenterol Rep. 2013;15:326. doi: 10.1007/s11894-013-0326-4. [DOI] [PubMed] [Google Scholar]
- 10.Parente JML. Inflammatory bowel disease in an underdeveloped region of Northeastern Brazil. World J Gastroenterol. 2015;21:1197. doi: 10.3748/wjg.v21.i4.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ng SC, Bernstein CN, Vatn MH, et al. Geographical variability and environmental risk factors in inflammatory bowel disease. Gut. 2013;62:630–49. doi: 10.1136/gutjnl-2012-303661. [DOI] [PubMed] [Google Scholar]
- 12.Adamiak T, Walkiewicz-Jedrzejczak D, Fish D, et al. Incidence, Clinical Characteristics, and Natural History of Pediatric IBD in Wisconsin: a Population-based Epidemiological Study. Inflamm Bowel Dis. 2013;19:1218–23. doi: 10.1097/MIB.0b013e318280b13e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lakatos L, Kiss LS, David G, et al. Incidence, disease phenotype at diagnosis, and early disease course in inflammatory bowel diseases in Western Hungary, 2002-2006. Inflamm Bowel Dis. 2011;17:2558–65. doi: 10.1002/ibd.21607. [DOI] [PubMed] [Google Scholar]
- 14.Gheorghe C, Pascu O, Gheorghe L, et al. Epidemiology of inflammatory bowel disease in adults who refer to gastroenterology care in Romania: a multicentre study. Eur J Gastroenterol Hepatol. 2004;16:1153–9. doi: 10.1097/00042737-200411000-00012. [DOI] [PubMed] [Google Scholar]
- 15.Burisch J, Munkholm P. The epidemiology of inflammatory bowel disease. Scand J Gastroenterol. 2015:1–10. doi: 10.3109/00365521.2015.1014407. [DOI] [PubMed] [Google Scholar]
- 16.Bernstein CN, Wajda A, Svenson LW, et al. The epidemiology of inflammatory bowel disease in Canada: A population-based study. Am J Gastroenterol. 2006;101:1559–68. doi: 10.1111/j.1572-0241.2006.00603.x. [DOI] [PubMed] [Google Scholar]
- 17.Schultz M, Butt AG. Is the north to south gradient in inflammatory bowel disease a global phenomenon? Expert Rev Gastroenterol Hepatol. 2012;6:445–7. doi: 10.1586/egh.12.31. [DOI] [PubMed] [Google Scholar]
- 18.Burisch J, Munkholm P. Inflammatory bowel disease epidemiology. Curr Opin Gastroenterol. 2013;29:357–62. doi: 10.1097/MOG.0b013e32836229fb. [DOI] [PubMed] [Google Scholar]
- 19.Holmes Ea, Xiang F, Lucas RM. Variation in Incidence of Pediatric Crohn's Disease in Relation to Latitude and Ambient Ultraviolet Radiation. Inflamm Bowel Dis. 2015;21:809–17. doi: 10.1097/MIB.0000000000000320. [DOI] [PubMed] [Google Scholar]
- 20.Sonnenberg A, Melton SD, Genta RM, et al. Absence of focally enhanced gastritis in macaques with idiopathic colitis. Inflamm Bowel Dis. 2011;17:2456–61. doi: 10.1002/ibd.21696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petronis A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature. 2010;465:721–7. doi: 10.1038/nature09230. [DOI] [PubMed] [Google Scholar]
- 22.Tjonneland A, Overvad K, Bergmann MM, et al. Linoleic acid, a dietary n-6 polyunsaturated fatty acid, and the aetiology of ulcerative colitis: a nested case-control study within a European prospective cohort study. Gut. 2009;58:1606–11. doi: 10.1136/gut.2008.169078. [DOI] [PubMed] [Google Scholar]
- 23.Octoratou M, Merikas E, Malgarinos G, et al. A prospective study of pre-illness diet in newly diagnosed patients with Crohn's disease. Rev medico-chirurgicală a Soc Medicişi Nat din Iaşi. 116:40–9. [PubMed] [Google Scholar]
- 24.Iablokov V, Sydora BC, Foshaug R, et al. Naturally occurring glycoalkaloids in potatoes aggravate intestinal inflammation in two mouse models of inflammatory bowel disease. Dig Dis Sci. 2010;55:3078–85. doi: 10.1007/s10620-010-1158-9. [DOI] [PubMed] [Google Scholar]
- 25.Nagy-Szakal D, Mir SA V, Harris RA, et al. Loss of n-6 fatty acid induced pediatric obesity protects against acute murine colitis. FASEB J. 2015 doi: 10.1096/fj.14-267690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hugot JP, Alberti C, Berrebi D, et al. Crohn's disease: the cold chain hypothesis. Lancet. 2003;362:2012–5. doi: 10.1016/S0140-6736(03)15024-6. [DOI] [PubMed] [Google Scholar]
- 27.Forbes A, Kalantzis T. Crohn's disease: the cold chain hypothesis. Int J Colorectal Dis. 2006;21:399–401. doi: 10.1007/s00384-005-0003-7. [DOI] [PubMed] [Google Scholar]
- 28.Goudie A, Cuff D, editors. Encyclopedia of Global Change. 1st. New York, New York: Oxford University Press Inc.; 2005. [Google Scholar]
- 29.Maslowski KM, Vieira AT, Ng A, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461:1282–6. doi: 10.1038/nature08530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagy-Szakal D, Mir Sa V, Ross MC, et al. Monotonous Diets Protect Against Acute Colitis in Mice. J Pediatr Gastroenterol Nutr. 2013;56:544–50. doi: 10.1097/MPG.0b013e3182769748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Atarashi K, Tanoue T, Shima T, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331:337–41. doi: 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olszak T, An D, Zeissig S, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012;336:489–93. doi: 10.1126/science.1219328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barreiro-de Acosta M, Alvarez Castro A, Souto R, et al. Emigration to western industrialized countries: A risk factor for developing inflammatory bowel disease. J Crohns Colitis. 2011;5:566–9. doi: 10.1016/j.crohns.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 34.Li X, Sundquist J, Hemminki K, et al. Risk of inflammatory bowel disease in first- and second-generation immigrants in Sweden: A nationwide follow-up study. Inflamm Bowel Dis. 2011;17:1784–91. doi: 10.1002/ibd.21535. [DOI] [PubMed] [Google Scholar]
- 35.Pinsk V, Lemberg DA, Grewal K, et al. Inflammatory bowel disease in the South Asian pediatric population of British Columbia. Am J Gastroenterol. 2007;102:1077–83. doi: 10.1111/j.1572-0241.2007.01124.x. [DOI] [PubMed] [Google Scholar]
- 36.Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–24. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo QS, Xia B, Jiang Y, et al. NOD2 3020insC frameshift mutation is not associated with inflammatory bowel disease in Chinese patients of Han nationality. World J Gastroenterol. 2004;10:1069–71. doi: 10.3748/wjg.v10.i7.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inoue N, Tamura K, Kinouchi Y, et al. Lack of common NOD2 variants in Japanese patients with Crohn's disease. Gastroenterology. 2002;123:86–91. doi: 10.1053/gast.2002.34155. [DOI] [PubMed] [Google Scholar]
