

Abstract
From July 19–24, 2015, 169 clinicians and basic scientists gathered in the vertiginous heights of Snowmass, Colorado (2,502 m) for the fourth FASEB summer research conference on the ‘Biology of Cilia and Flagella’. Organizers Maureen Barr (Rutgers University), Iain Drummond (Massachusetts General Hospital/Harvard Medical School), and Jagesh Shah (Brigham and Women’s Hospital/Harvard Medical School) assembled a program filled with new data and forward-thinking ideas documenting the ongoing growth of the field. Sixty oral presentations and 77 posters covered novel aspects of cilia structure, ciliogenesis, cilia motility, cilia-mediated signaling, and cilia-related disease. In this report, we summarize the meeting, highlight exciting developments and discuss open questions.
Keywords: Arl3, Arl13b, axoneme, Bardet-Biedl syndrome (BBS), calcium, ciliopathy, ciliary membrane, craniofacial defects, diabetes, dynein, GPCR, Hedgehog signaling, intraflagellar transport (IFT), motility, NPHP, obesity, phosphoinositide, polycystin, transition zone, tubulin
Open sesame – the ciliary gate
Located between the basal body and the axoneme is the transition zone (TZ), proposed to function both as a barrier preventing ciliary entry of proteins by diffusion and as a gate regulating the admission of proteins into the ciliary compartment. Opening the meeting with her keynote lecture, Susan Dutcher (Washington University) focused on the ciliary necklace and bracelet, rings of intramembrane particles of unknown composition and function in Chlamydomonas. Deep etch electron microcopy revealed that the necklace became progressively disorganized in the absence of IFT. Proteomics was used to test how a defective necklace affects the protein composition of cilia. Unexpectedly, numerous proteins were missing from cilia isolated from the conditional IFT mutant fla11-1, which at the restrictive temperature failed to maintain the necklace. These observations argue against a role of the necklace in preventing protein entry by diffusion; instead the necklace might function in the active transport of select proteins into the cilium. In the region of the TZ, the axonemal microtubules are decorated by a complex system of interconnected fibers, which contact the plasma membrane and, depending on the level of sectioning, appear as either Y-linkers in the TZ, proximal transitional fibers (TFs), or more distal cylindrical fibers; the latter might define the inversin compartment (Geimer and Melkonian, 2004). TZ proteins are hot spots for mutations resulting in human disease. Michel Leroux (Simon Fraser University, Canada) presented new insights into the modular and hierarchical organization and components of the TZ. Genetic interaction studies revealed that MKS5 and RPGRIP1L are central TZ organizers and are required for assembly of the multiprotein MKS and NPHP modules. During C. elegans development, the TZ assembles within a small time window and expression profiling was used to identify novel TZ components. These included transmembrane protein 218 (TMEM218), a novel component of the MKS module, and the cyclin-dependent kinase-like 1 (CDKL1), which controls ciliary length. Using dSTORM super-resolution imaging, Jung-Chi Liao (Institute of Atomic and Molecular Sciences, Academia Sinica, Taiwan) provided a particularly clear view of the distal appendages in mammalian cells, which may be homologous to the TFs. CEP83, CEP89, SCLT1, CEP164, and FBF1 were included into a molecular map of the distal appendages, which are just 120-nm in height. FBF1, for example, was visible as a ring of nine dots and is likely to be at the tip where the distal appendages contact the plasma membrane. A similar analysis of TZ using multiple markers (MKS1, TMEM67/MKS3, CEP290/MKS4, RPGRIP1L/MKS5, and TCTN2/MKS8) showed CEP290 at the proximal end, RPGRIP1L in the center and MKS1 more lateral (Fig. 1; Yang et al., 2015).
Fig. 1.

Superresolution STED imaging of proteins in the distal appendages and transition zone superimposed onto an electron micrograph. Shown are centrin (dark blue), CEP164 (light blue), CEP290 (magenta), TMEM67 (green), TCTN2 (yellow), and RPGRIP1L (red). Reprinted from Yang et al., 2015. Courtesy of Jung-Chi Liao.
A major challenge now is to link the increasingly detailed knowledge of TZ structure, organization, and composition to the mechanisms that govern protein gating and entry into the cilium. How, for example, does the TZ prevent relatively small non-ciliary proteins from entering the organelle while ensuring that multimega-dalton IFT trains loaded with ciliary components can pass? By screening for mutants defective in IFT entry in C. elegans, Jinghua Hu (Mayo Clinic, Rochester, MN) identified FBF-1, which localizes to the basal bodies in worm and mammalian TFs. fbf-1 mutants accumulate IFT particles at the ciliary base, suggesting FBF-1 facilitates docking and entry of IFT trains. FBF-1 localization depends on Hydrolethalus Syndrome-1 (HYLS-1), which is required for formation of the fiber structures at the ciliary base, and double mutants fail to assemble cilia. Although whether conventional TF structures exist in worm is a question of controversy, evidence obtained from studying fbf-1 and hyls-1 mutants suggests that the function of TF component(s) in actively loading IFT particles is conserved across ciliated species. Kristin Verhey (University of Michigan Medical School) used a pore-clogging assay to test a model in which active transport of cytosolic proteins into the ciliary compartment is regulated similarly to protein entry into the nucleus through the nuclear pore complex. Forced dimerization of NUP62 decreased the entry of IFT and soluble proteins into the ciliary compartment while the entry of membrane proteins was unaffected (Takao et al., 2014). Bimolecular fluorescence complementation (BiFC) was used to identify interactions between resident TZ proteins and proteins passing through the TZ. NUP62 and NPHP4 interacted with the kinesin-2 motor KIF17 whereas B9D1 interacted with the GPCR somatostatin receptor 3 (SSTR3), suggesting that there are distinct routes for entry of cytosolic and membrane proteins into the cilium.
