

Abstract
Prostate cancer, once it has progressed from its local to metastatic form, is a disease with poor prognosis and limited treatment options. Here we demonstrate an approach using nanoscale liposomes conjugated with E-selectin adhesion protein and Apo2L/TRAIL (TNF-related apoptosis-inducing ligand) apoptosis ligand that attach to the surface of leukocytes and rapidly clear viable cancer cells from circulating blood, both in human blood samples and in the living mouse. For the first time, it is shown that such an approach can be used to prevent the spontaneous formation and growth of metastatic tumors in an orthotopic xenograft model of prostate cancer, by greatly reducing the number of circulating tumor cells. We conclude that the use of circulating leukocytes as a carrier for the anti-cancer protein TRAIL could be an effective tool to directly target circulating tumor cells for the prevention of prostate cancer metastasis, and potentially other cancers that spread through the bloodstream.
Keywords: metastasis, circulating tumor cells, TRAIL, nanomedicine
Graphical abstract
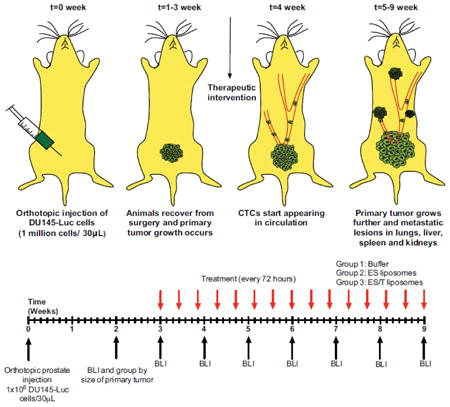
Introduction
Prostate cancer (PCa) is the second most common cancer in men, with an estimated 1.1 million new men diagnosed with prostate cancer and 307,000 deaths worldwide in 2012 [1]. When detected at an early stage, the 5-year survival rate for PCa is close to 100%. In contrast, if diagnosed at a late stage with advanced metastatic disease, the 5-year survival decreases to 33% [1].While effective therapies exist to treat primary tumors of the prostate, metastatic disease is generally considered incurable [2]. The primary tumor sheds tumor cells into the circulation, which can then transit through the bloodstream to form metastases in tissues of the bones, liver, lung, and other organs [1,3]. Clinically, the detection and quantification of circulating tumor cells (CTCs) in PCa patient blood samples before and after treatment is a strong predictor of patient survival – indeed, chemotherapies that cause a net reduction of CTC count from above, to below 5 cells per 7.5 mL of blood (as measured via CellSearch technology) are correlated with improved survival outcomes [4,5]. However, the direct targeting of CTCs as a means to interrupt the metastatic process has gone largely unexplored.
TRAIL is a promising cancer therapeutic because of its ability to induce apoptosis in tumor cells but not normal cells. Since the discovery of TRAIL there have been numerous preclinical trials that showed promising anticancer activity. These results led to the use of TRAIL-receptor agonists (TRAs) in dozens of clinical trials. Unfortunately, the outcomes failed to research their early promise. Primary reasons cited for this failure include the selection of TRAs that were well tolerated but tended to have lower agonist activity and the realization that many primary cancer cells exhibit resistance to TRAIL therapy. Consequently, there has been a push for developing higher-order configurations of TRAIL to increase potency and for developing chemical sensitizers.
Despite these early disappointing results in treating solid tumors, TRAIL has special potency for treating metastasis and in particular inducing apoptosis to circulating tumor cells within the bloodstream. Previous studies suggest that genetic expression of TRAIL is decreased in anchored tumor cells and further that detachment of tumor cells from the primary tumor sensitizes cells to apoptosis [6-8]. Moreover, blocking integrin-mediated cell adhesion has been found to cause increased sensitivity to TRAIL apoptosis [9]. When considering the physiologic conditions of the blood circulation, tumor cells show increased sensitivity to TRAIL-mediated apoptosis within the shear stress environment of the blood circulation. Tumor cells exhibited greater TRAIL-mediated apoptosis proportional to higher shear stress conditions in a manner that was not recapitulated when treating cells with doxorubicin [10]. Based on this collective evidence, technology that can maximize TRAIL capacity to bind to circulating tumor cells could prove to be quite beneficial.
Immune-based therapies are a promising alternative to radiation and chemotherapy treatment of prostate cancer. Currently, the standard of treatment includes radical prostatectomy, hormone injections, radiation therapy and chemotherapy [2,11]. The success of these therapies is often limited, as many patients become resistant to conventional therapies once the primary tumor has metastasized [12]. Prostate cancer lends itself to immunotherapies owing to the strong interaction of host immune cells with prostate cancer cells. Nanomedicine combined with immunotherapy approaches is becoming more attractive as formulations offer customizable functionality, improved pharmacokinetics, and targeting specificity. Functionalizing leukocytes to carry therapeutic nanoparticles has gained attention within the last decade. Largely, these have taken the form of ex vivo manipulations of immune cells. However, few if any studies have utilized immune cells themselves as a carrier for anti-tumor agents.
Recently, we described a unique nanomedicine approach to target and kill CTCs within the flowing blood [13]. Nanoscale liposomes were conjugated with two proteins: E-selectin (ES), a vascular adhesion molecule important in inflammation that binds to carbohydrate ligands on all leukocytes and many types of CTCs, and TRAIL, a protein produced by immune cells that induces apoptosis in cancer cells but has minimal effect on normal cells. When injected into the bloodstream, ES/TRAIL liposomes attach to the surface of peripheral blood leukocytes, which then become cytotoxic to any cancer cells present in the blood. Under physiological flow conditions, this results in near complete elimination of viable cancer cells within 2 h of shearing human blood samples ex vivo, or following liposome and cancer cell injection into the mouse circulation. The aim of the current study was to determine whether ES/TRAIL liposomes could be effective in preventing new metastatic tumor formation in a more realistic model of metastasis: one in which a primary tumor grows and then begins to shed CTCs into the bloodstream, which subsequently colonize distant organs. Orthotopic models of prostate cancer have been widely characterized [14-17] and used to investigate the effect of new therapeutics in mice. In this study, we demonstrated the prevention of metastatic tumor development in an orthotopic xenograft model of PCa, through the sustained delivery of ES/TRAIL liposomes designed to induce apoptosis in circulating tumor cells.