- 39.Ozen SC, Dagli U, Kiliç MY, et al. NOD2/CARD15, NOD1/CARD4, and ICAM-1 gene polymorphisms in Turkish patients with inflammatory bowel disease. J Gastroenterol. 2006;41:304–10. doi: 10.1007/s00535-005-1780-z. [DOI] [PubMed] [Google Scholar]
- 40.Juyal G, Prasad P, Senapati S, et al. An investigation of genome-wide studies reported susceptibility loci for ulcerative colitis shows limited replication in north Indians. PLoS One. 2011;6:e16565. doi: 10.1371/journal.pone.0016565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei SC, Tan YY, Weng MT, et al. SLCO3A1, A novel crohn's disease-associated gene, regulates nf-κB activity and associates with intestinal perforation. PLoS One. 2014;9:e100515. doi: 10.1371/journal.pone.0100515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brant SR. Update on the heritability of inflammatory bowel disease: The importance of twin studies. Inflamm Bowel Dis. 2011;17:1–5. doi: 10.1002/ibd.21385. [DOI] [PubMed] [Google Scholar]
- 43.Kellermayer R. Genetic drift. Physiologic noise obscures genotype-phenotype correlations. Am J Med Genet A. 2007;143A:1306–7. doi: 10.1002/ajmg.a.31825. [DOI] [PubMed] [Google Scholar]
- 44.Sejdić E, Lipsitz LA. Necessity of noise in physiology and medicine. Comput Methods Programs Biomed. 2013;111:459–70. doi: 10.1016/j.cmpb.2013.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kellermayer R. “Omics” as the filtering gateway between environment and phenotype: The inflammatory bowel diseases example. Am J Med Genet Part A. 2010;152 A:3022–5. doi: 10.1002/ajmg.a.33726. [DOI] [PubMed] [Google Scholar]
- 46.Petrosino JF, Highlander S, Luna RA, et al. Metagenomic pyrosequencing and microbial identification. Clin Chem. 2009;55:856–66. doi: 10.1373/clinchem.2008.107565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Turnbaugh PJ, Ley RE, Hamady M, et al. The human microbiome project. Nature. 2007;449:804–10. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clemente JC, Ursell LK, Parfrey LW, et al. The impact of the gut microbiota on human health: An integrative view. Cell. 2012;148:1258–70. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peterson J, Garges S, Giovanni M, et al. The NIH Human Microbiome Project. Genome Res. 2009;19:2317–23. doi: 10.1101/gr.096651.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gevers D, Kugathasan S, Denson LA, et al. The treatment-naive microbiome in new-onset Crohn's disease. Cell Host Microbe. 2014;15:382–92. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morgan XC, Tickle TL, Sokol H, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13:R79. doi: 10.1186/gb-2012-13-9-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fava F, Danese S. Intestinal microbiota in inflammatory bowel disease: friend of foe? World J Gastroenterol. 2011;17:557–66. doi: 10.3748/wjg.v17.i5.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Geremia A, Biancheri P, Allan P, et al. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun Rev. 2014;13:3–10. doi: 10.1016/j.autrev.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 54.Bruewer M, Luegering A, Kucharzik T, et al. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol. 2003;171:6164–72. doi: 10.4049/jimmunol.171.11.6164. [DOI] [PubMed] [Google Scholar]
- 55.Rieder F, Schleder S, Wolf A, et al. Association of the novel serologic anti-glycan antibodies anti-laminarin and anti-chitin with complicated Crohn's disease behavior. Inflamm Bowel Dis. 2010;16:263–74. doi: 10.1002/ibd.21046. [DOI] [PubMed] [Google Scholar]
- 56.Russell RK, Ip B, Aldhous MC, et al. Anti-Saccharomyces cerevisiae antibodies status is associated with oral involvement and disease severity in Crohn disease. J Pediatr Gastroenterol Nutr. 2009;48:161–7. doi: 10.1097/MPG.0b013e318183e112. [DOI] [PubMed] [Google Scholar]
- 57.Alenghat T, Osborne LC, Saenz Sa, et al. Histone deacetylase 3 coordinates commensal-bacteria-dependent intestinal homeostasis. Nature. 2013;504:153–7. doi: 10.1038/nature12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yatsunenko T, Rey FE, Manary MJ, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–7. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kellermayer R. Burdening Questions About Clostridium difficile in Pediatric Inflammatory Bowel Diseases. J Pediatr Gastroenterol Nutr. 2015;60:421–2. doi: 10.1097/MPG.0000000000000756. [DOI] [PubMed] [Google Scholar]
- 60.David LA, Maurice CF, Carmody RN, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–63. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Faith JJ, Guruge JL, Charbonneau M, et al. The long-term stability of the human gut microbiota. Science. 2013;341:1237439. doi: 10.1126/science.1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Carmody RN, Gerber GK, Luevano JM, et al. Diet Dominates Host Genotype in Shaping the Murine Gut Microbiota. Cell Host Microbe. 2014;17:72–84. doi: 10.1016/j.chom.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu GD, Chen J, Hoffmann C, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–8. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nagy-Szakal D, Hollister EB, Luna RA, et al. Cellulose supplementation early in life ameliorates colitis in adult mice. PLoS One. 2013;8:e56685. doi: 10.1371/journal.pone.0056685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Suez J, Korem T, Zeevi D, et al. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature. 2014;514:181–6. doi: 10.1038/nature13793. [DOI] [PubMed] [Google Scholar]
- 66.Vrieze A, Van Nood E, Holleman F, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. 2012;143:913–6.e7. doi: 10.1053/j.gastro.2012.06.031. [DOI] [PubMed] [Google Scholar]
