

Abstract
Mammalian orthoreoviruses (reoviruses) are members of the Reoviridae. Reoviruses contain 10 double-stranded (ds) RNA gene segments enclosed in two concentric protein shells, called outer capsid and core. These viruses serve as a versatile experimental system for studies of viral replication events at the virus-cell interface, including engagement of cell-surface receptors, internalization and disassembly, and activation of the innate immune response, including NF-κB-dependent cellular signaling pathways. Reoviruses also provide a model system for studies of virus-induced apoptosis and organ-specific disease in vivo.
Reoviruses attach to host cells via the filamentous attachment protein, σ1. The σ1 protein of all reovirus serotypes engages junctional adhesion molecule-A (JAM-A), an integral component of intercellular tight junctions. The σ1 protein also binds to cell-surface carbohydrate, with the type of carbohydrate bound varying by serotype. Following attachment to JAM-A and carbohydrate, reovirus internalization is mediated by β1 integrins, most likely via clathrin-dependent endocytosis. In the endocytic compartment, reovirus outer-capsid protein σ3 is removed by acid-dependent cysteine proteases in most cell types. Removal of σ3 results in the exposure of a hydrophobic conformer of the viral membrane-penetration protein, μ1, which pierces the endosomal membrane and delivers transcriptionally active reovirus core particles into the cytoplasm.
Reoviruses induce apoptosis in both cultured cells and infected mice. Perturbation of reovirus disassembly using inhibitors of endosomal acidification or protease activity abrogates apoptosis. The μ1-encoding M2 gene is genetically linked to strain-specific differences in apoptosis-inducing capacity, suggesting a function for μ1 in induction of death signaling. Reovirus disassembly leads to activation of transcription factor NF-κB, which modulates apoptotic signaling in numerous types of cells. Inhibition of NF-κB nuclear translocation using either pharmacologic agents or expression of transdominant forms of IκB blocks reovirus-induced apoptosis, suggesting an essential role for NF-κB activation in the death response. Multiple effector pathways downstream of NF-κB directed gene expression execute reovirus-induced cell death. This chapter will focus on the mechanisms by which reovirus attachment and disassembly activate NF-κB and stimulate the cellular proapoptotic machinery.
INTRODUCTION
For several virus families, significant progress has been made in understanding the molecular events associated with viral entry into host cells. Viral entry steps include stable attachment of the virus to the cell surface, penetration of the virus into the interior of the cell, disassembly of the viral capsid, and activation of the viral genetic program. These steps are essential for the virus to traverse the extracellular environment to the cellular compartment in which viral transcription and replication occur. Viral entry mechanisms also have relevance to viral pathogenesis as these events often determine target cell selection within the host, which dictates the site of virus-induced disease. Moreover, entry steps can induce intracellular signaling cascades that influence whether cells enter into an antiviral state or undergo apoptosis. In this chapter, we consider mechanisms of reovirus cell entry and describe our current understanding about how these entry events initiate proapoptotic signaling.
PATHOGENESIS OF REOVIRUS INFECTION
Members of the Reoviridae family are nonenveloped viruses containing genomes of 9-12 segments of double-stranded (ds) RNA1 (Fig. 1). This family includes mammalian orthoreoviruses (reoviruses), orbiviruses, and rotaviruses. For reoviruses, the viral proteins are designated with a Greek letter corresponding to the size of the encoding genome segment: sigma (σ) for proteins encoded by small genome segments, mu (μ) by medium segments, and lambda (λ) by large segments. Each of the genome segments encodes a single protein with the exception of the S1 gene, which encodes the viral attachment protein, σ1, and a small nonstructural protein, σ1s. Like other members of the Reoviridae, reovirus particles are formed from concentric protein shells. Two such shells exist for reoviruses, called outer capsid and core.1
Figure 1.
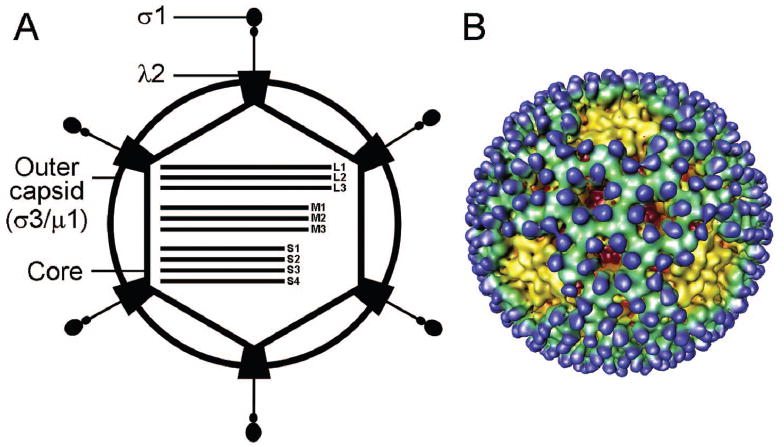
The reovirus virion. A) Schematic of a reovirus virion. Reovirus virions are composed of two concentric protein shells, outer capsid and core. The core contains the viral genome, which consists of 10 segments of double-stranded RNA. B) Cryo-EM image reconstruction of a reovirus virion at 23 Å resolution. Note the finger-like projections of σ3 (blue) that sit atop a layer of μ1 (green). The λ2 protein (yellow) forms a pentamer at each of the virion fivefold symmetry axes. Figure modified from: Nason E et al, J Virol 75:6625-6634; ©2001 with permission from the American Society for Microbiology.74
Reoviruses can infect many mammalian species, including humans, although they are rarely associated with disease.1,2 Three reovirus serotypes have been recognized based on neutralization and hemagglutination profiles. Each is represented by a prototype strain, type 1 Lang (T1L), type 2 Jones (T2J), and type 3 Dearing (T3D), which differ primarily in σ1 sequence.3,4 The pathogenesis of reovirus infections has been most extensively studied using newborn mice, in which serotype-specific patterns of disease have been identified.1,2 The best characterized of these models is reovirus pathogenesis in the murine central nervous system (CNS).
Following oral or intramuscular inoculation of newborn mice, strains of serotype 1 and serotype 3 reoviruses invade the CNS. However, these strains disseminate in the host by different routes and have distinct pathologic consequences. Serotype 1 reovirus spreads to the CNS hematogenously and infects ependymal cells,5,6 resulting in hydrocephalus.7 In contrast, serotype 3 reovirus spreads to the CNS by neural routes and infects neurons,5,6,8 causing lethal encephalitis.7,9 Studies using T1L × T3D reassortant viruses have shown that the pathways of viral spread5 and tropism for neural tissues6,10 segregate with the viral S1 gene. Since the S1 gene encodes attachment protein σ1,11,12 these studies suggest that σ1 dictates the CNS cell types that serve as targets for reovirus infection, presumably by its capacity to bind to receptors expressed by specific CNS cells.
ATTACHMENT RECEPTORS: CELL-SURFACE SIALIC ACID AND JUNCTIONAL ADHESION MOLECULE-A
The σ1 protein is a filamentous, trimeric molecule about 480 Å in length with distinct head-and-tail morphology13,14 (Fig. 2). Independent domains of the protein mediate binding to different types of cell-surface receptors. Sequences in the N-terminal σ1 tail bind to carbohydrate, which is known to be sialic acid in either α2,3 or α2,6 linkages for serotype 3 reoviruses.15-19 The C-terminal σ1 head binds to junctional adhesion molecule-A (JAM-A, previously called JAM or JAM1),20 a member of the immunoglobulin (Ig) superfamily that regulates formation of intercellular tight junctions.21-23 The σ1 tail partially inserts into the virion, while the head projects away from the virion surface.13,24
Figure 2.

Attachment protein σ1. Full-length model of σ1 generated by adding a trimeric α-helical coiled-coil to the N-terminus of the crystallized σ1 fragment.26 The three monomers of the crystallized fragment are shown in red, yellow, and blue; the model is shown in grey. Regions of the molecule that interact with sialic acid and JAM-A are indicated.
The crystal structure of the C-terminal half of T3D σ1 (residues 170-455) reveals a homotrimer with an unusual structural fold25,26 (Fig. 2). N-terminal residues in the crystallized fragment (170-309) form the body domain, which consists of seven β-spiral repeats interrupted by a short stretch of α-helix. β-spiral repeats are also observed in the adenovirus fiber27 and avian reovirus σC.28 C-terminal residues form the compact head domain (310-455), which consists of an 8-stranded β-barrel. Sequence analysis, coupled with the crystallographic data, has facilitated the development of a model of full-length σ125 (Fig. 2). the σ1 tail is predicted to contain ~20 heptad repeats of an N-terminal α-helical coiled-coil.3,4
Both murine (m) and human (h) homologs of JAM-A function as reovirus receptors.20 The crystal structure of the extracellular region of hJAM-A consists of two concatenated immunoglobulin domains (D1, membrane distal and D2, membrane proximal)29 (Fig. 3). Two monomers form a symmetrical dimer that is stabilized by extensive ionic and hydrophobic contacts between the D1 domains. Like the structures of reovirus σ1 and adenovirus fiber, the structures of JAM-A and the coxsackievirus and adenovirus receptor (CAR) are strikingly similar.30 These observations suggest that reovirus and adenovirus use similar mechanisms of attachment. In concordance with this prediction, the σ1 head binds to the membrane-distal D1 domain of monomeric JAM-A,31,32 analogous to the mechanism by which the adenovirus fiber binds to CAR.33,34
Figure 3.
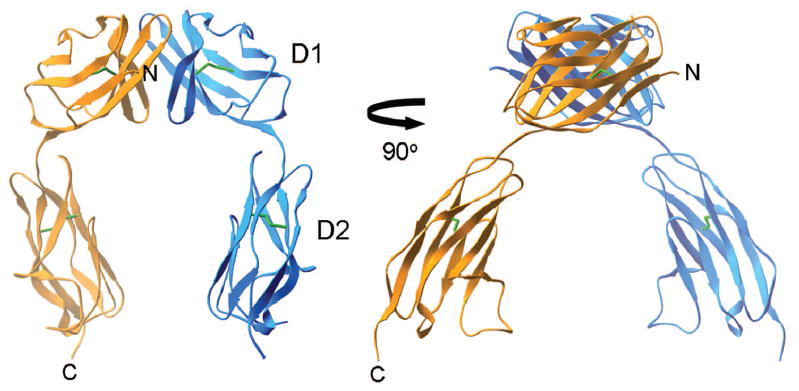
Crystal structure of the JAM-A extracellular region. Ribbon drawings of the hJAM-A dimer, with one monomer shown in yellow and the other in blue. Disulfide bonds are shown in green. The D1 domain is distal to the cell membrane. The two views differ by rotation of 90° along a vertical axis. Figure modified from: Prota AE et al, Proc Natl Acad Sci USA 100:5366-5371; ©2003 with permission from the National Academy of Sciences, USA.29
The presence of discrete receptor-binding domains in σ1 suggests that reoviruses employ a multiple-step binding process similar to that used by some herpesviruses35,36 and retroviruses.37,38 Binding studies using isogenic point-mutant viruses T3SA+ and T3SA-, which vary only in the capacity to engage sialic acid,39 support this hypothesis. Kinetic analyses using inhibitors of sialic acid and JAM-A binding demonstrate that sialic acid is engaged first in the adsorption process, as the inhibitory effect of sialic acid analogs on infection by T3SA+ occurs at early but not late timepoints.39 However, a σ1-specific monoclonal antibody (mAb) that blocks virus binding to JAM-A inhibits viral infectivity at both early and late times during adsorption.39 Thus, reovirus binding to sialic acid enhances virus attachment through rapid adhesion of the virus to the cell surface where access to JAM-A is thermodynamically favored.
INTERNALIZATION RECEPTORS: β1 INTEGRINS
Although engagement of JAM-A is required for high-affinity attachment of reovirus to cells, binding to this receptor does not appear to stimulate viral internalization. Expression of a JAM-A mutant lacking a cytoplasmic tail in nonpermissive cells confers full susceptibility to reovirus infection, suggesting that cell-surface molecules other than JAM-A mediate viral internalization following attachment.40 Outer-capsid protein λ2, which serves as the structural base for σ1,13,24 contains integrin-binding sequences,41 raising the possibility that integrins mediate reovirus endocytosis. Integrins are heterodimeric cell-surface molecules that consist of α and β subunits.42 Integrins function to mediate cellular adhesion to the extracellular matrix, regulate cellular trafficking, and transduce both outside-in and inside-out signaling events.43 Consistent with a role for integrins in reovirus internalization, a β1 integrin-specific antibody, but not antibodies specific for other integrin subunits, inhibits reovirus infection.40 In comparison to isogenic cells expressing β1 integrin, uptake of reovirus into β1-deficient mouse embryonic stem cells is substantially diminished40 (Fig. 4). This defect in uptake is associated with a parallel reduction in infectivity. Additionally, mutations in the NPXY motifs in the cytoplasmic tail of β1 integrin result in mislocalization of virions to lysosomes.44 These data provide strong evidence that β1 integrins facilitate reovirus internalization and suggest that viral entry occurs by interactions of reovirus virions with independent attachment and entry receptors on the cell surface.
Figure 4.
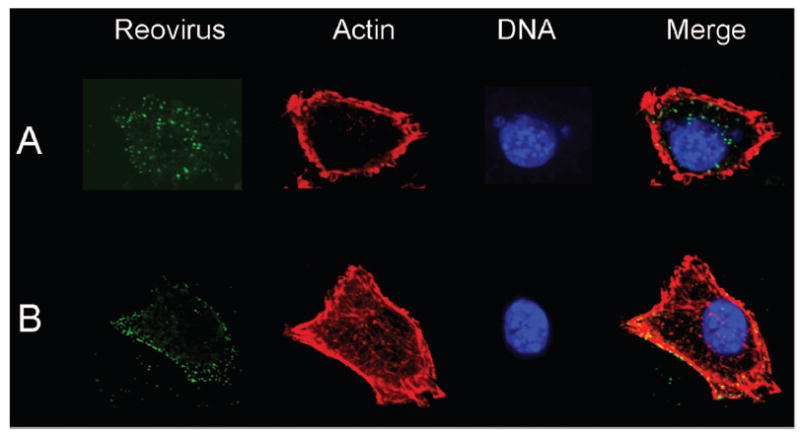
β1 integrin enhances reovirus entry into cells. (A) GD25β1A (β1 +/+) and (B) GD25 (β1 -/-) cells were chilled, adsorbed with T1L virions, and incubated at 4°C for 1 h. Nonadherent virus was removed, warm medium was added, and cells were incubated at 37°C for 30 min. Cells were fixed, stained for reovirus (green), actin (red), and DNA (blue), and imaged using confocal immunofluorescence microscopy. Representative digital fluorescence images of the same field are shown in each row. Figure modified from: Maginnis MS et al, J Virol 80:2760-2770; ©2006 with permission from the american Society for Microbiology.40
STEPWISE DISASSEMBLY IN THE ENDOCYTIC COMPARTMENT
Following attachment to cell-surface receptors, reovirus virions are delivered into the endocytic pathway (Fig. 5). Although conclusive evidence for the mechanism of internalization is lacking, current data support a role for clathrin-dependent endocytosis in reovirus cell entry. Thin-section EM images show virions in structures that resemble clathrin-coated pits on the cell surface and in clathrin-coated vesicles in the cytoplasm,45-48 suggesting clathrin-dependent uptake. Reovirus virions and clathrin colocalize during internalization,49 providing further evidence that reovirus entry is mediated by a mechanism involving clathrin.