Materials and Methods
Preparation of sterile ES/T liposomes
Multilamellar liposomes, composed of egg L-α-lysophosphatidylcholine (Egg PC), egg sphingomyelin (Egg SM), ovine wool cholesterol (Chol), and 1,2-dioleoyl-sn-glyc- ero-3-[(N-(5-amino-1-carboxypentyl) iminodiacetic acid) succinyl] (nickel salt) (DOGS NTA-Ni) at weight ratios 60-50%:30%:10%:10% (Egg PC/Egg SM/Chol/DOGS NTA-Ni), were prepared using a thin lipid film method. DOGS-NTA-Ni is a lipid conjugated to nickel-nitrilotriacetic acid (Ni-NTA) that allows for attachment to his-tagged proteins. Briefly, stock solutions of all lipids were prepared by dissolving powdered lipids in chloroform to produce a final concentration of 5 mg/mL Egg PC, 20 mg/mL Egg SM, 5 mg/mL Chol, and 20 mg/mL DOGS-NTA-Ni in glass containers and stored at -20°C. Appropriate volumes of the lipids were taken from the stock solution to make lipids in a glass tube and gently dried under nitrogen. To ensure complete removal of chloroform, the lipids were left under vacuum for an additional 12 h. The lipid film was hydrated with a liposome buffer composed of 150 mM NaCl, 10 mM Hepes, and 1 mM MgCl2 dissolved in nuclease-free water to create multilamellar liposomes. The resulting multilamellar liposomes were sized by repeated thawing and freezing, and then subjected to 15 extrusion cycles at 60°C through two different pore size (200 and 100 nm) polycarbonate membranes (Nucleopore; Whatman) to produce unilamellar nanoscale liposomes. A standard autoclaving cycle (15 min, 121°C) was used to sterilize the liposomes after pumping N2 gas into the liposomes in glass ampules to remove oxygen that can cause liquid oxidation. The ampules were then placed in a vacuum degasser to remove residual air and then transferred to autoclave chambers for sterilization. No change in pH or size of the liposomes was observed after autoclaving. The sterilized liposomes were allowed to cool to 4°C and then conjugated with recombinant human TRAIL and ES as described previously. ELISA was used to determine the final concentration of ES and TRAIL on liposomes (R&D Systems). To remove unbound TRAIL and ES, liposomes were diluted 1:6 with liposome buffer and subjected to ultracentrifugation at 100,000 × g for 6 h at 4°C. The mean particle diameter and surface charge (zeta potential) were measured by dynamic light scattering using a Malvern Zetasizer nano ZS (Malvern Instruments Ltd.), according to the manufacturer's protocols. Unconjugated liposomes were measured to be 103.4 ± 10.3 nm in diameter, with a zeta potential of -3.5 ± 2.3 mV before autoclaving and 104.7 ± 17.2 nm in diameter, with a zeta potential of -5.3 ± 1.1 mV after autoclaving. Conjugated liposomes were measured to be a 120.5 ± 5.7 nm in diameter with a zeta potential of -8.3 ± 4.0 mV. The negative zeta potential upon autoclaving could be attributed to the redistribution of phospholipids within the lipid bilayer [18]. The conjugation of proteins on the liposome surface further reduces the zeta potential, which could minimize their interaction with negative plasma proteins in circulation [19]. This change was expected, as the liposomes were not PEGylated and protein conjugation on non-PEGylated liposomes have been shown to reduce the surface charge by other groups [20].
ELISA was used to determine the final concentration of ES and TRAIL on liposomes. Unconjugated proteins were removed by ultracentrifugation and liposomes were dissociated using 0.1% Triton X-100. The dissociated liposomes were then diluted at 1:10, 1:100 and 1:1000 with liposome buffer and coated on 96-well ELISA plates, and a protocol supplied by the manufacturer was used to determine the concentration of ES and TRAIL using appropriate calibration curves created with standard ES and TRAIL provided with the ELISA kit.
Pharmacokinetics of ES/T liposomes
Freshly prepared nanoscale liposomes were conjugated with recombinant E-selectin (ES) (16 ng/mL final concentration) and TRAIL (8 ng/mL final concentration) for 30 min at 37°C and then overnight at 4°C, to ensure maximum protein binding via the interaction between his-tag on recombinant proteins and Ni-NTA on liposomes. To determine the circulation half-life of ES/TRAIL liposomes in the peripheral circulation of mouse, male NOD.CB17-Prkdcscid/J mice (6-8 weeks old, Jackson Laboratories) received retro-orbital injections of either ES/TRAIL liposomes or ES liposomes (120 μL per mouse). At t=3, 36 and 72 hr three mice were sacrificed to ensure maximum blood draw by cardiac puncture. Blood from three mice were pooled for each time point and peripheral polymorphonuclear and mononuclear cells were isolated by density centrifugation on Histopaque-1077 (Sigma Aldrich). About, 1×106 purified leukocytes were incubated for 45 minutes in dark at 4°C with APC conjugated anti-CD45 (eBioscience), a pan-leukocyte maker and its corresponding isotype control (eBioscience). To verify liposome binding to circulating leukocytes, leukocytes were labeled with FITC conjugated anti-human TRAIL (BD Biosciences) and analyzed using a Guava easyCyte™ flow cytometer. At 72 hr, labeled leukocytes were imaged using a Zeiss confocal microscope to verify liposome binding to circulating leukocytes. Appropriate isotype controls were used for all flow cytometry experiment to determine the gating procedure.
Prostate Orthotopic Implantation
Male NOD.CB17-Prkdcscid/J (Jackson Laboratory) mice 6-8 weeks old were used for this study. Animals were placed under anesthesia using 5% isoflurane. Isoflurane was reduced to 2% after the animal was completely anesthetized. Animals were shaved and cleaned using repeated iodine solution and 70% ethanol swabs. Using sterile scalpel, a low midline abdominal incision about 1-3mm wide was created through the skin and muscle layer. The ventral lobes of the prostate were located and 1 million D-luciferase labeled DU145 cells suspended in 50uL PBS were injected into the prostate gland using a 30G needle. Muscles and skin layers from the abdomen wound were closed separately using Vetbond Tissue Adhesive (3M). Animals were monitored every 8 hr and given analgesic medication for 3 days post surgery. Animals were housed in pathogen free conditions and all procedures are done under sterile conditions in approval from Cornell IACUC.
ES/TRAIL Liposome Treatment
Beginning at three-weeks post tumor implantation, animals received retro-orbital injection of, E-Selectin/TRAIL (ES/T), E-Selectin only (ES), or liposome buffer (Buffer). Treatment was administered once every three days and in alternating eye sockets. Injections were performed under 2% isoflurane anesthesia and the eyes were protected via application of eye ointment.
Circulating Tumor Cell (CTC) Enumeration
After 9 weeks post-implantation, blood was collected from three mice in each treatment group via cardiac puncture using a heparin-coated syringe. Blood samples were carefully layered over 3 mL Histopaque-1077 (Sigma Aldrich) and centrifuged at 480 × g for 50 min at room temperature. The buffy coat containing mononuclear cells and cancer cells was recovered and washed twice in resuspension buffer, collected, and placed into culture at 37°C and 5% CO2 under humidified conditions for 6 hr in a 24-well plate. The number of CTCs was determined for mCherry positive DU145 cells in culture. Fluorescent micrographs were taken at 100 randomly selected locations within each well, and total DU145 cells were estimated based on the total well area and reported as the number of cells per mL of blood in Figure 3.