- 67.Chassaing B, Koren O, Goodrich JK, et al. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature1 Chass B al Diet Emuls impact mouse gut microbiota Promot colitis Metab Syndr Nat 519, 92–96 (2015) 2015;519:92–6. doi: 10.1038/nature14232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Turnbaugh PJ, Ley RE, Mahowald Ma, et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–31. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 69.Borody TJ, Khoruts A. Fecal microbiota transplantation and emerging applications. Nat Rev Gastroenterol Hepatol. 2012;9:88–96. doi: 10.1038/nrgastro.2011.244. [DOI] [PubMed] [Google Scholar]
- 70.Kelly CR, Ihunnah C, Fischer M, et al. Fecal microbiota transplant for treatment of Clostridium difficile infection in immunocompromised patients. Am J Gastroenterol. 2014;109:1065–71. doi: 10.1038/ajg.2014.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weingarden AR, Chen C, Bobr A, et al. Microbiota transplantation restores normal fecal bile acid composition in recurrent Clostridium difficile infection. Am J Physiol Gastrointest Liver Physiol. 2014;306:G310–9. doi: 10.1152/ajpgi.00282.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Martino D, Loke YJ, Gordon L, et al. Longitudinal, genome-scale analysis of DNA methylation in twins from birth to 18 months of age reveals rapid epigenetic change in early life and pair-specific effects of discordance. Genome Biol. 2013;14:R42. doi: 10.1186/gb-2013-14-5-r42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kellermayer R, Balasa A, Zhang W, et al. Epigenetic maturation in colonic mucosa continues beyond infancy in mice. Hum Mol Genet. 2010;19:2168–76. doi: 10.1093/hmg/ddq095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Waterland RA, Kellermayer R, Laritsky E, et al. Season of conception in rural gambia affects DNA methylation at putative human metastable epialleles. PLoS Genet. 2010;6:1–10. doi: 10.1371/journal.pgen.1001252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nagy-Szakal D, Ross MC, Dowd SE, et al. Maternal micronutrients can modify colonic mucosal microbiota maturation in murine offspring. Gut Microbes. 2012;3:426–33. doi: 10.4161/gmic.20697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mir Sa, Nagy-Szakal D, Dowd SE, et al. Prenatal methyl-donor supplementation augments colitis in young adult mice. PLoS One. 2013;8:e73162. doi: 10.1371/journal.pone.0073162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cho CE, Pannia E, Huot PSP, et al. Methyl vitamins contribute to obesogenic effects of a high multivitamin gestational diet and epigenetic alterations in hypothalamic feeding pathways in Wistar rat offspring. Mol Nutr Food Res. 2015;59:476–89. doi: 10.1002/mnfr.201400663. [DOI] [PubMed] [Google Scholar]
- 78.Gao F, Zhang J, Jiang P, et al. Marked methylation changes in intestinal genes during the perinatal period of preterm neonates. BMC Genomics. 2014;15:716. doi: 10.1186/1471-2164-15-716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Voisin S, Almén MS, Moschonis G, et al. Dietary fat quality impacts genome-wide DNA methylation patterns in a cross-sectional study of Greek preadolescents. Eur J Hum Genet. 2014:1–9. doi: 10.1038/ejhg.2014.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schaible TD, Harris RA, Dowd SE, et al. Maternal methyl-donor supplementation induces prolonged murine offspring colitis susceptibility in association with mucosal epigenetic and microbiomic changes. Hum Mol Genet. 2011;20:1687–96. doi: 10.1093/hmg/ddr044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Heijmans BT, Tobi EW, Stein AD, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. 2008;105:17046–9. doi: 10.1073/pnas.0806560105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dauncey MJ. Recent advances in nutrition, genes and brain health. Proc Nutr Soc. 2012;71:581–91. doi: 10.1017/S0029665112000237. [DOI] [PubMed] [Google Scholar]
- 83.Schnabel RB, Baccarelli A, Lin H, et al. Next steps in cardiovascular disease genomic research - Sequencing, epigenetics, and transcriptomics. Clin Chem. 2012;58:113–26. doi: 10.1373/clinchem.2011.170423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Naguib M, Bie B, Ting AH. Fundamental concepts of epigenetics for consideration in anesthesiology. Curr Opin Anaesthesiol. 2012;25:434–43. doi: 10.1097/ACO.0b013e3283556211. [DOI] [PubMed] [Google Scholar]
- 85.Petronis A, Petroniene R. Epigenetics of inflammatory bowel disease. Gut. 2000;47:302–6. doi: 10.1136/gut.47.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Scarpa M, Stylianou E. Epigenetics: Concepts and relevance to IBD pathogenesis. Inflamm Bowel Dis. 2012;18:1982–96. doi: 10.1002/ibd.22934. [DOI] [PubMed] [Google Scholar]
- 87.Jenke AC, Zilbauer M. Epigenetics in inflammatory bowel disease. Curr Opin Gastroenterol. 2012;28:577–84. doi: 10.1097/MOG.0b013e328357336b. [DOI] [PubMed] [Google Scholar]
- 88.Rosen MJ, Frey MR, Washington MK, et al. STAT6 activation in ulcerative colitis: A new target for prevention of IL-13-induced colon epithelial cell dysfunction. Inflamm Bowel Dis. 2011;17:2224–34. doi: 10.1002/ibd.21628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Felice C, Lewis A, Armuzzi A, et al. Review article: selective histone deacetylase isoforms as potential therapeutic targets in inflammatory bowel diseases. Aliment Pharmacol Ther. 2015;41:26–38. doi: 10.1111/apt.13008. [DOI] [PubMed] [Google Scholar]
- 90.Wu F, Guo NJ, Tian H, et al. Peripheral blood MicroRNAs distinguish active ulcerative colitis and Crohn's disease. Inflamm Bowel Dis. 2011;17:241–50. doi: 10.1002/ibd.21450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chen Y, Ge W, Xu L, et al. miR-200b is involved in intestinal fibrosis of Crohn's disease. Int J Mol Med. 2012;29:601–6. doi: 10.3892/ijmm.2012.894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zahm AM, Thayu M, Hand NJ, et al. Circulating MicroRNA is a biomarker of pediatric Crohn disease. J Pediatr Gastroenterol Nutr. 2011;53:26–33. doi: 10.1097/MPG.0b013e31822200cc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Koukos G, Polytarchou C, Kaplan JL, et al. A MicroRNA Signature in Pediatric Ulcerative Colitis: Deregulation of the miR-4284/CXCL5 Pathway in the Intestinal Epithelium. Inflamm Bowel Dis. 2015;21:996–1005. doi: 10.1097/MIB.0000000000000339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wigler M, Levy D, Perucho M. The somatic replication of DNA methylation. Cell. 1981;24:33–40. doi: 10.1016/0092-8674(81)90498-0. [DOI] [PubMed] [Google Scholar]