Figure 5.
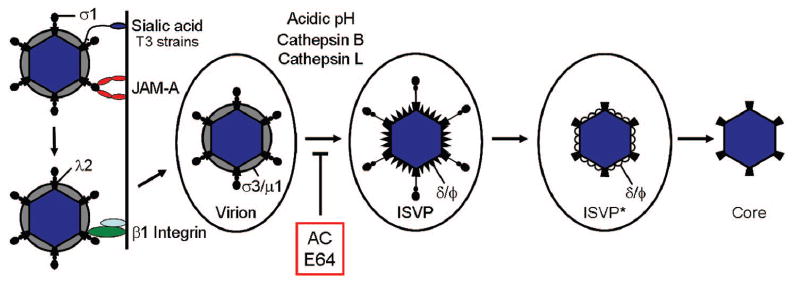
Stepwise disassembly of reovirus. Following attachment to cell-surface carbohydrate (sialic acid for serotype 3 [T3] strains) and JAM-A, reovirus uses β1 integrin to enter cells by receptor-mediated endocytosis. In the endocytic compartment, the viral outer capsid undergoes acid-dependent proteolysis. The first disassembly intermediate is the ISVP, which is characterized by loss of σ3 and cleavage of μ1 into particle-associated fragments δ and ϕ. The ISVP then undergoes further conformational changes to form the ISVP*. The ISVP* is characterized by loss of attachment protein σ1 and conformational rearrangements of the μ1 cleavage fragments to expose hydrophobic residues. The μ1 fragments mediate viral penetration of the endosomal membrane, releasing the transcriptionally active core into the cytoplasm. Treatment of cells with either AC or E64 blocks virion-to-ISVP conversion.
Vesicles containing internalized reovirus virions are transported via microtubules50 and accumulate in late endosomes.45-48,50,51 In the endocytic compartment, reovirus virions undergo stepwise disassembly forming sequential disassembly intermediates, the first of which is the infectious subvirion particle (ISVP) (Fig. 5). ISVPs are characterized by the loss of outer-capsid protein σ3, a conformational change in attachment protein σ1, and cleavage of outer-capsid protein μ1 to form particle-associated fragments, δ and ϕ. Following further processing, ISVP-like particles (called ISVP*s) penetrate endosomal membranes, leading to release of transcriptionally active core particles, which lack μ1 and σ1, into the cytoplasm. Thus, the disassembly process consists of a highly coordinated series of events that are dependent on host cell functions that act upon discrete components of the viral outer capsid.
Insight into mechanisms of reovirus disassembly was first provided by using pharmacologic inhibitors of endosomal acidification and protease function. Treatment of murine L929 (L) cells45,52,53 with the weak base ammonium chloride (AC), which raises the pH of endosomes and lysosomes,54,55 blocks replication of reovirus when infection is initiated with virions. However, ISVPs generated in vitro by treatment of virions with the intestinal serine proteases chymotrypsin or trypsin can infect AC-treated cells.45 This finding indicates that the block to reovirus replication mediated by AC occurs following internalization but prior to disassembly. Similarly, treatment of L cells with E64, an inhibitor of cysteine proteases,56 arrests infection by reovirus virions but not by ISVPs,57-60 suggesting that one or more endocytic cysteine proteases effects reovirus disassembly in host cells.
A CRITICAL ROLE FOR CATHEPSINS IN REOVIRUS ENDOSOMAL DISASSEMBLY
The major cysteine proteases in the endocytic compartment of fibroblasts such as L cells are cathepsins B, H, and L, with cathepsin L being the most abundant in several cell types.56,61-64 Infection of either L cells treated with the cathepsin L inhibitor A-Phe-Tyr(t-Bu)-diazomethyl ketone or cathepsin L-deficient mouse embryo fibroblasts results in inefficient proteolytic disassembly and decreased viral yields. In contrast, L cells treated with the cathepsin B inhibitor CA-074Me and cathepsin B-deficient mouse embryo fibroblasts support reovirus disassembly and growth. However, removal of both cathepsin B and cathepsin L activity completely abrogates disassembly and growth of reovirus. Concordantly, cathepsin L mediates reovirus disassembly more efficiently than cathepsin B in vitro.65 These results demonstrate that either cathepsin L or cathepsin B is required for reovirus entry into murine fibroblasts and indicate that cathepsin L is the primary mediator of reovirus disassembly in these cells. However, proteases other than cathepsin B and cathepsin L are capable of ISVP formation in other types of cells. In P388D cells, a macrophage-like cell line, cathepsin S, an acid-independent cysteine protease,66 mediates uncoating of some strains of reovirus.67 Titers of reovirus in mice lacking cathepsin B, L, or S are decreased at early times post-infection, indicating the importance of these proteases in reovirus replication in vivo.68 However, other proteases in the enteric tract or airway also facilitate reovirus infection.69
OUTER-CAPSID PROTEIN σ3, THE MAJOR TARGET FOR ENDOSOMAL PROTEASE ACTIVITY
The first step in the disassembly of reovirus virions is the proteolytic removal of outer-capsid protein σ3.45,57 The σ3 protein acts as a cap to protect μ1,24 which is the viral protein that mediates membrane penetration.70-72 Reovirus σ3 is a bi-lobed protein with its N-terminus in a virion-proximal smaller lobe bound to μ1 and its C-terminus in a virion-distal larger lobe73,74 (Fig. 6A). Treatment of reovirus virions in vitro with either cathepsin B or cathepsin L leads to an initial cleavage of σ3 most likely near the C-terminus of the protein.65 During proteolysis by cathepsin L, subsequent cleavages occur between residues 243-244 and 250-251,65 which are physically located near the σ3 C-terminus73 (Fig. 6A,B). Because of this proximity, the small end fragment released following initial cathepsin L cleavage likely exposes the other two sites, rendering them sensitive to subsequent cleavage events. The C-terminus therefore appears to act as a “safety latch” that controls access to internal, proteolytically sensitive sites in σ3. Because reovirus disassembly in some cell types is an acid-dependent process, the safety latch might be primed for movement at acidic pH. Numerous reovirus mutants, including those selected during persistent infection (PI viruses)75 and those selected for resistance to either AC (ACA-D viruses)76 or E64 (D-EA viruses)59 have mutations adjacent to the C-terminus of the protein (Fig. 6B). Residues at positions 198 and 354 are particularly important for regulating protease susceptibility.77 Changes at these positions may mediate structural alterations in the safety latch that provide enhanced access to the cleavage sites located more internally in the protein.
Figure 6.
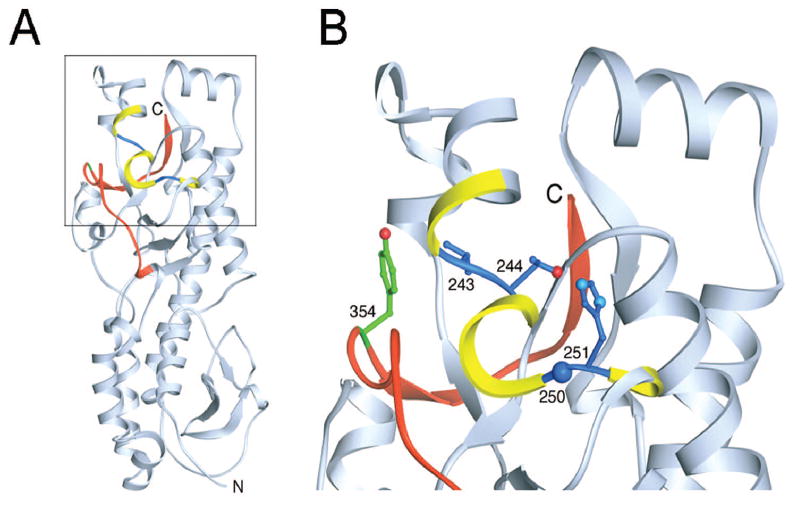
The σ3 protein. A) Ribbon diagram of the crystal structure of T3D σ3.73 The cathepsin L cleavage sites in T1L are depicted in blue between amino acids 243 and 244 and between 250 and 251.65 Surrounding residues, from amino acids 241 to 253, are shown in yellow. The C-terminal residues of σ3, from amino acids 340 to 365, are colored red. Tyrosine 354, which is altered in several PI,75 D-EA,59 and ACA-D viruses,76 is colored green. The virion-distal end of σ3 is at the top of the figure, and the virion proximal end and N-terminus are at the bottom. B) An enlarged view of the boxed region of σ3 indicated in panel B is shown using the same color scheme. Amino acids 243, 244, 250, 251, and 354 are depicted in ball-and-stick representation. Figure and legend modified from: Ebert DH et al, J Biol Chem 277:24609-24617; ©2002 with permission from the American Society for Biochemistry and Molecular Biology.65
MEMBRANE-PENETRATION PROTEIN μ1
The μ1 protein is genetically and biochemically linked to penetration of endosomal membranes by reovirus disassembly intermediates. Most of the μ1 protein on mature virions is autocatalytically cleaved near the N-terminus to generate two fragments, μ1N and μ1C78,79 (Fig. 7). This cleavage is not required for virion assembly80 and may occur physiologically during the transition from the ISVP to the ISVP*.81 In ISVPs, μ1C is further cleaved by either endocytic45,65 or intestinal82 proteases to form fragments δ and ϕ, which remain particle-associated.83 However, the role of this cleavage in viral penetration is not understood, as core particles recoated with mutant forms of μ1 incapable of δ/ϕ cleavage can establish productive infection.84 In addition, μ1 is not cleaved at the δ/ϕ junction in ISVPs generated in the presence of alkyl sulfate detergents (dpSVPs), yet dpSVPs are infectious.58
Figure 7.
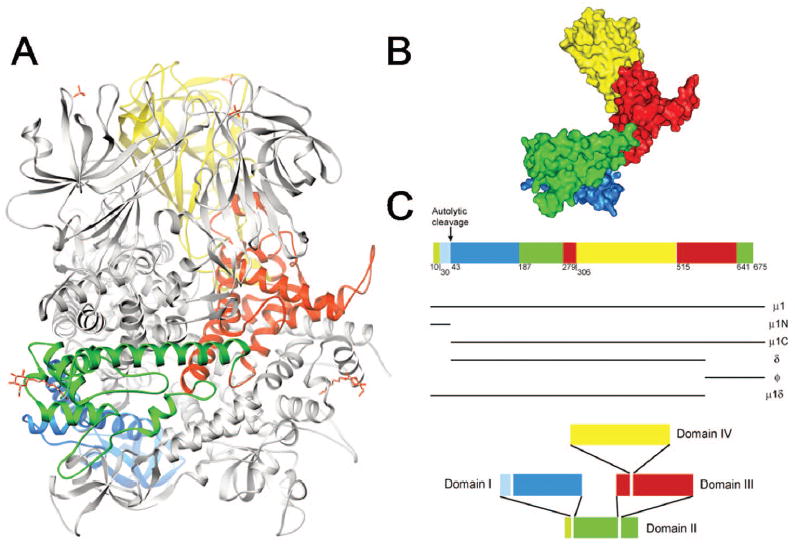
The μ1 protein. A) Ribbon diagram of the crystal structure of the T1L μ1 trimer without bound σ3.88 One μ1 subunit is colored by domain (domain I, light and dark blue [μ1N, μ1C]; domain II, light and dark green [μ1N, μ1C]; domain III, red; domain IV, yellow); the other two μ1 subunits are shown in gray. β-octyl glucosides and sulfate ions present in the structure are shown in red and yellow. B) Surface-shaded representation of an isolated μ1 subunit. Colors and orientation are as in (A). C) Domain segmentation of the amino acid sequence as determined from the three-dimensional structure. Domain color code as in (A) and (B). The central domain II contains domains I and III as “inserts,” and domain III similarly contains domain IV. Figure and legend modified from: Liemann S et al, Cell 108:283-295; ©2002 with permission from Elsevier.88
Transformation from the ISVP to the ISVP* in vitro is triggered by differential cationic concentration or interactions with membranes.85,87 In contrast to ISVPs, ISVP*s lack σ1 and have an altered conformer of μ1 in which internal hydrophobic residues are exposed. ISVP*s are capable of membrane penetration and transcription initiation.85,87 The conformational change in μ1 may be the driving force for both the loss of σ1 and the initiation of transcription.88 Mechanisms underlying these events are unknown, but it is possible that μ1 rearrangement induces a conformational change in λ2, the pentameric turret that anchors σ1, causing σ1 release.
Cleavage of intact μ1 to form μ1N and μ1C is required for the generation of the ISVP*.80,81 Particles recoated with mutant forms of μ1 incapable of μ1N/μ1C cleavage can facilitate each of the entry steps, including μ1 conformational changes and transcription initiation, but are deficient in membrane penetration.80 In addition to σ1, the N-terminal μ1 fragment μ1N is released from the ISVP* and forms membrane pores that recruit virus particles.85,86 The ϕ fragment may act as a μ1N chaperone. However, the full nature of the membrane-penetration complex is not completely understood.
The conformational changes in μ1 that accompany viral disassembly are thought to expose internal hydrophobic residues and release μ1N from the particle as a consequence of massive rearrangement in the μ1 structure.80,85,87,88 The cleavage of μ1 to form μ1N and μ1C is necessary for productive infection.80 However, it is not clear whether membrane penetration is accomplished by soluble or particle-associated μ1N, perhaps acting in concert with other regions of the molecule. For example, an anion-binding site in domain IV lies on the outermost, solvent-exposed surface of the ISVP88 (Fig. 7). This site may bind to phospholipid head groups bringing the virus particle into proximity with the endosomal membrane. This association also might trigger rearrangements in μ1 revealing the myristoylated μ1N and the internal hydrophobic residues.