Fig 3. Quantification of bioluminescence signal of DU145 cells following orthotopic tumor implantation.
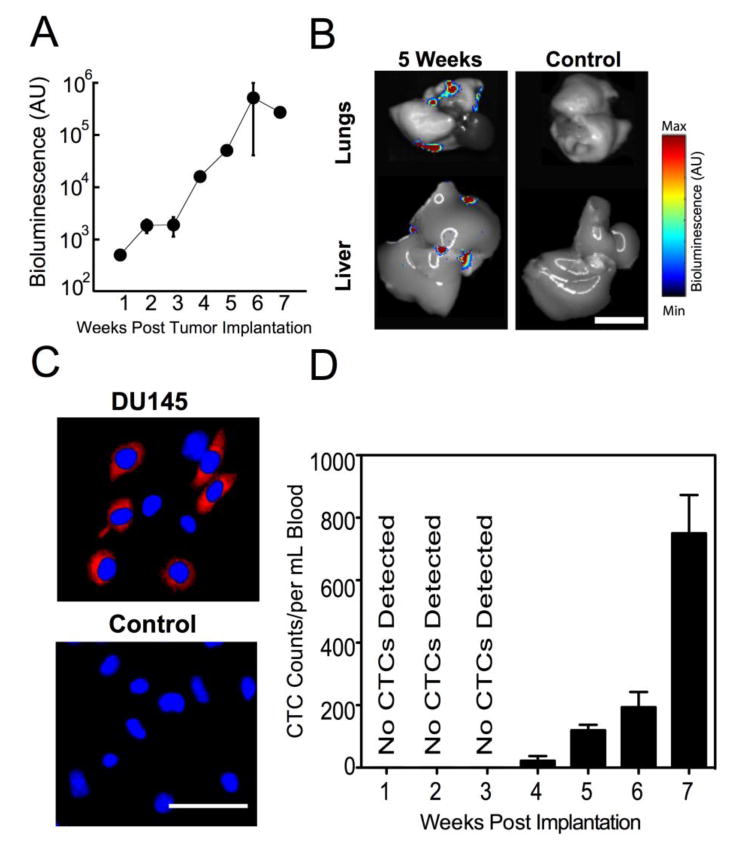
(A) Increased bioluminescence signal from tumor growth over time. (B) Bioluminescent signal from excised organs 5 weeks after tumor implantation (left) and age-matched control (right). (Scale bar = 1cm) (C) Confocal micrographs of buffy coat isolated from control mice (age-matched non-tumor bearing SCID mice) and mice bearing orthotopic DU145 tumor six weeks after implantation (blue, cell nuclei and red, mCherry expressing DU145 cells). (Scale bar = 100μm) (D) Number of CTCs per volume of blood, measured weekly after orthotopic implantation of tumor. n=3 mice were analyzed each week. Error bars represent the mean ± SD at each timepoint.
Bioluminescence Imaging
Post tumor implantation, animals were monitored weekly for bioluminescent activity. Luciferin was administered at 150mg/kg per animal via intraperitoneal injection using 30G insulin syringe needle. Animals are placed under anesthesia using 2% isoflurane and imaged 5 min post injection for maximum bioluminescence signal. Images were taken at 1 s exposure time using a Xenogen IVIS 200 Imaging System. For the quantitative measurements of average radiance, the area of the primary BLI was constant throughout all time points for each animal.
For the ex vivo organ quantitative measurements of BLI activity, luciferin was injected into the animal 5 minutes before euthanasia. After euthanasia, organs were immediately removed and placed in warm PBS containing luciferin. Once all organs were collected, the organs were arranged on a black sheet and imaged for 120 s for all samples. Each animal was imaged within 5 min post euthanasia for optimal detection of luciferase activity. Luminescence imaging data was acquired using a luminescence-imaging box custom-built by staff at the Cornell University Biotechnology Resource Center.
Individual bioluminescent organ intensities were summed across each image and divided by the number of pixels using a custom Matlab code. Background signal was subtracted from each image. Representative images from each treatment group were displayed.
Liver Enzyme Measurement
Human hepatocytes have shown sensitivity to TRAIL at sufficiently high dosages and we measured the serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) to assess liver function in treated mice. These proteins, if present in elevated levels in serum, indicate leakage from damaged cells due to inflammation or apoptosis. These enzymes normally reside inside the cells and any damage to the liver cells usually represents hepatocellular damage. Serum levels of ALT and AST were analyzed using a colorimetric assay kit (BioVision).
Hematocrit
Blood was drawn via cardiac puncture using a 28G heparin coated syringe and collected in a heparinized tube. Two drops per animal were placed in hematocrit tubes and spun for 2 minutes using CritSpin Microhematocrit Centrifuge. The ratio of red blood cells to total blood volume was used to calculate the hematocrit.
Weight
Every animal was weighted at least once per week throughout the duration of the experiment. Control animal weight was contributed from Jackson Laboratory website information about this specific strain. There were no significant differences in animal weight between the treatment groups.
Histology
After bioluminescence imaging, samples were transferred to 4% PFA, stored overnight and then transferred to 30% sucrose in PBS. Samples were then submitted to the Cornell Histology Lab for H&E staining and scoring by a veterinary pathologist. For scoring purposes the pathologist had no knowledge of which animals belonged to which treatment groups. Representative images were acquired for presentation.
TUNEL staining was performed on 20-micron thick sections that were incubated in TUNEL-mix (Roche) at 37°C from 60 min. A DAPI counterstain was applied for 3 min to label nuclei. Fluorescent images were acquired immediately after staining and the image exposure time was kept constant for each channel throughout each treatment group.
Results and Discussion
ES-TRAIL liposome coated leukocytes remain in circulation for greater than 96 hrs
To determine an appropriate timescale for ES-TRAIL treatment, we performed pharmacokinetic studies to determine the half-life of ES-TRAIL in circulation. Blood cells were isolated from mouse blood following injection of liposome treatment groups. Flow analysis was performed to identify the population of cells expressing CD45, a widespread marker for leukocytes, and human TRAIL. It was determined that within 30 min, nearly 100% of leukocytes stained positive for human TRAIL (Fig. 1). Since soluble TRAIL has been widely reported by other scientists as well as in our previous publications to have a half-life <30 minutes and ineffective against circulating tumor cells, a soluble TRAIL treatment group was not included. By 72 hr, only 10% of leukocytes maintained surface TRAIL (Fig. 1). The half-life was thus estimated to be ∼35hrs. Based on the pharmacokinetics presented, animals were treated every 72 hr.