- 95.Margueron R, Reinberg D. Chromatin structure and the inheritance of epigenetic information. Nat Rev Genet. 2010;11:285–96. doi: 10.1038/nrg2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Faulk C, Dolinoy DC. Timing is everything: The when and how of environmentally induced changes in the epigenome of animals. Epigenetics. 2011;6:791–7. doi: 10.4161/epi.6.7.16209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Adams AT, Kennedy Na, Hansen R, et al. Two-stage Genome-wide Methylation Profiling in Childhood-onset Crohn's Disease Implicates Epigenetic Alterations at the VMP1/MIR21 and HLA Loci. Inflamm Bowel Dis. 2014;0:1–10. doi: 10.1097/MIB.0000000000000179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Harris RA, Nagy-Szakal D, Pedersen N, et al. Genome-wide peripheral blood leukocyte DNA methylation microarrays identified a single association with inflammatory bowel diseases. Inflamm Bowel Dis. 2012;18:2334–41. doi: 10.1002/ibd.22956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kellermayer R. Hurdles for epigenetic disease associations from peripheral blood leukocytes. Inflamm Bowel Dis. 2013;19:E66–7. doi: 10.1097/MIB.0b013e318280eca4. [DOI] [PubMed] [Google Scholar]
- 100.Saito S, Kato J, Hiraoka S, et al. DNA methylation of colon mucosa in ulcerative colitis patients: Correlation with inflammatory status. Inflamm Bowel Dis. 2011;17:1955–65. doi: 10.1002/ibd.21573. [DOI] [PubMed] [Google Scholar]
- 101.Lin Z, Hegarty JP, Cappel JA, et al. Identification of disease-associated DNA methylation in intestinal tissues from patients with inflammatory bowel disease. Clin Genet. 2011;80:59–67. doi: 10.1111/j.1399-0004.2010.01546.x. [DOI] [PubMed] [Google Scholar]
- 102.Yuan B, Zhang J, Wang H, et al. 6-Thioguanine reactivates epigenetically silenced genes in acute lymphoblastic leukemia cells by facilitating proteasome-mediated degradation of DNMT. Cancer Res. 2011;71:1904–11. doi: 10.1158/0008-5472.CAN-10-3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Harris RA, Nagy-Szakal D, Mir SAV, et al. DNA methylation-associated colonic mucosal immune and defense responses in treatment-naïve pediatric ulcerative colitis. Epigenetics. 2014;9:1131–7. doi: 10.4161/epi.29446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Häsler R, Feng Z, Bäckdahl L, et al. A functional methylome map of ulcerative colitis. Genome Res. 2012;22:2130–7. doi: 10.1101/gr.138347.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Alenghat T. Epigenomics and the microbiota. Toxicol Pathol. 2015;43:101–6. doi: 10.1177/0192623314553805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Salvucci E. Microbiome, holobiont and the net of life. Crit Rev Microbiol. 2014:1–10. doi: 10.3109/1040841X.2014.962478. [DOI] [PubMed] [Google Scholar]
- 107.Bierne H, Hamon M, Cossart P. Epigenetics and bacterial infections. Cold Spring Harb Perspect Med. 2012;2:a010272. doi: 10.1101/cshperspect.a010272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tolg C, Sabha N, Cortese R, et al. Uropathogenic E. coli infection provokes epigenetic downregulation of CDKN2A (p16INK4A) in uroepithelial cells. Lab Invest. 2011;91:825–36. doi: 10.1038/labinvest.2010.197. [DOI] [PubMed] [Google Scholar]
- 109.Chernov AV, Reyes L, Xu Z, et al. Mycoplasma CG- and GATC-specific DNA methyltransferases selectively and efficiently methylate the host genome and alter the epigenetic landscape in human cells. Epigenetics. 2015 doi: 10.1080/15592294.2015.1020000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shenderov BA. Gut indigenous microbiota and epigenetics. Microb Ecol Health Dis. 2012;23:1–6. doi: 10.3402/mehd.v23i0.17195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Louis P, Flint HJ. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol Lett. 2009;294:1–8. doi: 10.1111/j.1574-6968.2009.01514.x. [DOI] [PubMed] [Google Scholar]
- 112.Lopez-Siles M, Martinez-Medina M, Busquets D, et al. Mucosa-associated Faecalibacterium prausnitzii and Escherichia coli co-abundance can distinguish Irritable Bowel Syndrome and Inflammatory Bowel Disease phenotypes. Int J Med Microbiol. 2014;304:464–75. doi: 10.1016/j.ijmm.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 113.Wang W, Chen L, Zhou R, et al. Increased proportions of Bifidobacterium and the Lactobacillus group and loss of butyrate-producing bacteria in inflammatory bowel disease. J Clin Microbiol. 2014;52:398–406. doi: 10.1128/JCM.01500-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Machiels K, Joossens M, Sabino J, et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut. 2014;63:1275–83. doi: 10.1136/gutjnl-2013-304833. [DOI] [PubMed] [Google Scholar]
- 115.Cao Y, Shen J, Ran ZH. Association between faecalibacterium prausnitzii reduction and inflammatory bowel disease: A meta-analysis and systematic review of the literature. Gastroenterol Res Pract. 2014;2014 doi: 10.1155/2014/872725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fujimoto T, Imaeda H, Takahashi K, et al. Decreased abundance of Faecalibacterium prausnitzii in the gut microbiota of Crohn's disease. J Gastroenterol Hepatol. 2013;28:613–9. doi: 10.1111/jgh.12073. [DOI] [PubMed] [Google Scholar]
- 117.Hamer HM, Jonkers D, Venema K, et al. Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther. 2008;27:104–19. doi: 10.1111/j.1365-2036.2007.03562.x. [DOI] [PubMed] [Google Scholar]
- 118.Berni Canani R, Di Costanzo M, Leone L. The epigenetic effects of butyrate: potential therapeutic implications for clinical practice. Clin Epigenetics. 2012;4:4. doi: 10.1186/1868-7083-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mali P, Chou BK, Yen J, et al. Butyrate greatly enhances derivation of human induced pluripotent stem cells by promoting epigenetic remodeling and the expression of pluripotency-associated genes. Stem Cells. 2010;28:713–20. doi: 10.1002/stem.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kumar A, Wu H, Collier-Hyams LS, et al. The bacterial fermentation product butyrate influences epithelial signaling via reactive oxygen species-mediated changes in cullin-1 neddylation. J Immunol. 2009;182:538–46. doi: 10.4049/jimmunol.182.1.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Boesmans L, Ramakers M, Arijs I, et al. Inflammation-induced down-regulation of butyrate uptake and oxidation is not caused by a reduced gene expression. J Cell Physiol. 2014:1–31. doi: 10.1002/jcp.24725. [DOI] [PubMed] [Google Scholar]
