

Abstract
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Factors produced within or administered directly into the tumor interstitium, such as cytokines, chemokines, proteases, exosomes, microvesicles, or therapeutic agents, play important and multifaceted roles in the regulation of malignant disease progression. Their bioavailability to mediate signaling in distributed tissues outside of the tumor microenvironment, however, has not been well described. We therefore sought to elucidate the relative extent to which factors from within the primary tumor disseminate to systemic tissues as well as how these distribution profiles are influenced by both hydrodynamic size and the remodeling tumor vasculature. To accomplish this goal, we intratumorally co-infused into the dermal lesions of B16F10 melanoma-bearing mice at prescribed times post tumor implantation a near infrared fluorescent tracer panel ranging from 5-500 nm in hydrodynamic diameter and compared the in vivo clearance and biodistribution profiles to that of naïve animals. Our results indicate that tumor growth reduces tumor-draining lymph node accumulation and alters the distribution of tumor-derived factors amongst systemic tissues. Despite these changes, previously developed principles of size-dependent lymph node drug targeting are conserved in melanomas, suggesting their applicability to sentinel lymph node-targeted drug delivery. Tumor progression was also found to result in a significant increase in the hydrodynamic size of factors originating from the tumor that accumulated within systemic tissues. This suggests that tumor vascular remodeling may redirect the organism-wide signaling activity of tumor-derived factors and may negatively contribute to disease progression by altering the bioavailability of molecules important to the regulation of pre-metastatic niche formation and the induction of anti-tumor immunity.
Keywords: Cancer, Vascular remodeling, Bioavailability, Clearance, Accumulation
Introduction
Tumor-secreted soluble factors (TSF) such as cytokines, chemokines, proteases, and microparticles (including microvesicles and exosomes) mediate intercellular signaling at the cell- and tissue-levels to regulate cellular proliferation [1], angiogenesis [2] and lymphangiogenesis [3], recruitment of regulatory immune cells [4, 5], as well as extracellular matrix remodeling [6] via their direct effects or nucleic and/or protein transfer. Accordingly, numerous TSF and their associated signaling pathways activated within the primary tumor have emerged as potential therapeutic targets given their implicated role in the promotion of malignant disease progression associated with poor prognosis including metastasis and anti-tumor immune suppression.
In addition to their function within the tumor microenvironment, TSF have reported activities in signaling to distant tissues, such as the lymph nodes, liver, lungs, kidneys, and spleen, which exacerbate disease progression. For example, intravenous (i.v.) infusion of melanoma-derived exosomes promotes metastasis by directing host progenitor cell differentiation toward a pro-metastatic phenotype [7]. Treatment with mammary tumor-conditioned media can also induce lung and lymph node remodeling to accelerate spontaneous metastasis [8]. Moreover, tumor antigen presentation in tumor-draining lymph nodes (TDLN) is implicated in directing anti-tumor immune suppression [3, 9]. These data therefore suggest mechanisms of signaling to distributed tissues that direct the activity of TSF in facilitating disease progression that are not restricted to the primary tumor.
As such, mechanisms of TSF clearance from the tumor microenvironment and distribution to disseminated tissues likely play important roles in the regulation of TSF signaling locality and resulting function in cancer. This has been widely considered in the context of tumor lymphatic transport function, in particular for its role in facilitating cellular invasion and metastasis [10-12] and as a conduit for immune modulatory TSF to reshape the TDLN immune microenvironment [3, 13]. The role of TSF clearance via the blood vasculature and resulting distribution in systemic tissues on the other hand has been largely overlooked. However, treatment with melanoma-conditioned media redirects Lewis lung carcinoma metastasis from the lung to the kidney, spleen, intestine, and oviduct [14], tissues more typical of melanoma dissemination, suggesting a potential role for systemically circulating TSF in directing organism-wide responses to growing tumors. Interestingly, TDLN-targeted, but not non-targeted, toll-like receptor ligand adjuvant therapy has been shown to be efficacious in reducing tumor burden [9] by exploiting localized depots of endogenously produced tumor antigen within TDLN delivered via tumor-draining lymphatics. Thus, the relative contribution of blood versus lymphatic-mediated TSF clearance and resulting accumulation in associated tissues may not only influence TSF signaling activity and role in disease progression but also susceptibility to therapeutic interventions neutralizing or exploiting TSF function.
An added level of complexity in this problem is the influence of vascular remodeling that occurs within growing tumors. Hyper-angiogenic signaling during tumor formation and growth causes haphazard tissue remodeling and a tortuous, dilated, and leaky [15, 16] tumor blood vascular network that is accompanied by lymphatic hyperplasia [17]. These characteristics are well recognized to manifest in the enhanced permeability and retention effect in which systemically circulating particulates at the nano- and microscales accumulate to a greater extent within malignant relative to healthy tissues [18]. Yet while this is often exploited for tumor-targeted drug delivery applications, its impact on the clearance and biodistribution of TSF and therapeutic agents administered directly to the tumor remains as-of-yet unknown.
Hence, although TSF and intratumorally administered therapeutic agents have been regarded for their role in local signaling within the tumor microenvironment, their bioavailability in distributed tissues and the impact of disease course on these biodistribution profiles has been overlooked. Unfortunately, detection and quantification of endogenously produced TSF are limited by significant dilutional effects as well as uncertainty in the spatial and temporal source of constitutive or inducible reporter systems. Furthermore, biodistribution analysis of specific exogenously supplied TSF is challenging due to degradation in vivo. To circumvent these limitations, we established a panel of near-infrared fluorescent tracers comprised of inert polymers that are resistant to hydrolysis and proteolytic degradation. Since TSF and therapeutic agents such as small molecule drugs and engineered drug delivery vehicles vary widely in hydrodynamic diameter and the rate and extent of blood versus lymphatic clearance from healthy tissues are acutely size-dependent [19, 20], tracers over a physiological biomolecule size-matched range of 5-500 nm in hydrodynamic diameter were chosen. We examined in a temporal- and tissue-resolved manner the clearance and biodistribution of fluorescent tracers after infusion into the B16F10 melanomas of C57Bl6 mice at prescribed tumor growth phases or in the skin of naïve animals. Our findings support the hypothesis that lymphatic drainage significantly enriches levels of tumor-derived factors in draining lymph nodes (dLN), that melanoma progression attenuates lymphatic-mediated transport but not the predominant molecular size regime accumulating within TDLN, and that vascular remodeling within advanced melanomas increases the access of factors derived from the tumor interstitium to systemic tissues.
Materials and Methods
Cell culture
B16F10 murine melanoma cells were cultured in Dulbecco's Modified Eagle Medium supplemented with 10% heat-inactivated fetal bovine serum and 1% penicillin/streptomycin/amphotericin B. Media, serum, and antibiotics were obtained from Life Technologies (Carlsbad, CA). Cell lines were routinely checked and confirmed to be negative for mycoplasma infection and rodent pathogens.
TSF analysis in tumor cell conditioned media
48 hour B16F10 cell-conditioned serum-free medium, concentrated 20× by lyophilization after removing any possible cells by centrifugation at 300×g for 5 minutes, was fractionated in a size-resolved manner on a Sepharose CL-6B (GE Healthcare, Pittsburgh, PA) gravity chromatography column. The protein and deoxyribonucleic acid (DNA) content in fractionated media was analyzed using the bicinchoninic acid assay (Thermo Fisher Scientific Pierce, Waltham, MA) or by incubation with a 0.5× solution of GelRed (Biotium Inc., Hayward, CA) on an orbital shaker followed by absorbance or fluorescence measurements (BioTek Instruments Inc, Winooski, VT), respectively.