VIRAL DETERMINANTS OF APOPTOSIS INDUCTION BY REOVIRUS
Reovirus elicits the morphological and biochemical features of apoptosis in both cultured cells89-91 and in the murine CNS92,93 and heart.93-95 Insights into mechanisms by which reovirus induces apoptosis first emerged from studies of strain-specific differences in the efficiency of apoptosis induction. Reovirus strain T3D induces apoptosis to a much greater extent than T1L in a variety of cell types.89,90,96 Experiments using T1L × T3D reassortant viruses implicated the S1 and M2 genes as the key determinants of differences in the capacity of reovirus strains to induce apoptosis in L cells89 and Madin-Darby canine kidney (MDCK) cells.90 Linear regression analysis of these data pointed to a primary role for the S1 gene in apoptosis induction with a minor contribution from the M2 gene. These findings were corroborated by studies analyzing genetic determinants of differences in apoptosis efficiency displayed by T1L and another serotype 3 strain, type 3 Abney (T3A).97 Since no other viral gene segments are significantly associated with differences in apoptosis induction by serotype 1 and serotype 3 reovirus strains, these studies collectively pinpoint important functions for the S1 and M2 genes in the induction of this cellular response.
The S1 gene encodes viral attachment protein σ1 and nonstructural protein σ1s from distinct but overlapping reading frames.98-100 The role of both proteins in apoptosis induction has been investigated. Reovirus strains T3C84 and T3C84-MA, which differ in the expression of σ1s,101 do not differ in apoptosis-inducing capacity following infection of L cells or MDCK cells,101 indicating that the σ1s protein is not required for apoptosis induction following infection of cultured cells. In contrast to those in vitro results, σ1s appears to influence the kinetics and severity of apoptosis induction in both the heart and CNS of infected mice.102 However, since the viral strains compared in that study were not isogenic, it is not possible to exclude the involvement of other viral determinants in the observed differences in viral pathology. Studies using σ1s-null viruses recovered by reverse genetics identified an essential role for σ1s in establishing viremia and promoting viral dissemination to sites of secondary replication.103,104 However, the precise role of σ1s in apoptosis induction in vivo remains unknown.
The σ1 protein binds to two cell-surface receptors, the proteinaceous receptor JAM-A20 and a carbohydrate receptor. Strains of all three reovirus serotypes bind to JAM-A,20,29,31,105 but only serotype 3 strains bind to sialic acid.16,18,106 Sialic-acid-binding strain T3SA+ induces significantly higher levels of apoptosis than isogenic nonsialic-acid-binding strain T3SA-.96 Consistent with these findings, removal of cell-surface sialic acid with neuraminidase, or blockade of virus binding to cell-surface sialic acid using a soluble competitor, sialyllactose, abolishes the capacity of T3SA+ to induce apoptosis.96 These data indicate that the capacity to bind to sialic acid enhances the efficiency of apoptosis induced by reovirus infection. However, sialic acid binding is not sufficient to induce apoptosis. Blockade of σ1 binding to JAM-A by using either σ1- or JAM-A-specific mAbs also diminishes the apoptosis-inducing capacity of sialic-acid-binding reoviruses.20,89 Thus, attachment of σ1 to both sialic acid and JAM-A is required for efficient induction of apoptosis.
Despite the role of the reovirus receptors in influencing apoptosis efficiency, receptor binding alone is not sufficient for apoptosis induction. Blockade of viral disassembly using either AC or E64 diminishes reovirus-induced apoptosis107 (Fig. 8A). On the other hand, inhibition of de novo viral RNA synthesis using ribavirin does not affect apoptosis induced by reovirus107 (Fig. 8B). Thus, in addition to sialic acid- and JAM-A-mediated attachment of reovirus to cells, replication steps that occur during or after viral disassembly but before the cytoplasmically delivered core becomes transcriptionally active also contribute to reovirus-induced apoptosis. Since the M2-encoded μ1 protein functions in virus-induced endosomal membrane penetration following disassembly but prior to synthesis of viral RNA,80,83,88 the deleterious effects of reovirus disassembly inhibitors on apoptosis induction suggest a functional link between the M2 gene and differences in the efficiency of apoptosis exhibited by different reovirus strains in previous genetic studies.89,90,97
Figure 8.
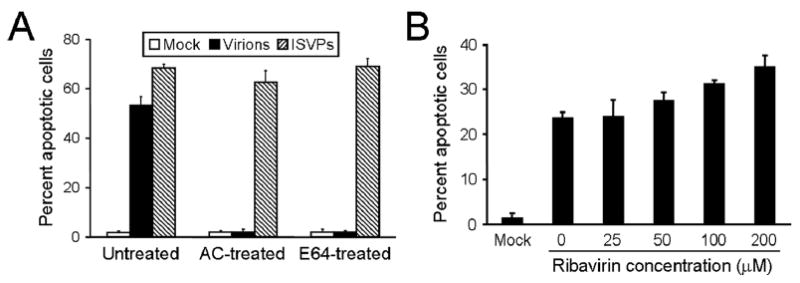
Reovirus-induced apoptosis in cells treated with inhibitors of viral replication. A) HeLa cells were either mock infected or infected with T3SA+ virions or ISVPs at an MOI of 100 PFU/cell and incubated in the absence or presence of 10 mM AC or 200 μM E64. B) HeLa cells were either mock infected or infected with T3SA+ at an MOI of 1,000 PFU/cell prior to incubation in the absence or presence of ribavirin at the concentrations shown. Mock-infected cells were incubated in the presence of 200 μM ribavirin. After incubation for 48 h (A and B), cells were stained with acridine orange. The results are expressed as the mean percentage of cells undergoing apoptosis for three independent experiments. Error bars indicate SD. Figure modified from: Connolly JL, Dermody TS, J Virol 76:1632-1641; ©2002 with permission from the American Society for Microbiology.107
Further evidence for a role of the M2-encoded μ1 protein in apoptosis induction was gathered in a study in which productive reovirus infection was initiated in JAM-A-negative, Fc receptor-expressing Chinese hamster ovary cells (CHO-B1) using reovirus preincubated with capsid-specific mAbs.108 Fc mediated infection of CHO-B1 cells was found to induce apoptosis in an antibody dose-dependent manner108 (Fig. 9). Furthermore, antibody-directed binding of reovirus to Fc receptors expressed on CHO-B1 cells was not sufficient for reovirus-induced apoptosis. Analogous to apoptosis initiated following uptake via cognate receptors, apoptosis induced following the Fc-receptor dependent pathway is sensitive to inhibitors of viral disassembly but is not perturbed by inhibitors of viral replication.108 These data suggest that regardless of the receptor used to initiate infection, reovirus-induced apoptosis requires events that occur during or after viral disassembly but prior to viral RNA synthesis. Uptake via Fc receptors also allows nonsialic acid-binding reovirus strains to efficiently induce apoptosis,108 suggesting that σ1-mediated differences in the efficiency of apoptosis induction are eliminated following Fc-receptor mediated uptake. Collectively, these findings suggest that signaling pathways initiated as a result of ligation of σ1 to sialic acid and JAM-A are dispensable for reovirus-induced apoptosis.
Figure 9.
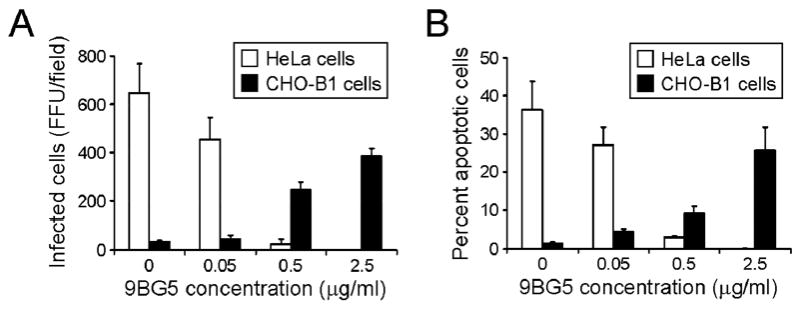
JAM-A-independent uptake of reovirus via Fc receptors leads to apoptosis. A) Reovirus T3D virions were incubated overnight with the indicated concentration of mAb 9BG5 and adsorbed to either HeLa cells or CHO-B1 cells at an MOI of 100 PFU/cell. After incubation at 37°C for 18 h, cells were fixed using methanol. Infected cells were visualized by immunostaining with polyclonal rabbit anti-reovirus serum. Reovirus-infected cells were quantified by counting fluorescent cells. The results are presented as mean fluorescent focus units (FFU)/field. B) HeLa cells or CHO-B1 cells were adsorbed with 100 PFU/cell of either virus or virus-antibody complex, harvested 48 h after infection, and stained with acridine orange. The results are expressed as the mean percentage of cells undergoing apoptosis for three independent experiments. Error bars indicate SD. Figure modified from: Danthi P et al, J Virol 80:1261-1270; ©2006 with permission from the American Society for Microbiology.108
Differences in the capacity of T1L × T3D reassortant viruses to induce apoptosis following Fc-mediated uptake segregate strictly with the μ1-encoding M2 gene,108 providing further support for a role of the μ1 protein in apoptosis induction. In addition, temperature-sensitive reovirus mutant tsA279.64, which bears a lesion in the M2 gene,109 is defective in apoptosis induction108 (Fig. 10). Particles of this virus strain when grown at nonpermissive temperature contain a misfolded, membrane penetration-defective μ1 protein.109 In comparison to particles assembled at permissive temperature, those assembled at nonpermissive temperature are less efficient inducers of apoptosis. Moreover, reovirus μ1 mutants recovered by reverse genetics induce apoptosis less efficiently than does wild-type virus.110,111 These findings suggest that reovirus membrane-penetration protein μ1 induces proapoptotic signaling events during or after endosomal membrane penetration.
Figure 10.
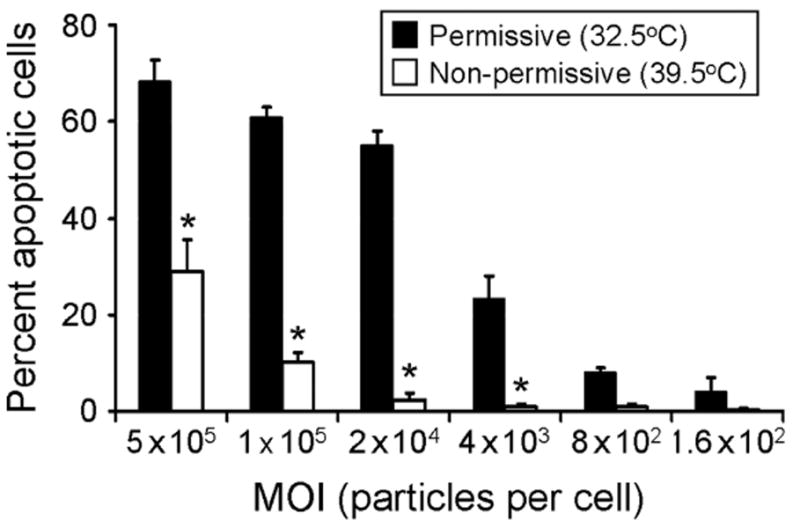
Apoptosis induced by temperature-sensitive μ1 mutant virus tsA279.64. HeLa cells were adsorbed with tsA279.64 grown at permissive or nonpermissive temperatures at the MOIs shown. After incubation at 37°C for 48 h, cells were harvested and stained with acridine orange. The results are expressed as the mean percentage of cells undergoing apoptosis for three independent experiments. Error bars indicate SD. *, P < 0.05 by Student’s t-test relative to virions grown at permissive temperature at an equivalent MOI. Figure modified from: Danthi P et al, J Virol 80:1261-1270; ©2006 with permission from the American Society for Microbiology.108
SIGNALING PATHWAYS ACTIVATED BY REOVIRUS INFECTION
NF-κB
A critical component of the intracellular signal transduction apparatus activated following reovirus infection is nuclear factor-κB (NF-κB), a family of structurally related transcription factors that play important roles in cell growth and survival. Reovirus activates NF-κB in cell culture beginning at 2-4 h post infection and achieves maximal levels of activation at 8-10 h post-infection91 (Fig. 11A). Electrophoretic mobility shift assays using antisera specific for p50 and p65 identified both of these subunits in the NF-κB complexes activated during reovirus infection (Fig. 11B). Concordantly, cells devoid of either p50 or p65 do not activate NF-κB following reovirus infection,91 supporting the involvement of these NF-κB subunits in the complexes activated by reovirus. A second phase of NF-κB regulation occurs in some cell types following viral RNA synthesis and involves downregulation of NF-κB signaling through a mechanism dependent on the inhibition of IκBα degradation.112 This sophisticated manipulation of a central cell fate-determining transcription factor emphasizes the importance of NF-κB activation status in the reovirus replication cycle.
Figure 11.
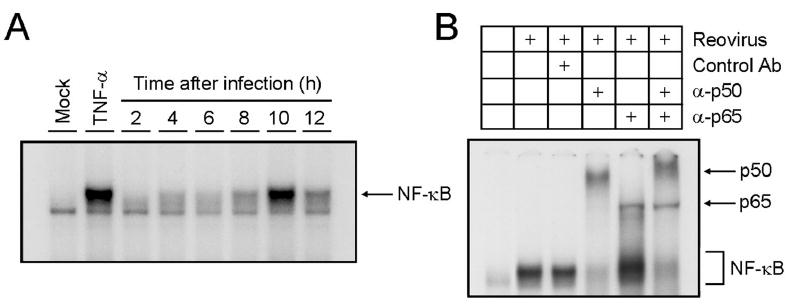
Reovirus activates NF-κB. A) NF-κB DNA binding activity following reovirus infection. HeLa cells were either mock-infected or infected with T3D at an MOI of 100 PFU/cell and incubated at 37°C for the times shown. Uninfected cells also were treated with 20 ng/ml TNF-α for 1 h. Nuclear extracts were prepared and incubated with a [32P]-labeled oligonucleotide consisting of the NF-κB consensus binding sequence. Incubation mixtures were resolved by acrylamide gel electrophoresis, dried, and exposed to film. NF-κB containing complexes are indicated. B) NF-κB complexes activated by reovirus contain p50 and p65/RelA subunits. Nuclear extracts were prepared 10 h after viral adsorption. Extracts were incubated with no antibody, a control antibody, p50-specific antiserum (α-p50), p65-specific antiserum (α-p65), or both p50- and p65-specific antisera. NF-κB complexes unaffected by antibody and complexes demonstrating retarded mobility with antibodies to p50 or p65 are indicated. Figure modified from: Connolly JL et al, J Virol 74:2981-2989; ©2000 with permission from the American Society for Microbiology.91
Mechanisms leading to NF-κB activation following reovirus infection are not completely understood. The efficiency with which reovirus activates NF-κB is influenced by viral attachment to both sialic acid96 and JAM-A.39 Viral disassembly also is required for NF-κB activation, but subsequent events in viral replication are dispensable.107 This finding suggests that replication steps following formation of ISVPs but before commencement of RNA synthesis are responsible for activating NF-κB. Intriguingly, NF-κB activation following reovirus infection occurs over a much longer time course than that elicited by other NF-κB agonists such as TNF-α,113 suggesting that the viral agonist is constitutively active, similar to Epstein-Barr virus latent membrane protein 1114 or human T-cell leukemia virus Tax.115
NF-κB activation can either potentiate116-118 or inhibit apoptosis119-121 depending on the nature of the NF-κB agonist. Three independent lines of evidence suggest that activation of NF-κB by reovirus is proapoptotic in cultured cells.91 First, treatment of HeLa cells with proteasome inhibitors to block NF-κB activation following reovirus infection substantially diminishes reovirus-induced apoptosis. Second, transient expression of a dominant-negative form of IκB that constitutively represses NF-κB activation significantly reduces levels of apoptosis induced by reovirus. Third, apoptosis by reovirus is substantially diminished in mouse embryonic fibroblasts (MEFs) lacking either of the NF-κB subunits p50 or p65 (Fig. 12). Together, these data indicate that NF-κB plays an essential role in the mechanism by which reovirus induces apoptosis of host cells.