- 122.De Preter V, Arijs I, Windey K, et al. Impaired butyrate oxidation in ulcerative colitis is due to decreased butyrate uptake and a defect in the oxidation pathway. Inflamm Bowel Dis. 2012;18:1127–36. doi: 10.1002/ibd.21894. [DOI] [PubMed] [Google Scholar]
- 123.Santhanam S, Venkatraman A, Ramakrishna BS. Impairment of mitochondrial acetoacetyl CoA thiolase activity in the colonic mucosa of patients with ulcerative colitis. Gut. 2007;56:1543–9. doi: 10.1136/gut.2006.108449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Thibault R, Blachier F, Darcy-Vrillon B, et al. Butyrate utilization by the colonic mucosa in inflammatory bowel diseases. Inflamm Bowel Dis. 2010;16:684–95. doi: 10.1002/ibd.21108. [DOI] [PubMed] [Google Scholar]
- 125.Vernia P, Annese V, Bresci G, et al. Topical butyrate improves efficacy of 5-ASA in refractory distal ulcerative colitis: results of a multicentre trial. Eur J Clin Invest. 2003;33:244–8. doi: 10.1046/j.1365-2362.2003.01130.x. [DOI] [PubMed] [Google Scholar]
- 126.Rossi M, Amaretti A, Raimondi S. Folate production by probiotic bacteria. Nutrients. 2011;3:118–34. doi: 10.3390/nu3010118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sartor RB. Microbial Influences in Inflammatory Bowel Diseases. Gastroenterology. 2008;134:577–94. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- 128.Chassaing B, Darfeuillemichaud A. The commensal microbiota and enteropathogens in the pathogenesis of inflammatory bowel diseases. Gastroenterology. 2011;140:1720–8. doi: 10.1053/j.gastro.2011.01.054. [DOI] [PubMed] [Google Scholar]
- 129.Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453:620–5. doi: 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- 130.Nelson DB, Murdoch M, Sandozi IK, et al. Increased levels of homocysteine in patients with Crohn's disease are related to folate levels. Am J Gastroenterol. 2000;95:3498–502. doi: 10.1111/j.1572-0241.2000.03367.x. [DOI] [PubMed] [Google Scholar]
- 131.Heyman M, Garnett E. Folate concentrations in pediatric patients with newly diagnosed inflammatory bowel disease. Am J …. 2009:545–50. doi: 10.3945/ajcn.2008.26576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Alkhouri RH, Hashmi H, Baker RD, et al. Vitamin and mineral status in patients with inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 2013;56:89–92. doi: 10.1097/MPG.0b013e31826a105d. [DOI] [PubMed] [Google Scholar]
- 133.Vagianos K, Bernstein CN. Homocysteinemia and B vitamin status among adult patients with inflammatory bowel disease: A one-year prospective follow-up study. Inflamm Bowel Dis. 2012;18:718–24. doi: 10.1002/ibd.21785. [DOI] [PubMed] [Google Scholar]
- 134.Stappenbeck TS, Hooper LV, Gordon JI. Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc Natl Acad Sci U S A. 2002;99:15451–5. doi: 10.1073/pnas.202604299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hill DA, Artis D. Intestinal bacteria and the regulation of immune cell homeostasis. Annu Rev Immunol. 2010;28:623–67. doi: 10.1146/annurev-immunol-030409-101330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cerf-Bensussan N, Gaboriau-Routhiau V. The immune system and the gut microbiota: friends or foes? Nat Rev Immunol. 2010;10:735–44. doi: 10.1038/nri2850. [DOI] [PubMed] [Google Scholar]
- 137.Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A. 2010;107:12204–9. doi: 10.1073/pnas.0909122107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Adkins B. Development of neonatal Th1/Th2 function. Int Rev Immunol. 2000;19:157–71. doi: 10.3109/08830180009088503. [DOI] [PubMed] [Google Scholar]
- 139.McLoughlin RM, Mills KHG. Influence of gastrointestinal commensal bacteria on the immune responses that mediate allergy and asthma. J Allergy Clin Immunol. 2011;127:1097–107. doi: 10.1016/j.jaci.2011.02.012. quiz 1108–9. [DOI] [PubMed] [Google Scholar]
- 140.Wang Y, Devkota S, Musch MW, et al. Regional mucosa-associated microbiota determine physiological expression of TLR2 and TLR4 in murine colon. PLoS One. 2010;5:e13607. doi: 10.1371/journal.pone.0013607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Takahashi K, Sugi Y, Nakano K, et al. Epigenetic Control of the Host Gene by Commensal Bacteria in Large Intestinal Epithelial Cells. J Biol Chem. 2011;286:35755–62. doi: 10.1074/jbc.M111.271007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Takahashi K, Sugi Y, Hosono A, et al. Epigenetic regulation of TLR4 gene expression in intestinal epithelial cells for the maintenance of intestinal homeostasis. J Immunol. 2009;183:6522–9. doi: 10.4049/jimmunol.0901271. [DOI] [PubMed] [Google Scholar]
- 143.Patel RM, Lin PW. Developmental biology of gut-probiotic interaction. Gut Microbes. 2010;1:186–95. doi: 10.4161/gmic.1.3.12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kumar H, Lund R, Laiho A, et al. Gut Microbiota as an Epigenetic Regulator: Pilot Study Based on Whole-Genome Methylation Analysis. MBio. 2014;5:e02113–4. doi: 10.1128/mBio.02113-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chu KM, Watermeyer G, Shelly L, et al. Childhood helminth exposure is protective against inflammatory bowel disease: a case control study in South Africa. Inflamm Bowel Dis. 2013;19:614–20. doi: 10.1097/MIB.0b013e31827f27f4. [DOI] [PubMed] [Google Scholar]
- 146.Weinstock JV, Elliott DE. Helminths and the IBD hygiene hypothesis. Inflamm Bowel Dis. 2009;15:128–33. doi: 10.1002/ibd.20633. [DOI] [PubMed] [Google Scholar]