Near-infrared fluorescent tracers
500 and 50 nm fluorescent (580/610 nm and 660/680 nm excitation/emission, respectively) carboxylate-modified microspheres were purchased from Life Technologies. 500 kDa or 10 kDa amine-dextrans (Sigma-Aldrich, St. Louis, MO) were covalently labeled by incubation for 4 hours in 0.1 M NaHCO3 buffer at pH 8.4 on a shaker with Alexa Fluor 700 NHS-Ester or Alexa Fluor 610-X NHS-Ester dye (Life Technologies), respectively. Individual fluorescent dextran conjugates were purified from unreacted free dye by Sepharose CL-6B gravity column chromatography. Purified dextran-fluorophore conjugates were confirmed to be free of unconjugated dye by Sepharose CL-6B gravity column chromatography analysis. The size and zeta potentials of 500 and 50 nm fluorescent microspheres as well as 500 and 10 kDa dextran-AF700 conjugates suspended in Dulbecco's Phosphate Buffered Saline (D-PBS, Life Technologies) were confirmed using a Zetasizer Nano ZS (Malvern Instruments Ltd, WR14 1XZ, United Kingdom). All reagents were used and maintained under sterile conditions.
In vivo mouse melanoma model
C57Bl6 mice were purchased from Jackson Laboratories. All protocols were approved by the Institutional Animal Care and Use Committee. 0.5×106 B16F10 murine melanoma cells were intradermally implanted into the left dorsal skin of 6-8 weeks old mice on day 0. Tumors were monitored in anesthetized mice by caliper measurements of melanoma width, length, and depth and reported as an ellipsoidal volume. For intradermal injections, a depth-marked 27 gauge needle (Becton Dickinson, Franklin Lakes, NJ) was inserted perpendicularly into the center of the tumor of mice anesthetized with isofluorane and 10 μL of the fluorescent tracers (1.2×108 500 nm spheres, 7.9×1011 50 nm spheres, 23.8 μg 500 kDa AF700-dextran, and 3 μg of 10 kDa AF610-dextran) were co-infused as a saline solution by syringe pump at a rate of ∼300 nL per sec for a slow infusion rate that resulted in minimal alterations in interstitial pressure. The day post B16F10 implantation indicates the day on which tracer injections were made. Alternatively, naïve mice were injected in the same manner with the same fluorescent tracer panel solution in the dermal layer of the left dorsal skin. For i.v. injections, 3 μg of 10kDa AF610-dextran in 100 μL saline was injected with a 29 gauge insulin syringe into the jugular vein of mice. The syringe was aspirated to withdraw a small amount of blood before and after injection to confirm that the entire volume was administered i.v.
Micro-computed tomographic imaging for vascular measurements
Animals were perfused with neutral buffered formalin for 10 minutes, a saline wash for five more minutes, and MicroFil (Flow Tech Inc., Carver, MA) catalyzed at a viscosity appropriate for small vessels (5 mL lead-based contrast agent: 2.5 mL diluent: 0.25 mL curing agent) by syringe with the application of constant pressure. Afterwards, perfused mice were carefully stored at 4°C overnight to cure the contrast agent. The following day, skin or tumor samples were harvested and stored in D-PBS. Micro-computed tomographic imaging was performed using a SCANCO Medical μCT50 (SCANCO USA, Inc., Wayne, PA, USA). μCT image slices were constrained using manual selection of the sample outline and processed with a Gaussian filter at a global threshold via the SCANCO Medical μCT Evaluation Program before 3-dimensional reconstruction. In-house designed algorithms were used to calculate the vascular volume, vascular surface area, and mean vessel diameter within tissue samples [21].
Biodistribution analysis
At prescribed times post injection (p.i.), the tumor-draining and non-tumor-draining axillary and brachial lymph nodes, tumor or skin at the injection site, along with the spleen, lungs, liver, and kidneys were harvested and homogenized in D-PBS using 1.4 mm acid washed zirconium grinding beads (OPS Diagnostics LLC, Lebanon, NJ) with a FastPrep-24 Automated Homogenizer (MP Biomedicals, Santa Ana, CA). Tissue homogenate fluorescence was measured using a Synergy H4 BioTek plate reader and fluorescent tracer standard curves were made in individual tissue homogenates. Prism 6 (GraphPad, LaJolla, CA) was used to calculate exposure (as area under the curve from 1-72 hr post injection, AUC) from either the measured % injected or concentration of tracer in each tissue as well as the fluorescent tracer half-life of residence within the site of injection using one-phase decay least-squares regression. Error propagation analysis was used to calculate the standard error of AUC measurements. Concentrations are presented as the percent of tracer injection amount per tissue volume calculated from the respective tissue's weight and assuming a density of water.
Statistical analysis
Data are represented as the mean with S.E.M. and statistics were calculated using Prism 6. Statistical significance was defined as p < 0.05 following two-way ANOVA and post-hoc analysis with Tukey tests or one-way ANOVA followed by Fisher's Least Significant Difference (LSD) test. One, two, three, and four symbols denoting statistical significance represent p<0.05, 0.01, 0.001, and 0.0001, respectively, unless otherwise specified.
Results
TSF and fluorescent tracer surrogates
As a proxy for TSF produced in vivo, we first analyzed the size distribution of protein- and DNA-containing species within B16F10 cell-conditioned media. Using gravity column chromatography, we found protein and DNA to be contained in fractions in both the large (fractions 6-10 corresponding to hydrodynamic diameters ranging from ∼10-1000 nm) and small (fractions 15-24 corresponding to sizes < 10 nm) hydrodynamic size regimes (Fig. 1A). We thus chose to model this large size range of physiological TSF that also corresponds to the broad size range of various anti-cancer therapeutic agents using a panel of near-infrared fluorescently labeled tracers (Supplementary Table S1) verified free of unconjugated dye (Fig. S1) with similar physiological levels of negative charge (Supplementary Table S1) spanning from 5 to 500 nm in hydrodynamic diameter (Fig. 1B): 10kDa (5 nm) and 500kDa (30 nm) chainlike dextrans were chosen to represent small peptides and larger protein complexes while 50 and 500 nm polystyrene spheres were selected to model exosomes or nanoparticles and microvesicles or microparticles, respectively (Fig. 1B-C). Hence, tracer features other than size, such as differences in formulation as well as shape (e.g. spherical particles versus chain-like molecules), may also influence the measured profiles of clearance and biodistribution.
Figure 1. Analysis of B16F10 conditioned media and fluorescent tracers.
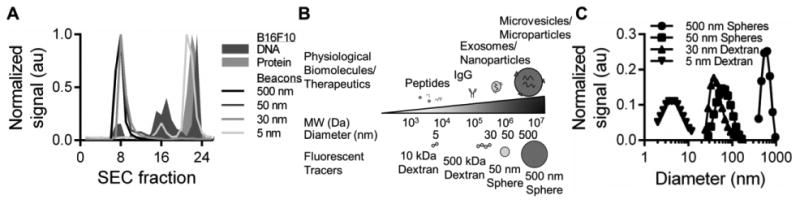
(A) Size exclusion chromatography analysis of protein and DNA containing B16F10 conditioned media fractions and fluorescent tracers. (B) Physiological TSF, typical anti-cancer therapeutic agents, and their size-matched tracers used for this study. (C) Dynamic light scattering measurements of tracers.
Rates and extents of tracer clearance from the naïve skin
To determine the rate of clearance from the skin and resulting distribution into disseminated tissues, we co-infused the fluorescent tracer cocktail into the lateral dorsal skin of naïve animals and analyzed at timescales over which passive drainage (1-24 hr) versus active cell mediated trafficking (72 hr) from peripheral tissues transpire [22, 23]. This approach thus provides an attractive internal consistency that all tracers are tested in exactly the same microenvironment of each tumor. Tracer fluorescence was quantified via endpoint analysis of homogenized individual tissues rather than commonly used whole animal imaging techniques, providing superior spatial resolution and limits of detection (Supplementary Table S2). As such, the amount and resulting concentrations of infused tracers within various tissue compartments could be quantified with high sensitivity.