Fig 1. Pharmacokinetic dynamics of TRAIL-coated leukocytes.
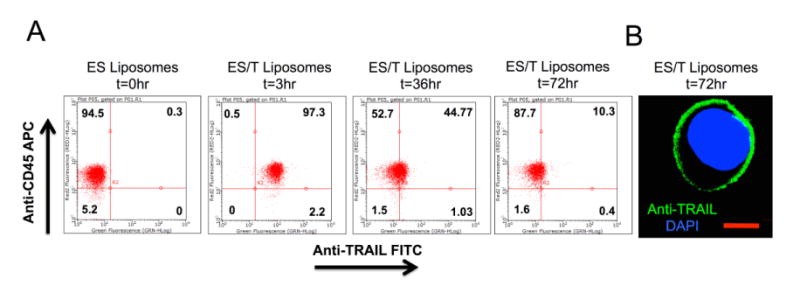
(A) Scatter plots showing percentage of circulating mouse leukocytes staining positive for surface bound human TRAIL protein. (B) Confocal micrograph of a mouse leukocyte presenting human TRAIL protein on its surface at t=72 hours (Scale bar = 10μm).
In a separate experiment, confocal imaging was used to confirm that 72 hr post injection ES/TRAIL liposomes remained bound to the leukocyte surface and had not been internalized (Fig. 1). In addition, the confocal images suggest that the exponential decrease in ES/TRAIL coated leukocytes is not due to the shedding or unbinding of ES/TRAIL liposomes from the leukocyte membrane (Fig.1B). Rather, the half-life is likely linked to the lifespan of circulating leukocytes themselves. As an example, neutrophils are the most abundant leukocyte population and have a short half-lifeof 6-8 hours [21]. Thus, we expect that the emergence of new cells and the death of older cells contribute to the decrease in populations of ES/TRAIL liposome labeled leukocytes.
ES/TRAIL liposome coated leukocytes result in reduction in size of the primary tumor
Dual labeled DU145-luc-mcherry prostate cells were orthotopically injected into the prostate of NOD-SCID animals (Fig. 2A). We observed that CTC populations were detectable after 4 weeks (Fig. 3) and decided to begin treatment in the proceeding week (Fig. 2B). Mice were sorted by primary tumor size at week 2, and three tumor size-matched groups of mice (n=8 each) were formed. The experiment was terminated at week 9 as a humane endpoint, as several of the mice in the two control groups (buffer treatment, ES liposomes without TRAIL) showed visible signs of distress such as skin lesions and lethargy. At week 9, whole animal BLI showed the widespread presence of metastatic tumors throughout the abdominal cavity in the two control groups, with no macroscopic metastases visible in the ES/TRAIL treatment group (Fig. 2C). Remarkably, ES/TRAIL liposomes also caused significant reduction in the growth rate of the primary tumor (Fig. 2D), with no BLI signal detectable in treated mice when imaged from the dorsal side (Fig. 2C). This difference in treatment group is likely due to ES-TRAIL liposome exposure to the cells in the primary tumor due to leukocyte infiltration as is typically associated with cancer inflammation [22]. Our previously published data indicated that ES liposomes may keep CTCs in circulation by blocking the ES-mediated adhesion of CTCs to the endothelium [23]. The difference in the average radiance between the ES-only and Buffer treatment groups, though not significantly different, suggests that leukocytes functionalized with ES liposomes may also be interacting with cells within the primary tumor. In this light, the stark difference in primary tumor growth between ES and ES/TRAIL treated animals corroborates that ES binding facilitates adhesion to cancer cells and, when combined with the apoptosis inducing TRAIL, leads to efficient cancer cell death.
Fig 2. Test of ES/TRAIL liposomes to prevent metastasis in an orthotopic model of prostate cancer.
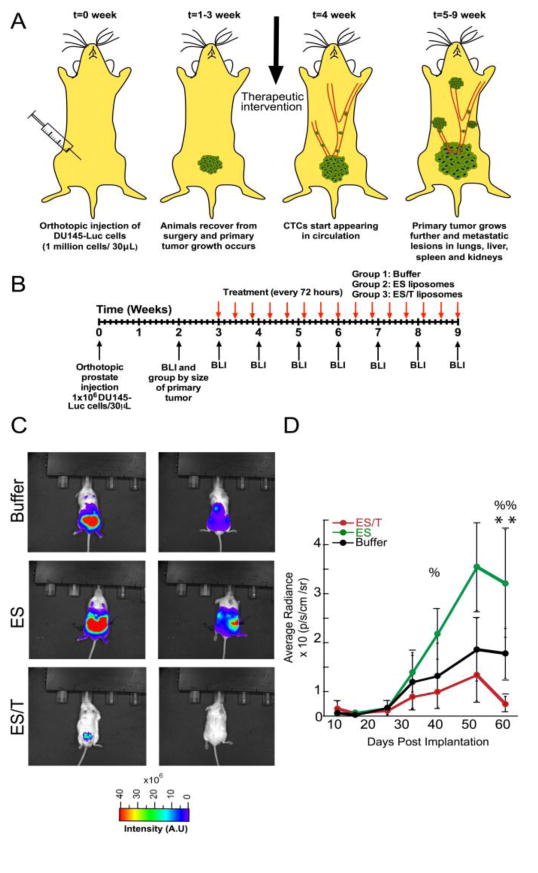
(A) Schematic of orthotopic xenograft model for metastatic prostate cancer progression. (B) Timeline for ES/T liposome efficacy trial in tumor-bearing mice. (C) Whole animal BLI of the ventral (left) and dorsal (right) side of representative animals from each treatment group at the end of the trial (week 9) (D) Average radiance from the primary tumor. Comparisons were made via one-way ANOVA with Tukey posttest. Error bars represent the mean ± SD at each timepoint. ES vs ES/TRAIL: %=(p < 0.05) %%=(p<0.01). Buffer vs ES/TRAIL: **=(p<0.05).
ES/TRAIL functionalized leukocytes reduce the number of circulating tumor cells in a prostate orthotopic tumor model
In a previous publication, we showed that ES/TRAIL functionalized leukocytes were able to kill over 90% of CTCs within 2 hrs of circulation time using COLO 205 human cancer cells [23]. In the current study, we seek to determine whether ES/TRAIL liposomes were successful in eliminating CTCs over a sustained period of time where there was already an established primary tumor. For this study we used a different cancer cell line and found that DU145 cells also undergo apoptosis when exposed to ES/TRAIL liposomes in blood (Supplementary Fig. 2). Blood was collected from all mice at the end of the in vivo study to quantify number of CTCs. Quantitative comparison of the CTC count in buffer-treated mice (∼1,600 CTCs/mL blood) vs. ES/TRAIL liposome-treated mice (∼100 CTCs/mL blood) shows a difference of about 94% (Fig. 4A). Representative fluorescent images reveal dramatic decrease in collected CTCs from ES/TRAIL liposome-treated mice (Fig. 4B).