- 147.Broadhurst MJ, Ardeshir A, Kanwar B, et al. Therapeutic helminth infection of macaques with idiopathic chronic diarrhea alters the inflammatory signature and mucosal microbiota of the colon. PLoS Pathog. 2012;8:e1003000. doi: 10.1371/journal.ppat.1003000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lee SC, Tang MS, Lim YAL, et al. Helminth colonization is associated with increased diversity of the gut microbiota. PLoS Negl Trop Dis. 2014;8:e2880. doi: 10.1371/journal.pntd.0002880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Deaton AM, Cook PC, De Sousa D, et al. A unique DNA methylation signature defines a population of IFN-γ/IL-4 double-positive T cells during helminth infection. Eur J Immunol. 2014;44:1835–41. doi: 10.1002/eji.201344098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Rodríguez LAG, Ruigómez A, Panés J. Acute Gastroenteritis Is Followed by an Increased Risk of Inflammatory Bowel Disease. Gastroenterology. 2006;130:1588–94. doi: 10.1053/j.gastro.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 151.Shaw SY, Blanchard JF, Bernstein CN. Association between the use of antibiotics in the first year of life and pediatric inflammatory bowel disease. Am J Gastroenterol. 2010;105:2687–92. doi: 10.1038/ajg.2010.398. [DOI] [PubMed] [Google Scholar]
- 152.Shaw SY, Blanchard JF, Bernstein CN. Association between the use of antibiotics and new diagnoses of Crohn's disease and ulcerative colitis. Am J Gastroenterol. 2011;106:2133–42. doi: 10.1038/ajg.2011.304. [DOI] [PubMed] [Google Scholar]
- 153.Virta L, Auvinen A, Helenius H, et al. Association of repeated exposure to antibiotics with the development of pediatric crohn's disease - A nationwide, register-based Finnish case-control study. Am J Epidemiol. 2012;175:775–84. doi: 10.1093/aje/kwr400. [DOI] [PubMed] [Google Scholar]
- 154.Porter CK, Tribble DR, Aliaga PA, et al. Infectious Gastroenteritis and Risk of Developing Inflammatory Bowel Disease. Gastroenterology. 2008;135:781–6. doi: 10.1053/j.gastro.2008.05.081. [DOI] [PubMed] [Google Scholar]
- 155.Green C, Elliott L, Beaudoin C, et al. A population-based ecologic study of inflammatory bowel disease: Searching for etiologic clues. Am J Epidemiol. 2006;164:615–23. doi: 10.1093/aje/kwj260. [DOI] [PubMed] [Google Scholar]
- 156.Modi SR, Collins JJ, Relman DA. Antibiotics and the gut microbiota. J Clin Invest. 2014;124:4212–8. doi: 10.1172/JCI72333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zeh JA, Bonilla MM, Adrian AJ, et al. From father to son: transgenerational effect of tetracycline on sperm viability. Sci Rep. 2012;2:375. doi: 10.1038/srep00375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Munyaka PM, Khafipour E, Ghia JE. External Influence of Early Childhood Establishment of Gut Microbiota and Subsequent Health Implications. Front Pediatr. 2014;2:1–9. doi: 10.3389/fped.2014.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Soon IS, Molodecky NA, Rabi DM, et al. The relationship between urban environment and the inflammatory bowel diseases: a systematic review and meta-analysis. BMC Gastroenterol. 2012;12:51. doi: 10.1186/1471-230X-12-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Castiglione F, Diaferia M, Morace F, et al. Risk factors for inflammatory bowel diseases according to the “hygiene hypothesis”: a case-control, multi-centre, prospective study in Southern Italy. J Crohns Colitis. 2012;6:324–9. doi: 10.1016/j.crohns.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 161.Timm S, Svanes C, Janson C, et al. Place of upbringing in early childhood as related to inflammatory bowel diseases in adulthood: A population-based cohort study in Northern Europe. Eur J Epidemiol. 2014;29:429–37. doi: 10.1007/s10654-014-9922-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kaplan GG, Hubbard J, Korzenik J, et al. The Inflammatory Bowel Diseases and Ambient Air Pollution: A Novel Association. Am J Gastroenterol. 2010;105:2412–9. doi: 10.1038/ajg.2010.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ananthakrishnan AN, McGinley EL, Binion DG, et al. Ambient air pollution correlates with hospitalizations for inflammatory bowel disease: An ecologic analysis. Inflamm Bowel Dis. 2011;17:1138–45. doi: 10.1002/ibd.21455. [DOI] [PubMed] [Google Scholar]
- 164.Tyakht AV, Kostryukova ES, Popenko AS, et al. Human gut microbiota community structures in urban and rural populations in Russia. Nat Commun. 2013;4:2469. doi: 10.1038/ncomms3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Tyakht AV, Alexeev DG, Popenko AS, et al. Rural and urban microbiota. Gut Microbes. 2014;5:351–6. doi: 10.4161/gmic.28685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Boyko EJ, Perera DR, Koepsell TD, et al. Effects of cigarette smoking on the clinical course of ulcerative colitis. Scand J Gastroenterol. 1988;23:1147–52. doi: 10.3109/00365528809090183. [DOI] [PubMed] [Google Scholar]
- 167.Calabrese E, Yanai H, Shuster D, et al. Low-dose smoking resumption in ex-smokers with refractory ulcerative colitis. J Crohn's Colitis. 2012;6:756–62. doi: 10.1016/j.crohns.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 168.Beaugerie L, Massot N, Carbonnel F, et al. Impact of cessation of smoking on the course of ulcerative colitis. Am J Gastroenterol. 2001;96:2113–6. doi: 10.1111/j.1572-0241.2001.03944.x. [DOI] [PubMed] [Google Scholar]
- 169.Van der Heide F, Dijkstra A, Weersma RK, et al. Effects of active and passive smoking on disease course of Crohn's disease and ulcerative colitis. Inflamm Bowel Dis. 2009;15:1199–207. doi: 10.1002/ibd.20884. [DOI] [PubMed] [Google Scholar]
- 170.Cosnes J, Beaugerie L, Carbonnel F, et al. Smoking cessation and the course of Crohn's disease: an intervention study. Gastroenterology. 2001;120:1093–9. doi: 10.1053/gast.2001.23231. [DOI] [PubMed] [Google Scholar]
- 171.Seksik P, Nion-Larmurier I, Sokol H, et al. Effects of light smoking consumption on the clinical course of Crohn's disease. Inflamm Bowel Dis. 2009;15:734–41. doi: 10.1002/ibd.20828. [DOI] [PubMed] [Google Scholar]
- 172.Biedermann L, Zeitz J, Mwinyi J, et al. Smoking Cessation Induces Profound Changes in the Composition of the Intestinal Microbiota in Humans. PLoS One. 2013;8 doi: 10.1371/journal.pone.0059260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Biedermann L, Brulisauer K, Zeitz J, et al. Smoking cessation alters intestinal microbiota: insights from quantitative investigations on human fecal samples using FISH. Inflamm Bowel Dis. 2014;20:1496–501. doi: 10.1097/MIB.0000000000000129. [DOI] [PubMed] [Google Scholar]