IFT carries the heavy load: Protein transport in cilia
Ciliary assembly requires intraflagellar transport (IFT), a bidirectional motility of large protein arrays (IFT trains) along the axonemal microtubules. A more in-depth knowledge of train architecture, assembly, and protein binding sites is a prerequisite to understanding how cells ensure that the right proteins are transported in the correct ratio and in a timely manner into cilia. IFT trains are polymers of IFT particles each consisting of IFT-A and IFT-B subcomplexes. John Wallingford (UT-Austin/HHMI) described CPLANE (ciliogenesis and planar polarity effectors), a novel basal body-associated complex required for assembly of the IFT-A subcomplex. In the absence of CPLANE in Xenopus, a 3-subunit IFT-A core complex, consisting of IFT144, IFT140, and IFT122, assembles, enters cilia, and undergoes bidirectional transport, while the IFT-A periphery complex, comprised of IFT139, IFT43 and IFT121, fails to assemble disrupting transport of IFT-B. Thus, the IFT-A core is capable to interact with both the retrograde and anterograde IFT motors. CPLANE is composed of the Joubert syndrome linked protein, JBTS17, together with Fuz, Intu, Wdpcp, and Rsg1; mutations in several of the subunits have been linked to cilia-related disease in humans. Malavika Raman (Wade Harper’s lab, Harvard University) showed that IFT assembly also requires the AAA-ATPase VCP and its adapter UBXD3/UBXN10 (Raman et al., 2015). VCP functions in numerous cellular pathways by extracting polyubiquitinated proteins from multiprotein complexes, and UBX proteins aid in identifying VCP targets. UBXD3 pull-downs identified ~200 novel interactors, which included numerous IFT-B proteins. VCP and UBXD3 show IFT-like particle motility that is likely directed by the binding of UBXD3 to IFT38/FAP22/qilin/cluap1. The absence of functional VCP/UBXD3 causes ciliogenesis defects in zebrafish, and loss of functional VCP/UBXD3 in cells causes the IFT trains to disassemble as reflected by the different trafficking velocities of IFT-A and – B particles and the disappearance of BBSomes from cilia. BBSomes are 8-subunit protein complexes which cycle through cilia via IFT, and when defective, cause Bardet-Biedl syndrome (BBS). VCP/UBXD3 apparently function in assembling IFT-A, IFT-B, and BBSomes into IFT trains at the ciliary base and/or in the remodeling of IFT trains at the tip. Using recombinant IFT proteins, Esben Lorentzen (MPI of Biochemistry, Martinsried, Germany) assembled a 15-subunit IFT-B complex. IFT-B consists of a salt-stable IFT-B core (now IFT-B1 complex) and an IFT-B2 complex composed of IFT38, 57, 54, 20, 80, and 172, previously known as peripheral IFT-B proteins. IFT52 and IFT88 are critical for the association of B1 and B2 subcomplexes. IFT80 is unique among IFT-B proteins in forming a dimer and IFT80 dimerization might dimerize two entire IFT-B complexes potentially explaining the appearance of IFT trains as a double row of particles in electron tomograms (Pigino et al., 2009). Binding studies further showed that IFT54 possesses a tubulin-binding motif, in addition to the previously characterized IFT81/IFT74 tubulin-binding module (Bhogaraju et al., 2013). Tomohiro Kubo (Witman lab, University of Massachusetts Medical School) used null mutants in Chlamydomonas to rigorously test the in vivo role of IFT81 and 74 in tubulin binding and ciliary assembly. After replacing all five lysine and arginine residues critical for the binding of tubulin by the IFT81 CH-domain with glutamate, cells still assembled cilia albeit they grew more slowly and were shorter. Similarly, cilia were shorter and assembly delayed in a strain expressing an N-terminally truncated IFT74. Double mutants, however, had only very short 9+2 cilia confirming the importance of IFT81-CH and IFT74N for tubulin transport and cilia assembly. The CH-domains of IFT81 and IFT54 and the N-terminal domain of IFT74 could form a three-pronged tubulin binding motif, which tolerates the removal of one but not two of the involved domains. Using TIRF microscopy, Karl Lechtreck (University of Georgia) showed that numerous axonemal proteins including tubulin move on IFT trains in Chlamydomonas flagella. The frequency of tubulin transport by IFT was greatly elevated in the growing cilia suggesting that the cells regulate the amount of cargo transported by IFT in a ciliary length-dependent manner (Craft et al., 2015). The microtubule plus-end binding protein EB1, in contrast, rapidly diffuses into cilia and accumulates at the ciliary tip in an IFT-independent manner. IFT transport could be required to move small protein, which can pass freely through the ciliary gate, against cellular concentration gradients in and out of cilia.
Up and down the cilium ex transition zone
Some protists and multicellular organisms possess structurally distinct and functionally specialized cilia. Using electron tomography and high resolution imaging of various basal body and ciliary components in Drosophila, Swadhin Jana (Bettencourt-Dias lab, Instituto Gulbenkian de Ciencia, Portugal) uncovered that this diversity is reflected at the ciliary base too. Core ciliary base components, e.g., Cep290 and SAS6, displayed distinct localization in different cilia type. These differences in the ciliary base organization are likely to contribute to the structural and functional specialization of cilia and could explain the tissue-specific phenotypes of certain ciliopathies. In contrast to the structural complexity of the basal body and TZ, the axoneme with its essentially identical 96-nm repeats appears monotonous. Daniela Nicastro (UT Southwestern Medical Center) exploits this repetitive structure of the axoneme for subtomogram averaging, which has led to an ever more detailed picture of the axoneme with now routinely 3–4 nm resolution, and with up to 1.5 nm using a newly developed imaging approach. To determine how individual proteins are oriented within the axoneme, clonable tagging followed by gold-labeling was used to visualize the position of the N- and C-termini of several nexin-dynein regulatory complex (N-DRC) components. Comparing the structure of the outer dynein arms (ODAs) in straight and bending segments of the axoneme suggests that the default state of the ODAs is active and bound to the axoneme, which is also evident by the straight and stiff appearance of cilia in most motility mutants; the N-DRC and other structures regulating ODA activity will locally inactivate the dyneins to allow for ciliary bending. Establishing proper length of cilia is critical for both motile and sensory functions. Pete Lefebvre (University of Minnesota) reported on conserved MAP- and CDK-like protein kinases, which result in excessively long cilia when mutated. A novel player, the CDKL5-kinase LF5p, typically resides at the proximal end of the cilium; in mutants defective in a length regulating complex (LRC) consisting of the CDK-like kinase LF2p and two associated proteins, LF5p moves to the ciliary tip. A kinase dead version of LF5p cannot achieve its proper position in the proximal cilium, even when wild-type LF5p is present. How this network of length-regulating kinases actually measures the length of cilia and adjusts it when necessary remains to be determined.
Ciliary tubulin is subjected to numerous posttranslational modifications, which are referred to as the tubulin code. Acetylation of α-tubulin is unique as it occurs on the inside of the microtubular cylinder. Antonina Roll-Mecak (Cell Biology and Biophysics Unit, NIH) showed that tubulin acetyl transferase (TAT) stochastically acetylates tubulin while diffusing in the microtubule lumen. Rather than TAT diffusion, TAT catalytic rate is likely to be rate-limiting in tubulin acetylation suggesting that TAT activity could function as a biological timer for the microtubule lifetime. Tubulin tyrosine ligase-like (TTLL) proteins add glycine and glutamate residues to the flexible tail domains of α- and β-tubulin. X-ray, cryoelectron microscopy and single molecule fluorescence revealed a three-step substrate recognition strategy of TTLL7, which can explain how the various TTLLs discriminate between the α- and β-tubulin tails (Fig. 2A; Garnham et al., 2015). TTLLs also catalyze glycylation, the addition of one or several glycine residues to glutamates in the tubulin tail. Carsten Janke (Institut Curie, Orsay, France) showed that glycylation is required for the stability of motile and primary cilia in mice and that defects in tubulin glycylation can lead to disease (Fig. 2B). Studies in Ttll3−/− mice showed fewer primary cilia in the colon epithelium resulting in faster proliferation and accelerated progression of colon cancers. Up to now, neither the ciliary tubulin detyrosinase nor the de-glycylating enzymes have been identified. Contrasting with the biochemically well-characterized TZ, the ciliary tip has remained a ciliary terra incognita, and only a handful of proteins have been shown to localize to the ultrastructurally complex ciliary tip compartment. Jacek Gaertig (University of Georgia) identified Spef1 as a novel tip protein, which binds to microtubule-ends via an EB1-like CH-domain. To establish a proteome for the ciliary tip, the promiscuous protein biotinylase BirA* was fused to bona fide tip proteins such as FAP256, which can now be used to identify novel proteins in their vicinity. The biochemical characterization of the tip is a prerequisite to understanding the role of tip structures in ciliary motility, length control, IFT remodeling, and ectosome formation.
