

Abstract
Detection of endogenous nucleic acids by cytosolic receptors, dependent on STING, and endosomal sensors, dependent on Unc93b1, can provoke inflammatory responses that contribute to a variety of autoimmune and autoinflammatory diseases. In DNase II deficient mice, the excessive accrual of undegraded DNA leads to both a STING-dependent inflammatory arthritis and additional Unc93b1-dependent autoimmune manifestations, including splenomegaly, extramedullary hematopoiesis, and autoantibody production. Here we utilize bone marrow chimeras to show that clinical and histological inflammation in the joint depends upon DNase II deficiency in both donor hematopoietic cells and host radioresistant cells. Additional features of autoimmunity in these mice, known to depend on Unc93b1 and therefore endosomal Toll-like receptors (TLRs), also require DNase II deficiency in both donor and host compartments, but only require functional TLRs in the hematopoietic cells. Collectively, our data demonstrate a major role of both stromal and hematopoietic cells in all aspects of DNA-driven autoimmunity. These findings further point to the importance of cytosolic nucleic acid sensors in creating an inflammatory environment that facilitates the development of Unc93b1-dependent autoimmunity.
Introduction
Cytosolic DNA sensors were first identified in the context of host defense but, similar to endosomal nucleic acid detecting Toll-like receptors (TLRs), these sensors can also detect endogenous ligands and thereby promote sterile inflammation. A number of cytosolic DNA receptors have now been identified, including cGAS and Ifi16, among others (1, 2). These sensors converge on the ER-associated protein STING to activate downstream pathways leading to the expression of both IFN-stimulated genes and proinflammatory cytokines (3). Importantly, gain of function mutations in STING have recently been linked to a clinical syndrome called SAVI, associated with upregulation of type I IFN, severe vasculopathy, arthritis, pulmonary fibrosis, and in some cases autoantibody production (4–9). In addition, loss of function mutations in a variety of cellular nucleases can lead to the accumulation of self-DNA and also contribute to inflammatory disease. For example, loss of function mutations in Trex1 (a cytosolic DNase), SAMHD1 (a cytosolic RNase), RNaseH2A, and ADAR1 have been linked to both the neuroinflammatory disease Aicardi-Goutieres syndrome (10–13), as well as different forms of lupus (6). Trex1 deficiency can also lead to systemic inflammation in mice, initially evident as myocarditis with subsequent progression to other organs (14, 15). Furthermore, SNPs in the promoter region of the DNase II gene have been identified as risk factors for rheumatoid arthritis (16).
Mice lacking the phagolysosomal nuclease, DNase II, are embryonic lethal due to excessive type I IFN production downstream of STING-dependent pathways. These mice can be rescued by intercrossing with mice that lack the type I IFN receptor (IFNaR). DNase II−/− IFNaR−/− double knockout (DKO) mice survive to adulthood but then develop an inflammatory arthritis not seen in the DNase II+/− IFNaR−/− (Het) control group. The development of DKO arthritis is STING-dependent (17, 18). DKO mice also produce anti-nuclear antibodies (ANAs) and develop splenomegaly and extramedullary hematopoiesis, and these aspects of disease turn out to require a functional form of Unc93b1, and by inference, signaling by endosomal TLRs (18, 19). Thus, both cytosolic and endosomal nucleic acid sensing receptors contribute to the clinical manifestations of DKO mice.
Previous studies involving radiation chimeras have indicated that radioresistant Trex1−/− endocardial cells are sufficient for lymphocyte activation and the development myocarditis (20), while the arthritic phenotype of DNase II−/− mice was found to depend entirely on hematopoietic cells (21). However, the DKO chimeric mice in the latter study were evaluated at a relatively early stage in the disease process, and the Unc93b1-associated manifestations were not examined. As considerable data now demonstrate a proinflammatory role for STING in non-hematopoietic cells, we reasoned that it was important to re-evaluate the contribution of hematopoietic cells and non-hematopoietic cells to the various DKO disease parameters. Our data reveal a major contribution of both bone marrow-derived hematopoietic and radioresistant host cells to all aspects of the DKO phenotype. Moreover, our data further point to a critical interplay between endosomal and cytosolic nucleic acid receptors in the development of systemic autoimmunity.