Fig 4. ES/TRAIL liposome treatment is effective in significantly reducing the number of circulating tumor cells in blood.
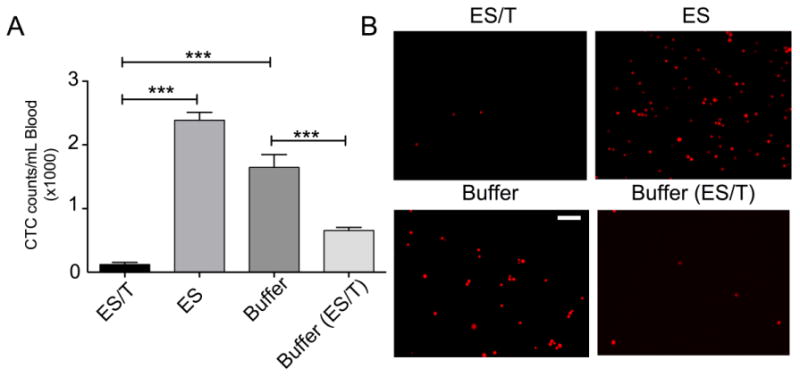
(A) Average number of CTCs in the peripheral circulation post treatment. n=3 mice for each group. Bars represent mean ± SD for each group. (B) Representative fluorescent micrographs showing mCherry positive DU145 cells isolated from whole blood of animals from different treatment groups at the end of the study. Scale bar = 100μm. n= 6, 6, 6, and 3 for treatment groups ES/T, ES, Buffer, Buffer(ES/T), respectively. Error bars represent the mean ± SD in each group. ***p<0.0001
To determine the effect of ES/TRAIL liposomes on untreated blood with high tumor burden, 3 animals from the Buffer-treatment group were treated with a single dose of ES/TRAIL (indicated as Buffer (ES/T)). Remarkably, this single dose resulted in a greater than 50% reduction in CTC count (Fig. 4A). Furthermore, the dramatic effect of a single late-stage dose of ES/TRAIL suggests that CTC targeting could yield therapeutic benefit to patients even at later stages associated with high CTC count. Taken together with the absence of metastatic tumors observed in ES/TRAIL liposome-treated mice (Figs. 5 and Supplementary Fig. 1), this indicates that CTC targeting need not be 100% effective in order to achieve the clinically significant outcome of metastasis prevention. That is, by tipping the scales and reducing the metastatic load in the peripheral circulation to a fraction of its untreated numbers, the inherent inefficiency of the metastatic process could take over to benefit the patient. Likely, these remaining CTCs undergo anoikis and immune clearance and lack sufficient numbers to successfully colonize distant organs.
Fig 5. Ex vivo BLI analysis reveals that ES/TRAIL liposomes block widespread metastasis.
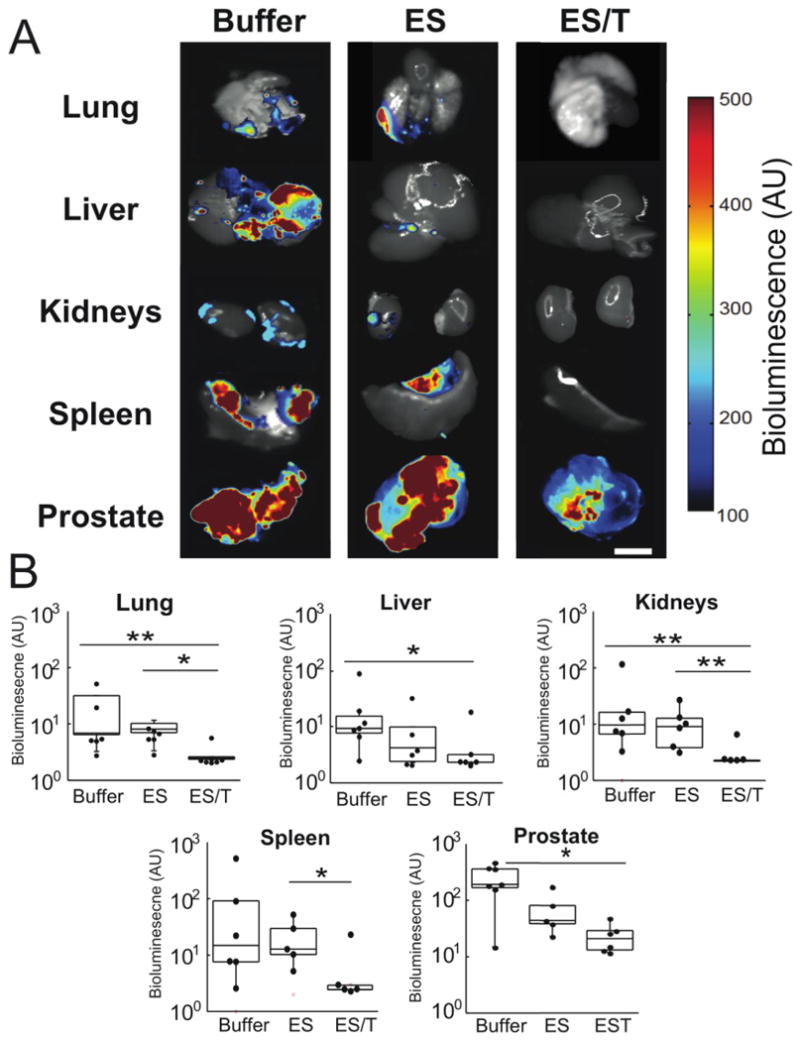
(A) Representative BLI images reveal the spread of DU145-Luc cells to lung, liver, kidney and spleen across Buffer, ES, and ES/T treatment groups (left-to-right). Scale bar=1cm. (B) Bioluminescent signal from each organ was quantified for each treatment groups: Buffer (n=8), ES-liposomes (n=6), and ES/T (n=6). Data are plotted on a log scale. Individual data points represent total organ signal from an individual animal. Superimposed box plots bound the 25th to 75th percentage of all data points and the whiskers extend 1.5 times the interquartile range beyond boxes. *P<0.05, **P<0.01 (one-way ANOVA with Tukey posttest).
Ex-vivo organ analysis reveals reduction in metastasis in ES-TRAIL treated animals
To determine whether ES-TRAIL was successful in reducing metastases, we assayed growth of tumors in distant organs. Organs removed for ex vivo BLI analysis revealed widespread proliferation of luciferase-expressing DU145 cancer cells in the lungs and liver, kidneys and spleen of the buffer-treated and ES liposome treated control mice (Fig. 5A) with virtually no signal above background in ES/TRAIL liposome treated mice (Fig. 5B). All images were taken at the same exposure. Each image is a composite of the black and white camera photo and bioluminescence signal. Difference in organ coloration is due to natural organ pigmentation in addition to variations in the presence of blood at the time of organ harvesting.