Figure 12.
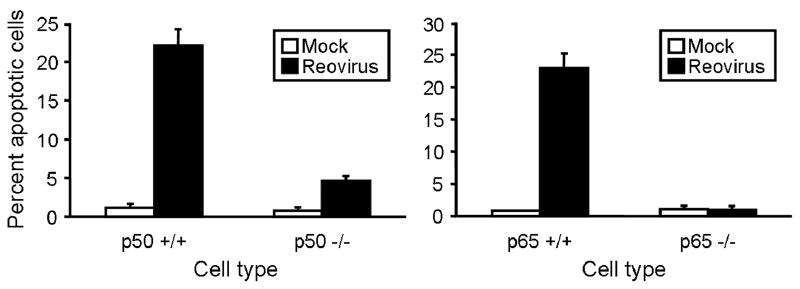
Apoptosis induced by reovirus is diminished in cells lacking NF-κB subunits p50 and p65. Cells were either mock-infected or infected with T3D at an MOI of 100 PFU/cell. After incubation at 37°C for 48 h, cells were stained with acridine orange. The results are expressed as the mean percentage of cells undergoing apoptosis for three independent experiments. Error bars indicate SD. Figure modified from: Connolly JL et al, J Virol 74:2981-2989; ©2000 with permission from the American Society for Microbiology.91
MAPKs
Mitogen-activated protein kinases (MAPKs) are important signal transducers that respond to a wide variety of stimuli. Several MAPKs transduce signals initiated by reovirus infection. Reovirus activates c-Jun NH2-terminal kinase (JNK) by 10-12 h post-infection, and this activation is sustained for at least 20-30 h.122 Strain-specific differences in the capacity of reovirus to activate JNK and its downstream effector c-Jun correlate with the capacity of those strains to induce apoptosis, suggesting that JNK activation is required for apoptotic signaling. Additionally, cells lacking MEK kinase 1 (MEKK1), an upstream activator of JNK, or engineered to express a kinase-inactive form of MEKK1 do not phosphorylate JNK or undergo apoptosis in response to reovirus infection.123 Pharmacologic inhibitors of JNK inhibit reovirus-induced apoptosis123 but do not block reovirus growth,123,124 indicating that JNK activity is not required for reovirus replication. Interestingly, although JNK phosphorylates and activates c-Jun in response to reovirus infection, c-Jun activation is not required for apoptosis. These data indicate that JNK contributes to apoptosis induction via a mechanism independent of its activation of c-Jun,123 possibly through its effect on mitochondrial signaling pathways.
The p38 MAPK is activated by reovirus between 4-8 h post-infection; 125 however, p38 becomes downregulated at late times (24-48 h) post-infection.122 Pharmacologic inhibitors of p38 MAPK reduce reovirus growth in cells that express an activated Ras pathway, indicating that this pathway is important for replication of the virus.124 Inhibition of p38 also blocks reovirus-induced secretion of the proinflammatory cytokines IL-1β and TNF-α.125 However, pharmacologic inhibitors of p38 have no effect on reovirus-induced apoptosis,122 indicating that this pathway is distinct from NF-κB mediated death signaling during reovirus infection.91 Finally, although reovirus infection activates the MAPK extracellular signal-regulated kinase (ERK) at early (10-30 min) and late (24 h) times post-infection, pharmacological inhibitors of ERK do not inhibit reovirus-induced apoptosis.122 The role of ERK activation in reovirus replication is unknown.
Ras
Reovirus replication is potentiated in transformed cells containing an activated Ras pathway.126,127 Expression of constitutively active signal transducers in the Ras pathway, including the vErbB oncogene and v-Ras-h, render nonpermissive cells permissive for reovirus replication.128,129 This activity is mediated by the small G protein Ral and its guanine nucleotide exchange factors (RalGEFs) and p38 MAPK.124 Activated Ras also can inhibit the antiviral protein kinase R (PKR),130 which blocks protein synthesis and inhibits reovirus growth.127 These observations suggest that the increased yield of reovirus following infection of transformed cells is due in part to suppression of PKR. The capacity of reovirus to preferentially infect and kill transformed cells has led to several studies evaluating its potential as an oncolytic agent.129,131-133
IRF-3
Innate immune responses are stimulated following reovirus infection, leading to the activation of transcription factor IRF-3 and the subsequent induction of a type I interferon (IFN) response. Reovirus activates IRF-3 in HeLa cells by 2-4 h post-infection and stimulates maximal transcription factor activity by 8-12 h.134 IRF-3 activation depends on the cellular dsRNa sensors RIG-I, Mda-5, and IPS-1.134-136 Importantly, decreased expression of RIG-I or IPS-1 by RNA interference does not inhibit NF-κB activation,134 suggesting that reovirus engages the NF-κB pathway via an alternative mechanism. IRF-3 is required for efficient induction of apoptosis in reovirus-infected cells and elaboration of IFN-β in cultured cells and in vivo.134 However, signals emanating from the type I interferon receptor are dispensable for apoptosis induction, indicating that cell death is mediated through an IRF-3-dependent, interferon-independent mechanism.137
CELLULAR GENE EXPRESSION PROFILES FOLLOWING REOVIRUS INFECTION
The role of transcription factors such as NF-κB and c-Jun/AP1 in the inductive proapoptotic signaling pathway elicited by reovirus led several groups to investigate cellular genes regulated by reovirus using oligonucleotide microarrays.138-140 Strikingly, these studies identified few classical proapoptotic Bcl-2 family members (such as Bid, Bax, or Bak) or components of death receptor-mediated pathways (such as Fas, Fas ligand, or FADD) upregulated in response to reovirus infection. Additionally, few proapoptotic genes regulated by reovirus were noted to be NF-κB dependent, indicating that the cell-death response downstream of NF-κB may represent secondary or tertiary events initiated by NF-κB activation at early times post-infection.139 Despite the lack of apoptosis effectors identified in the microarray studies, three major functional categories of genes that may influence apoptotic processes stand out as significantly regulated in response to reovirus infection: the DNA damage response, the endoplasmic reticulum (ER) stress response, and the host innate antiviral immune response. Analysis of these gene networks may provide clues about the mechanisms by which inductive signaling events elicited by reovirus lead to a widespread apoptotic response in host cells.
DNA damage response genes were found to be significantly downregulated by reovirus infection in two microarray studies.138,139 Reovirus-regulated DNA damage response genes include damage-specific DNA binding protein 2 (DDB2), excision repair cross-complementing group 4 (ERCC4), and fusion involved in t(12; 16) in malignant liposarcoma (FUS). The physiological role of this response in viral replication or cell death is unknown. However, interactions between the NF-κB pathway and the proapoptotic tumor suppressor protein p53 pathway141,142 may link these responses in reovirus-infected cells. In support of this hypothesis, the NF-κB dependent genes PLK3143 and IER3 (IEX-1128), both of which are induced by reovirus infection as rapidly as 2 h post-infection,139 can regulate p53-mediated apoptosis.
Genes involved in the ER stress response are also regulated following reovirus infection. These include growth and DNA-damage-inducible 45 α and β (GADD45α and GADD45β) and heat shock protein 70 (Hsp70), which were identified in all three microarray studies.138-140 Interestingly, expression of those genes was significantly greater following infection with reovirus strains that potently inhibit host-cell protein synthesis (T3C8 and T3C87) in comparison to a strain that does not (T3D), suggesting that the ER stress response may be potentiated by mechanisms of translation inhibition.140 One functional effect of ER stress is the activation of PERK,145 a kinase that phosphorylates translation initiation factor eIF2α to inhibit translation.146 Despite inhibition of cellular protein synthesis, reovirus replication is enhanced in the presence of PERK, suggesting that reovirus may benefit from the ER stress response, perhaps by preferentially allowing synthesis of viral proteins.140
A large number of genes regulated by reovirus infection identified in previous microarray studies belong to the cellular innate immune response to viral infection, particularly IFN-stimulated genes (ISGs) upregulated downstream of NF-κB dependent production of type I IFN. Secreted type I IFNs activate the Janus-activated kinase (JAK)-signal transducer and activator of transcription (STAT) signal-transduction pathway leading to transcription of ISGs via a STAT1/STAT2/IRF9 complex, known as ISGF3.147,148 ISGs induce an antiviral state in uninfected cells through a variety of mechanisms including inhibition of protein synthesis.149 type I IFNs also greatly sensitize tissue-culture cells to apoptosis in response to dsRNA and influenza virus.150 Reovirus infection upregulates STAT1, STAT 2, and many of the classical ISGs, including 2’5’ oligoadenylate synthase (OAS1), double-stranded RNA-activated protein kinase (PKR), ISG15, and myxoma resistance 1 and 2 (M×1/M×2) (Table 1). The type I IFN response is significantly enhanced following infection with two strains that are potent apoptosis inducers (T3D and T3C87) in comparison to a strain that is not (T3C8).140 However, apoptosis following reovirus infection does not require type I IFN signaling,137 suggesting that other mechanisms dependent on IRF-3 and NF-κB are responsible.
Table 1.
Interferon-inducible genes regulated by reovirus infection in 2 out of 3 microarray analysesa
| Gene Symbol- Human (Mouse)b | Gene Name/Alternate Designationc | Reference
|
||
|---|---|---|---|---|
| DeBiasi et al138 | O’Donnell et al139 | Smith et al140 | ||
| CXCL10 | Interferon-inducible cytokine IP-10, IP-10 | x | x | x |
| CXCL11 | Interferon-induced T-cell chemoattractant, I-TAC | x | x | |
| GBP1 | Guanylate binding protein 1, interferon-inducible, 67kDa | x | x | |
| HSPA1A | Heat shock protein 70kD 1A | x | x | |
| HSPA1B | Heat shock protein 70kD 1B | x | x | |
| IFI16 (Ifi204) | Interferon, gamma-inducible protein 16 | x | x | x |
| IFI35 | Interferon-induced protein 35 | x | x | x |
| IFI44 | Microtubule associated protein, 44 kDa, MTAp44 | x | x | |
| IFIT1 | IFI-56k | x | x | x |
| IFIT2 | IFI-54k | x | x | x |
| IFIT3 | Interferon-induced protein with tetratricopeptide repeats 3 | x | x | x |
| IFIT5 | Interferon-induced protein with tetratricopeptide repeats 5 | x | x | |
| IFITM1 | 9-27 | x | x | |
| IFITM3 | 1-8U | x | x | |
| IFNB1 | Interferon-β | x | x | |
| IRF1 | Interferon regulatory factor 1 | x | x | |
| IRF7 | Interferon regulatory factor 7 | x | x | x |
| ISG15 | ISG15 ubiquitin-like modifier, G1P2 | x | x | x |
| ISG20 | Interferon stimulated exonuclease gene, 20kDa | x | x | |
| ISGF3G | Interferon regulatory factor 9, IRF9 | x | x | x |
| MX2 | Myxovirus resistance 2 | x | x | x |
| OAS1 | 2′,5′-oligoadenylate synthetase 1, 40/46kDa | x | x | |
| OAS2 | 2′,5′-oligoadenylate synthetase-like, 59kDa | x | x | |
| PLAUR | Plasminogen activator, urokinase receptor, CD87 | x | x | |
| PSMB9 | Proteasome subunit, beta type 9 | x | x | |
| RSAD2 | Radical S-adenosyl methionine domain containing 2 | x | x | |
| SP100 | SP100 nuclear antigen | x | x | |
| SP110 (5830484A20Rik) | IFI41, SP110 nuclear body protein | x | x | |
| TAP1 | Transporter 1, ATP-binding cassette, sub-family B | x | x | x |
) Genes were identified as reovirus-regulated and interferon-inducible by the criteria stated in the respective reference.
) Gene symbol is given for human genes. If the symbol differs between human and mouse, the mouse symbol is given in parentheses.
) If different from gene symbol, the full name or common designation is given.
Several proteins involved in the intrinsic, mitochondrial-dependent apoptosis pathway are also regulated by reovirus infection,138,139 including MCL1,151 PAWR (Par-4152), BNIP3L,153 and MOAP1.154 These genes are NF-κB independent, suggesting that they may function in concert with, or in parallel to, NF-κB dependent genes to modulate the apoptotic response in reovirus-infected cells. Proteins in the extrinsic, death receptor-mediated apoptosis pathway regulated by reovirus infection as determined by either microarray or RT-PCR include TNF-related apoptosis-inducing ligand (TRAIL), death receptors DR4, DR5, and DR6, and the Fas death domain-associated protein DAXX (refs. 140, 155 and O’Donnell, Holm, and Dermody, unpublished). While TRAIL and DR5 have been shown to be involved in reovirus-induced apoptosis,155 upregulation of both of these genes was not observed in the same microarray study, indicating that either these genes are under differential temporal regulation or endogenous mRNA or protein levels are sufficient to bring about an apoptotic response. DAXX is a particularly intriguing candidate, as it is a multifunctional protein that associates with the Fas death domain to potentiate Fas-induced apoptosis.156 DAXX also joins with two other IFN-induced proteins upregulated by reovirus, PML and Sp100, to direct stress responses in the nucleus.157 Together, these discovery-based approaches have provided a number of potential proapoptotic candidates that warrant further examination.