Materials and Methods
Mouse Strains
DNase II+/− C57BL/6 embryos were kindly provided by Dr. S. Nagata (Osaka Medical School) through the RIKEN Institute, and mice were intercrossed with Ifnar−/− C57BL/6 or Igha/a C57BL/6 mice to produce DNase II−/− IFNaR−/− double knockout (DKO), DNase II+/− IFNaR−/− heterozygous (Het), and Igha DKO mice. C57BL/6 mice expressing GFP under the MHC I class promoter were kindly provided by Dr. R. Gerstein (UMMS), and were crossed to Ifnar−/− C57BL/6 mice to generate DNase II+/− IFNaR−/− GFP donor mice for bone marrow chimera studies. Unc93b13d/3d mice on a C57BL/6J background were kindly provided by Bruce Beutler (UTSW) (22). STING-deficient mice were generated on a 129SvEvxC57BL/6J background by Dr. G. Barber (UMiami) (23), backcrossed to C57BL/6J mice and kindly provided by Dr. D. Stetson (UWashington), and then further backcrossed to C57/BL6J at UMMS. The Unc93b13d/3d and STING-deficient mice were then crossed to the DKO strain to yield Unc93 TKO and STING TKO lines, respectively, as described previously (18). All animal procedures were approved by and performed in accordance with the Institutional Animal Care and Use Committee at the University of Massachusetts Medical School.
Generation of Bone Marrow Chimeras
Lethally irradiated (850R) 8–12 week-old recipients were reconstituted by i.v. injection of 107 total bone marrow cells from 8–10 week-old mice. For the Het/DKO chimeras, female Het (Ighb) or DKO (Ighb) hosts were reconstituted with cells from female Het (GFP) or DKO (Igha) mice. For the Unc93 TKO/DKO chimeras, the Unc93 TKO mice were Ighb and the DKO mice were Igha. Clinical monitoring for the development of arthritis was performed until the mice were euthanized for analysis at 10 months post transplant (Het/DKO chimeras) or 4 months post transplant (Unc93 TKO/DKO chimeras). The extent of reconstitution was determined by flow cytometry for GFP+ or IgD allotype markers in total peripheral blood or mature B cells, respectively, and confirmed by FACS analysis of the spleen at the time of euthanasia.
Clinical and Histologic Inflammation Scores
Clinical arthritis was measured using a previously described scoring system (24). Histologic inflammation was assessed in decalcified, paraffin-embedded left hind limbs. Blocks were sectioned at 5 μm, deparaffinized, and stained with H&E. 40 sections were cut from each block and sections 10, 20, 30, and 40 were scored using a modification of a previously described system (24) on a scale from 0–4. Cellular infiltrates in the distal tibias were scored on a scale from 0–3 (0=no infiltrate, 1= slight infiltrate, 2= moderate infiltrate, 3=severe infiltrate).
Antinuclear Antibodies
Mouse sera diluted 1:50 was incubated on HEp-2 antigen substrate slides (MBL BION), and bound Abs were detected with DyLight 488–coupled detecting reagents. ANA fluorescent intensity was scored on a scale from 0–4 per the manufacturer’s instructions.
MMP-3 Quantification
MMP-3 protein levels were measured in the sera of mice per the manufacturer’s (R&D) instructions.
Flow Cytometry
Spleen and bone marrow cell suspensions were stained with the following antibodies: Ter119, CD11b, Ly6G, Ly6C, B220, CD95 and GL-7 (eBioscience or BD Biosciences). Multicolor flow cytometry was performed using an LSR II with DIVA software (BD Biosciences), and analysis was conducted with FlowJo software (TreeStar, Ashland, OR).
Statistical analysis
Data are reported as mean ± SEM. Statistical significance was analyzed with the unpaired, two-tailed Student’s t test using Prism software (GraphPad). Statistical significance is represented by the following notation in the figures: p<0.05=*, p<0.01=**, p<0.001=***.
Results
DNase II deficiency in both hematopoietic and radioresistant cells is required for the development of inflammatory arthritis and bone marrow (BM) hypercellularity in DKO mice
To investigate the relative contribution of hematopoietic vs. radioresistant host cells to the various autoimmune manifestations of DKO mice, lethally irradiated (850R) Het (DNase II+/− IFNaR−/−) or DKO (DNase II−/− IFNaR−/−) mice were reconstituted with Het or DKO stem cells to generate four experimental groups: Het→Het, DKO→DKO, Het→DKO and DKO→Het. We then assessed arthritis severity by clinical and histologic evaluation. As expected, Het→Het chimeras showed no evidence of clinical arthritis while DKO→DKO mice showed significant inflammation in the distal joints and paws (Figure 1A). Furthermore, serum levels of matrix metalloproteinase 3 (MMP3), a surrogate marker for inflammation, reflected the arthritis scores and further confirmed the absence of inflammation in the Het→DKO and DKO→Het chimeras (Figure 1B). Histologic scoring of ankle joints (Figure 1C) also confirmed the presence of arthritis only in DKO→DKO mice. Remarkably, neither the Het→DKO nor the DKO→Het mice developed any clinical or histological evidence of arthritis over a 10-month period (Figure 1A–C). Therefore both DKO donor hematopoietic cells and DKO host radioresistant cells are required for the development of arthritis.