While the present study was principally focused on suppressing the development of bloodborne metastasis, a significant decrease in primary tumor size was also observed (Supplementary Fig. 1), together with a reduction in the presence of abdominal visceral metastases observed at the organ boundaries in untreated mice. This suggests that TRAIL-coated leukocytes, such as lymphocytes, enter into the visceral fluid of the peritoneal cavity and the primary tumor. The presence of lymphocytes is consistent with the well-documented leaky phenotype associated with the NOD.CB17-Prkdcscid/J mouse line and is confirmed with our previous characterization of ES liposome binding to nearly all leukocyte subtypes [23]. Hence, decorating leukocytes with ES/TRAIL liposomes can provide the additional benefit of drug delivery to tumors by attaching drug to cell populations that will intrinsically aggregate in the region of disease.
Histology was performed on all organs and interpreted by an independent veterinary pathologist, with results found to be consistent with the BLI analysis (Supplementary Fig. 3). Specifically, metastatic prostate carcinoma was found in the lung, diaphragm, liver and peri-adipose tissue of the kidneys and spleen in buffer and ES liposome treated animals, with some minimal evidence of scattered neoplastic cells in the lungs and liver of ES/TRAIL treated animals.
Toxicity studies indicate little negative effects with ES-TRAIL liposome treatment
The dosage of TRAIL used in the ES/TRAIL liposome formulation represents ∼1.0% of the concentrations that were well tolerated in previous animal and human trials with soluble TRAIL protein [24,25], and so consistent with our prior study we did not expect to see evidence of toxicity [23]. We examined for common signs of TRAIL toxicity observed in previous rodent trials featuring extremely high doses of TRAIL [26,27]. Indeed, mice treated with 15 consecutive doses of ES/TRAIL liposomes showed no evidence of: (Fig. 6A) elevated liver enzymes in the serum; (Fig. 6B) significant difference in hematocrit relative to untreated mice; (Fig. 6C) loss of appetite or body weight or behavioral distress compared to untreated mice; (Fig. 6D); or enlarged kidney mass (Fig. 6E). It should be noted that two mice in the treatment group died within hours of liposome injection in week 5, which we attribute to imperfect liposome sterilization since local retro-orbital inflammation was immediately evident and no characteristic signs of TRAIL toxicity were noted in these animals. In a previous pilot study, 100% of liposome-treated animals experienced such acute sepsis before our liposome sterilization procedures were improved to include an autoclaving step prior to protein conjugation. Future work with these materials should utilize current Good Manufacturing Processes that are not currently available to our laboratory.
Fig 6. ES/TRAIL treatment does not induce toxicity.
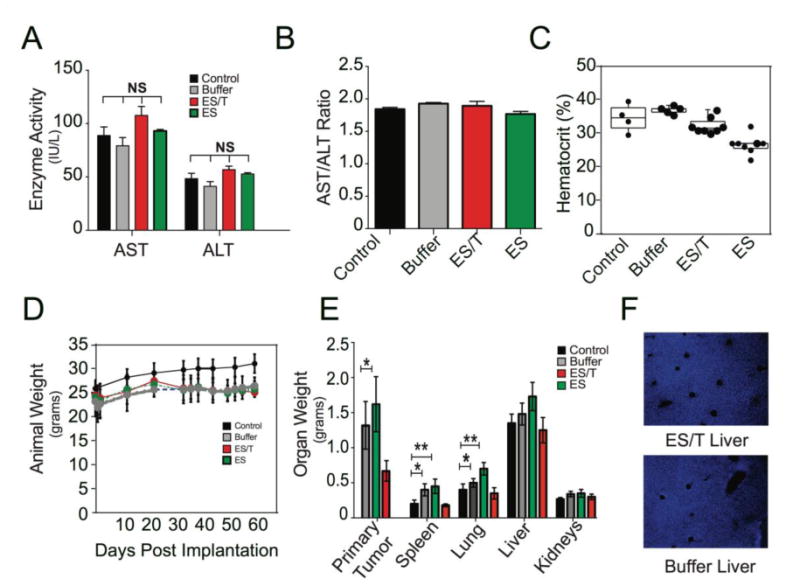
(A) Serum levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) liver enzymes in mice from different treatment groups and aged matched controls at the end of the study. Bars represent mean ± SD for enzyme levels from three mice in each treatment group. (B) AST/ALT ratio for mice from different treatment groups and aged matched controls. AST/ALT >2 indicates liver toxicity. (C) Systemic hematocrit of mouse blood. Each dot represents the hematocrit measurement from an individual animal. Blood was drawn prior to euthanasia. Control group (Control) represents age-matched and strain-matched animals that received no tumor or drug. (D) Weight of animals throughout the experiment. Each dot represents the average weight (grams) of each animal. Control group represents age-matched and strain-matched animals that received no tumor or drug. (E) Weight of excised organs post-mortem. (F) TUNEL staining of liver from ES/T liposome and Buffer treated animals. Error bars represent mean ± SD of each treatment group. NS= non-significant difference. Comparisons made using one way-ANOVA. *p<0.01, **p<0.001.
Discussion
In this paper, we present an innovative scheme for TRAIL presentation to treat metastatic disease by specifically targeting circulating tumor cells. Past preclinical studies and clinical trials have focused on delivery of TRAIL variants to treat solid tumors, rather than metastatic cells within the bloodstream [28,29]. In fact, CTCs are mostly valued as predictors of disease progression rather than a bottleneck of metastasis that are available for targeting. Our study explicitly demonstrates that directly eliminating CTCs can reduce metastasis disease in secondary organs.
The therapeutic effect is due to the liposomal-bound TRAIL presentation on the surface of leukocytes and amplified binding opportunities between TRAIL and CTCs mediated by margination in the bloodstream. TRAIL is a membrane bound protein expressed on the surface of natural killer cells. Liposomal carriers increase therapeutic efficacy by presenting TRAIL in its natural, membrane-bound state. No further cleavage or activation of TRAIL is necessary for this formulation. We have also shown that ES/TRAIL liposomes are stable in whole blood isolated from healthy volunteers. In our previous work we established that ES/TRAIL liposomes can effectively target cancer cells spiked into whole blood collected from healthy volunteers under physiologically relevant shear stress conditions, recapitulated in a cone-and-plate viscometer [23]. The ES/TRAIL liposomes were effective under different hematocrit levels, indicating their stability in whole blood.