Fig. 2.

Posttranslational modifications of tubulin. A) Side view of TTLL7 atomic models docked to a tubulin protofilament. Reprinted from Garnham et al., 2015. Courtesy of Antonina Roll-Mecak. B) Multiciliated ependymal cells stained for glycylated (green) and acetylated (red) tubulin. Courtesy of Carsten Janke.
The more the merrier: multiciliated cells
Multiciliated cells (MCCs) perform a wide range of tasks from mucociliary clearance in airways to the locomotion of entire organisms such as Xenopus embryos (Fig. 3). Typically, the formation of new centrioles/basal bodies is tightly linked to the cell cycle, but non-cycling MCCs produce dozens or hundreds of basal bodies. The program underlying MCC development was analyzed by Sudipto Roy (Institute of Molecular and Cell Biology, A*STAR, Singapore). Taking advantage of the zebrafish embryonic nephron, in which multi- and monociliated cells are interspersed, he identified Gmnc as a novel factor required for MCC development. gmnc is induced by FoxJ1a, the master regulator of motile cilia differentiation, and conversely, is suppressed by Notch signalling, a suppressor of MCC development. GMNC acts upstream of the transcription factor Multicilin, which regulates centriole amplification. MCCs are also prominent in the epithelium lining the ventricular cavities in the brain; these ependymal multiciliated cells are derived from neuronal stem cells (NSC) in the underlying tissue. Issei Shimada (Morisson lab, UTSW) showed that the absence of the transcription factor Prdm16 in NSC of postnatal mice resulted in reduced NSC self-renewal and decreased FoxJ1 expression in Prdm16-null NSC. The latter led to a decline in normal multiciliated ependymal cells, reduced flow, and the development of hydrocephalus. Analyzing the mechanisms of centriole amplification in Xenopus epithelial cells, Brian Mitchell (Northwestern University) identified CCDC78 as a specific marker of deuterosomes, the organizing centers of centrioles in MCCs. CCDC78 recruits CEP152 to deuterosomes, which then allows for the binding of polo-like kinase 4 (PLK4), a known regulator of centriole duplication during the cell cycle. Pharmacological inhibition of PLK4 reduced centriole number whereas over-expression of CEP152 drastically increased centriole number from ~150/cell to 450/cell. Before assembling cilia, the centrioles have to dock to the plasma membrane. While still in the cytoplasm, vesicles are recruited to the distal appendices of the centriole. Christopher Westlake (National Institute of Child Health and Human Development, NIH) analyzed centriolar docking in monociliated cells and reported that the EPS15 homology (EH) domain-containing proteins EHD1 and EHD3, known for their presence in the ciliary pocket and caveolae, are key players in this process (Lu et al., 2015). The EHDs recruit the SNARE SNAP29 to the distal centriolar appendices to initiate the fusion of the many small centriolar vesicles into a single vesicle, which will later fuse to the plasma membrane. The EHDs are further required for the removal of CP110 from centrioles, which is thought to be a negative regulator of ciliary assembly. Data presented by Sharda Yadav (Swaroop lab, National Eye Institute, NIH) showed now that CP110 is also required for ciliary assembly: CP110 knock-out mice display various features typical of ciliopathies and die shortly after birth. Cilia were reduced in number or missing in various tissues, and in CP110−/− MEFs, centriole maturation was affected resulting in mitotic defects and polyploidy, and basal bodies failed to dock at the plasma membrane. Once centrioles are docked to the plasma membrane, cilia will be assembled. The production of a directed fluid flow requires that cilia beat in a coordinated fashion within the individual multiciliated cell as well as across the entire epithelium. Establishing a stereotyped arrangement of cells within epithelia requires the planar cell polarity (PCP) pathway. Jeff Axelrod (Stanford University School of Medicine) analyzed the feedback between PCP and ciliogenesis and reported that robust planar cell polarization in the airway requires initiation of a multiciliated differentiation path, whereas centriole docking, ciliogenesis, and the unidirectional orientation of cilia are dispensable. The unicellular ciliate Tetrahymena uses hundreds of cilia for cell locomotion. Chad Pearson (University of Colorado) developed imaging software to determine basal body number and patterns and used this approach to identify mutations resulting in the loss of basal bodies. Progressive loss of basal bodies was observed in bld10 and poc1 mutants. Interestingly, basal body loss was suppressed after chemical inhibition of cilia motility suggesting a feedback of ciliary motion onto basal body integrity. Ultrastructural analysis showed that basal body degeneration started at specific triplets that likely bear a major share of the force generated by the ciliary beat; with FOP1, a novel biochemical marker for those triplets was identified.
Fig. 3.

Scanning EM micrograph of ciliated ependymal cells in the fourth ventricle. Courtesy of Karl Lechtreck.
Primary Ciliary Dyskinesia (PCD)
Much of what we have learned about cilia has originated from studies in Chlamydomonas. This is especially true for cilia motility, which is conferred by outer and inner dynein arms associated with each of the nine outer microtubule doublets. Neighboring outer microtubule doublets connect to each other by a nexin link, which constitutes the dynein regulatory complex (N-DRC). Using iTRAQ-based proteomics, Mary Porter (University of Minnesota) has identified novel components of the BOP2 (bypass of paralysis) and MBO2 (move backwards only) regulatory complexes, which she hypothesizes interconnect the I1 dynein/Modifier of Inner Arms (MIA) complex, the N-DRC, and several inner arm dyneins to coordinate dynein activity. Indeed, bop2 and mbo2 mutants display defective ciliary waveform and lack several axonemal polypeptides. Identification of evolutionarily conserved novel components of these regulatory complexes such as FAP57 will help identify new loci involved in PCD. Nick Berbari (Indiana University Purdue University Indianapolis) has identified mutations in Growth Arrest Specific 8 (GAS8/GAS11), a homologue of Chlamydomonas DRC4, in two patients with PCD (Yoder et al., FASEB J, 2015). Gas8GT mutant mice exhibited hydrocephalus and situs inversus and decreased ependymal and tracheal cilia-mediated flow. Transmission electron microscopy of mutant motile cilia revealed structural defects indicating a role for GAS8 in the assembly of mammalian inner dynein arms and the N-DRC. Accordingly, GAS8 localizes to axonemes of motile cilia, but also to the base of primary cilia, where its function is yet to be discovered. Marion Failler (Saunier lab, INSERM U1163) demonstrated additive effects of mutations in two genes, WDR19 and TEKT1, in a patient with Mainzer-Saldino Syndrome (MZSDS) and pulmonary ciliary dyskinesia. Mutations in IFT-A WDR19/IFT144 caused accumulation of IFT-B proteins in bulbous tips of primary cilia, while mutations in TEKT1, an uncharacterized member of the tektin family of outer microtubule doublet structural components, resulted in the absence of tektin-1 from axonemes of motile cilia associated with a strong motility defect. In addition to localizing to the axonemes of motile cilia (Fig. 4), tektin-1 localized to the centrosome. This centrosomal localization was decreased in patient fibroblasts and TEKT1 siRNA decreased primary cilia length, suggesting tektin-1 may have a role in both motile and primary cilia function. Nicolas Morante (Burdine lab, Princeton University) reported on three zebrafish genetic mutants of PCD displaying ventral body curvature and kidney cysts. A DYX1C1-null generated using CRISPR genome editing recapitulating a DYX1C1 loss-of-function human mutation, showed immotile cilia lacking dynein arms. The ENU-derived braciole mutant, encoded by another dyx1c1 allele, has well-formed but immotile cilia. Intriguingly, a novel ENU-derived mutant, mew, shows well-formed cilia that beat regularly, suggesting a potential novel regulator of motile cilia function.