Effectors of Reovirus-Induced Apoptosis
Apoptosis induced by reovirus requires both extrinsic death receptor pathways and mitochondrial damage. Activation of the extrinsic pathway following reovirus infection is mediated by TRAIL and its receptors DR4 and DR5.155 TRAIL is released from cells following reovirus infection with maximal levels detected at 48 h post-infection. DR5 is upregulated in response to reovirus beginning at 4 h and increases to maximal levels at 24 h post-infection.155 Treatment of cells with soluble TRAIL receptors or TRAIL-specific antibodies decreases apoptosis following reovirus infection of HEK293 cells155 and neuroblastoma cells.158 Death receptors engaged by TRAIL mediate apoptotic signaling through the initiator caspase, caspase 8,159which is activated following reovirus infection.160 Expression of a dominant-negative FADD mutant, which blocks the capacity of caspase 8 to engage death receptors,161 decreases reovirus-induced activation of caspase 3,155 which serves as the executioner caspase.159 Mechanisms by which reovirus upregulates the release of TRAIL and DR5 expression are not fully understood. Maximal levels of TRAIL are detected following NF-κB activation, suggesting that TRAIL release is mediated by NF-κB.91,155 However, TRAIL sensitivity is potentiated by blockade of NF-κB activation observed later in infection,112 which might be mediated by downregulation of the antiapoptotic protein, cellular FLICE (caspase 8) inhibitory protein (cFLIP).162 TRAIL and death receptor expression are regulated by type I IFN,163,164 providing further support for the hypothesis that extrinsic apoptotic pathways initiated by reovirus infection are activated downstream of type I IFN action.
Reovirus infection also activates the intrinsic apoptosis pathway, which is initiated by mitochondrial injury and release of cytochrome c and Smac/DIABLO.165-168 Release of these mediators leads to activation of caspase 9, which in turn activates caspase 3.166,167,169 Smac/DIABLO can be detected in the cytoplasm of HEK293 cells approximately 4 h following reovirus infection,170 which is subsequent to the activation of NF-κB.91 Identification of Noxa as an NF-κB and IRF-3 dependent protein that is upregulated following reovirus infection and required for efficient apoptosis induction provides a mechanistic link between the initial signaling events and the intrinsic, mitochondrial-dependent apoptotic pathways.137 These findings suggest a direct connection between initial signaling events and mitochondrial damage. Another link could be Bid, a proapoptotic Bcl-2 family member that, following an activating cleavage event, translocates to mitochondria and mediates cytochrome c release and activation of caspase 9.169,172 Bid cleavage can be detected at approximately 10 h post infection of HEK293 cells or 24 h post infection in murine fibroblasts.160,170,171 Bid is required for apoptosis induction following reovirus infection and potentiates reovirus disease in the central nervous system of newborn mice.171 Together, these data suggest that mitochondrial factors and the overall stability of mitochondria play important contributing roles in the induction of apoptosis by reovirus.
REOVIRUS-INDUCED APOPTOSIS IN VIVO
Determinants of reovirus apoptosis in vivo show some similarity to those observed in cell-culture models, as well as some surprising differences. Markers of apoptosis, such as caspase 3 activation and cleavage of cellular DNA, are detected in both the brain and heart of newborn mice infected with reovirus.92-94 Reovirus induces apoptosis in neurons following either intracranial92 or peroral93 inoculation. Differences in the capacity of reovirus strains to induce encephalitis are associated with the capacity of these strains to induce neuronal apoptosis.173 Moreover, as judged from studies using primary neuronal cultures, apoptosis induction may enhance viral replication in those tissues.173 Accordingly, inhibition of neuronal apoptosis with minocycline delays reovirus encephalitis and reduces virus growth following intracranial inoculation.174 Similar to its role in cell-culture models, NF-κB is required for apoptosis of neurons following reovirus infection in vivo, as mice deficient in the NF-κB p50 subunit are protected from neuronal injury.93
In keeping with the pathogenesis of reovirus encephalitis, myocarditis following reovirus infection also is mediated by apoptosis, which can be attenuated by pharmacologic inhibitors of either caspases95 or calpain.94 The innate antiviral immune response plays a key role in determining the extent of myocardial injury following reovirus infection. Nonmyocarditic reovirus strains induce higher levels of type IIFN and are more sensitive to its effects than myocarditic strains.175 Additionally, the ISGs IRF-1 and PKR play protective roles in reovirus myocarditis.176,177 Type IIFN production in the heart or in cardiac myocyte cultures is downstream of both IRF-3178 and NF-κB.93 Intriguingly, the lack of NF-κB p50 markedly enhances reovirus myocarditis, but this disease manifestation can be attenuated by treatment with IFN-β.93 These findings demonstrate a tissue-specific role for NF-κB following reovirus infection in vivo: NF-κB is required for reovirus-induced apoptosis in the CNS, whereas it protects the heart from viral damage via activation of type I IFN.
CONCLUSION AND FUTURE DIRECTIONS
Despite the accumulated knowledge about reovirus attachment to cell-surface receptors and internalization into host cells, a precise understanding of the role of the viral attachment and internalization receptors in reovirus disease is not available. Serotype 1 and serotype 3 reovirus strains vary in the types of cell-surface carbohydrate used as coreceptors,19 but both serotypes bind to JAM-A.20,105 These observations make it unlikely that JAM-A is the sole determinant of reovirus tropism. It is possible that JAM-A serves as a serotype-independent reovirus receptor at some sites within the host and other as yet unidentified receptors, perhaps carbohydrate in nature, confer serotype-dependent differences in growth in other tissues. It is also possible that reovirus serotypes engage JAM-A with different affinities, which influences tissue tropism in infected animals.
Little is known about the signaling events that are initiated by the binding of reovirus to its cell-surface receptors. JAM-A contains a cytoplasmic domain that is approximately 45 amino acids in length, includes 13 potential phosphorylation sites, and interacts with several PDZ domain-containing proteins, suggesting a role in ligand-induced cell signaling.179,180 Although signaling through JAM-A is not required for reovirus infection40,108 or apoptosis,108 the role of JAM-A signaling, if any, in the reovirus infectious cycle remains unknown. The cytoplasmic domains of the β1 integrin heterodimers that function as reovirus internalization receptors also are involved in a number of signaling pathways.43 In particular, the β1 integrin cytoplasmic tail is linked to cytoskeletal proteins such as talin181 and α-actinin,182 in addition to signaling molecules like paxillin and focal adhesion kinase.183 Furthermore, the β1 integrin cytoplasmic domain contains two NPXY motifs,184 which serve as recognition sites for the cellular endocytic machinery185 and are required for transport of reovirus to late endosomes for viral disassembly.44,51 The mechanisms by which signaling pathways elicited by β1 integrins promote reovirus infection have not been resolved.
As with most nonenveloped viruses, it is unclear how reovirus overcomes the physiological barrier posed by cell membranes during viral entry. Although structural features of the μ1 protein that contribute to membrane penetration have been identified, the precise role of specific μ1 domains in delivery of the viral core into the cytoplasm is not known. The μ1 protein is a key regulator of reovirus-induced apoptosis,108,110,111 but it is not clear how the viral disassembly events culminating in μ1-mediated membrane penetration elicit proapoptotic signaling. It is possible that endosomal disruption by μ1 leads to the release of hydrolytic enzymes such as cathepsins, which in turn damage mitochondria and stimulate death signaling.186-188 Interestingly, mitochondrial injury has been reported as early as 4 h following reovirus adsorption, suggesting the involvement of an early viral replication event.160,170 It is also possible that release of these enzymes causes apoptosis via their action on death regulators such as Bid.189 Alternatively, fragments of μ1 produced during proteolytic viral disassembly are known to gain access to the cytoplasm,85 and peptides derived from the μ1 ϕ domain can destabilize membranes and induce cell death.190,191,192 These fragments may activate other cellular sensors of viral infection or directly injure mitochondria to activate proapoptotic signaling pathways.
Events in reovirus replication following viral disassembly in endosomes but prior to viral RNA synthesis result in the activation of NF-κB, which is required for apoptosis following reovirus infection.91 The signaling pathways that connect the μ1-mediated events during viral disassembly to the activation of NF-κB are not known. Furthermore, although NF-κB activation is required for apoptosis induction by reovirus, the activation of this transcription factor alone is not sufficient.162 These results suggest a role for other cellular signal transducers in the initiation of the apoptotic response. Since MEKK1 and its downstream target JNK also are required for reovirus-induced apoptosis,122,123 it is possible that the MAP kinase cascade acts together with NF-κB to trigger the apoptotic response. However, neither the mechanism of activation of the MAP kinase cascade nor the relationship between MAP kinases and NF-κB during reovirus infection is understood.
Insights into how the activation of intracellular signaling pathways results in the execution of the cell-death response are also being elucidated. Both classical extrinsic and intrinsic apoptotic pathways are activated following reovirus infection.160 In some cell types, these pathways appear to be activated by the release of TRAIL from infected cells and the upregulation of TRAIL receptors DR4 and DR5.155 However, mechanisms by which reovirus infection results in secretion of TRAIL or upregulation of DR4 and DR5 remain unknown. Microarray studies comparing gene expression profiles of cells infected with reovirus strains that differ significantly in apoptotic potential138 or cells blocked in apoptosis due to functional absence of NF-κB139 do not demonstrate upregulation of any classical apoptosis effector pathway components. Rather, they show an upregulation of several ISGs in an NF-κB dependent manner,138,139 suggesting a requirement for the expression of type I-IFNs in the apoptotic response to reovirus. However, type I IFNs are not required for the induction of apoptosis following infection by reovirus,137 in at least in some cell types. Nonetheless, these cytokines are clearly crucial for cell fate decisions as part of the innate immune response150 and may contribute to reovirus-induced cell death in some tissues.
Studies describing the pathogenesis of reovirus in mice lacking the p50 subunit of NF-κB demonstrate that p50 serves two distinctly different functions in reovirus pathogenesis, inducing apoptosis in the brain, while mediating survival in the heart.93 At least two nonmutually exclusive possibilities may account for these different outcomes following reovirus-induced NF-κB activation. It is possible that NF-κB activation by reovirus leads to expression of the same constellation of genes regardless of cell type, and proapoptotic or prosurvival signaling pathways are dictated by the cellular response to these expression patterns. Alternatively, reovirus-induced NF-κB activation may activate different signaling pathways depending on the cell type and tissue microenvironment. In the CNS, NF-κB signaling may upregulate expression of apoptosis-inducing genes, whereas in the heart, NF-κB signaling may lead to expression of prosurvival genes. Further studies are required to precisely define the basis for the differences in reovirus virulence in different tissues. Such studies will reveal new mechanisms by which viral attachment and disassembly regulate prodeath signaling responses and extend an understanding of how viruses cause tissue-specific injury.
Acknowledgments
We thank Karl Boehme and Kristen Ogden for review of the manuscript and members of our laboratories for many helpful discussions. We acknowledge support from public health Service awards T32 AI49824 (G.H.H.), F32 AI071440 (G.H.H.), R15 AI94440 (G.H.H.), R01 AI32539 (T.S.D.), R37 AI38296 (T.S.D.), R01 AI50080 (T.S.D.), and R01 AI76983 (T.S. and T.S.D.) and the Elizabeth B. Lamb Center for Pediatric Research.