- 174.Benjamin JL, Hedin CRH, Koutsoumpas A, et al. Smokers with active Crohn's disease have a clinically relevant dysbiosis of the gastrointestinal microbiota. Inflamm Bowel Dis. 2012;18:1092–100. doi: 10.1002/ibd.21864. [DOI] [PubMed] [Google Scholar]
- 175.Breton CV, Byun HM, Wenten M, et al. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am J Respir Crit Care Med. 2009;180:462–7. doi: 10.1164/rccm.200901-0135OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Guerrero-Preston R, Goldman LR, Brebi-Mieville P, et al. Global DNA hypomethylation is associated with in utero exposure to cotinine and perfluorinated alkyl compounds. Epigenetics. 2010;5:539–46. doi: 10.4161/epi.5.6.12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lee KWK, Pausova Z. Cigarette smoking and DNA methylation. Front Genet. 2013;4 doi: 10.3389/fgene.2013.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Drake AJ, Shaughnessy PJO, Bhattacharya S, et al. In utero exposure to cigarette chemicals induces sex-specific disruption of one-carbon metabolism and DNA methylation in the human fetal liver. BMC Med. 2015;13:1–12. doi: 10.1186/s12916-014-0251-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Novakovic B, Ryan J, Pereira N, et al. Postnatal stability and tissue- and time-specific effects of AHRR methylation change in response to maternal smoking in pregnancy. Epigenetics. 2013;9 doi: 10.4161/epi.27248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Philibert Ra, Beach SRH, Lei MK, et al. Changes in DNA methylation at the aryl hydrocarbon receptor repressor may be a new biomarker for smoking. Clin Epigenetics. 2013;5:19. doi: 10.1186/1868-7083-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Zhang Y, Yang R, Burwinkel B, et al. F2RL3 methylation as a biomarker of current and lifetime smoking exposures. Environ Health Perspect. 2014;122:131–7. doi: 10.1289/ehp.1306937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zeilinger S, Kühnel B, Klopp N, et al. Tobacco Smoking Leads to Extensive Genome-Wide Changes in DNA Methylation. PLoS One. 2013;8 doi: 10.1371/journal.pone.0063812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sonntag B, Stolze B, Heinecke A, et al. Preterm birth but not mode of delivery is associated with an increased risk of developing inflammatory bowel disease later in life. Inflamm Bowel Dis. 2007;13:1385–90. doi: 10.1002/ibd.20206. [DOI] [PubMed] [Google Scholar]
- 184.Gritz EC, Bhandari V. The Human Neonatal Gut Microbiome: A Brief Review. Front Pediatr. 2015;3:17. doi: 10.3389/fped.2015.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Cruickshank MN, Oshlack A, Theda C, et al. Analysis of epigenetic changes in survivors of preterm birth reveals the effect of gestational age and evidence for a long term legacy. Genome Med. 2013;5:96. doi: 10.1186/gm500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kuss SK, Best GT, Etheredge CA, et al. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science. 2011;334:249–52. doi: 10.1126/science.1211057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Robinson CM, Jesudhasan PR, Pfeiffer JK. Bacterial lipopolysaccharide binding enhances virion stability and promotes environmental fitness of an enteric virus. Cell Host Microbe. 2014;15:36–46. doi: 10.1016/j.chom.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Juste RA, Elguezabal N, Pavón A, et al. Association between Mycobacterium avium subsp. paratuberculosis DNA in blood and cellular and humoral immune response in inflammatory bowel disease patients and controls. Int J Infect Dis. 2009;13:247–54. doi: 10.1016/j.ijid.2008.06.034. [DOI] [PubMed] [Google Scholar]
- 189.Lee A, Griffiths TA, Parab RS, et al. Association of Mycobacterium avium subspecies paratuberculosis with Crohn Disease in pediatric patients. J Pediatr Gastroenterol Nutr. 2011;52:170–4. doi: 10.1097/MPG.0b013e3181ef37ba. [DOI] [PubMed] [Google Scholar]
- 190.Kirkwood CD, Wagner J, Boniface K, et al. Mycobacterium avium subspecies paratuberculosis in children with early-onset Crohn's disease. Inflamm Bowel Dis. 2009;15:1643–55. doi: 10.1002/ibd.20967. [DOI] [PubMed] [Google Scholar]
- 191.Pennini ME, Pai RK, Schultz DC, et al. Mycobacterium tuberculosis 19-kDa lipoprotein inhibits IFN-gamma-induced chromatin remodeling of MHC2TA by TLR2 and MAPK signaling. J Immunol. 2006;176:4323–30. doi: 10.4049/jimmunol.176.7.4323. [DOI] [PubMed] [Google Scholar]
- 192.Gardenbroek TJ, Eshuis EJ, Ponsioen CIJ, et al. The effect of appendectomy on the course of ulcerative colitis: a systematic review. Colorectal Dis. 2012;14:545–53. doi: 10.1111/j.1463-1318.2011.02600.x. [DOI] [PubMed] [Google Scholar]
- 193.Radford-Smith GL, Edwards JE, Purdie DM, et al. Protective role of appendicectomy on onset and severity of ulcerative colitis and Crohn's disease. Gut. 2002;51:808–13. doi: 10.1136/gut.51.6.808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Russel MG, Dorant E, Brummer RJ, et al. Appendectomy and the risk of developing ulcerative colitis or Crohn's disease: results of a large case-control study. South Limburg Inflammatory Bowel Disease Study Group. Gastroenterology. 1997;113:377–82. doi: 10.1053/gast.1997.v113.pm9247453. [DOI] [PubMed] [Google Scholar]
- 195.López-Serrano P, Pérez-Calle JL, Pérez-Fernández MT, et al. Environmental risk factors in inflammatory bowel diseases. Investigating the hygiene hypothesis: a Spanish case-control study. Scand J Gastroenterol. 2010;45:1464–71. doi: 10.3109/00365521.2010.510575. [DOI] [PubMed] [Google Scholar]
- 196.Reif S, Lavy A, Keter D, et al. Appendectomy is more frequent but not a risk factor in Crohn's disease while being protective in ulcerative colitis: a comparison of surgical procedures in inflammatory bowel disease. Am J Gastroenterol. 2001;96:829–32. doi: 10.1111/j.1572-0241.2001.03529.x. [DOI] [PubMed] [Google Scholar]