Fig. 4.
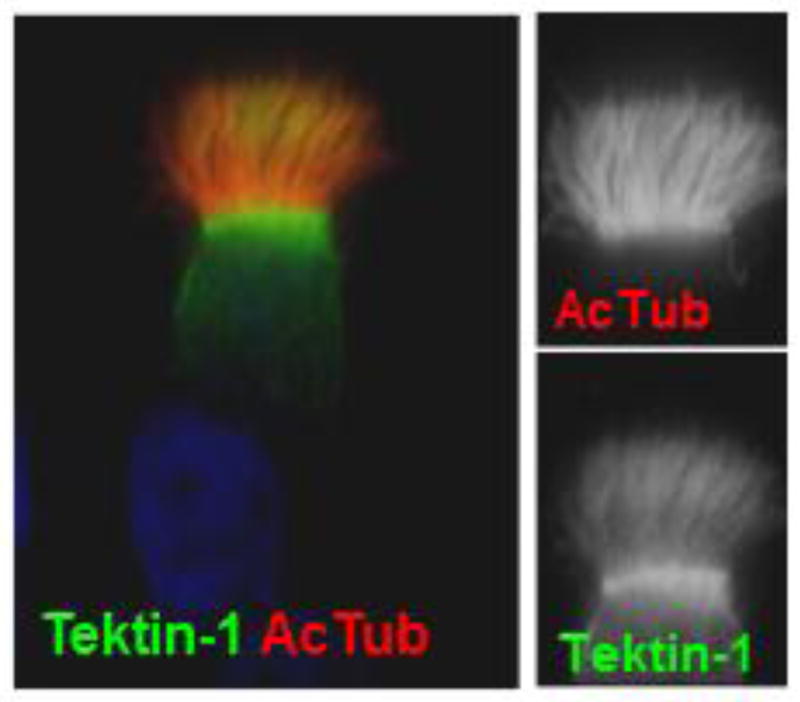
Immunolocalization of Tektin-1 to axonemes of motile cilia. Courtesy of Marion Failler.
Knowing left from right – nodal cilia
Loss of ciliary motility will not only cause chronic airway infections and male sterility, but in about half of the patients also results in situs anomalies. Establishing a proper left-right (L/R) axis requires the motility of the embryonic nodal cilia, which typically have a 9+0 structure lacking the radial spokes and the central pair microtubules, which regulate dynein motility (Fig. 5). Not surprisingly, mutations in radial spokes or central pair proteins result in PCD without situs anomalies. In a new twist, Dominic Norris (MRC Harwell, Oxford, UK) reported on a mutation in the third AAA domain of dynein motor DNAH11 that affects only the motility of nodal cilia while tracheal cilia beat normally. The mutant nodal cilia move faster but with reduced amplitude impairing the leftward fluid flow that is required for establishing a proper L/R axis. In humans, L/R asymmetry defects also result from mutations in CCDC11, yet the molecular and cellular functions of the protein remain unknown. Moe Mahjoub (Washington University School of Medicine) reported that CCDC11 localizes to the centriolar satellites, a network of ill-structured granules surrounding centrioles and basal bodies. Satellites are thought to function in the transport of precursors to the basal bodies and loss of CCDC11 interferes with this process. Indeed, depletion of CCDC11 affected primary ciliogenesis in mammalian cells and reduced the number, length, and motility of multiciliated epithelial cells in Xenopus and zebrafish. Ccdc11 mutant fish display aberrant L/R axis determination, recapitulating the human phenotype.
Fig. 5.

Drawing of monociliated cells as they are characteristic for the embryonic node. Courtesy of Sudipto Roy.
The leftward fluid flow generated by nodal cilia leads to elevated cytoplasmic calcium concentration on the left side of the node triggering a cascade of asymmetric gene expression. How this flow is sensed and the role of polycystin-2 (PC2), a transient receptor potential (TRP) channel family member that localizes to cilia, in sensing flow are questions of ongoing debate. Martina Brueckner (Yale University) used in vivo calcium imaging in Kupffer’s vesicle (KV), the zebrafish equivalent of the mouse node, to identify calcium (Ca2+) oscillations in the non-motile cilia (Yuan et al., 2015). The oscillations were more pronounced on the left side of KV suggesting that these cilia function as mechanosensors in which the leftward-directed flow triggers a Ca2+ influx. Further, knock-down of pkd2 affected L/R asymmetry suggesting that these cilia-localized TRP calcium-channels open in response to flow-induced ciliary bending. How are Ca2+ oscillations linked to the morphogenetic events leading to L/R asymmetry? Alexis Hofherr (Köttgen lab, Medical Center - University of Freiburg, Germany) performed a forward genetic screen in Drosophila melanogaster to elucidate novel factors required for processing of PC2-dependent Ca2+ influx and identified an evolutionarily conserved Ca2+-regulated mitochondrial metabolite carrier (MMC). Loss-of-MMC phenocopied loss-of-PC2 in flies and morphant zebrafish suggesting that MMC might functions to translate PC2 Ca2+ signals into changes of intracellular metabolite concentration and ultimately L/R gene expression.
In contrast to the mechanical hypothesis of L/R axis formation stands the chemical hypothesis, in which cilia at the rim of the node sense a morphogen asymmetrically distributed by the flow. Julien Vermot (IGBMC, France) took a hydrodynamics approach to the question: After methodically analyzing cilia density, angles, and beat frequencies in the Kupffer’s vesicle, the flow and the resulting forces within this spherical organ were modeled. The data indicate that the flow force is in the order of one magnitude too small to trigger a mechanosensory response from cilia. However, flow could function in chemical signaling but only under the assumption that the morphogen is secreted in a restricted area of the node and will disappear once it hits its ciliary target. Using in vivo Ca2+ imaging, David Clapham (Harvard University) measured the calcium response to mechanical stimuli of primary cilia in the kidney, node and inner ear. In the ear, a 9+0 kinocilium is physically attached to a neighboring microvillus (‘stereocilium’), and bending of the cilium increased Ca2+ levels only in the microvillus. Similarly, ciliary calcium levels remained constant after flow stimulation in the node and kidney tubules, arguing against a mechanosensory function of primary cilia, at least one involving changes in ciliary Ca2+ levels. Using optical tweezers, Christoph Schmidt (Georg-August-Universitaet, Goettingen, Germany) analyzed the mechanical properties of primary cilia. In contrast to the cross-linked 9+2 axoneme, the 9+0 axoneme of primary cilia has little rigidity and behaves like a bundle of sticks. However, primary cilia vibrate with frequencies of 1–100 Hz, which is driven by the cortical actin cytoskeleton. The hinge point of the shaking cilia appears to be located on the cell nucleus raising the question whether mechanical forces applied to the cilium could be directly transmitted to the nucleus. Addressing the question of how mechanical properties of cells contribute to negative gravitaxis in Chlamydomonas, Azusa Kage (Assistant Professor with Takuji Ishikawa, Tohoku University, Japan) studied the sinking properties of fixed cells with and without flagella. The presence of flagella increased the upward rotational velocity generating a passive upward torque which will rotate the cells in a way that the flagella point away from the bottom, a feature which is likely to contribute to the negative gravitaxis of the cells.