We found the 5 nm tracer to be most extensively cleared, with less than 10% of the injected amount retained after 1 hour p.i. whereas the tracers of larger sizes experienced much more restricted clearance (Fig. 2A). In terms of the percent remaining 72 hours after injection (Fig. 2B) and tissue half-life (Fig. 2C), we found a linear increase with respect to tracer size up to 50 nm. For example, the amount remaining in the dermis after 72 hours was only 3% of the initial injection for the 5 nm tracer, but approximately 50 and 80% for the 30 versus 50 and 500 nm tracers, respectively (Fig. 2B). The 5 nm tracer was also cleared most quickly, with a calculated retention half-life of only about 10 minutes, while the larger tracers were cleared more slowly with retention half-lives on the order of tens to hundreds of hours (Fig. 2C).
Figure 2. Tracer-dependent clearance and biodistribution into local versus systemic tissues after intradermal injection in naïve mice.
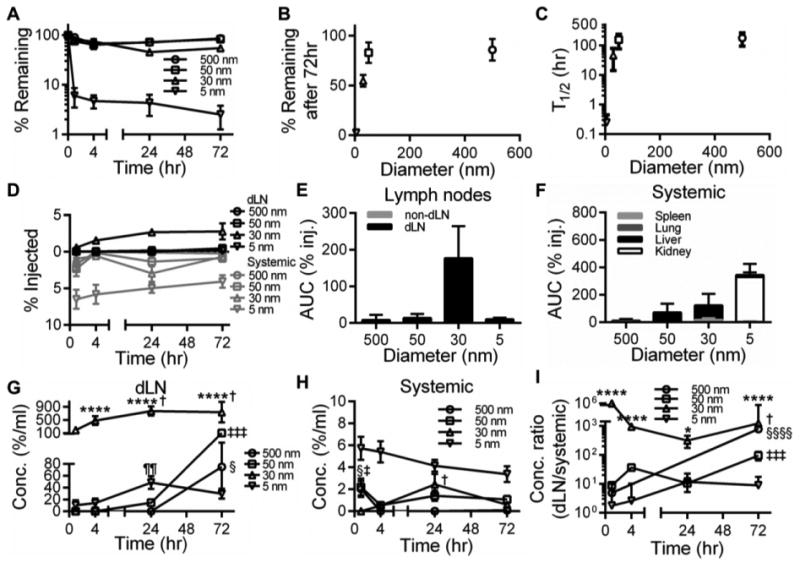
Time-resolved retention profiles (A), % remaining after 72 hr (B), and retention half-lives (T1/2, C) of tracers within skin. Time-resolved accumulation (D) and exposure (E-F) of tracers in dLN and non-dLN (E) versus systemic tissues including the spleen, lungs, liver, and kidneys (F). E, *** indicates significance relative to all other tracers by one-way ANOVA and post-hoc Fisher's LSD tests. F, *** indicates significance relative to all other tracers and kidneys vs all other tissues for 5 nm tracer by two-way ANOVA and post-hoc Tukey's tests. AUC was calculated from measured levels of % of total tracer injected in individual tissues 1-72 hr p.i. Tracer concentrations in dLN (G) and systemic tissues (H). (I) Ratio of accumulating tracer concentrations within dLN to systemic tissues. G-I, * indicates significance relative to all other tracers at the same time point by two-way ANOVA and post-hoc Tukey's tests. § indicates significance for 500 nm tracer relative to all other time points (H-I), ‡ for 50 nm tracer relative to all other time points in (G,I) but only vs 4 hr in (H), † for 30 nm tracer vs 1 hr in (G) but relative to all other time points in (H,I), and ¶ for 5 nm tracer vs 1 and 4 hr in (G) by one-way ANOVA and post-hoc Fisher's LSD tests; results representative of n=9 mice per group performed in three independent experiments.
Tracer biodistribution after clearance from naïve skin
Concurrent with analyzing their clearance from naïve skin, we measured the accumulation of infused tracers within dLN or systemic tissues, including the lungs, spleen, liver, and kidneys, in excised tissue homogenates via fluorescence. We found that 30 nm dextran accumulated most appreciably within dLN at all measured time points (Fig. 2D). This resulted in a dramatically higher exposure (% injected) of the 30 nm tracer within dLN relative to both smaller (5 nm) and larger tracers (50 and 500 nm) tested here (Fig. 2E). No exposure (% injected) was seen in non-dLN (Fig. 2E), confirming this clearance was lymphatic mediated. Conversely, 5 nm dextran was most appreciably found within systemically distributed tissues (lungs, spleen, liver, and kidneys) (Fig. 2D) resulting in the highest levels of total systemic exposure (% injected) of all tracers (Fig. 2F). Exposure (% injected) in these tissues for the 5 nm tracer was highest in the kidneys whereas larger tracer sizes accumulated predominantly in the liver (Fig. 2F). When accounting for tissue volumes, these biodistribution profiles resulted in appreciable levels of dLN accumulation for 50 and 500 nm tracers only after 72 hr p.i., suggesting their transport was primarily via cell-mediated uptake and migration to dLN (Fig. 2G). 5 nm dextran also accumulated in dLN, though to a more modest extent, peaking at 24 hr p.i., suggesting a mechanism of passive lymphatic drainage (Fig. 2G). Corroborating previous reports [24], 30 nm dextran, on the other hand, demonstrated sustained accumulation within dLN at significantly higher levels (5-20 fold) relative to all other tested tracer sizes at all tested time points p.i. (Fig. 2G). By comparison, all tracer sizes exhibited dramatically lower concentrations in systemic tissues (Fig. 2H). Over the size regimes tested, we consequently found a significant enrichment in tracer concentrations within dLN relative to systemic tissues (Fig. 2I). Most strikingly, the 30 nm tracer exhibited a three to six order of magnitude increase in dLN concentration relative that seen in systemic tissues at all measured time points and 50 and 500 nm tracers were found to be enriched by two to three orders of magnitude at 72 hours p.i. (Fig. 2I).
Remodeling of the tumor vascular plexus with tumor growth
In order to evaluate the effect of melanoma growth and remodeling on tracer clearance and resulting biodistribution from the dermis, we next evaluated the changes in the dorsolateral dermal vasculature induced by growth of the B16F10 melanoma in immune-competent C57Bl6 mice (Fig. 3A). The melanoma vascular plexus exhibited branching irregularities as early as day 5 post tumor implantation and by day 7 exhibited a striking hyper-vascularized expansion (Fig. 3B). This was reflected in a more than doubling of vascular volume and surface area between days 5 and 7 post tumor implantation (Fig. 3C). The mean vessel diameter also increased throughout the course of tumor growth (p<0.05) to grow by as much as 75% by day 9 post tumor implantation (Fig. 3D). The blood vasculature of growing B16F10 melanomas also exhibited a significant enhancement in permeability that increased with time post tumor implantation (p<0.01), as demonstrated by the accumulation of i.v. infused 5 nm dextran within the tumor interstitium at 4 hours p.i. (Fig. 3E).
Figure 3. B16F10 melanoma vascular remodeling.
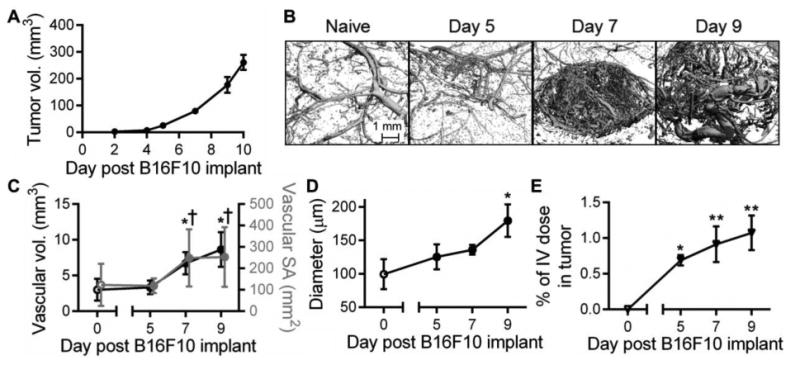
Representative B16F10 melanoma growth curve (A) and micro-computed tomography 3D reconstructions of the tumor blood vasculature (B). Mean vascular volume (C), vascular surface area (SA, C), and vessel diameter (D). (E) Intratumoral accumulation of i.v. injected dextran. * indicates significance relative to day 0 (naïve) and † relative to day 5 by ANOVA and post-hoc Tukey's tests; n=3-5 mice per group.