Past and present works support these findings that the therapeutic potential of TRAIL is increased even in TRAIL resistant cancer cell lines—without the use of sensitization agents—when presented on lipid bilayers [30]. Furthermore, the fluid dynamics of blood flow cause an effect called margination that results in increased binding/contact opportunities between ES/TRAIL coated leukocytes and CTCs. In the fluid shear stress microenvironment of the blood circulation, larger, less deformable objects are deflected away from the center and towards the vascular wall. Both CTCs and leukocytes are both relatively large cells and bind to ES ligands to roll along the vascular endothelium. It is in this environment that these cells are able to undergo repeated collisions and TRAIL that is bound to leukocytes has more opportunities to bind to CTCs. It should also be noted that TRAIL liposomes tethered to the surface of circulating leukocytes exhibited a circulation half-life (Fig. 1) significantly longer than any previously described nanoparticle formulations of TRAIL [31-33]. Nanomedicine-based therapeutic approaches such as the one demonstrated here may hold the key to exploiting the potency of TRAIL within the circulation.
In addition, many cell-based therapies are rising in popularity due to their natural ability to migrate toward and penetrate inflamed regions that are often difficult to achieve with drug delivery mechanisms. The mechanism presented here is unique because it does not require any ex vivo-based manipulations of immune cells for enhanced therapeutic effect. In this way, one is able to avoid donor harvesting, specific activation and production challenges associated with adoptive T cell therapies and cell-mediated drug delivery, which carries drug nanoparticles internally. Since many other common cancers such as those derived from breast, lung, colon and skin are known to metastasize through the bloodstream [34] and given the dramatic decrease in the number of CTCs after only one dose, ES/TRAIL liposomes could be administered clinically as a preventative measure for newly diagnosed patients. Most cell types exhibit some level of apoptosis response to the TRAIL ligand [35], nonetheless, should a specific tumor prove to be completely resistant to TRAIL therapy, scores of studies have been published over the past 15 years that report new chemical agents (chemotherapeutics, inhibitors, natural products) that are able to sensitize cancer cells to TRAIL-mediated apoptosis [36].
Future work should focus on testing the efficacy of ES/TRAIL liposomes in other models of bloodborne metastatic cancer, and the ex vivo testing of patient-derived CTCs under physiologic blood flow conditions. While E-Selectin is a broad target for leukocytes, further specificity can be gained by using ligands specific for leukocyte subpopulations such as NK killer cells, B-cells and T-cells that are current promising targets in cancer immunotherapy. Finally, it should be noted that while our focus has been on cancer metastasis, this drug delivery mechanism could be applied to other diseases where there is substantial leukocyte infiltration such as neurodegenerative diseases.
Supplementary Material
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Crehange G, Roach MI, Martin E, Cormier L, Peiffert D, Cochet A, et al. Salvage reirradiation for locoregional failure after radiation therapy for prostate cancer: Who, when, where and how? Cancer Radiotherapie. 2014 Oct;18(5-6):524–34. doi: 10.1016/j.canrad.2014.07.153. [DOI] [PubMed] [Google Scholar]
- 3.Wilt TJ, Ahmed HU. Prostate cancer screening and the management of clinically localized disease. BMJ. 2013;346:f325. doi: 10.1136/bmj.f325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delacruz A. Using circulating tumor cells as a prognostic indicator in metastatic castration-resistant prostate cancer. Clin J Oncol Nurs. 2012 Apr;16(2):E44–7. doi: 10.1188/12.CJON.E44-E47. [DOI] [PubMed] [Google Scholar]
- 5.Ma X, Xiao Z, Li X, Wang F, Zhang J, Zhou R, et al. Prognostic role of circulating tumor cells and disseminated tumor cells in patients with prostate cancer: a systematic review and meta-analysis. Tumour Biol. 2014 Jun;35(6):5551–60. doi: 10.1007/s13277-014-1731-5. [DOI] [PubMed] [Google Scholar]
- 6.Grosse-Wilde A, Kemp CJ. Metastasis Suppressor Function of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand-R in Mice: Implications for TRAIL-Based Therapy in Humans? Cancer Research. 2008 Aug 1;68(15):6035–7. doi: 10.1158/0008-5472.CAN-08-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldberg GS, Jin Z, Ichikawa H, Naito A, Ohki M, El-Deiry WS, et al. Global effects of anchorage on gene expression during mammary carcinoma cell growth reveal role of tumor necrosis factor-related apoptosis-inducing ligand in anoikis. Cancer Research. 2001 Feb 15;61(4):1334–7. [PubMed] [Google Scholar]
- 8.Lane D, Cartier A, Rancourt C, Piché A. Cell detachment modulates TRAIL resistance in ovarian cancer cells by downregulating the phosphatidylinositol 3-kinase/Akt pathway. Int J Gynecol Cancer. 2008 Jul;18(4):670–6. doi: 10.1111/j.1525-1438.2007.01062.x. [DOI] [PubMed] [Google Scholar]
- 9.Phipps LE, Hino S, Muschel RJ. Targeting Cell Spreading: A Method of Sensitizing Metastatic Tumor Cells to TRAIL-Induced Apoptosis. Molecular Cancer Research. 2011 Mar 15;9(3):249–58. doi: 10.1158/1541-7786.MCR-11-0021. [DOI] [PubMed] [Google Scholar]
- 10.Mitchell MJ, King MR. Fluid shear stress sensitizes cancer cells to receptor-mediated apoptosis via trimeric death receptors. New J Phys. 2013 Jan 1;15(1):015008. doi: 10.1088/1367-2630/15/1/015008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wallace TJ, Torre T, Grob M, Yu J, Avital I, Bruecher B, et al. Current Approaches, Challenges and Future Directions for Monitoring Treatment Response in Prostate Cancer. Journal of Cancer. 2014;5(1):3–24. doi: 10.7150/jca.7709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fernández-García EM, Vera-Badillo FE, Perez-Valderrama B, Matos-Pita AS, Duran I. Immunotherapy in prostate cancer: review of the current evidence. Clin Transl Oncol. 2015 May;17(5):339–57. doi: 10.1007/s12094-014-1259-6. [DOI] [PubMed] [Google Scholar]