Fine-tuning Hedgehog signaling
The Hedgehog (Hh) signaling pathway was the first mammalian signaling pathway discovered to occur at the primary cilium. Upon receival of the Hh ligand by the Patched receptor at the cilium, the signal transducer Smoothened (SMO) is enriched in the primary cilium where it is activated. Full-length Glioblastoma transcription factors 2 and 3 (GLI2 and GLI3) localize to the ciliary distal tip, which leads to their activation. The ciliary events that intricately tune this developmental pathway continue to emerge and include modulating ciliary Ca2+ and cyclic AMP (cAMP) levels and regulating ciliary localization and exit of signaling components by the BBSome, IFT particles and phospholipid content of the ciliary membrane.
By developing a ratiometric cAMP sensor targeted to the primary cilium, Bryn Moore (Mirshahi lab, Geisinger Clinic) demonstrated approximately 5-fold higher cAMP concentrations in the cilium than in the cytosol of primary mouse embryonic fibroblasts. SHH stimulation decreased ciliary cAMP and increased ciliary Ca2+. These SHH-induced changes were prevented by gadolinium, which is known to block Ca2+ channels. These data extend previous findings by David Clapham (Harvard Medical School), who showed that ciliary Ca2+ concentrations are 5-fold higher than cytosolic concentrations and identified PC1-L1 and PC2-L1 as the ciliary channels that enable elevated ciliary Ca2+ levels in response to SHH (Delling et al., 2013).
Concomitant with Smo activation, Gpr161, a G-protein coupled receptor (GPCR) and negative regulator of the Hh pathway, is removed from the cilium in a 2-step process that was elucidated by Kasturi Pal (Mukhopadhyay lab, UT Southwestern). In the first step, Smo activation enhances Gpr161 binding to β-arrestins, which mediates exit of Gpr161 from the cilium. In the second step, clathrin-mediated endocytosis in the ciliary pocket completes Gpr161 ciliary removal. Inappropriate retention of Gpr161 in cilia of β-arrestin1/2 double knock out mice is associated with central polydactyly and hypoplastic fifth digits.
While most IFT complex B proteins are required for ciliary assembly, IFT25/IFT27 dimers function as a subcomplex dispensible for ciliogenesis but essential for Hh pathway activation and for ciliary exit of the BBSome and its cargoes, PTCH1 and SMO (Eguether et al, 2014). Consistent with this, Max Nachury (Stanford University) used proteomics (Apex Technology) and biochemical reconstitution to reveal that IFT27 interacts with the Arf-like GTPase, ARL6, to regulate ciliary removal of the BBSome, which in turn, mediates ciliary exit of its cargoes, PTCH1, SMO, GPR161, SSTR3 and melanin concentrating hormone receptor 1 (MCHR1) (Liew et al, 2014). Thibaut Eguether (Pazour lab, University of Massachusetts Medical School) further showed that IFT27 is required at several steps in the Hh signaling cascade, in the cilium both upstream and downstream of SMO activation as well as in the cytoplasm upstream of GLI transcriptional activity. An activator form of GLI2 (GLI2ΔN), but not constitutively active SMO-M2, activated the pathway in Ift27−/− cells, and cells null for both Ift27 and Suppressor of Fused (Sufu), which suppresses GLI activation, showed greater activation of the pathway, than Sufu-null cells alone.
Mutations in the phophoinositide phosphatase, INPP5E, cause ciliopathies and Jeremy Reiter (UCSF) delved deeper into the underlying molecular mechanism. He showed that INPP5E generates a distinct phosphoinositide distribution in the ciliary membrane, whereby phosphoinositide 4-phosphate (PI(4)P) is present in the membrane enclosing the axoneme and phosphoinositide 4, 5-bisphosphate (PI(4,5)P2) comprises the membrane at the ciliary base (Garcia-Gonzalo et al., Dev. Cell, 2015). Loss of INPP5E caused ciliary localization of PI(4,5)P2, which increased ciliary localization of Hh negative modulators, PI(4,5)P2-binding Tulp3, Gpr161 and IFT140, which inhibited Hh signaling (Figure 6). Reducing Tulp3 in Inpp5e−/− cells decreased ciliary localization of Gpr161 and restored Hh signaling, indicating that Tulp3 monitors the ciliary membrane phosphoinositide content to control Gpr161 and IFT-A localization in cilia (Garcia-Gonzalo et al., 2015). Using a chemically-inducible dimerization technique targeted to the cilium, Takanari Inoue (Johns Hopkins University) also showed that loss of INPP5E caused ectopic localization of PI(4,5)P2 in the distal cilium. He further showed that this was accompanied by the ectopic ciliary presence of actin and PI(4,5)P2–associated actin remodeling factors. He showed that serum stimulation induced a form of ciliary disassembly, cilia shaft separation, which coincided with a transient burst of F-actin at the separation site (Figure 7). Cilium shaft separation was enhanced by loss of INPP5E, and conversely, was reduced by inhibition of actin polymerization. Thus, INPP5E is essential for inhibiting F-actin assembly in primary cilia and maintaining cilia stability.
Fig. 6.

Schematic of phosphoinositide distribution in ciliary membrane. Reprinted from Garcia-Gonzalo et al. 2015. Courtesy of Jeremy Reiter.
Fig. 7.

Schematic of cilia shaft separation. Courtesy of Takanari Inoue.
Cilia in sickness and in health
The essential role of cilia in development and maintenance of multiple organs is reflected in the diverse clinical manifestations of ciliopathies. A prominent clinical feature of ciliopathies is renal cystic disease, and the most common is Autosomal Dominant Polycystic Kidney Disease (ADPKD), which enlarges the kidney, and is caused by deficient PC1 or PC2 function in intact primary cilia. In mice, loss of IFT proteins causes a milder PKD-like disease, while loss of proteins of the inversin compartment, transition zone or centrosomes causes nephronophthisis (NPHP), a fibrocystic renal disease that does not enlarge the kidney. The role of primary cilia in renal cystogenesis remains elusive, particularly since loss of IFT-B/primary cilia, together with either PC1 or PC2 ameliorated the PC-mediated PKD phenotype in mice (Ma et al., 2013).