Malignancy increases locoregional tracer exposure
Given these remodeling responses, we evaluated the rate of clearance from the growing melanomas at prescribed time points p.i. We found exposure (% injected) of 30-500, but not 5, nm tracers in all analyzed tumors was higher relative to that of naïve skin tissues (Fig. 4A). When analyzed with respect to tumor stage, 50 and 500 nm tracers exhibited higher levels of tumor exposure (% injected) relative to naïve skin as early as 5 days post tumor implantation whereas increased tumor exposure (% injected) began only at day 7 post tumor implantation for 30 nm dextran (Fig. 4B). Although not statistically significant, as a result of the slower clearance profiles from malignant tissues, the exposure (% injected) within tumors increased by only 20% with malignant progression for the 500 and 50 nm tracers for days 5, 7 and 9 post tumor implantation, but increased up to 50% more for the 30 nm tracer at days 7 and 9 (Fig. 4C). 5 nm dextran exposure (% injected) in the tumor also increased by ∼60% at day 5 post tumor implantation but then decreased thereafter, reaching ∼40% of that seen in naïve skin in day 9 tumors (Fig. 4C).
Figure 4. Tracer exposure is modestly increased in the tumor interstitium.
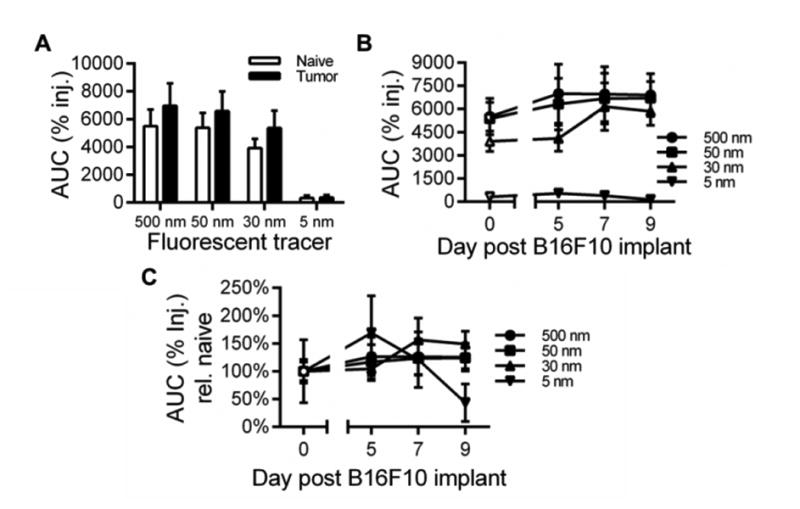
(A-B) Tracer exposure within the skin or melanoma site of injection. (C) Tracer exposure resolved by tumor day normalized to levels in naïve skin. AUC was calculated from measured levels of % of total tracer injected in individual tissues 1-72 hr p.i. Results representative of n=6-9 mice per group performed in two to three independent experiments.
Tumor progression reduces dLN accumulation and increases systemic exposure to tumor-derived factors
We next analyzed the biodistribution of systemically distributed tracers after infusion. At day 5 and 7 post tumor implantation we noted a surprising increase in exposure (% injected) in systemic tissues, which increased further by day 9 post tumor implantation for 5 nm dextran but not for other larger tracers (Fig. 5A). Similar to the biodistribution seen after clearance from naïve skin (Fig. 2F), the tissue subjected to the most significant exposure (% injected) to 30, 50 and 500 nm tracers was the liver (Fig. 5B). Contrastingly, whereas the 5 nm dextran disseminated primarily to the kidneys when cleared from the naïve skin tissue (Fig. 2F), 5 nm dextran was found to also accumulate in tumor-bearing animals in the liver (Fig. 5B). These results corresponded to increases in the maximum concentrations of tracer accumulating in the spleen, lungs, liver, and kidneys (Fig. 5C). Notably, the maximum concentrations of 500 and 50 nm tracers were significantly non-zero in the lung tissue of day 9 melanoma-bearing animals in distinct contrast to naïve and day 5 and 7 post tumor implantation animals (Fig. 5C). These concentration profile changes resulted in an initial increase in exposure (% injected) of 500 and 50 nm tracers in the spleen and 500 nm tracer exposure (% injected) in the liver days 5 and 7 post tumor implantation that was greatly diminished in day 9 tumors (Fig. 5D). Exposure (% injected) in day 9 tumor bearing animals was reduced relative to naïve animals with respect to 30 and 5 nm tracers in the spleen and the 30 nm tracer in the liver, whereas liver exposure (% injected) to the 5 nm tracer was elevated relative to that seen in naïve animals at all tumor stages analyzed (Fig. 5D). Accumulation of 30 nm dextran in the spleen and liver also occurred at earlier times post-injection in day 7 and 9 melanoma bearing animals, shifting from a 24 hour p.i. concentration peak in naïve animals to a peak in concentration at 1-4 hour p.i. (Fig. 5E).
Figure 5. Fluorescent tracers exhibit increased accumulation in systemic tissues with tumor progression.
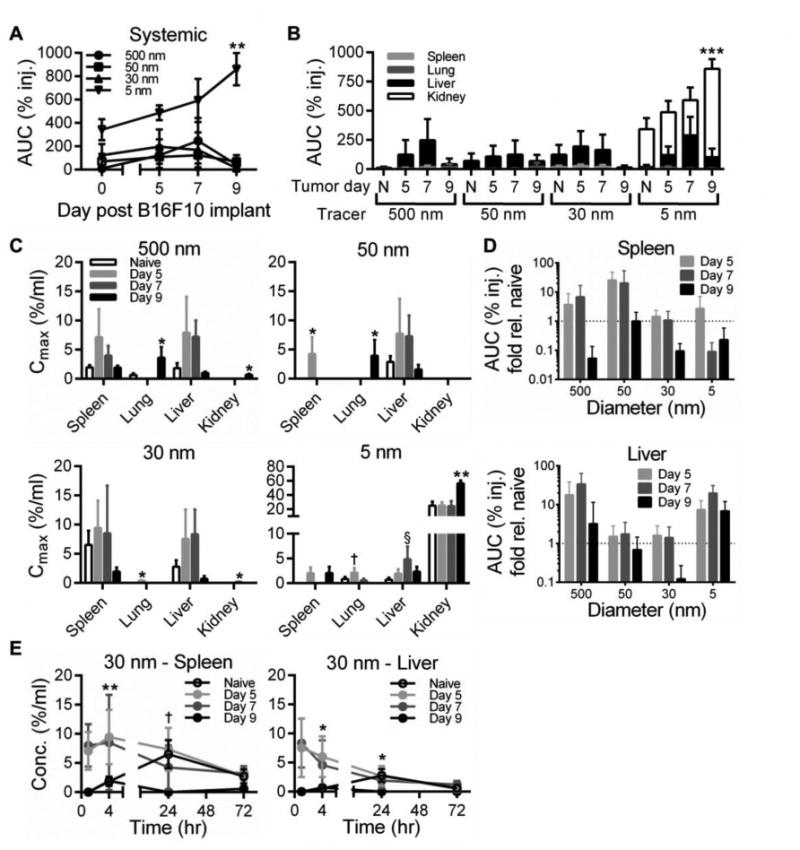
(A-B) Total tracer exposure in systemic tissues including the spleen, lungs, liver, and kidneys. ** indicates significance for day 9 vs naïve for same tracer. *** indicates significance for 5 nm tracer at day 9 in kidney vs all other groups by two-way ANOVA and post-hoc Tukey's tests. (C) Maximum tracer concentrations measured over 72 hr experimental timeframe. * indicates significance for day 9 vs all other groups of same tissue and tracer, † p<0.001 for day 5 vs all other groups of same tissue and tracer, and § p<0.001 for day 7 vs all other groups of same tissue and tracer by one-way ANOVA with post-hoc Fisher's LSD tests. (D) Fold change in tracer exposure within spleen and liver tissues resulting from melanoma growth with respect to exposure measured after injection in naïve skin. AUC was calculated from measured levels of % of total tracer injected in individual tissues 1-72 hr p.i. (E) Time-resolved concentration of 30 nm tracer in spleen and liver tissues. † p<0.05 for naïve 24 hr time point vs 1 and 4 hr naïve time points, ** p<0.01 for day 9 4 hr time point vs all other day 9 time points, * p<0.05 for naïve 24 hr time point and day 9 four hr time point vs all other by one-way ANOVA with post-hoc Fisher's LSD tests. Results representative of n=6-9 mice per group performed in two to three independent experiments.