Figure 1. Arthritis in DKO mice depends on DNase II deficiency in both donor-derived hematopoietic and radioresistant host cells.

Donor and recipient strains indicated by donor→recipient or by D (donor) and R (recipient). A) Representative images of arthritis in forepaws (top) and hindpaws (bottom) of chimeric mice and summary of clinical inflammation scores. B) Serum MMP-3 protein levels. C) Representative histologic images of inflammation in the ankle (upper panel) and midfoot (lower panel) and quantitation of histologic inflammation. Arrow designates synovium and asterisk marks bone marrow cavity. Final magnification 4x. D/E) Representative images of bone marrow cellularity in the distal tibiae and quantitation of degree of cellularity (4 sections/tibia were analyzed). Final magnification 4x. All analyses were performed on 10 month-old female chimeric mice (n=4–7 mice/genotype). F) Clinical inflammation scores of 6–12 month-old male and female mice (n=8–16 mice/genotype). Values are the mean ± SEM; *=p<0.05, **=p<0.01, ***=p<0.001 compared to Het→Het. G) Engraftment in bone marrow chimeras. Reconstituted mice were analyzed by flow cytometry 5 months after bone marrow transplant for the %GFP+ cells in total peripheral blood mononuclear cells or for the %IgD donor allotype in mature B cells. Representative data are shown (n=4–7 mice/genotype). Left: Het (GFP+)→Het (GFP−): blue line; Het (GFP+)→DKO (GFP−): green line; GFP positive control: dashed red line; GFP negative control: grey fill. Right: Representative image of DKO donors, including DKO (IgDa)→DKO (IgDb) and DKO (IgDa)→Het (IgDb). Data is representative of 2–3 individual experiments.
Histological examination of the tibiae of DKO→DKO mice also revealed a dense accumulation of cells within the marrow space (Figure 1D), also evident in marrow cavities of the ankle sections (Figure 1C). This infiltrate included a high proportion of neutrophils as well as engorged erythropoietic island macrophages. This infiltrate was not detected in Het mice, STING−/− DNase II−/− IFNaR−/− triple knockout (STING TKO) mice, or Unc93b13d/3d DNase II−/− IFNaR−/− triple knockout (Unc93 TKO) mice (Figure 1E). Furthermore, as evident from clinical inflammation scores, arthritis still develops in the Unc93b1 TKO mice (Figure 1F) whereas arthritis is abrogated in STING TKO mice (18). Altogether our data indicate that the arthritic phenotype is completely dependent on STING and not Unc93b1 (17, 18), BM hypercellularity is dependent on both STING and Unc93b1, and both the arthritic and BM hypercellularity depend on a combination of hematopoietic and radioresistant cell types. These outcomes are unlikely to be due to residual host hematopoietic cells as complete hematopoietic repopulation of all groups by donor stem cells was verified through the use of transgene or congenic markers (Figure 1G).
DNase II deficiency in both hematopoietic and radioresistant cells contributes to the development of additional TLR-dependent features of autoimmunity
DKO mice develop massive splenomegaly, clearly apparent by as early as 2 wks of age, which depends upon functional Unc93b1 and not STING (19). Since endosomal TLRs are preferentially expressed in hematopoietic cells, we expected TLR-mediated splenomegaly to track with the Unc93b1-sufficient DKO hematopoietic compartment. As expected, the DKO→DKO chimeras developed splenomegaly, and the Het→DKO did not. However, surprisingly, the DKO→Het mice also failed to develop splenomegaly (Figure 2A). Along with splenomegaly, normal splenic architecture, associated with the presence of organized follicles, is disrupted in DKO but not in Het mice. From H&E staining of splenic sections, it was clear that a similar loss of defined T and B cell regions occurred in the DKO→DKO spleens but not in the Het→DKO or DKO→Het spleens (Figure 2B). These data demonstrate that a host component is required for the TLR-dependent splenic abnormalities characteristic of DKO mice.