References
- 1.Dermody TS, Parker JS, Sherry B. Orthoreoviruses. In: Knipe DM, Howley PM, editors. Fields Virology. Sixth edition. Philadelphia: Lippincott Williams & Wilkins; In press. [Google Scholar]
- 2.Virgin HW, Tyler KL, Dermody TS. Reovirus. In: Nathanson N, editor. Viral pathogenesis. New York: Lippincott-Raven; 1997. pp. 669–699. [Google Scholar]
- 3.Duncan R, Horne D, Cashdollar LW, et al. Identification of conserved domains in the cell attachment proteins of the three serotypes of reovirus. Virology. 1990;174:399–409. doi: 10.1016/0042-6822(90)90093-7. [DOI] [PubMed] [Google Scholar]
- 4.Nibert ML, Dermody TS, Fields BN. Structure of the reovirus cell attachment protein: A model for the domain organization of σ1. J Virol. 1990;64:2976–2989. doi: 10.1128/jvi.64.6.2976-2989.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tyler KL, McPhee DA, Fields BN. Distinct pathways of viral spread in the host determined by reovirus S1 gene segment. Science. 1986;233:770–774. doi: 10.1126/science.3016895. [DOI] [PubMed] [Google Scholar]
- 6.Weiner HL, Powers ML, Fields BN. Absolute linkage of virulence and central nervous system tropism of reoviruses to viral hemagglutinin. J Infect Dis. 1980;141:609–616. doi: 10.1093/infdis/141.5.609. [DOI] [PubMed] [Google Scholar]
- 7.Weiner HL, Drayna D, Averill DR, Jr, et al. Molecular basis of reovirus virulence: role of the S1 gene. Proc Natl Acad Sci USA. 1977;74:5744–5748. doi: 10.1073/pnas.74.12.5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morrison LA, Sidman RL, Fields BN. Direct spread of reovirus from the intestinal lumen to the central nervous system through vagal autonomic nerve fibers. Proc Natl Acad Sci USA. 1991;88:3852–3856. doi: 10.1073/pnas.88.9.3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tardieu M, Powers ML, Weiner HL. Age-dependent susceptibility to reovirus type 3 encephalitis: role of viral and host factors. Ann Neurol. 1983;13:602–607. doi: 10.1002/ana.410130604. [DOI] [PubMed] [Google Scholar]
- 10.Dichter MA, Weiner HL. Infection of neuronal cell cultures with reovirus mimics in vitro patterns of neurotropism. Ann Neurol. 1984;16:603–610. doi: 10.1002/ana.410160512. [DOI] [PubMed] [Google Scholar]
- 11.Weiner HL, Ault KA, Fields BN. Interaction of reovirus with cell surface receptors. I. Murine and human lymphocytes have a receptor for the hemagglutinin of reovirus type 3. J Immunol. 1980;124:2143–2148. [PubMed] [Google Scholar]
- 12.Leep W, Hayes EC, Joklik WK. Protein σ1 is the reovirus cell attachment protein. Virology. 1981;108:156–163. doi: 10.1016/0042-6822(81)90535-3. [DOI] [PubMed] [Google Scholar]
- 13.Furlong DB, Nibert ML, Fields BN. Sigma 1 protein of mammalian reoviruses extends from the surfaces of viral particles. J Virol. 1988;62:246–256. doi: 10.1128/jvi.62.1.246-256.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fraser RDB, Furlong DB, Trus BL, et al. Molecular structure of the cell attachment protein of reovirus: Correlation of computer-processed electron micrographs with sequence-based predictions. J Virol. 1990;64:2990–3000. doi: 10.1128/jvi.64.6.2990-3000.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gentsch JR, Pacitti AF. Effect of neuraminidase treatment of cells and effect of soluble glycoproteins on type 3 reovirus attachment to murine L cells. J Virol. 1985;56:356–364. doi: 10.1128/jvi.56.2.356-364.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paul RW, Choi AH, Lee PWK. The α-anomeric form of sialic acid is the minimal receptor determinant recognized by reovirus. Virology. 1989;172:382–385. doi: 10.1016/0042-6822(89)90146-3. [DOI] [PubMed] [Google Scholar]
- 17.Dermody TS, Nibert ML, Bassel-Duby R, et al. Sequence diversity in S1 genes and S1 translation products of 11 serotype 3 reovirus strains. J Virol. 1990;64:4842–4850. doi: 10.1128/jvi.64.10.4842-4850.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chappell JD, Gunn VL, Wetzel JD, et al. Mutations in type 3 reovirus that determine binding to sialic acid are contained in the fibrous tail domain of viral attachment protein sigma1. J Virol. 1997;71:1834–1841. doi: 10.1128/jvi.71.3.1834-1841.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chappell JD, Duong JL, Wright BW, et al. Identification of carbohydrate binding domains in the attachment proteins of type 1 and type 3 reoviruses. J Virol. 2000;74:8472–8479. doi: 10.1128/jvi.74.18.8472-8479.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barton ES, Forrest JC, Connolly JL, et al. Junction adhesion molecule is a receptor for reovirus. Cell. 2001;104:441–451. doi: 10.1016/s0092-8674(01)00231-8. [DOI] [PubMed] [Google Scholar]
- 21.Martin-Padura I, Lostaglio S, Schneemann M, et al. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol. 1998;142:117–127. doi: 10.1083/jcb.142.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williams LA, Martin-Padura I, Dejana E, et al. Identification and characterisation of human junctional adhesion molecule (JAM) Mol Immunol. 1999;36:1175–1188. doi: 10.1016/s0161-5890(99)00122-4. [DOI] [PubMed] [Google Scholar]
- 23.Liu Y, Nusrat A, Schnell FJ, et al. Human junction adhesion molecule regulates tight junction resealing in epithelia. J Cell Sci. 2000;113:2363–2374. doi: 10.1242/jcs.113.13.2363. [DOI] [PubMed] [Google Scholar]
- 24.Dryden KA, Wang G, Yeager M, et al. Early steps in reovirus infection are associated with dramatic changes in supramolecular structure and protein conformation: analysis of virions and subviral particles by cryoelectron microscopy and image reconstruction. J Cell Biol. 1993;122:1023–1041. doi: 10.1083/jcb.122.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chappell JD, Prota A, Dermody TS, et al. Crystal structure of reovirus attachment protein σ1 reveals evolutionary relationship to adenovirus fiber. EMBO J. 2002;21:1–11. doi: 10.1093/emboj/21.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reiter DM, Frierson JM, Halvorsonee ET, et al. Crystal structure of reovirus attachment protein σ1 in complex with sialylated oligosaccharides. PLoS Pathog. 2011;7:e1002166. doi: 10.1371/journal.ppat.1002166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Raaij MJ, Mitraki A, Lavigne G, et al. A triple β-spiral in the adenovirus fibre shaft reveals a new structural motif for a fibrous protein. Nature. 1999;401:935–938. doi: 10.1038/44880. [DOI] [PubMed] [Google Scholar]
- 28.Guardado CP, Fox GC, Hermo-parrado XL, et al. Structure of the carboxy-terminal receptor-binding domain of avian reovirus fibre sigmaC. J Mol Biol. 2005;354:137–149. doi: 10.1016/j.jmb.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 29.Prota AE, Campbell JA, Schelling P, et al. Crystal structure of human junctional adhesion molecule 1: Implications for reovirus binding. Proc Natl Acad Sci USA. 2003;100:5366–5371. doi: 10.1073/pnas.0937718100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stehle T, Dermody TS. Structural similarities in the cellular receptors used by adenovirus and reovirus. Viral Immunol. 2004;17:129–143. doi: 10.1089/0882824041310621. [DOI] [PubMed] [Google Scholar]
- 31.Forrest JC, Campbell JA, Schelling P, et al. Structure-function analysis of reovirus binding to junctional adhesion molecule 1. Implications for the mechanism of reovirus attachment. J Biol Chem. 2003;278:48434–48444. doi: 10.1074/jbc.M305649200. [DOI] [PubMed] [Google Scholar]
- 32.Kirchner E, Guglielmi KM, Strauss H, et al. Structure of reovirus σ1 in complex with its receptor junctional adhesion molecule-A. PLoS Pathog. 2008;4:e1000235. doi: 10.1371/journal.ppat.1000235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bewley MC, Springer K, Zhang YB, et al. Structural analysis of the mechanism of adenovirus binding to its human cellular receptor, Car. Science. 1999;286:1579–1583. doi: 10.1126/science.286.5444.1579. [DOI] [PubMed] [Google Scholar]
- 34.van Raaij MJ, Chouin E, van Der Zandt H, et al. Dimeric structure of the coxsackievirus and adenovirus receptor D1 domain at 1.7 a resolution. Structure. 2000;8:1147–1155. doi: 10.1016/s0969-2126(00)00528-1. [DOI] [PubMed] [Google Scholar]
- 35.Spear PG. Viral interactions with receptors in cell junctions and effects on junctional stability. Dev Cell. 2002;3:462–464. doi: 10.1016/s1534-5807(02)00298-8. [DOI] [PubMed] [Google Scholar]
- 36.Compton T. Receptors and immune sensors: the complex entry path of human cytomegalovirus. Trends Cell Biol. 2004;14:5–8. doi: 10.1016/j.tcb.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 37.Ugolini S, Mondor I, Sattentau QJ. HIV-1 attachment: another look. Trends Microbiol. 1999;7:144–149. doi: 10.1016/s0966-842x(99)01474-2. [DOI] [PubMed] [Google Scholar]
- 38.Berger EA, Murphy PM, Farber JM. Chemokine receptors as HIV-1 coreceptors: Roles in viral entry, tropism, and disease. Annu Rev Lmmunol. 1999;17:657–700. doi: 10.1146/annurev.immunol.17.1.657. [DOI] [PubMed] [Google Scholar]
- 39.Barton ES, Connolly JL, Forrest JC, et al. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J Biol Chem. 2001;276:2200–2211. doi: 10.1074/jbc.M004680200. [DOI] [PubMed] [Google Scholar]
- 40.Maginnis MS, Forrest JC, Kopecky-Bromberg SA et al. β1 integrin mediates internalization of mammalian reovirus. J Virol. 2006;80:2760–2770. doi: 10.1128/JVI.80.6.2760-2770.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Breun LA, Broering TJ, McCutcheon AM, et al. Mammalian reovirus L2 gene and λ2 core spike protein sequences and whole-genome comparisons of reoviruses type 1 Lang, type 2 Jones, and type 3 Dearing. Virology. 2001;287:333–348. doi: 10.1006/viro.2001.1052. [DOI] [PubMed] [Google Scholar]
- 42.Hynes RO. Integrins: Versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 43.Hynes R. Integrins: Bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 44.Maginnis MS, Mainou BA, Derdowski AM, et al. NPXY motifs in the β1 integrin cytoplasmic tail are required for functional reovirus entry. J Virol. 2008;82:3181–3191. doi: 10.1128/JVI.01612-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sturzenbecker LJ, Nibert ML, Furlong DB, et al. Intracellular digestion of reovirus particles requires a low ph and is an essential step in the viral infectious cycle. J Virol. 1987;61:2351–2361. doi: 10.1128/jvi.61.8.2351-2361.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borsa J, Morash BD, Sargent MD, et al. Two modes of entry of reovirus particles into L cells. J Gen Virol. 1979;45:161–170. doi: 10.1099/0022-1317-45-1-161. [DOI] [PubMed] [Google Scholar]
- 47.Borsa J, Sargent MD, Lievaart PA, et al. Reovirus: Evidence for a second step in the intracellular uncoating and transcriptase activation process. Virology. 1981;111:191–200. doi: 10.1016/0042-6822(81)90664-4. [DOI] [PubMed] [Google Scholar]
- 48.Rubin DH, Weiner DB, Dworkin C, et al. Receptor utilization by reovirus type 3: Distinct binding sites on thymoma and fibroblast cell lines result in differential compartmentalization of virions. Microb pathog. 1992;12:351–365. doi: 10.1016/0882-4010(92)90098-9. [DOI] [PubMed] [Google Scholar]
- 49.Ehrlich M, Boll W, Van Oijen A, et al. Endocytosis by random initiation and stabilization of clathrin-coated pits. Cell. 2004;118:591–605. doi: 10.1016/j.cell.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 50.Georgi A, Mottola-hartshorn C, Warner A, et al. Detection of individual fluorescently labeled reovirions in living cells. Proc Natl Acad Sci USA. 1990;87:6579–6583. doi: 10.1073/pnas.87.17.6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mainou BA, Dermody TS. Transport to late endosomes is required for efficient reovirus infection. J Virol. 2012;86:8346–8358. doi: 10.1128/JVI.00100-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Canning WM, Fields BN. Ammonium chloride prevents lytic growth of reovirus and helps to establish persistent infection in mouse L cells. Science. 1983;219:987–988. doi: 10.1126/science.6297010. [DOI] [PubMed] [Google Scholar]
- 53.Maratos-Flier E, Goodman MJ, Murray AH, et al. Ammonium inhibits processing and cytotoxicity of reovirus, a nonenveloped virus. J Clin Invest. 1986;78:617–625. doi: 10.1172/JCI112653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maxfield Fr. Weak bases and ionophores rapidly and reversibly raise the pH in endocytic vesicles in cultured mouse fibroblasts. J Cell Biol. 1982;95:676–681. doi: 10.1083/jcb.95.2.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ohkuma S, Poole B. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc Natl Acad Sci USA. 1978;75:3327–3331. doi: 10.1073/pnas.75.7.3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barrett AJ, Kembhavi AA, Brown MA, et al. L-Trans-Epoxysuccinyl-leucylamido(4-guanidino)butane (E-64) and its analogues as inhibitors of cysteine proteinases including cathepsins B, H and L. Biochem J. 1982;201:189–198. doi: 10.1042/bj2010189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baer GS, Dermody TS. Mutations in reovirus outer-capsid protein σ3 selected during persistent infections of L cells confer resistance to protease inhibitor E64. J Virol. 1997;71:4921–4928. doi: 10.1128/jvi.71.7.4921-4928.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chandran K, Nibert ML. Protease cleavage of reovirus capsid protein μ1/μ1C is blocked by alkyl sulfate detergents, yielding a new type of infectious subvirion particle. J Virol. 1998;762:467–475. doi: 10.1128/jvi.72.1.467-475.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ebert DH, Wetzel JD, Brumbaugh DE, et al. Adaptation of reovirus to growth in the presence of protease inhibitor E64 segregates with a mutation in the carboxy terminus of viral outer-capsid protein σ3. J Virol. 2001;75:3197–3206. doi: 10.1128/JVI.75.7.3197-3206.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jané-Valbuena J, Nibert ML, Spencer SM, et al. Reovirus virion-like particles obtained by recoating infectious subvirion particles with baculovirus-expressed σ3 protein: an approach for analyzing σ3 functions during virus entry. J Virol. 1999;73:2963–2973. doi: 10.1128/jvi.73.4.2963-2973.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bond JS, Butler PE. Intracellular proteases. Annu Rev Biochem. 1987;56:333–364. doi: 10.1146/annurev.bi.56.070187.002001. [DOI] [PubMed] [Google Scholar]
- 62.Gal S, Gottesman MM. The major excreted protein (Mep) of transformed mouse cells and cathepsin L have similar protease specificity. Biochem Biophys Res Commun. 1986;139:156–162. doi: 10.1016/s0006-291x(86)80093-6. [DOI] [PubMed] [Google Scholar]
- 63.Gottesman MM, Sobel ME. Tumor promoters and Kirsten sarcoma virus increase synthesis of a secreted glycoprotein by regulating levels of translatable mRNA. Cell. 1980;19:449–455. doi: 10.1016/0092-8674(80)90519-x. [DOI] [PubMed] [Google Scholar]
- 64.Kirschke H, Langner J, Wiederanders B, Cathepsin L, et al. A new proteinase from rat liver lysosomes. Eur J Biochem. 1977;74:293–301. doi: 10.1111/j.1432-1033.1977.tb11393.x. [DOI] [PubMed] [Google Scholar]