When analyzed in a time-resolved fashion, as in naïve animals (Fig. 2G), clearance through active cell-mediated trafficking via the lymphatics remained appreciable in day 5 tumor bearing animals, as indicated by significant concentrations of the 500 and 50 nm tracers within dLN only 72 hours p.i (Fig. 6A-B). This transport activity, however, was abolished in day 7 and 9 tumors (Fig. 6A-B). Similarly, passive lymphatic drainage was also decreased with tumor progression, as indicated by reduced levels of sustained 5 and 30 nm tracer concentrations in dLN at all times p.i. (Fig. 6C-D). As a result, dLN exposure (concentration) to all tracer sizes was significantly attenuated in tumor-bearing animals relative to naïve animals (Fig. 6E-F). Specifically, relative dLN exposure (concentration) to all tracer sizes decreased rapidly with tumor progression, reaching near zero levels by day 7 for 5 and 50 nm tracers and for all tracer sizes at day 9 post tumor implantation (Fig. 6F). Notably, however, exposure (concentration) of 30 nm tracers at even their most attenuated levels (day 9 post tumor implantation) remained significantly higher (nearly two-fold) than that of all other tracer sizes in even naïve animals (Fig. 6E).
Figure 6. Tumor progression reduces tracer accumulation within TDLN.
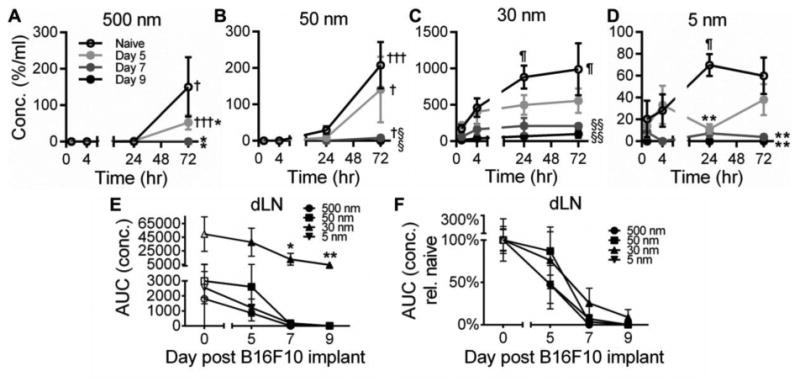
(A-D) Time-resolved tracer concentrations within TDLN. † indicates significance relative to all other time points for same group, ¶ relative to 1 hr time point for same group, * relative to naïve, and § relative to naïve and day 5 groups by one-way ANOVA with post-hoc Fisher's LSD test. Tracer exposure within dLN is attenuated by tumor growth (E) relative to naïve skin (F). * indicates significance relative to all other tracers within same tumor day group by one-way ANOVA with post-hoc Fisher's LSD test. AUC was calculated from measured levels of tracer concentrations in individual tissues 1-72 hr p.i. Results representative of n=6-9 mice per group performed in two to three independent experiments.
When comparing the relative levels of exposure (concentration) of different tracer sizes with respect to tumor stage in dLN versus systemic tissues, we found an increase in the relative extents of transport of tracers 500, 50, and 5 nm in size into systemic tissues as indicated by a decreasing ratio of dLN to systemic concentration AUC (Fig. 7A-C). While the 30 nm tracer exhibited a reduced level of accumulation within dLN post tumor injection (Fig. 6E-F), exposure (concentration) in dLN still remained two to three orders of magnitude higher relative to that seen in systemic tissues (Fig. 7A). When normalized relative to the measured ratios of naïve animals, the 500 nm tracer experienced the most significant redirection into systemic relative to dLN tissues at all tumor stages analyzed (Fig. 7B). However, both the 50 and 5 nm tracers also exhibited a bias towards increased accumulation in systemic tissues rather than dLN by days 7 and 9 post tumor implantation (Fig. 7B). Changes in the accumulation profile of 30 nm dextran, however, were less pronounced, even in day 9 tumor-bearing animals (Fig. 7B). This is because whereas for 500, 50, and 5 nm tracers an increasing proportion accumulates in systemic tissues as the disease progresses, the 30 nm tracer primarily accumulates (>99%) instead in dLN at all tumor growth stages analyzed (Fig. 7C).
Figure 7. Melanoma disease progression has no effect on the proportion of 30 nm tracer exposure in TDLN but increases the exposure of other tracer sizes within systemic tissues.
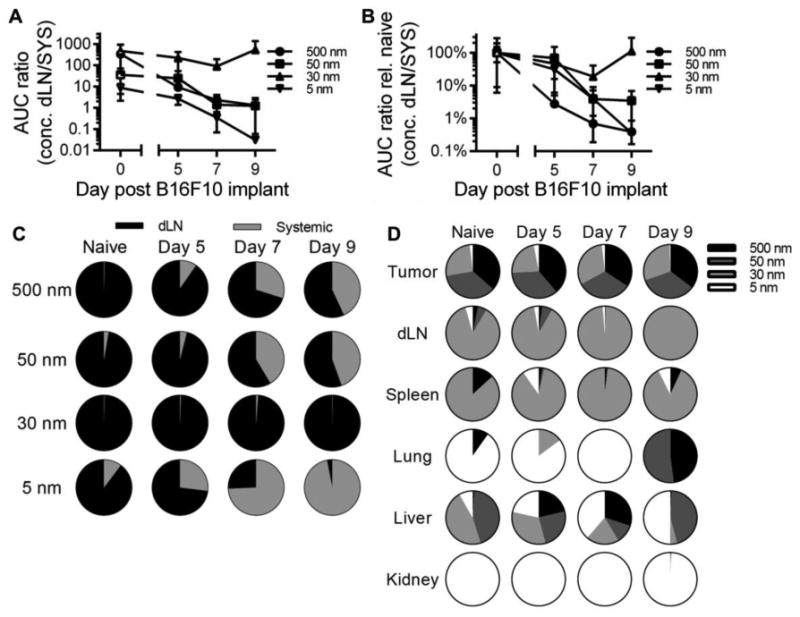
(A) Ratio of accumulating tracer concentrations within dLN to systemic tissues with respect to tumor stage and (B) normalized to naïve skin. (C) Proportion of dLN versus systemic tissue exposure (concentration). (D) Proportion of accumulating tracers with respect to hydrodynamic size. AUC was calculated from measured levels of tracer concentrations in individual tissues 1-72 hr p.i. Results representative of n=6-9 mice per group performed in two to three independent experiments.