- 13.Mitchell MJ, King MR. Leukocytes as carriers for targeted cancer drug delivery. Expert Opinion on Drug Delivery Informa UK, Ltd. 2015;12(3):000–0. doi: 10.1517/17425247.2015.966684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rembrink K, Romijn JC, vanderKwast TH, Rubben H, Schroder FH. Orthotopic implantation of human prostate cancer cell lines: A clinically relevant animal model for metastatic prostate cancer. Prostate. 1997;31(3):168–74. doi: 10.1002/(sici)1097-0045(19970515)31:3<168::aid-pros4>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 15.Bastide C, Bagnis C, Mannoni P, Hassoun J, Bladou F. A Nod Scid mouse model to study human prostate cancer. Prostate Cancer Prostatic Dis. 2002;5(4):311–5. doi: 10.1038/sj.pcan.4500606. [DOI] [PubMed] [Google Scholar]
- 16.Scatena CD, Hepner MA, Oei YA, Dusich JM, Yu S-F, Purchio T, et al. Imaging of bioluminescent LNCaP-luc-M6 Tumors: A new animal model for the study of metastatic human prostate cancer. Prostate. 2004 May 15;59(3):292–303. doi: 10.1002/pros.20003. [DOI] [PubMed] [Google Scholar]
- 17.Waters DJ, Janovitz EB, Chan TCK. Spontaneous metastasis of PC-3 cells in athymic mice after implantation in orthotopic or ectopic microenvironments. Prostate. 1995 May;26(5):227–34. doi: 10.1002/pros.2990260502. [DOI] [PubMed] [Google Scholar]
- 18.Chaturvedi PR, Patel NM, Lodhi SA. Effect of terminal heat sterilization on the stability of phospholipid-stabilized submicron emulsions. Acta Pharm Nord. 1992;4(1):51–5. [PubMed] [Google Scholar]
- 19.Honary S, Zahir F. Effect of Zeta Potential on the Properties of Nano-Drug Delivery Systems - A Review (Part 1) Trop J Pharm Res. 2013 May 9;12(2) [Google Scholar]
- 20.Blenke EO, Klaasse G, Merten H, Plückthun A, Mastrobattista E, Martin NI. J Control Release. C. Vol. 202. Elsevier B.V; 2015. Mar 28, Journal of Controlled Release; pp. 14–20. [DOI] [PubMed] [Google Scholar]
- 21.Gabelloni ML, Trevani AS, Sabatté J, Geffner J. Mechanisms regulating neutrophil survival and cell death. Semin Immunopathol. 2013 Feb 1;35(4):423–37. doi: 10.1007/s00281-013-0364-x. [DOI] [PubMed] [Google Scholar]
- 22.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008 Jul 24;454(7203):436–44. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 23.Mitchell MJ, Wayne E, Rana K, Schaffer CB, King MR. TRAIL-coated leukocytes that kill cancer cells in the circulation Proceedings of the. 3. Vol. 111. National Academy of Sciences; 2014. Jan 21, pp. 930–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ashkenazi A, Holland P, Eckhardt SG. Ligand-Based Targeting of Apoptosis in Cancer: The Potential of Recombinant Human Apoptosis Ligand 2/Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (rhApo2L/TRAIL) Journal of Clinical Oncology. 2008 Jul 20;26(21):3621–30. doi: 10.1200/JCO.2007.15.7198. [DOI] [PubMed] [Google Scholar]
- 25.Herbst RS, Eckhardt SG, Kurzrock R, Ebbinghaus S, O'Dwyer PJ, Gordon MS, et al. Phase I Dose-Escalation Study of Recombinant Human Apo2L/TRAIL, a Dual Proapoptotic Receptor Agonist, in Patients With Advanced Cancer. Journal of Clinical Oncology. 2010 Jun 8;28(17):2839–46. doi: 10.1200/JCO.2009.25.1991. [DOI] [PubMed] [Google Scholar]
- 26.Zhang D, Hu X, Qian L, Chen SH, Zhou H. Microglial MAC1 receptor and PI3K are essential in mediating b-amyloid peptide-induced microglial activation and subsequent neurotoxicity. Journal of Neuroinflammation. 2011;8(3):1–14. doi: 10.1186/1742-2094-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zou YX, Zhang XD, Mao Y, Lu GC, Huang M, Yuan BJ. Acute toxicity of a single dose DATR, recombinant soluble human TRAIL mutant, in rodents and crab-eating macaques. Hum Exp Toxicol. 2010 Aug;29(8):645–52. doi: 10.1177/0960327109357214. [DOI] [PubMed] [Google Scholar]
- 28.Tolcher AW, Mita M, Meropol NJ, Mehren von M, Patnaik A, Padavic K. Phase I pharmacokinetic and biologic correlative study of mapatumumab, a fully human monoclonal antibody with agonist activity to tumor necrosis factor-related apoptosis-inducing ligand receptor-1. Journal of Clinical Oncology. 2007 Apr 10;25(11):1390–5. doi: 10.1200/JCO.2006.08.8898. [DOI] [PubMed] [Google Scholar]
- 29.Zhang H-Y, Man J-H, Liang B, Zhou T, Wang C-H, Li T, et al. Cancer Gene Therapy. 5. Vol. 17. Nature Publishing Group; 2010. Jan 15, Tumor-targeted delivery of biologically active TRAIL protein; pp. 334–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nair PM, Flores H, Gogineni A, Marsters S, Lawrence DA, Kelley RF, et al. Enhancing the antitumor efficacy of a cell-surface death ligand by covalent membrane display Proceedings of the. National Academy of Sciences; 2015. Apr 20, p. 201418962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seifert O, Pollak N, Nusser A, Steiniger F, Rüger R, Pfizenmaier K, et al. Immuno-LipoTRAIL: Targeted delivery of TRAIL-functionalized liposomal nanoparticles. Bioconjug Chem. 2014 May 21;25(5):879–87. doi: 10.1021/bc400517j. [DOI] [PubMed] [Google Scholar]
- 32.Lim SM, Kim TH, Jiang HH, Park CW, Lee S, Chen X, et al. Improved biological half-life and anti-tumor activity of TNF-related apoptosis-inducing ligand (TRAIL) using PEG-exposed nanoparticles. Biomaterials. 2011 May;32(13):3538–46. doi: 10.1016/j.biomaterials.2011.01.054. [DOI] [PubMed] [Google Scholar]
- 33.Chae SY, Kim TH, Park K, Jin CH, Son S, Lee S, et al. Improved Antitumor Activity and Tumor Targeting of NH2-Terminal-Specific PEGylated Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand. Molecular Cancer Therapeutics. 2010 Jun 9;9(6):1719–29. doi: 10.1158/1535-7163.MCT-09-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MacDonald IC, Groom AC, Chambers AF. Cancer spread and micrometastasis development: quantitative approaches forin vivo models. Bioessays. 2002 Sep 7;24(10):885–93. doi: 10.1002/bies.10156. [DOI] [PubMed] [Google Scholar]
- 35.Stuckey DW, Shah K. TRAIL on trial: preclinical advances in cancer therapy. Trends Mol Med. 2013 Nov;19(11):685–94. doi: 10.1016/j.molmed.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ashkenazi A, Herbst RS. To kill a tumor cell: the potential of proapoptotic receptor agonists. J Clin Invest. 2008 Jun 2;118(6):1979–90. doi: 10.1172/JCI34359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.