Figure 2. Splenomegaly and disrupted splenic architecture depend on DNase II deficiency in both donor-derived hematopoietic and radioresistant host cells.
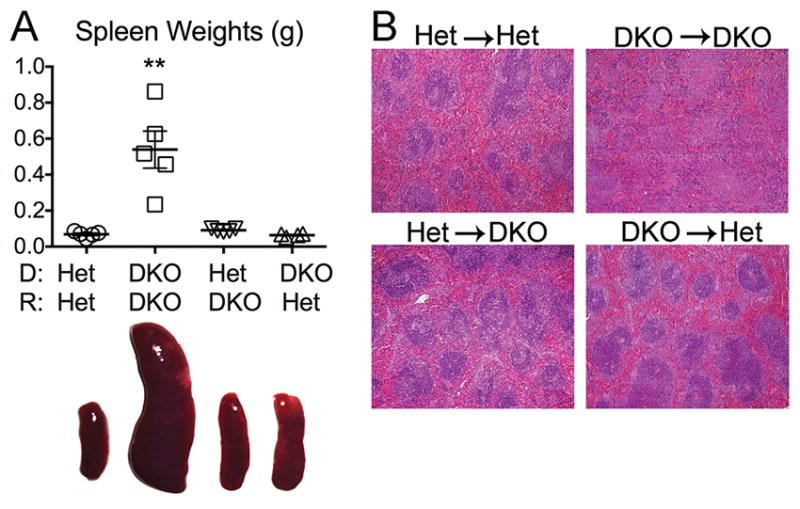
A) Splenic weights (upper panel) and representative images of spleens (lower panel). B) Splenic histology in 10 month-old female chimeric mice (n=4–7 mice/genotype; H&E-stained, final magnification 4x). Data is representative of 4 individual experiments. Values are the mean ± SEM; **=p<0.01 compared to Het→Het.
Other Unc93b1-dependent abnormalities of DKO mice include disruption of bone marrow (BM) erythropoiesis and ensuing extramedullary hematopoiesis in the spleen (19). To assess the contribution of the radioresistant host and hematopoietic elements to these aspects of hematopoiesis, the chimeric mice were evaluated for the frequency of BM and spleen cells expressing the RBC lineage marker Ter119. Similar to the DKO strain, the percentage and overall number of Ter119+ cells in the DKO→DKO chimeras was decreased in the BM and dramatically increased in the spleen. In contrast, the frequency of Ter119+ cells in the BM of the Het→DKO and DKO→Het chimeras was not significantly different from the Het→Het control, and the frequency of Ter119+ cells in the spleen of the Het→DKO and DKO→Het chimeras was only slightly increased relative to the Het→Het controls (Figure 3A). These data point to a requirement for both hematopoietic and non-hematopoietic elements in the overall disruption of erythropoiesis in DKO mice.
Figure 3. Extramedullary hematopoiesis and myeloid cell expansion depend on DNase II deficiency in both donor-derived hematopoietic and radioresistant host cells.

A) Percentage of total bone marrow (BM) and spleen (Spl) cell suspensions expressing the erythroid lineage marker Ter119. B) Percentage of total bone marrow and spleen cell suspensions expressing the myeloid lineage marker CD11b. C) Percentage of CD11b+ cells expressing the granulocyte phenotype Ly6C+ Ly6Ghi. Representative FACS plots for Het→Het and DKO→DKO are shown to the right of the compiled data figures in A, B, and C. Data is representative of 2 individual experiments. Values are the mean ± SEM; *=p<0.05, **=p<0.01, ***=p<0.001 compared to Het→Het.
DKO mice also develop an increased frequency of CD11b+ myeloid cells, and especially Ly6C+ Ly6Ghi granulocytes, in both the BM and spleen. Comparable increases were only found in the DKO→DKO chimeras (Figure 3B,C). However, both the Het→DKO and DKO→Het chimeras tended toward a greater frequency of granulocytes in the spleen, consistent with the notion that hematopoietic and radioresistant DKO cells independently provoke modest inflammatory responses, while more severe inflammation depends on donor and host cell synergy.