When assessing the relative exposure of each tracer size in individual tissues, we found significant differences in tracer accumulation profiles that result from tumor formation and growth, despite no substantial differences in the relative levels of exposure within the skin versus tumor after injection (Fig. 7D). Specifically, 500, 50, and 30 nm tracers each accounted for approximately a third of the total exposure at the site of injection irrespective of tumor stage (Fig. 7D). The 5 nm tracer on the other hand exhibited little to no local exposure (Fig. 7D), consistent with its rapid clearance (Fig. 2A). Irrespective of tumor stage, 30 nm was the predominant tracer size that accumulated within dLN and the proportion of 30 nm tracer of total accumulating within the dLN increased with tumor growth (Fig. 7D). Strikingly, the livers of melanoma-bearing animals day 5 and 7 post tumor implantation exhibited profiles of exposure strikingly similar to those seen within the tumor (albeit with higher 5 nm tracer levels) rather than naïve liver (Fig. 7D). Day 9, but not 5 or 7, melanoma-bearing animals also exhibited lung exposure profiles more similar to that of the tumor rather than naïve lung, with exposure to only higher molecular weight tracers (equally shared between 500 and 50 nm tracers) versus predominantly 5 nm, respectively (Fig. 7D).
Discussion
The deleterious effects of cancer are not limited to the primary tumor itself, as evidenced by the sentinel lymph node's role in tumor immune suppression [3, 9] as well as the high number of cancer-associated complications and mortalities associated with both metastasis [25, 26] and thrombosis [27, 28]. Distant intercellular signaling can occur even prior to metastasis to precipitate such effects, for example via remodeling of sentinel lymph nodes [29], VEGFR1+ haematopoietic progenitor cell recruitment to pre-metastatic niches [14], and secretion of tumor-derived pro-coagulants and cytokines driving thrombosis [30]. Paracellular communication between cancer cells, their secreted factors, and the endothelium within the malignant melanoma microenvironment that regulate vascular permeability thus have a putative role in a myriad of pathways implicated in disease progression.
In numerous cancer types including melanoma [7], breast carcinoma [31-33], and prostate carcinoma [34], amongst others [35, 36], circulating inflammatory cytokine [34], chemokine [32, 37], enzyme [31], exosome [7], and/or microvesicle [35] levels and qualities are altered relative to healthy controls and with respect to disease stage in both patients and pre-clinical tumor models. These changes have been attributed to both higher production of such factors within the tumor [14, 31, 37] and extratumoral host tissue response to tumor growth [38]. Our data support the notion that increased systemic exposure to TSF may accompany tumor growth (Fig. 5) in a manner independent of TSF abundance since higher levels of exposure in systemic tissues were noted in animals with late stage malignancies despite the same amount of tracer being infused into each animal. These changes occurred before formation of apparent metastases, suggesting that altered rates, magnitudes, and mechanisms of TSF clearance arise as a result of tissue remodeling within the primary tumor microenvironment.
Increased systemic exposure to intratumorally infused tracers (Fig. 5) accompanied enhanced intratumoral accumulation of a systemically administered tracer (Fig. 3E). For example, in as early as day 5 lesions, the exposure of spleen and liver tissues to large 500 and 50 nm tracers was elevated (Fig. 5B) approximately two- to 30fold (Fig. 5D). Reduced exposure was seen in animals with more advanced (day 9) lesions (Fig. 5B-D) and is likely the result of interstitial matrix remodeling within the tumor [39, 40] leading to larger tracer entrapment since levels of systemic exposure to 5 nm dextran continued to increase at this tumor stage (Fig. 5A-B). Liver and spleen accumulation of large (30-500 nm) tracers peaking at 1-4 hours p.i. in animals bearing early stage (day 5 and 7) malignancies rather than 24 hr in naïve animals (Fig. 5E) also supports the concept that these increases in systemic exposure were the result of direct tracer uptake into the blood rather than enhanced lymphatic clearance and return to the systemic circulation. Increased tumor vascular permeability, as has been previously reported for this and other tumor models [41-43], thus may also enhance blood-mediated clearance that manifests as increased tumor-derived factor exposure in systemic tissues. Injection resulted in a small (∼ 1 mmHg) increase in interstitial pressure that was similar between naïve and tumor tissues that returned to pre-injection levels after removal of the needle (data not shown). However, it should be noted that measured tracer pharmacokinetics could differ from those of native tissues in vivo.
Our data support the concept that tumor growth and remodeling confer the potential of TSF to mediate signaling in distant tissues that is absent in healthy animals. Tumor-secreted microparticles, including exosomes and microvesicles, which range in hydrodynamic diameter from 30-100 nm and 100-1000 nm [44], respectively, have reported capacities to support neovascularization [45], extracellular matrix remodeling [6], and immunomodulation [33]. Here we demonstrate 500 and 50 nm tracers cleared from the tumor to accumulate at elevated levels in the liver and spleen (Fig. 5D). Moreover, we found these tracers to accumulate appreciably within the lungs of animals with advanced (day 9) malignancies (Fig. 5C), a redirection in their biodistribution that occurred concurrently with attenuated transport to TDLN (Fig. 6). TSF signaling that would take place within dLN in naïve animals appears to instead be redirected to lungs of tumor-bearing animals. Small to intermediate size biomolecules secreted by tumors such as peptides, proteins, and other biopolymers have also been implicated in a multitude of roles that exacerbate disease progression [33, 37]. We provide evidence that exposure of small molecules ∼5 nm in hydrodynamic size is also appreciably increased in systemic relative to dLN tissues in tumor-bearing animals (Fig. 5). Overall, accumulation of 500, 50 and 5 nm tracers shifted significantly from dLN to systemic tissues over the time course of disease progression evaluated here (Fig. 7). As a result, profiles of TSF tracer accumulation in the liver and lung diverged from that seen in naïve animals and instead more closely mirrored those of the primary tumor (Fig. 7D). This suggests that signaling processes active within the tumor microenvironment have the potential to be operational in systemic tissues of melanoma-bearing animals and may significantly regulate and negatively influence the course of disease progression. They also indicate that therapeutic agents administered intralesionally, for example oncolytic immunotherapy talimogene laherparepavec [46], may also result in significant signaling activity outside of the primary tumor in distributed systemic tissues.
The effect of melanoma growth on tracer clearance and biodistribution diverged with respect to its effects on blood versus lymphatic mediated transport. As opposed to increased levels of uptake via the blood vasculature, melanomas at day 7 and later post implantation demonstrated a dramatic attenuation in lymphatic function, which is regulated by local inflammatory [47] and biomechanical [48] signaling, as evidenced by reduced levels of tracer accumulation within dLN relative to that seen in naïve animals (Fig. 6C-F). Parallel reductions in lymphatic-mediated cellular migration to dLN, which are regulated by flow-organized chemokine gradients and vascular signaling [49-51], were also observed (Fig. 6A-B, E-F). These reductions coincided with tumor vascular volume and surface area doubling relative to naïve skin tissues at day 7 and 9 post tumor implantation (Fig. 3C). In line with previous reports [24], the predominant lymph-draining tracers were found to be 30 nm in size in both healthy (Fig. 2D-E,G) and malignant (Fig. 6E) skin. Interestingly, despite reduced total levels of dLN accumulation, the relative accumulation of 30 nm tracer in dLN relative to systemic tissues remained relatively unchanged. This indicates that despite reductions in lymphatic transport function (Fig. 6), TSF approximately 30 nm in diameter such as exosomes and large protein complexes continue to primarily signal extratumorally within TDLN (Fig. 7C-D). These results also underscore the potential utility of drug formulations ∼30 nm in size to accumulate substantially in sentinel lymph nodes when intralesionally administered.