Kidney
PC1 is an 11-transmembrane spanning protein that undergoes autoproteolytic cleavage at a G protein-coupled receptor proteolytic site (GPS). Stefan Somlo (Yale University) showed that 4 out of 5 PKD1 missense mutations disrupted ciliary localization of PC1, and half of these were due to a primary defect in GPS cleavage. Use of a BAC transgenic mouse model demonstrated that ciliary localization of PC1 is essential for PC1 function (Cai et al., 2014). He proposes that GPS cleavage mutants comprise a “class” of variants that could be coordinately targeted for therapy and that high throughput screening for molecules that reconstitute GPS cleavage or cilia trafficking may identify novel therapeutic strategies. PCs also localize to extracellular vesicles (EVs), which are emerging as a mode of intercellular signaling. Juan Wang (Barr lab, Rutgers University) performed RNA sequencing on purified EV-releasing ciliated sensory neurons of adult C. elegans and identified novel modulators of PC signaling networks and of EV biogenesis and signaling. P38 MAPK, PMK-1, and tumor necrosis associated factor (TRAF) homologues, TRF-1 and TRF-2, were found to be necessary for PC-mediated male mating behaviors. Further, P38 MAPK is also required for ciliary-mediated EV biogenesis, while TRF-1 and TRF-2 appear to retain EVs in the extracellular lumen surrounding cilia, revealing a novel regulatory mechanism for EV signaling. Peter Czarnecki (Jagesh Shah’s lab, Harvard Medical School) showed that ANKS6, a member of the inversin compartment protein complex, is a substrate and activator of the NEK8 kinase (Czarnecki et al., Nat Comm, 2015). Ethylnitrosourea (ENU)-mutagenized mouse mutants harboring homozygous NEK8I124T or ANKS6M187K mutations displayed heterotaxy and renal cysts. Both mutations ultimately disrupted the NEK8 kinase activity. The similar phenotypes of INVS and NPHP3-null mice suggest NEK8-mediated phosphorylation may be a final common inversin compartment signaling output. Mutations in Centrosomal protein 290 (Cep290)/Nphp6 cause Joubert Syndrome, which manifests NPHP. Rachel Giles (University Medical Center Utrecht) demonstrated that loss of CEP290 in mouse kidneys and cells increased DNA damage signaling, DNA replication stress and cyclin-dependent kinase (CDK) activity. Importantly, treatment of Cep290-deficient cells with CDK1/2 inhibitors rescued the DNA damage as well as the loss of primary cilia (Slaats et al., 2015). Brad Yoder (University of Alabama) presented in vivo imaging of renal cilia, using the CiliaGFP mouse, which globally expresses a SSTR3:GFP fusion protein causing cilia to fluoresce (O’Connor et al., 2013). Ciliary axonemes shorten by almost 50% from embryogenesis to adulthood, but interestingly, ciliary length changes did not coincide with the P12-P14 developmental window that determines severity of cystic kidney disease. In healthy renal tubules, cilia deflected continuously lying almost parallel to the renal tubules in the direction of fluid flow. In contrast, cilia of cyst-lining epithelial cells did not deflect due to an absence of flow in cysts. The CiliaGFP mouse and novel in vivo imaging techniques will be used to elucidate the role of primary cilia in PKD.
Vasculature and Heart
Primary cilia also play essential roles in development of the vasculature and heart. Zhaoxia Sun (Yale University) showed that zebrafish IFT-B mutants, Ift172−/−, ift81−/− and qilin−/−, develop intracranial hemorrhage (ICH), a severe form of stroke with unknown genetic etiology in the human population (Kallakuri et al., 2015). These cilia mutants were sensitized to brain hemorrhage induced by treatment with Hh inhibitor, cyclopamine, suggesting that loss of Hh signaling underlies ICH. In contrast to IFT mutants, Pkd2 mutants did not develop ICH, even following treatment with cyclopamine.
To identify novel genes required for heart development, Cecilia Lo (University of Pittsburgh) performed a large-scale forward genetic ENU mutagenesis screen for congenital heart defects (CHD) in mice (Li et al., 2015). From more than 250 mutant mouse lines, all made accessible through the Mouse Genomics Informatics (MGI) database, sixty-four genes not previously associated with CHD were identified and half of these genes are cilia-related. Mutations in 40% of cilia-related genes caused non-laterality defects, while 60% affected motile cilia. Other identified genes played a role in cilia-transduced signaling, revealing ciliary dysfunction as a major cause of CHD.
Face
Craniofacial defects occur in over 30% of ciliopathies. Samantha Brugmann (Cincinnati Children’s Hospital) deleted Kif3a in neural crest cells in mice, which caused severe midfacial widening, aglossia and micrognathia. Intriguingly, despite loss of cilia, midfacial phenotypes were caused by a gain-of-Hh activity and were partially rescued by genetic restoration of GLI3 repressor. Conversely, aglossia/micrognathia phenotypes were caused by a loss-of-Hh function and were partially rescued by genetic increase of GLI2 activator. Thus, development of the face requires a prominence-specific, cilium-dependent GLI activator:GLI repressor ratio.
Blindness and Anosmia
The sensory cilia axoneme marks the base of the outer segment of photoreceptors, which is renewed every 10 days requiring steady protein transport to maintain the phototransduction machinery. Wolfgang Baehr (University of Utah) described the chaperone proteins that traffic lipidated cargo to the outer segment. Prenylation or acylation of proteins is required for membrane anchoring. The major chaperones, cGMP phosphodiesterase (PDEδ) and Uncoordinated 119 (UNC119), bind to prenylated and acylated proteins, respectively, which is controlled by the Arf-like small GTPase, ARL3. Mice null for PDEδ or lacking Arl3 in the photoreceptors show photoreceptor degeneration and mistrafficking of prenylated rhodopsin kinase (GRK1) and phosphodiesterase 6 (PDE6). Recently, INPP5E was shown to be a cargo of PDEδ as well (Thomas et al., 2014). Combined deletion of Unc119a and Unc119b in mice causes mistrafficking of acylated transducin-α. The GTPase activating protein (GAP) of ARL3GTP is Retinitis Pigmentosa protein 2 (RP2). Loss of RP2 causes X-linked retinitis pigmentosa in the human population and has been shown to disrupt PDE6δ-cargo interactions and trafficking of PDE6 and GRK1 to the outer segments in mice. Katja Gotthardt (Wittinghofer lab, MPI, Dortmund, Germany) used a yeast 2 hybrid screen to search for Arl3 binding partners and identified Arl13b. This well-known ciliary GTPase was convincingly shown to function as the Arl3 guanine nucleotide exchange factor (GEF). Arl13b is limited to cilia in distribution, which will ensure that Arl3 assumes its active GTP-bound state when inside cilia spatially restricting the release of myristoylated and prenylated cargoes from the carriers UNC119 and PDE6D. Judith Bergboer (Drummond lab, Massachusetts General Hospital/Harvard Medical School) developed a novel live imaging assay for cilia-dependent sensory neuronal activity in zebrafish. Responses of ciliated olfactory sensory neurons (OSN) to various odorants were examined in zebrafish expressing a pan-neuronal genetically encoded fluorescent calcium biosensor. oval/Ift88 mutants and Ift172 morphants showed a decreased Ca2+ response to bile acids and food, while response to amino acids, which are sensed by non-ciliated OSN, was unaffected. This assay will be used to further explore mechanisms of neuronal sensory cilia function and for in vivo screens for therapeutic compounds. Jeff Martens (University of Florida) demonstrated the utility of gene therapy to rescue ciliation defects in OSNs. In OSN-specific Ift88 knock-out (ko) mice, adenovirus-mediated re-expression of Ift88 prevented aberrant OSN reorganization in the brain and restored cilia and olfaction. Bbs1 OSN conditional ko mice and Bbs4 ko mice showed a reduction in ciliature in mature differentiated OSNs. Gene therapeutic rescue by adenovirus gene delivery restored cilia and odor detection in Bbs mutants. In residual Bbs mutant olfactory cilia, the velocity of retrograde IFT was increased; changes in IFT velocity could be part of the pathomechanism of BBS in the olfactory system.