In addition, B cell differentiation is compromised in DKO mice, as evidenced by a markedly reduced frequency of immature and mature B cells in both the BM and spleen (19). Here again the DKO phenotype was recapitulated by the DKO→DKO chimeras, as shown by the overall percent of B220+ cells, but B cell development appeared relatively normal in Het→DKO and DKO→Het mice (Figure 4A,B). Normal B cell development was restored in the Unc93b1 TKO mice (19). Despite the reduced frequency of mature B220+ lymphocytes, the frequency of CD95+ germinal center (GC) cells, an indication of autoreactive B cell activation, was increased in the DKO→DKO chimeras, and not in the other chimera groups (Figure 4C).
Figure 4. Defective B cell development and autoantibody production depend on DNase II deficiency in both donor-derived hematopoietic and radioresistant host cells.

Percentage of B220+ cells in A) bone marrow (BM) and B) total spleen (Spl) by FACS analysis. C) Percentage of B220+ germinal center (GC) B cells in spleens. Representative FACS plots for Het→Het and DKO→DKO are shown to the right of the compiled data figures in A, B, and C. D) Quantitation of anti-nuclear antibody (ANA) fluorescent intensity. The samples marked in green correspond to the ANA patterns shown in panel E. E) ANA staining patterns from sera by immunofluorescence (original magnification x 200). Het→Het chimeras: negative; DKO→DKO: speckled nuclear; Het→DKO: homogeneous nuclear; DKO→Het: speckled nuclear, weak, 1/5 mice). All analyses performed on 10 month-old female chimeric mice (n=5–7 mice/genotype). Data is representative of 2–3 individual experiments. Values are the mean ± SEM; **=p<0.01, ***=p<0.001 compared to Het→Het.
As further evidence of B cell activation, DKO mice produce high titers of autoantibodies. Endogenous dsDNA associated with cell debris cannot activate TLR9 in DKO mice, because in the absence of DNase II the DNA is not degraded sufficiently to generate a functional TLR9 ligand (19, 25). Therefore sera from DKO mice normally show HEp2 immunofluorescent staining patterns consistent with BCR/TLR7 driven autoreactive B cell activation (e.g. speckled nuclear or cytoplasmic). As expected, Het→Het chimeras failed to make anti-nuclear antibodies and all the DKO→DKO mice developed high ANA titers with speckled nuclear or cytoplasmic staining patterns (Figure 4D,E). Remarkably, 4 out 7 Het→DKO mice also developed ANA titers, despite the fact that they failed to exhibit most other indications of systemic autoimmunity. However, in contrast to the speckled nuclear staining pattern characteristic of the DKO mouse sera and the DKO→DKO chimeric sera, the ANA+ Het→DKO chimeric sera showed homogeneous nuclear staining patterns (Figure 4E). This homogeneous nuclear pattern most likely reflects the expression of functional DNase II by the Het-derived B cells, and therefore the ability of these B cells to degrade dsDNA and generate endogenous TLR9 ligands. The Het B cells in the ANA+ Het→DKO chimeras are presumably responding to the excessive DNA accrual that occurs in radioresistant DKO host cells, and becoming activated through a TLR9-dependent mechanism. One (out of 5) ANA+ DKO→Het chimera sera showed a modest speckled nuclear pattern, again indicative of the inability of DNase II−/− B cells to respond to endogenous DNA ligands. The limited number of ANA+ Het→DKO and DKO→Het mice represent one exception to the overall requirement for DNase II deficiency in both the recipient and host for clinical manifestations of disease. In all cases the B cells are presumably responding to an external source of nucleic acid-associated ligand. Overall the defects in B cell development (reduced number of B220+ cells) and increased numbers of GC B cells are only apparent in DKO→DKO chimeras.
Expression of functional Unc93b1 in hematopoietic cells is sufficient for the development of Unc93b1-dependent clinical manifestations
One possible explanation for the failure of DKO→Het chimeras to develop Unc93b1-dependent clinical manifestations was a requirement for a TLR-expressing host component. To test this possibility, we used Unc93 TKO mice to generate DKO→Unc93 TKO and Unc93 TKO→DKO chimeras and compared them to DKO→Het chimeras. Since synovial inflammation is Unc93b1-independent, it was not surprising that both the DKO→Unc93 TKO and Unc93 TKO→DKO chimeras developed arthritis as determined by clinical examination (Figure 5A) and joint histology (Figure 5B). Furthermore, as expected, the Unc93 TKO→DKO chimeras had less severe BM inflammation (Figure 5C), failed to develop splenomegaly (Figure 5D), and failed to make ANAs (Figure 5E). Thus, the Unc93b1-dependent clinical manifestations require Unc93b1 expression in hematopoietic cells. However, the DKO→Unc93 TKO chimeras were essentially indistinguishable from DKO→DKO chimeras by all criteria evaluated. Thus, the hematopoietic expression of endosomal TLRs is necessary but not sufficient for SLE-like clinical manifestations, and there must be an additional TLR-independent host component(s) that promotes the onset of TLR-dependent autoimmunity.