Conclusions
In summary, we have demonstrated that tracer clearance from primary melanomas is concurrently altered with tumor vascular remodeling and results in distinct biodistribution profiles relative to those seen in healthy animals. These are the first measurements juxtaposing the size-resolved biodistribution profiles of tumor-derived factors in naïve versus tumor-bearing animals to elucidate the influence of disease. Our results highlight the role lymphatic drainage function plays in enriching TSF accumulation in dLN. Furthermore, these data suggest that melanoma growth redistributes TSF signaling to systemic tissues by compromising transport barriers in healthy tissues and that despite attenuation of total levels of accumulation, previously developed principles of size-dependent lymph node drug targeting are conserved in melanomas over the course of disease progression. Our findings contribute evidence that tumor vascular remodeling not only manifests in the enhanced permeability and retention effect in solid tumors, but also increased access of factors from the tumor interstitium to systemically distributed tissues. These results will inform the rational design of sentinel lymph node-targeted drug delivery strategies as well as future studies on how angioplastic therapy might be used to mitigate the pathological effects of TSF in disease progression.
Supplementary Material
Acknowledgments
This work was supported by institutional funds from the Georgia Institute of Technology and the National Institutes of Health Cell and Tissue Engineering Training Grant T32 GM008433. We thank Dr. Selwyn Hurwitz for helpful discussions.
Footnotes
Conflicts of Interest: The authors declare no conflict of interest.
References
- 1.Vetvicka V, Vetvickova J. Procathepsin D and cytokines influence the proliferation of lung cancer cells. Anticancer Res. 2011;31:47–51. [PubMed] [Google Scholar]
- 2.Pepper MS, Ferrara N, Orci L, Montesano R. Potent synergism between vascular endothelial growth factor and basic fibroblast growth factor in the induction of angiogenesis in vitro. Biochemical and Biophysical Research Communications. 1992;189:824–831. doi: 10.1016/0006-291x(92)92277-5. [DOI] [PubMed] [Google Scholar]
- 3.Lund AW, Duraes FV, Hirosue S, Raghavan VR, Nembrini C, Thomas SN, Issa A, Hugues S, Swartz MA. VEGF-C promotes immune tolerance in B16 melanomas and cross-presentation of tumor antigen by lymph node lymphatics. Cell reports. 2012;1:191–199. doi: 10.1016/j.celrep.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 4.Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science (New York, NY) 2010;328:749–752. doi: 10.1126/science.1185837. [DOI] [PubMed] [Google Scholar]
- 5.Crane CA, Ahn BJ, Han SJ, Parsa AT. Soluble factors secreted by glioblastoma cell lines facilitate recruitment, survival, and expansion of regulatory T cells: implications for immunotherapy. Neuro-oncology. 2012;14:584–595. doi: 10.1093/neuonc/nos014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mu W, Rana S, Zöller M. Host Matrix Modulation by Tumor Exosomes Promotes Motility and Invasiveness. Neoplasia. 2013;15:875–IN874. doi: 10.1593/neo.13786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peinado H, Alečković M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, García-Santos G, Nitadori-Hoshino A, Hoffman C, Badal K, Garcia BA, Callahan MK, Yuan J, Martins VR, Skog J, Kaplan RN, Brady MS, Wolchok JD, Chapman PB, Kang Y, Bromberg J, Lyden D. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nature medicine. 2012;18:883–891. doi: 10.1038/nm.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee E, Pandey NB, Popel AS. Pre-treatment of mice with tumor-conditioned media accelerates metastasis to lymph nodes and lungs: a new spontaneous breast cancer metastasis model. Clinical & experimental metastasis. 2014;31:67–79. doi: 10.1007/s10585-013-9610-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas SN, Vokali E, Lund AW, Hubbell JA, Swartz MA. Targeting the tumor-draining lymph node with adjuvanted nanoparticles reshapes the anti-tumor immune response. Biomaterials. 2014;35:814–824. doi: 10.1016/j.biomaterials.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 10.Skobe M, Hawighorst T, Jackson DG, Prevo R, Janes L, Velasco P, Riccardi L, Alitalo K, Claffey K, Detmar M. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med. 2001;7:192–198. doi: 10.1038/84643. [DOI] [PubMed] [Google Scholar]
- 11.Karpanen T, Egeblad M, Karkkainen MJ, Kubo H, Yla-Herttuala S, Jaattela M, Alitalo K. Vascular endothelial growth factor C promotes tumor lymphangiogenesis and intralymphatic tumor growth. Cancer research. 2001;61:1786–1790. [PubMed] [Google Scholar]
- 12.Harrell MI, Iritani BM, Ruddell A. Tumor-Induced Sentinel Lymph Node Lymphangiogenesis and Increased Lymph Flow Precede Melanoma Metastasis. The American Journal of Pathology. 2007;170:774–786. doi: 10.2353/ajpath.2007.060761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mansfield AS, Holtan SG, Grotz TE, Allred JB, Jakub JW, Erickson LA, Markovic SN. Regional immunity in melanoma: immunosuppressive changes precede nodal metastasis. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2011;24:487–494. doi: 10.1038/modpathol.2010.227. [DOI] [PubMed] [Google Scholar]
- 14.Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, Zhu Z, Hicklin D, Wu Y, Port JL, Altorki N, Port ER, Ruggero D, Shmelkov SV, Jensen KK, Rafii S, Lyden D. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Less JR, Skalak TC, Sevick EM, Jain RK. Microvascular Architecture in a Mammary Carcinoma: Branching Patterns and Vessel Dimensions. Cancer research. 1991;51:265–273. [PubMed] [Google Scholar]
- 16.Yuan F, Chen Y, Dellian M, Safabakhsh N, Ferrara N, Jain RK. Time-dependent vascular regression and permeability changes in established human tumor xenografts induced by an anti-vascular endothelial growth factor/vascular permeability factor antibody. Proceedings of the National Academy of Sciences. 1996;93:14765–14770. doi: 10.1073/pnas.93.25.14765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoshida T, Isaka N, Hagendoorn J, di Tomaso E, Chen YL, Pytowski B, Fukumura D, Padera TP, Jain RK. Imaging steps of lymphatic metastasis reveals that vascular endothelial growth factor-C increases metastasis by increasing delivery of cancer cells to lymph nodes: therapeutic implications. Cancer research. 2006;66:8065–8075. doi: 10.1158/0008-5472.CAN-06-1392. [DOI] [PubMed] [Google Scholar]
- 18.Matsumura Y, Maeda H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer research. 1986;46:6387–6392. [PubMed] [Google Scholar]
- 19.Sarin H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. Journal of angiogenesis research. 2010;2:14. doi: 10.1186/2040-2384-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thomas SN. A. Schudel, Overcoming transport barriers for interstitial-, lymphatic-, and lymph node-targeted drug delivery. Current Opinion in Chemical Engineering. 2015;7:65–74. doi: 10.1016/j.coche.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duvall CL, Taylor WR, Weiss D, Guldberg RE. Quantitative microcomputed tomography analysis of collateral vessel development after ischemic injury, American journal of physiology. Heart and circulatory physiology. 2004;287:H302–310. doi: 10.1152/ajpheart.00928.2003. [DOI] [PubMed] [Google Scholar]