Brain
Unlike the kidney, which is relatively resilient to disruption of ciliary genes once fully matured, both developing and adult brains are susceptible to cilia-mediated, severe neurological disease. Tamara Caspary (Emory University) investigated the underlying mechanism by which loss of Arl13B-mediated SHH signaling causes the “molar tooth sign”, a cerebellar developmental defect caused by axon misguidance in Joubert Syndrome. While ciliary-mediated, GLI transcription-dependent SHH signaling is required for morphogenesis and mitogenesis, GLI-independent SHH signaling, in which SMO localizes outside the cilium to mediate cytoskeletal changes, is required for cell migration and chemotaxis. Yet expression of an Arl13BV358A mutant, which blocks ciliary localization of ARL13B, failed to rescue the cell migration defect of ARL13B-null fibroblasts, suggesting that SHH-mediated cell migration requires ARL13B function in cilia. Thus disease phenotypes proposed to be cilia-independent may actually require ciliary protein function. Edwin Oh (Duke University) showed that disruption of the pericentriolar matrix (PCM) leads to degeneration of the adult brain in mice. Pcm1−/− adult mice, but not young pups, showed enlarged brain ventricles and reduced cortical structures and hippocampus. Shortened cilia with bulbous distal tips were observed in the brain, commencing at P21 and increasing in number by P40. Dopamine receptor 2 ectopically localized around the base of the cilium in Pcm1−/− mutants but was not present near the cilium in control mice. Pcm1−/− adult mice showed sensorimotor gating defects, a phenotype of schizophrenia, which was not rescued by treatment with anti-psychotic drugs. Further, sequencing of patients with treatment-resistant schizophrenia revealed an enrichment of loss-of-function PCM1 alleles, suggesting that PCM1 and dopamine signaling deficits may underlie severe psychosis.
Obesity and Diabetes
In mice, loss of BBS and IFT-B ciliary components and of centrosomal ALMS1 causes hyperphagia and obesity. Using Thm1 conditional ko mice, Pamela Tran (University of Kansas Medical Center) showed that IFT-A dysfunction also causes hyperphagia and obesity, with females showing greater tendency to increase adipose tissue weight and males more susceptible to developing diabetes. Misregulated neuropeptide gene expression and phospho-STAT3 in the hypothalamic arcuate nucleus, which integrates peripheral signals to regulate feeding behavior, suggests that the IFT-A ciliary defect disrupts sensing of changes in feeding signals. Kirk Mykytyn (Ohio State University) showed in multiple contexts that neuronal cilia are essential for GPCR signaling. Ablation of cilia on gonadotropin-releasing hormone neurons reduces kisspeptin receptor 1 signaling (Koemeter-Cox et al., 2014). On Bbs−/− neurons, SSTR3 and MCHR1 fail to localize to cilia, while Dopamine receptor 1 (D1) accumulates in cilia. Additionally, β-arrestin 2 localizes to neuronal cilia in response to SSTR3 activation and is required for SSTR3 ciliary export. Further, Kirk Mykytyn showed that disrupting Bbs1 specifically in D1-expressing neurons leads to reduced locomotor activity and obesity in mice. Norann Zaghloul (University of Maryland) showed that BBS4 is required to activate brain-derived neurotrophic factor (BDNF) signaling and to localize the TrkB receptor to the ciliary axoneme in the presence of BDNF. Suppression of BBS4 expression in zebrafish decreased pro-opiomelanocortin neuronal projections, which regulate appetite. Importantly, these projections were restored by BDNF treatment (Figure 8). Thus misregulated BDNF signaling may contribute to hyperphagia in BBS. Moreover, BDNF appears to regulate BBS4-mediated neuronal specification. Whether the BBSome functions in the import of proteins into cilia, or exports proteins from cilia as reported in Chlamydomonas, or mediates bidirectional transport of ciliary membrane proteins is still unclear. Jantje Gerdes (Helmholtz Zentrum München) showed a direct role for primary cilia in β-cell function and susceptibility to type 2 diabetes (Gerdes et al., 2014). Pre-obese Bbs4−/− mice displayed impaired glucose handling and first phase insulin response. In β-cells stimulated with insulin, the insulin receptor localized to primary cilia, which was essential for insulin signaling, which occurs through the AKT pathway. Stimulated Bbs4-deficient β-cells showed low phospho-AKT levels, indicating impaired insulin signaling. Further, in Bbs4-deficient islets, SNARE components, SNAP25 and Syntaxin 1a, which complex to mediate insulin release, were reduced.
Fig. 8.

Rescue of bbs4 morphant POMC neuronal projections by BDNF. Courtesy of Norann Zaghloul.
Aging
Fascinatingly, Piali Sengupta (Brandeis University) has found that C. elegans Osm-6/Ift52 hypomorphic mutants improved their sensory behaviors during midlife due to a transient elongation of shortened cilia. This aging-dependent cilia elongation required IFT and did not occur in the absence of kinesin-2 or in IFT-null mutants. Middle-aged IFT hypomorphs also showed improved proteostasis, and cell-autonomous activity of heat shock transcription factor (HSF-1) and Hsp90 was essential for ciliary elongation. These findings suggest temporal and tissue-specific regulation of proteostasis may control ciliary homeostasis and suggest novel avenues for targeting ciliopathies.
Perspectives and Future Directions
State-of-the-art imaging and proteomics techniques, unbiased genetic screens and generation of novel models across different ciliated species have contributed to the many new discoveries presented. A few highlights include the role of the phospholipid content of the ciliary membrane in signaling and axonemal stability, the role of aging in ciliary homeostasis and the impact of post-translational modification of ciliary tubulin in colon cancer. These and other new findings have led to provocative new questions. The role of cilia as mechanosensory organelles and the regulation of ciliary versus cytosolic calcium signaling, which is involved in a range of cilia functions from left-right axis formation to the pathomechanism of cystic kidney disease, have been questioned. Tissue-specific regulation of cilia-mediated Hh signaling needs further exploration given that inappropriate activation of the pathway can occur in the absence of primary cilia. Incorporating the newly identified components and structures of the TZ into the mechanisms that regulate selective ciliary protein entry is now required. Further, the full assembly of the IFT-B subcomplex should accelerate understanding of how IFT interacts with numerous distinct proteins and with its own components to assemble into IFT trains. With an increased knowledge of the function of different classes of ciliary proteins (eg. IFT, TZ, BB), discovery of EVs, and an increased understanding of the regulation of signaling pathways, how far away are we to developing therapies for more ciliopathies? The first decade of the 21st century was considered a renaissance period for cilia during which these organelles were finally recognized to be important. The current questions suggest a new era for cilia - an age of enlightenment - that should unveil exciting and thought-provoking answers.
Alternative end
During the first decade of the 21st century cilia were finally recognized to be important for human health and disease. Perhaps the field is moving now from the cilia renaissance to an age of enlightenment were increasing knowledge of the organelles biology will bring us closer to developing therapies for more ciliopathies.