- 65.Ebert DH, Deussing J, Peters C, et al. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J Biol Chem. 2002;277:24609–24617. doi: 10.1074/jbc.M201107200. [DOI] [PubMed] [Google Scholar]
- 66.Riese RJ, Wolf PR, Bromme D, et al. Essential role for cathepsin S in MHC class II-associated invariant chain processing and peptide loading. Immunity. 1996;4:357–366. doi: 10.1016/s1074-7613(00)80249-6. [DOI] [PubMed] [Google Scholar]
- 67.Golden JW, Bahe JA, Lucas WT, et al. Cathepsin S supports acid-independent infection by some reoviruses. J Biol Chem. 2004;279:8547–8557. doi: 10.1074/jbc.M309758200. [DOI] [PubMed] [Google Scholar]
- 68.Johnson EM, Wetzel JD, Doyle JD, et al. Genetic and pharmacologic alteration of cathepsin expression influences reovirus pathogenesis. J Virol. 2009;83:9630–9640. doi: 10.1128/JVI.01095-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nygaard RM, Golden JW, Schiff LA. Impact of host proteases on reovirus infection in the respiratory tract. J Virol. 2012;86:1238–1243. doi: 10.1128/JVI.06429-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tosteson MT, Nibert ML, Fields BN. Ion channels induced in lipid bilayers by subvirion particles of the nonenveloped mammalian reoviruses. Proc Natl Acad Sci USA. 1993;90:10549–10552. doi: 10.1073/pnas.90.22.10549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lucia-Jandris P, Hooper JW, Fields BN. Reovirus M2 gene is associated with chromium release from mouse L cells. J Virol. 1993;67:5339–5345. doi: 10.1128/jvi.67.9.5339-5345.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hooper JW, Fields BN. Role of the μ1 protein in reovirus stability and capacity to cause chromium release from host cells. J Virol. 1996;70:459–467. doi: 10.1128/jvi.70.1.459-467.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Olland AM, Jané-Valbuena J, Schiff LA, et al. Structure of the reovirus outer capsid and dsRNA-binding protein σ3 at 1.8 Å resolution. EMBO J. 2001;20:979–989. doi: 10.1093/emboj/20.5.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nason E, Wetzel J, Mukherjee S, et al. A monoclonal antibody specific for reovirus outer-capsid protein σ3 inhibits σ1-mediated hemagglutination by steric hindrance. J Virol. 2001;75:6625–6634. doi: 10.1128/JVI.75.14.6625-6634.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wetzel JD, Wilson GJ, Baer GS, et al. Reovirus variants selected during persistent infections of L cells contain mutations in the viral S1 and S4 genes and are altered in viral disassembly. J Virol. 1997;71:1362–1369. doi: 10.1128/jvi.71.2.1362-1369.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Clark KM, Wetzel JD, Bayley J, et al. Reovirus variants selected for resistance to ammonium chloride have mutations in viral outer-capsid protein σ3. J Virol. 2006;80:671–681. doi: 10.1128/JVI.80.2.671-681.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Doyle JD, Danthi P, Kendall EA, et al. Molecular determinants of proteolytic disassembly of the reovirus outer capsid. J Biol Chem. 2012;287:8029–8038. doi: 10.1074/jbc.M111.334854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nibert ML, Schiff LA, Fields BN. Mammalian reoviruses contain a myristoylated structural protein. J Virol. 1991;65:1960–1967. doi: 10.1128/jvi.65.4.1960-1967.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Smith RE, Zweerink HJ, Joklik WK. Polypeptide components of virions, top component and cores of reovirus type 3. Virology. 1969;39:791–810. doi: 10.1016/0042-6822(69)90017-8. [DOI] [PubMed] [Google Scholar]
- 80.Odegard AL, Chandran K, Zhang X, et al. Putative autocleavage of outer capsid protein μ1, allowing release of myristoylated peptide μ1N during particle uncoating, is critical for cell entry by reovirus. J Virol. 2004;78:8732–8745. doi: 10.1128/JVI.78.16.8732-8745.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nibert ML, Odegard AL, Agosto MA, et al. Putative autocleavage of reovirus μ1 protein in concert with outer capsid disassembly and activation for membrane permeabilization. J Mol Biol. 2005;345:461–474. doi: 10.1016/j.jmb.2004.10.026. [DOI] [PubMed] [Google Scholar]
- 82.Bodkin DK, Nibert ML, Fields BN. Proteolytic digestion of reovirus in the intestinal lumens of neonatal mice. J Virol. 1989;63:4676–4681. doi: 10.1128/jvi.63.11.4676-4681.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nibert ML, Fields BN. A carboxy-terminal fragment of protein μ1/μ1C is present in infectious subvirion particles of mammalian reoviruses and is proposed to have a role in penetration. J Virol. 1992;66:6408–6418. doi: 10.1128/jvi.66.11.6408-6418.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chandran K, Walker SB, Chen Y. In vitro recoating of reovirus cores with baculovirus expressed outer-capsid proteins μ1 and σ3. J Virol. 1999;73:3941–3950. doi: 10.1128/jvi.73.5.3941-3950.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chandran K, Parker JS, Ehrlich Metal. The delta region of outer-capsid protein μ1 undergoes conformational change and release from reovirus particles during cell entry. J Virol. 2003;77:13361–13375. doi: 10.1128/JVI.77.24.13361-13375.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ivanovic T, Agosto MA, Zhang L, et al. Peptides released from reovirus outer capsid form membrane pores that recruit virus particles. EMBO J. 2008;27:1289–1298. doi: 10.1038/emboj.2008.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chandran K, Farsetta DL, Nibert ML. Strategy for nonenveloped virus entry: a hydrophobic conformer of the reovirus membrane penetration protein μ1 mediates membrane disruption. J Virol. 2002;76:9920–9933. doi: 10.1128/JVI.76.19.9920-9933.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liemann S, Chandran K, Baker TS, et al. Structure of the reovirus membrane penetration protein, μ1, in a complex with its protector protein, σ3. Cell. 2002;108:283–295. doi: 10.1016/s0092-8674(02)00612-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tyler KL, Squier MK, Rodgers SE, et al. Differences in the capacity of reovirus strains to induce apoptosis are determined by the viral attachment protein σ1. J Virol. 1995;69:6972–6979. doi: 10.1128/jvi.69.11.6972-6979.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rodgers SE, Barton ES, Oberhaus SM, et al. Reovirus-induced apoptosis of MDCK cells is not linked to viral yield and is blocked by Bcl-2. J Virol. 1997;71:2540–2546. doi: 10.1128/jvi.71.3.2540-2546.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Connolly JL, Rodgers SE, Clarke P, et al. Reovirus-induced apoptosis requires activation of transcription factor NF-κB. J Virol. 2000;74:2981–2989. doi: 10.1128/jvi.74.7.2981-2989.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Oberhaus SM, Smith RL, Clayton GH, et al. Reovirus infection and tissue injury in the mouse central nervous system are associated with apoptosis. J Virol. 1997;71:2100–2106. doi: 10.1128/jvi.71.3.2100-2106.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.O’Donnell SM, Hansberger MW, Connolly JL, et al. Organ-specific roles for transcription factor NF-κB in reovirus-induced apoptosis and disease. J Clin Invest. 2005;115:2341–2350. doi: 10.1172/JCI22428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.DeBiasi R, Edelstein C, Sherry B, et al. Calpain inhibition protects against virus-induced apoptotic myocardial injury. J Virol. 2001;75:351–361. doi: 10.1128/JVI.75.1.351-361.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.DeBiasi RL, Robinson BA, Sherry B, et al. Caspase inhibition protects against reovirus-induced myocardial injury in vitro and in vivo. J Virol. 2004;78:11040–11050. doi: 10.1128/JVI.78.20.11040-11050.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Connolly JL, Barton ES, Dermody TS. Reovirus binding to cell surface sialic acid potentiates virus-induced apoptosis. J Virol. 2001;75:4029–4039. doi: 10.1128/JVI.75.9.4029-4039.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tyler KL, Squier MKT, Brown AL, et al. Linkage between reovirus-induced apoptosis and inhibition of cellular DNA synthesis: role of the S1 and M2 genes. J Virol. 1996;70:7984–7991. doi: 10.1128/jvi.70.11.7984-7991.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ernst H, Shatkin AJ. Reovirus hemagglutinin mRNA codes for two polypeptides in overlapping reading frames. Proc Natl Acad Sci USA. 1985;82:48–52. doi: 10.1073/pnas.82.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jacobs BL, Atwater JA, Munemitsu SM, et al. Biosynthesis of reovirus-specified polypeptides. The S1 mRNA synthesized in vivo is structurally and functionally indistinguishable from in vitro-synthesized S1 mRNA and encodes two polypeptides, σ1a and σ1bNS. Virology. 1985;147:9–18. doi: 10.1016/0042-6822(85)90222-3. [DOI] [PubMed] [Google Scholar]
- 100.Sarkar G, Pelletier J, Bassel-Duby R, et al. Identification of a new polypeptide coded by reovirus gene S1. J Virol. 1985;54:720–725. doi: 10.1128/jvi.54.3.720-725.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rodgers SE, Connolly JL, Chappell JD, et al. Reovirus growth in cell culture does not require the full complement of viral proteins: Identification of a σ1s-null mutant. J Virol. 1998;72:8597–8604. doi: 10.1128/jvi.72.11.8597-8604.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hoyt CC, Richardson-Burns SM, Goody RJ, et al. Nonstructural protein sigma1s is a determinant of reovirus virulence and influences the kinetics and severity of apoptosis induction in the heart and central nervous system. J Virol. 2005;79:2743–2753. doi: 10.1128/JVI.79.5.2743-2753.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Boehme KW, Guglielmi KM, Dermody TS. Reovirus nonstructural protein σ1s is required for establishment of viremia and systemic dissemination. Proc Natl Acad Sci USA. 2009;106:19986–19991. doi: 10.1073/pnas.0907412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Boehme KW, Frierson JM, Konopka JL, et al. The reovirus σ1s protein is a determinant of hematogenous but not neural viral dissemination in mice. J Virol. 2011;85:11781–11790. doi: 10.1128/JVI.02289-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Campbell JA, Shelling P, Wetzel JD, et al. Junctional adhesion molecule-a serves as a receptor for prototype and field-isolate strains of mammalian reovirus. J Virol. 2005;79:7967–7978. doi: 10.1128/JVI.79.13.7967-7978.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dermody TS, Nibert ML, Bassel-Duby R. A sigma 1 region important for hemagglutination by serotype 3 reovirus strains. J Virol. 1990;64:5173–5176. doi: 10.1128/jvi.64.10.5173-5176.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Connolly JL, Dermody TS. Virion disassembly is required for apoptosis induced by reovirus. J Virol. 2002;76:1632–1641. doi: 10.1128/JVI.76.4.1632-1641.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Danthi P, Hansberger MW, Campbell JA, et al. JAM-A-independent, antibody-mediated uptake of reovirus into cells leads to apoptosis. J Virol. 2006;80:1261–1270. doi: 10.1128/JVI.80.3.1261-1270.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hazelton PR, Coombs KM. The reovirus mutant tsA279 has temperature-sensitive lesions in the M2 and L2 genes: the M2 gene is associated with decreased viral protein production and blockade in transmembrane transport. Virology. 1995;207:46–58. doi: 10.1006/viro.1995.1050. [DOI] [PubMed] [Google Scholar]
- 110.Danthi P, Kobayashi T, Holm GH, et al. Reovirus apoptosis and virulence are regulated by host cell membrane penetration efficiency. J Virol. 2008;82:161–172. doi: 10.1128/JVI.01739-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Danthi P, Kobayashi T, Coffey CM, et al. Independent regulation of reovirus membrane penetration and apoptosis by the μ1 f domain. PLoS Pathog. 2008;4:e1000248. doi: 10.1371/journal.ppat.1000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Clarke P, Meintzer SM, Moffitt LA, et al. Two distinct phases of virus-induced nuclear factor kappa B regulation enhance tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in virus-infected cells. J Biol Chem. 2003;278:18092–18100. doi: 10.1074/jbc.M300265200. [DOI] [PubMed] [Google Scholar]
- 113.Beg A, Finco T, Nantermet P, et al. Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of IκBα: a mechanism for NF-κB activation. Mol Cell Biol. 1993;13:3301–3310. doi: 10.1128/mcb.13.6.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cahir McFarland ED, Izumi KM, Mosialos G. Epstein-barr virus transformation: Involvement of latent membrane protein 1-mediated activation of NF-kappaB. Oncogene. 1999;18:6959–6964. doi: 10.1038/sj.onc.1203217. [DOI] [PubMed] [Google Scholar]
- 115.McKinsey TA, Brockman JA, Scherer DC, et al. Inactivation of IkappaBbeta by the tax protein of human T-cell leukemia virus type 1: A potential mechanism for constitutive induction of NF-kappaB. Mol Cell Biol. 1996;16:2083–2090. doi: 10.1128/mcb.16.5.2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Abbadie C, Kabrun N, Bouali F, et al. High levels of c-rel expression are associated with programmed cell death in the developing avian embryo and in bone marrow cells in vitro. Cell. 1993;75:899–912. doi: 10.1016/0092-8674(93)90534-w. [DOI] [PubMed] [Google Scholar]
- 117.Grimm S, Bauer MKA, Baeuerle PA. Bcl-2 down-regulates the activity of transcription factor NF-κB induced upon apoptosis. J Cell Biol. 1996;134:13–23. doi: 10.1083/jcb.134.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jung M, Zhang Y, Lee S, et al. Correction of radiation sensitivity in ataxia telangiectasia cells by a truncated IκB-α. Science. 1995;268:1619–1621. doi: 10.1126/science.7777860. [DOI] [PubMed] [Google Scholar]
- 119.Beg A, Baltimore D. An essential role for NF-κB in preventing TNF-α-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 120.Liu ZG, Hsu H, Goeddel D, et al. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 121.Van Antwerp D, Martin S, Kafri T, et al. Suppression of TNF-α-induced apoptosis by NF-κB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 122.Clarke P, Meintzer SM, Widmann C. Reovirus infection activates JNK and the JNK-dependent transcription factor c-Jun. J Virol. 2001;75:11275–11283. doi: 10.1128/JVI.75.23.11275-11283.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Clarke P, Meintzer SM, Wang Y, et al. JNK regulates the release of proapoptotic mitochondrial factors in reovirus-infected cells. J Virol. 2004;78:13132–13138. doi: 10.1128/JVI.78.23.13132-13138.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Norman KL, Hirasawa K, Yang AD, et al. Reovirus oncolysis: The Ras/RalGEF/p38 pathway dictates host cell permissiveness to reovirus infection. Proc Natl acad Sci USa. 2004;101:11099–11104. doi: 10.1073/pnas.0404310101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Meusel TR, Imani F. Viral induction of inflammatory cytokines in human epithelial cells follows a p38 mitogen-activated protein kinase-dependent but NF-kappa B-independent pathway. J Immunol. 2003;171:3768–3774. doi: 10.4049/jimmunol.171.7.3768. [DOI] [PubMed] [Google Scholar]