- 22.Kissenpfennig A, Henri S, Dubois B, Laplace-Builhé C, Perrin P, Romani N, Tripp CH, Douillard P, Leserman L, Kaiserlian D, Saeland S, Davoust J, Malissen B. Dynamics and Function of Langerhans Cells In Vivo: Dermal Dendritic Cells Colonize Lymph Node AreasDistinct from Slower Migrating Langerhans Cells. Immunity. 2005;22:643–654. doi: 10.1016/j.immuni.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 23.Thomas SN, Rutkowski JM, Pasquier M, Kuan EL, Alitalo K, Randolph GJ, Swartz MA. Impaired humoral immunity and tolerance in K14-VEGFR-3-Ig mice that lack dermal lymphatic drainage. Journal of immunology (Baltimore, Md : 1950) 2012;189:2181–2190. doi: 10.4049/jimmunol.1103545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reddy ST, Rehor A, Schmoekel HG, Hubbell JA, Swartz MA. In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. J Control Release. 2006;112:26–34. doi: 10.1016/j.jconrel.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 25.Kontani K, Hashimoto S, Murazawa C, Norimura S, Tanaka H, Ohtani M, Fujiwara-Honjo N, Date M, Teramoto K, Houchi H, Yokomise H. Factors responsible for long-term survival in metastatic breast cancer. World journal of surgical oncology. 2014;12:344. doi: 10.1186/1477-7819-12-344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolchok JD, Hodi FS, Weber JS, Allison JP, Urba WJ, Robert C, O'Day SJ, Hoos A, Humphrey R, Berman DM, Lonberg N, Korman AJ. Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Annals of the New York Academy of Sciences. 2013;1291:1–13. doi: 10.1111/nyas.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Timp JF, Braekkan SK, Versteeg HH, Cannegieter SC. Epidemiology of cancer-associated venous thrombosis. Blood. 2013;122:1712–1723. doi: 10.1182/blood-2013-04-460121. [DOI] [PubMed] [Google Scholar]
- 28.Walker AJ, Card TR, West J, Crooks C, Grainge MJ. Incidence of venous thromboembolism in patients with cancer – A cohort study using linked United Kingdom databases. European Journal of Cancer. 2013;49:1404–1413. doi: 10.1016/j.ejca.2012.10.021. [DOI] [PubMed] [Google Scholar]
- 29.Rohner NA, McClain J, Tuell SL, Warner A, Smith B, Yun Y, Mohan A, Sushnitha M, Thomas SN. Lymph node biophysical remodeling is associated with melanoma lymphatic drainage. FASEB journal : official publication of the Federation of American Societies for Experimental Biology, ( 2015 doi: 10.1096/fj.15-274761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rickles FR. Mechanisms of cancer-induced thrombosis in cancer. Pathophysiology of haemostasis and thrombosis. 2006;35:103–110. doi: 10.1159/000093551. [DOI] [PubMed] [Google Scholar]
- 31.Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, Le QT, Giaccia AJ. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15:35–44. doi: 10.1016/j.ccr.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jafarzadeh A, Fooladseresht H, Minaee K, Bazrafshani MR, Khosravimashizi A, Nemati M, Mohammadizadeh M, Mohammadi MM, Ghaderi A. Higher circulating levels of chemokine CCL22 in patients with breast cancer: evaluation of the influences of tumor stage and chemokine gene polymorphism. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2015;36:1163–1171. doi: 10.1007/s13277-014-2739-6. [DOI] [PubMed] [Google Scholar]
- 33.Pitteri SJ, Kelly-Spratt KS, Gurley KE, Kennedy J, Buson TB, Chin A, Wang H, Zhang Q, Wong CH, Chodosh LA, Nelson PS, Hanash SM, Kemp CJ. Tumor Microenvironment–Derived Proteins Dominate the Plasma Proteome Response during Breast Cancer Induction and Progression. Cancer research. 2011;71:5090–5100. doi: 10.1158/0008-5472.CAN-11-0568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pfitzenmaier J, Vessella R, Higano CS, Noteboom JL, Wallace D, Corey E. Elevation of cytokine levels in cachectic patients with prostate carcinoma. Cancer. 2003;97:1211–1216. doi: 10.1002/cncr.11178. [DOI] [PubMed] [Google Scholar]
- 35.Baran J, Baj-Krzyworzeka M, Weglarczyk K, Szatanek R, Zembala M, Barbasz J, Czupryna A, Szczepanik A, Zembala M. Circulating tumour-derived microvesicles in plasma of gastric cancer patients. Cancer immunology, immunotherapy : CII. 2010;59:841–850. doi: 10.1007/s00262-009-0808-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bergmann C, Strauss L, Wieckowski E, Czystowska M, Albers A, Wang Y, Zeidler R, Lang S, Whiteside TL. Tumor-derived microvesicles in sera of patients with head and neck cancer and their role in tumor progression. Head & neck. 2009;31:371–380. doi: 10.1002/hed.20968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tichet M, Prod'Homme V, Fenouille N, Ambrosetti D, Mallavialle A, Cerezo M, Ohanna M, Audebert S, Rocchi S, Giacchero D, Boukari F, Allegra M, Chambard JC, Lacour JP, Michiels JF, Borg JP, Deckert M, Tartare-Deckert S. Tumour-derived SPARC drives vascular permeability and extravasation through endothelial VCAM1 signalling to promote metastasis. Nat Commun. 2015;6 doi: 10.1038/ncomms7993. [DOI] [PubMed] [Google Scholar]
- 38.Hiratsuka S, Watanabe A, Aburatani H, Maru Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nature cell biology. 2006;8:1369–1375. doi: 10.1038/ncb1507. [DOI] [PubMed] [Google Scholar]
- 39.Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer research. 2000;60:2497–2503. [PubMed] [Google Scholar]
- 40.Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7:653–664. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seymour LW, Ulbrich K, Steyger PS, Brereton M, Subr V, Strohalm J, Duncan R. Tumour tropism and anti-cancer efficacy of polymer-based doxorubicin prodrugs in the treatment of subcutaneous murine B16F10 melanoma. British journal of cancer. 1994;70:636–641. doi: 10.1038/bjc.1994.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, Jain RK. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer research. 1995;55:3752–3756. [PubMed] [Google Scholar]
- 43.Maeda H, Fang J, Inutsuka T, Kitamoto Y. Vascular permeability enhancement in solid tumor: various factors, mechanisms involved and its implications. International Immunopharmacology. 2003;3:319–328. doi: 10.1016/S1567-5769(02)00271-0. [DOI] [PubMed] [Google Scholar]
- 44.Lee TH, D'Asti E, Magnus N, Al-Nedawi K, Meehan B, Rak J. Microvesicles as mediators of intercellular communication in cancer--the emerging science of cellular ‘debris’. Seminars in immunopathology. 2011;33:455–467. doi: 10.1007/s00281-011-0250-3. [DOI] [PubMed] [Google Scholar]
- 45.Kim CW, Lee HM, Lee TH, Kang C, Kleinman HK, Gho YS. Extracellular membrane vesicles from tumor cells promote angiogenesis via sphingomyelin. Cancer research. 2002;62:6312–6317. [PubMed] [Google Scholar]
- 46.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS, Milhem M, Cranmer L, Curti B, Lewis K, Ross M, Guthrie T, Linette GP, Daniels GA, Harrington K, Middleton MR, Miller WH, Jr, Zager JS, Ye Y, Yao B, Li A, Doleman S, VanderWalde A, Gansert J, Coffin RS. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 47.Liao S, Cheng G, Conner DA, Huang Y, Kucherlapati RS, Munn LL, Ruddle NH, Jain RK, Fukumura D, Padera TP. Impaired lymphatic contraction associated with immunosuppression. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:18784–18789. doi: 10.1073/pnas.1116152108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Munn LL. Mechanobiology of lymphatic contractions. Semin Cell Dev Biol. 2015;38:67–74. doi: 10.1016/j.semcdb.2015.01.010. [DOI] [PubMed] [Google Scholar]
- 49.Fleury ME, Boardman KC, Swartz MA. Autologous morphogen gradients by subtle interstitial flow and matrix interactions. Biophysical journal. 2006;91:113–121. doi: 10.1529/biophysj.105.080192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miteva DO, Rutkowski JM, Dixon JB, Kilarski W, Shields JD, Swartz MA. Transmural flow modulates cell and fluid transport functions of lymphatic endothelium. Circulation research. 2010;106:920–931. doi: 10.1161/CIRCRESAHA.109.207274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haessler U, Pisano M, Wu M, Swartz MA. Dendritic cell chemotaxis in 3D under defined chemokine gradients reveals differential response to ligands CCL21 and CCL19. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:5614–5619. doi: 10.1073/pnas.1014920108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.