- 197.Jackson HT, Mongodin EF, Davenport KP, et al. Culture-independent evaluation of the appendix and rectum microbiomes in children with and without appendicitis. PLoS One. 2014;9:e95414. doi: 10.1371/journal.pone.0095414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Barclay AR, Russell RK, Wilson ML, et al. Systematic review: the role of breastfeeding in the development of pediatric inflammatory bowel disease. J Pediatr. 2009;155:421–6. doi: 10.1016/j.jpeds.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 199.Rogier EW, Frantz AL, Bruno MEC, et al. Secretory antibodies in breast milk promote long-term intestinal homeostasis by regulating the gut microbiota and host gene expression. Proc Natl Acad Sci U S A. 2014;111:3074–9. doi: 10.1073/pnas.1315792111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Thompson AL, Monteagudo-Mera A, Cadenas MB, et al. Milk- and solid-feeding practices and daycare attendance are associated with differences in bacterial diversity, predominant communities, and metabolic and immune function of the infant gut microbiome. Front Cell Infect Microbiol. 2015;5:1–15. doi: 10.3389/fcimb.2015.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Sävendahl L, Underwood LE, Haldeman KM, et al. Fasting prevents experimental murine colitis produced by dextran sulfate sodium and decreases interleukin-1β and insulin-like growth factor I messenger ribonucleic acid. Endocrinology. 1997;138:734–40. doi: 10.1210/endo.138.2.4941. [DOI] [PubMed] [Google Scholar]
- 202.Singh UP, Singh NP, Guan H, et al. The emerging role of leptin antagonist as potential therapeutic option for inflammatory bowel disease. Int Rev Immunol. 2014;33:23–33. doi: 10.3109/08830185.2013.809071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Matarese G, La Cava A, Sanna V, et al. Balancing susceptibility to infection and autoimmunity: A role for leptin? Trends Immunol. 2002;23:182–7. doi: 10.1016/s1471-4906(02)02188-9. [DOI] [PubMed] [Google Scholar]
- 204.Barbier M, Cherbut C, Aubé AC, et al. Elevated plasma leptin concentrations in early stages of experimental intestinal inflammation in rats. Gut. 1998;43:783–90. doi: 10.1136/gut.43.6.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Siegmund B, Lehr HA, Fantuzzi G. Leptin: a pivotal mediator of intestinal inflammation in mice. Gastroenterology. 2002;122:2011–25. doi: 10.1053/gast.2002.33631. [DOI] [PubMed] [Google Scholar]
- 206.De Filippo C, Cavalieri D, Di Paola M, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A. 2010;107:14691–6. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Kohl KD, Amaya J, Passement CA, et al. Unique and shared responses of the gut microbiota to prolonged fasting: a comparative study across five classes of vertebrate hosts. FEMS Microbiol Ecol. 2014;90:883–94. doi: 10.1111/1574-6941.12442. [DOI] [PubMed] [Google Scholar]
- 208.Damms-Machado A, Mitra S, Schollenberger AE, et al. Effects of surgical and dietary weight loss therapy for obesity on gut microbiota composition and nutrient absorption. Biomed Res Int. 2015;2015:806248. doi: 10.1155/2015/806248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Zhang C, Li S, Yang L, et al. Structural modulation of gut microbiota in life-long calorie-restricted mice. Nat Commun. 2013;4:2163. doi: 10.1038/ncomms3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Tobi EW, Goeman JJ, Monajemi R, et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat Commun. 2014;5:5592. doi: 10.1038/ncomms6592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Dominguez-Salas P, Moore SE, Baker MS, et al. Maternal nutrition at conception modulates DNA methylation of human metastable epialleles. Nat Commun. 2014;5:3746. doi: 10.1038/ncomms4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Hughes LAE, van den Brandt PA, de Bruïne AP, et al. Early life exposure to famine and colorectal cancer risk: a role for epigenetic mechanisms. PLoS One. 2009;4:e7951. doi: 10.1371/journal.pone.0007951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Ananthakrishnan AN, Khalili H, Konijeti GG, et al. Long-term intake of dietary fat and risk of ulcerative colitis and Crohn's disease. Gut. 2014;63:776–84. doi: 10.1136/gutjnl-2013-305304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Goldsmith JR, Sartor RB. The role of diet on intestinal microbiota metabolism: downstream impacts on host immune function and health, and therapeutic implications. J Gastroenterol. 2014 doi: 10.1007/s00535-014-0953-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Hou JK, Abraham B, El-Serag H. Dietary intake and risk of developing inflammatory bowel disease: a systematic review of the literature. Am J Gastroenterol. 2011;106:563–73. doi: 10.1038/ajg.2011.44. [DOI] [PubMed] [Google Scholar]
- 216.Paparo L, di Costanzo M, di Scala C, et al. The Influence of Early Life Nutrition on Epigenetic Regulatory Mechanisms of the Immune System. Nutrients. 2014;6:4706–19. doi: 10.3390/nu6114706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Johnson IT, Belshaw NJ. The effect of diet on the intestinal epigenome. Epigenomics. 2014;6:239–51. doi: 10.2217/epi.14.8. [DOI] [PubMed] [Google Scholar]
- 218.Andersen V, Olsen A, Carbonnel F, et al. Diet and risk of inflammatory bowel disease. Dig Liver Dis. 2012;44:185–94. doi: 10.1016/j.dld.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 219.Ooi JH, Li Y, Rogers CJ, et al. Vitamin D regulates the gut microbiome and protects mice from dextran sodium sulfate-induced colitis. J Nutr. 2013;143:1679–86. doi: 10.3945/jn.113.180794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Ananthakrishnan AN, Cagan A, Gainer VS, et al. Higher plasma vitamin D is associated with reduced risk of Clostridium difficile infection in patients with inflammatory bowel diseases. Aliment Pharmacol Ther. 2014;39:1136–42. doi: 10.1111/apt.12706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Sahay T, Ananthakrishnan AN. Vitamin D deficiency is associated with community-acquired clostridium difficile infection: a case-control study. BMC Infect Dis. 2014;14:661. doi: 10.1186/s12879-014-0661-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Yu S, Cantorna MT. Epigenetic reduction in invariant NKT cells following in utero vitamin D deficiency in mice. J Immunol. 2011;186:1384–90. doi: 10.4049/jimmunol.1002545. [DOI] [PMC free article] [PubMed] [Google Scholar]