- 126.Duncan MR, Stanish SM, Cox DC. Differential sensitivity of normal and transformed human cells to reovirus infection. J Virol. 1978;28:444–449. doi: 10.1128/jvi.28.2.444-449.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Strong JE, Coffey MC, Tang D, et al. The molecular basis of viral oncolysis: Usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998;17:3351–3362. doi: 10.1093/emboj/17.12.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Strong JE, Lee PW. The v-erbB oncogene confers enhanced cellular susceptibility to reovirus infection. J Virol. 1996;70:612–616. doi: 10.1128/jvi.70.1.612-616.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Coffey MC, Strong JE, Forsyth PA, et al. Reovirus therapy of tumors with activated Ras pathway. Science. 1998;282:1332–1334. doi: 10.1126/science.282.5392.1332. [DOI] [PubMed] [Google Scholar]
- 130.Mundschau LJ, Faller DV. Oncogenic ras induces an inhibitor of double-stranded RNA-dependent eukaryotic initiation factor 2 alpha-kinase activation. J Biol Chem. 1992;267:23092–23098. [PubMed] [Google Scholar]
- 131.Williams ME, Cox DC, Stevenson JR. Rejection of reovirus-treated L1210 leukemia cells by mice. Cancer Immunol Immunother. 1986;23:87–92. doi: 10.1007/BF00199812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wilcox ME, Yang W, Senger D, et al. Reovirus as an oncolytic agent against experimental human malignant gliomas. J Natl Cancer Inst. 2001;93:903–912. doi: 10.1093/jnci/93.12.903. [DOI] [PubMed] [Google Scholar]
- 133.Norman KL, Coffey MC, Hirasawa K, et al. Reovirus oncolysis of human breast cancer. Hum Gene Ther. 2002;13:641–652. doi: 10.1089/10430340252837233. [DOI] [PubMed] [Google Scholar]
- 134.Holm GH, Zurney J, Tumilasci V, et al. Retinoic acid-inducible gene-I and interferon-β promoter stimulator-1 augment proapoptotic responses following mammalian reovirus infection via interferon regulatory factor-3. J Biol Chem. 2007;282:21953–21961. doi: 10.1074/jbc.M702112200. [DOI] [PubMed] [Google Scholar]
- 135.Kato H, Takeuchi O, Mikamo-Satoh E, et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med. 2008;205:1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Loo YM, Fornek J, Crochet N, et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol. 2008;82:335–345. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Knowlton JJ, Dermody TS, Holm GH. Apoptosis induced by mammalian reovirus is interferon-β-independent and enhanced by IRF-3- and NF-κB dependent-expression of Noxa. J Virol. 2012;86:1650–1660. doi: 10.1128/JVI.05924-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.DeBiasi RL, Clarke P, Meintzer SM, et al. Reovirus-induced alteration in expression of apoptosis and DNa repair genes with potential roles in viral pathogenesis. J Virol. 2003;77:8934–8947. doi: 10.1128/JVI.77.16.8934-8947.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.O’Donnell SM, Holm GH, Pierce JM, et al. Identification of an NF-κB dependent gene network in cells infected by mammalian reovirus. J Virol. 2006;80:1077–1086. doi: 10.1128/JVI.80.3.1077-1086.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Smith JA, Schmechel SC, Raghavan A, et al. Reovirus induces and benefits from an integrated cellular stress response. J Virol. 2006;80:2019–2033. doi: 10.1128/JVI.80.4.2019-2033.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Webster GA, Perkins ND. Transcriptional cross talk between NF-κ B and p53. Mol Cell Biol. 1999;19:3485–3495. doi: 10.1128/mcb.19.5.3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Dreyfus D, Nagasawa M, Gelfand E, et al. Modulation of p53 activity by IκBα: evidence suggesting a common phylogeny between NF-κB and p53 transcription factors. BMC Immunol. 2005;6:12. doi: 10.1186/1471-2172-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Li Z, Niu J, Uwagawa T, et al. Function of polo-like kinase 3 in NF-κB mediated proapoptotic response. J Biol Chem. 2005;280:16843–16850. doi: 10.1074/jbc.M410119200. [DOI] [PubMed] [Google Scholar]
- 144.Huang YH, WU JY, Zhang Y, et al. Synergistic and opposing regulation of the stress-responsive gene IEX-1 by p53, c-Myc, and multiple NF-kappaB/rel complexes. Oncogene. 2002;21:6819–6828. doi: 10.1038/sj.onc.1205854. [DOI] [PubMed] [Google Scholar]
- 145.Liu CY, Schröder M, Kaufman RJ. Ligand-independent dimerization activates the stress response kinases IRE1 and PERK in the lumen of the endoplasmic reticulum. J Biol Chem. 2000;275:24881–24885. doi: 10.1074/jbc.M004454200. [DOI] [PubMed] [Google Scholar]
- 146.Harding HP, Zhang Y, Bertolotti A, et al. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 147.Sen GC. Viruses and interferons. Annu Rev Microbiol. 2001;55:255–281. doi: 10.1146/annurev.micro.55.1.255. [DOI] [PubMed] [Google Scholar]
- 148.Stark GR, Kerr IM, Williams BR, et al. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 149.Kunzi MS, Pitha PM. Interferon targeted genes in host defense. Autoimmunity. 2003;36:457–461. doi: 10.1080/08916930310001605855. [DOI] [PubMed] [Google Scholar]
- 150.Tanaka N, Sato M, Lamphier MS, et al. Type I interferons are essential mediators of apoptotic death in virally infected cells. Genes Cells. 1998;3:29–37. doi: 10.1046/j.1365-2443.1998.00164.x. [DOI] [PubMed] [Google Scholar]
- 151.Bingle CD, Craig RW, Swales BM, et al. Exon skipping in Mcl-1 results in a Bcl-2 homology domain 3 only gene product that promotes cell death. J Biol Chem. 2000;275:22136–22146. doi: 10.1074/jbc.M909572199. [DOI] [PubMed] [Google Scholar]
- 152.Gurumurthy S, Goswami A, Vasudevan KM, et al. Phosphorylation of Par-4 by protein kinase a is critical for apoptosis. Mol Cell Biol. 2005;25:1146–1161. doi: 10.1128/MCB.25.3.1146-1161.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Imazu T, Shimizu S, Tagami S, et al. Bcl-2/E1B 19 kDa-interacting protein 3-like protein (Bnip3L) interacts with Bcl-2/Bcl-xL and induces apoptosis by altering mitochondrial membrane permeability. Oncogene. 1999;18:4523–4529. doi: 10.1038/sj.onc.1202722. [DOI] [PubMed] [Google Scholar]
- 154.Tan KO, Tan KML, Chan SL, et al. MAP-1, a novel proapoptotic protein containing a BH3-like motif that associates with bax through its Bcl-2 homology domains. J Biol Chem. 2001;276:2802–2807. doi: 10.1074/jbc.M008955200. [DOI] [PubMed] [Google Scholar]
- 155.Clarke P, Meintzer SM, Gibson S, et al. Reovirus-induced apoptosis is mediated by TRAIL. J Virol. 2000;74:8135–8139. doi: 10.1128/jvi.74.17.8135-8139.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Torii S, Egan DA, Evans RA, et al. Human Daxx regulates Fas-induced apoptosis from nuclear PML oncogenic domains (PODs) EMBO J. 1999;18:6037–6049. doi: 10.1093/emboj/18.21.6037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Takahashi Y, Lallemand-Breitenbach V, Zhu J, et al. PML nuclear bodies and apoptosis. Oncogene. 2004;23:2819–2824. doi: 10.1038/sj.onc.1207533. [DOI] [PubMed] [Google Scholar]
- 158.Richardson-Burns SM, Kominsky DJ, Tyler KL. Reovirus-induced neuronal apoptosis is mediated by caspase 3 and is associated with the activation of death receptors. J Neurovirol. 2002;8:365–380. doi: 10.1080/13550280260422677. [DOI] [PubMed] [Google Scholar]
- 159.Blatt NB, Glick GD. Signaling pathways and effector mechanisms preprogrammed cell death. Bioorg Med Chem. 2001;9:1371–1384. doi: 10.1016/s0968-0896(01)00041-4. [DOI] [PubMed] [Google Scholar]
- 160.Kominsky DJ, Bickel RJ, Tyler KL. Reovirus-induced apoptosis requires both death receptor- and mitochondrial-mediated caspase-dependent pathways of cell death. Cell Death Differ. 2002;9:926–933. doi: 10.1038/sj.cdd.4401045. [DOI] [PubMed] [Google Scholar]
- 161.Wajant H, Johannes FJ, Haas E, et al. Dominant-negative FADD inhibits TNFR60-, Fas/Apo1- and TRAIL-R/Apo2-mediated cell death but not gene induction. Curr Biol. 1998;8:113–116. doi: 10.1016/s0960-9822(98)70042-9. [DOI] [PubMed] [Google Scholar]
- 162.Clarke P, Debiasi RL, Meintzer SM, et al. Inhibition of NF-kappa B activity and cFLIP expression contribute to viral-induced apoptosis. Apoptosis. 2005;10:513–524. doi: 10.1007/s10495-005-1881-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chawla-Sarkar M, Lindner DJ, Liu YF, et al. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis. 2003;8:237–249. doi: 10.1023/a:1023668705040. [DOI] [PubMed] [Google Scholar]
- 164.Shigeno M, Nakao K, Ichikawa T, et al. Interferon-alpha sensitizes human hepatoma cells to TRAIL-induced apoptosis through DR5 upregulation and NF-kappa B inactivation. Oncogene. 2003;22:1653–1662. doi: 10.1038/sj.onc.1206139. [DOI] [PubMed] [Google Scholar]
- 165.Li P, Nijhawan D, Budhardjo I, et al. Cytochrome c and dATP dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 166.Verhagen AM, Ekert PG, Pakusch M, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing Iap proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 167.Du C, Fang M, Li Y, et al. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating Iap inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 168.Joza N, Susin SA, Daugas E, et al. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature. 2001;410:549–554. doi: 10.1038/35069004. [DOI] [PubMed] [Google Scholar]
- 169.Li H, Zhu H, Xu CJ, et al. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 170.Kominsky DJ, Bickel RJ, Tyler KL. Reovirus-induced apoptosis requires mitochondrial release of Smac/DIABLO and involves reduction of cellular inhibitor of apoptosis protein levels. J Virol. 2002;76:11414–11424. doi: 10.1128/JVI.76.22.11414-11424.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Danthi P, Pruijssers AJ, Berger AK, et al. Bid regulates the pathogenesis of neurotropic reovirus. PLoS Pathog. 2010;6:e1000980. doi: 10.1371/journal.ppat.1000980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Luo X, Budihardjo I, Zou H, et al. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 173.Richardson-Burns SM, Tyler KL. Regional differences in viral growth and central nervous system injury correlate with apoptosis. J Virol. 2004;78:5466–5475. doi: 10.1128/JVI.78.10.5466-5475.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Richardson-Burns SM, Tyler KL. Minocycline delays disease onset and mortality in reovirus encephalitis. Exp Neurol. 2005;192:331–339. doi: 10.1016/j.expneurol.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 175.Sherry B, Torres J, Blum MA. Reovirus induction of and sensitivity to beta interferon in cardiac myocyte cultures correlate with induction of myocarditis and are determined by viral core proteins. J Virol. 1998;72:1314–1323. doi: 10.1128/jvi.72.2.1314-1323.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Azzam-Smoak K, Noah DL, Stewart MJ, et al. Interferon regulatory factor-1, interferon-beta, and reovirus-induced myocarditis. Virology. 2002;298:20–29. doi: 10.1006/viro.2002.1470. [DOI] [PubMed] [Google Scholar]
- 177.Stewart MJ, Blum MA, Sherry B. PKR’s protective role in viral myocarditis. Virology. 2003;314:92–100. doi: 10.1016/s0042-6822(03)00414-8. [DOI] [PubMed] [Google Scholar]
- 178.Noah DL, Blum MA, Sherry B. Interferon regulatory factor 3 is required for viral induction of beta interferon in primary cardiac myocyte cultures. J Virol. 1999;73:10208–10213. doi: 10.1128/jvi.73.12.10208-10213.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Bazzoni G, Martinez-Estrada OM, Orsenigo F, et al. Interaction of junctional adhesion molecule with the tight junction components ZO-1, cingulin, and occludin. J Biol Chem. 2000;275:20520–20526. doi: 10.1074/jbc.M905251199. [DOI] [PubMed] [Google Scholar]
- 180.Ebnet K, Schulz CU, Meyer ZU Brickwedde MK, et al. Junctional adhesion molecule interacts with the PDZ domain-containing proteins AF-6 and ZO-1. J Biol Chem. 2000;275:27979–27988. doi: 10.1074/jbc.M002363200. [DOI] [PubMed] [Google Scholar]
- 181.Pfaff M, Liu S, Erle DJ, et al. Integrin beta cytoplasmic domains differentially bind to cytoskeletal proteins. J Biol Chem. 1998;273:6104–6109. doi: 10.1074/jbc.273.11.6104. [DOI] [PubMed] [Google Scholar]
- 182.Otey CA, Pavalko FM, Burridge K. An interaction between alpha-actinin and the beta 1 integrin subunit in vitro. J Cell Biol. 1990;111:721–729. doi: 10.1083/jcb.111.2.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Schaller MD, Otey CA, Hildebrand JD, et al. Focal adhesion kinase and paxillin bind to peptides mimicking beta integrin cytoplasmic domains. J Cell Biol. 1995;130:1181–1187. doi: 10.1083/jcb.130.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Reszka AA, Hayashi Y, Horwitza F. Identification of amino acid sequences in the integrin beta 1 cytoplasmic domain implicated in cytoskeletal association. J Cell Biol. 1992;117:1321–1330. doi: 10.1083/jcb.117.6.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Chen WJ, Goldstein JL, Brown MS. NPXY, a sequence often found in cytoplasmic tails, is required for coated pit-mediated internalization of the low density lipoprotein receptor. J Biol Chem. 1990;265:3116–3123. [PubMed] [Google Scholar]
- 186.Deiss LP, Galinka H, Berissi H, et al. Cathepsin D protease mediates programmed cell death induced by interferon-gamma, Fas/APO-1 and TNF-alpha. EMBO J. 1996;15:3861–3870. [PMC free article] [PubMed] [Google Scholar]
- 187.Guicciardi ME, Deussing J, Miyoshi H, et al. Cathepsin B contributes to TNF-alpha-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J Clin Invest. 2000;106:1127–1137. doi: 10.1172/JCI9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Roberg K. Relocalization of cathepsin D and cytochrome c early in apoptosis revealed by immunoelectron microscopy. Lab Invest. 2001;81:149–158. doi: 10.1038/labinvest.3780222. [DOI] [PubMed] [Google Scholar]
- 189.Stoka V, Turk B, Schendel SL, et al. Lysosomal protease pathways to apoptosis. Cleavage of bid, not pro caspases, is the most likely route. J Biol Chem. 2001;276:3149–3157. doi: 10.1074/jbc.M008944200. [DOI] [PubMed] [Google Scholar]
- 190.Coffey CM, Sheh A, Kim IS, et al. Reovirus outer capsid protein μ1 induces apoptosis and associates with lipid droplets, endoplasmic reticulum, and mitochondria. J Virol. 2006;80:8422–38. doi: 10.1128/JVI.02601-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kim JW, Lyi SM, Parrish CR, Parker JS. A proapoptotic peptide derived from reovirus outer capsid protein μ1 has membrane-destabilizing activity. J Virol. 2011;85:1507–16. doi: 10.1128/JVI.01876-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Wisniewski ML, Werner BG, Hom LG et al. Reovirus infection or ectopic expression of outer capsid protein μ1 induces apoptosis independently of the cellular proapoptotic proteins Bax and Bak. J Virol. 2011;85:296–304. doi: 10.1128/JVI.01982-10. [DOI] [PMC free article] [PubMed] [Google Scholar]