Acknowledgments
We thank the many researchers who granted permission to cite their work. This meeting was supported by the FASEB SRC, the NIH/NIDDK, the PKD foundation, the EMBO, Nikon, Acetylon Pharmaceuticals Inc., cells, and Bruker. Related work in the authors’ labs is supported by NIH grants 1R01GM110413 (K.F.L.) and 1R01DK103033 (P.V.T.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bhogaraju S, Cajanek L, Fort C, Blisnick T, Weber K, Taschner M, Mizuno N, Lamla S, Bastin P, Nigg EA, Lorentzen E. Molecular basis of tubulin transport within the cilium by IFT74 and IFT81. Science. 2013;341:1009–1012. doi: 10.1126/science.1240985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Fedeles SV, Dong K, Anyatonwu G, Onoe T, Mitobe M, Gao JD, Okuhara D, Tian X, Gallagher AR, Tang Z, Xie X, Lalioti MD, Lee AH, Ehrlich BE, Somlo S. Altered trafficking and stability of polycystins underlie polycystic kidney disease. The Journal of clinical investigation. 2014;124:5129–5144. doi: 10.1172/JCI67273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft JM, Harris JA, Hyman S, Kner P, Lechtreck KF. Tubulin transport by IFT is upregulated during ciliary growth by a cilium-autonomous mechanism. The Journal of cell biology. 2015;208:223–237. doi: 10.1083/jcb.201409036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delling M, DeCaen PG, Doerner JF, Febvay S, Clapham DE. Primary cilia are specialized calcium signalling organelles. Nature. 2013;504:311–314. doi: 10.1038/nature12833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gonzalo FR, Phua SC, Roberson EC, Garcia G, 3rd, Abedin M, Schurmans S, Inoue T, Reiter JF. Phosphoinositides Regulate Ciliary Protein Trafficking to Modulate Hedgehog Signaling. Developmental cell. 2015;34:400–409. doi: 10.1016/j.devcel.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnham CP, Vemu A, Wilson-Kubalek EM, Yu I, Szyk A, Lander GC, Milligan RA, Roll-Mecak A. Multivalent Microtubule Recognition by Tubulin Tyrosine Ligase-like Family Glutamylases. Cell. 2015;161:1112–1123. doi: 10.1016/j.cell.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geimer S, Melkonian M. The ultrastructure of the Chlamydomonas reinhardtii basal apparatus: identification of an early marker of radial asymmetry inherent in the basal body. Journal of cell science. 2004;117:2663–2674. doi: 10.1242/jcs.01120. [DOI] [PubMed] [Google Scholar]
- Gerdes JM, Christou-Savina S, Xiong Y, Moede T, Moruzzi N, Karlsson-Edlund P, Leibiger B, Leibiger IB, Ostenson CG, Beales PL, Berggren PO. Ciliary dysfunction impairs beta-cell insulin secretion and promotes development of type 2 diabetes in rodents. Nature communications. 2014;5:5308. doi: 10.1038/ncomms6308. [DOI] [PubMed] [Google Scholar]
- Kallakuri S, Yu JA, Li J, Li Y, Weinstein BM, Nicoli S, Sun Z. Endothelial cilia are essential for developmental vascular integrity in zebrafish. Journal of the American Society of Nephrology : JASN. 2015;26:864–875. doi: 10.1681/ASN.2013121314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koemeter-Cox AI, Sherwood TW, Green JA, Steiner RA, Berbari NF, Yoder BK, Kauffman AS, Monsma PC, Brown A, Askwith CC, Mykytyn K. Primary cilia enhance kisspeptin receptor signaling on gonadotropin-releasing hormone neurons. Proc Natl Acad Sci U S A. 2014;111:10335–40. doi: 10.1073/pnas.1403286111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Klena NT, Gabriel GC, Liu X, Kim AJ, Lemke K, Chen Y, Chatterjee B, Devine W, Damerla RR, Chang C, Yagi H, San Agustin JT, Thahir M, Anderton S, Lawhead C, Vescovi A, Pratt H, Morgan J, Haynes L, Smith CL, Eppig JT, Reinholdt L, Francis R, Leatherbury L, Ganapathiraju MK, Tobita K, Pazour GJ, Lo CW. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature. 2015;521:520–524. doi: 10.1038/nature14269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q, Insinna C, Ott C, Stauffer J, Pintado PA, Rahajeng J, Baxa U, Walia V, Cuenca A, Hwang YS, Daar IO, Lopes S, Lippincott-Schwartz J, Jackson PK, Caplan S, Westlake CJ. Early steps in primary cilium assembly require EHD1/EHD3-dependent ciliary vesicle formation. Nature cell biology. 2015;17:228–240. doi: 10.1038/ncb3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M, Tian X, Igarashi P, Pazour GJ, Somlo S. Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nature genetics. 2013;45:1004–1012. doi: 10.1038/ng.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor AK, Malarkey EB, Berbari NF, Croyle MJ, Haycraft CJ, Bell PD, Hohenstein P, Kesterson RA, Yoder BK. An inducible CiliaGFP mouse model for in vivo visualization and analysis of cilia in live tissue. Cilia. 2013;2:8. doi: 10.1186/2046-2530-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigino G, Geimer S, Lanzavecchia S, Paccagnini E, Cantele F, Diener DR, Rosenbaum JL, Lupetti P. Electron-tomographic analysis of intraflagellar transport particle trains in situ. The Journal of cell biology. 2009;187:135–148. doi: 10.1083/jcb.200905103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman M, Sergeev M, Garnaas M, Lydeard JR, Huttlin EL, Goessling W, Shah JV, Harper JW. Systematic proteomics of the VCP-UBXD adaptor network identifies a role for UBXN10 in regulating ciliogenesis. Nature cell biology. 2015 doi: 10.1038/ncb3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaats GG, Saldivar JC, Bacal J, Zeman MK, Kile AC, Hynes AM, Srivastava S, Nazmutdinova J, Ouden K, Zagers MS, Foletto V, Verhaar MC, Miles C, Sayer JA, Cimprich KA, Giles RH. DNA replication stress underlies renal phenotypes in CEP290-associated Joubert syndrome. The Journal of clinical investigation. 2015;125:3657–3666. doi: 10.1172/JCI80657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takao D, Dishinger JF, Kee HL, Pinskey JM, Allen BL, Verhey KJ. An assay for clogging the ciliary pore complex distinguishes mechanisms of cytosolic and membrane protein entry. Current biology. 2014;24:2288–2294. doi: 10.1016/j.cub.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S, Wright KJ, Le Corre S, Micalizzi A, Romani M, Abhyankar A, Saada J, Perrault I, Amiel J, Litzler J, Filhol E, Elkhartoufi N, Kwong M, Casanova JL, Boddaert N, Baehr W, Lyonnet S, Munnich A, Burglen L, Chassaing N, Encha-Ravazi F, Vekemans M, Gleeson JG, Valente EM, Jackson PK, Drummond IA, Saunier S, Attie-Bitach T. A homozygous PDE6D mutation in Joubert syndrome impairs targeting of farnesylated INPP5E protein to the primary cilium. Human mutation. 2014;35:137–146. doi: 10.1002/humu.22470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Su J, Wang WJ, Craige B, Witman GB, Bryan Tsou MF, Liao JC. Superresolution Pattern Recognition Reveals the Architectural Map of the Ciliary Transition Zone. Sci Rep. 2015;5:14096. doi: 10.1038/srep14096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S, Zhao L, Brueckner M, Sun Z. Intraciliary calcium oscillations initiate vertebrate left-right asymmetry. Current biology. 2015;25:556–567. doi: 10.1016/j.cub.2014.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]