Figure 5. Expression of Unc93b1 in hematopoietic cells is required for the development of Unc93b1-dependent clinical manifestations.

A) Clinical inflammation scores. B) Representative histologic images of inflammation in the ankle (upper panel) and midfoot (lower panel). Arrow designates synovium and arrowhead marks cellular infiltrate in the talus. Final magnification 4x. C) Quantitation of degree of bone marrow (BM) cellularity. D) Spleen weights. E) Representative ANA staining patterns of chimeric sera (original magnification x 200). All analyses performed on female chimeric mice. Data is representative of 2 individual experiments. Values are the mean ± SEM; *=p<0.05, ***=p<0.001 compared to DKO→Het.
Discussion
The main message that emerges from the current study, together with previous reports, is that the accrual of excessive undegraded DNA in multiple cell types of DKO mice promotes the activation of both cytosolic and endosomal nucleic acid sensors, leading to a type I IFN-independent disease spectrum that incorporates features of both inflammatory arthritis and SLE. Cytosolic sensors are responding to DNA and the endosomal sensors are most likely responding to RNA-associated cell debris internalized by a cell surface receptor. Our Het/DKO chimera data further show that both hematopoietic and radioresistant host cells are required for all the various clinical manifestations.
The importance of radioresistant cells in autoinflammation has been previously reported by Stetson and colleagues in their analysis of mice lacking Trex-1, a model of STING-dependent autoinflammation. By using a Trex1−/− IFN-reporter line, they identified cardiac endothelial cells as the initial site of IFN-driven inflammation. As a result, these mice first develop myocarditis although additional tissues subsequently became inflamed. In contrast to the DKO mice described in the current report, Trex1 deficiency in radioresistant host cells was sufficient to activate WT bone marrow derived cells and trigger a systemic response where organ damage depended on activated T and B lymphocytes. However, a role for hematopoietic cells in the DKO model is not surprising since DNase II is a lysosomal DNase required for the degradation of cell debris/DNA phagocytosed by myeloid lineage phagocytes.
Another difference between the Trex1−/− and DNase II−/− IFNaR−/− DKO models is the dependency on type I IFNs. IFNaR−/− x Trex1−/− mice fail to develop clinical manifestations of disease (20), while both STING-dependent and Unc93-dependent aspects of our DKO mice are type I IFN independent due to the absence of a functional type I IFN receptor. The strong autoantibody response, presumably dependent on TLR7, is particularly unexpected, especially since we have previously shown that TLR7-dependent B cell responses are highly type I IFN dependent (26). Exactly how cytokine production by DKO mice circumvents a need for type I IFNs as far as RNA-dependent B cell activation and other Unc93-dependent outcomes is not clear. Experiments are in progress to determine whether IFN-inducible genes downstream of RNA-sensing TLRs will exacerbate the Unc93-dependent SLE-like aspects of the “DKO” phenotype in IFNaR-sufficient DNase II−/− STING−/− mice (17).
The nature of the radioresistant cell(s) activated in DKO mice remains to be determined. Previous studies have documented an important role for STING dependent pathways in fibroblast lineage cells (4, 27). Therefore it is possible that STING promotes arthritis through its capacity to activate synovial fibroblasts. These cells have long been known to play a key role in the pathogenesis of rheumatoid arthritis (28) and are a source of proinflammatory cytokines and other factors that promote and perpetuate chronic inflammation and joint damage. Studies in early rheumatoid arthritis have demonstrated changes in the stromal compartments of synovium as early as the first months of disease (29). With disease progression, there is expansion of the synovial fibroblast compartment and these cells secrete proteinases that destroy cartilage, as well as factors that promote chronicity of inflammation through recruitment and retention of inflammatory cells (30). Therefore, it is likely that synovial fibroblasts play a critical role in the initiation and perpetuation of disease in our model. Although we assume that the radioresistant cell is a stromal component, we cannot at this stage rule out a role for embryonally-derived macrophages. These are radioresistant tissue resident macrophages that develop from precursors that seed peripheral tissues during fetal development, and could reside in synovial tissues as well (31).
The distinct overall phenotype of the DKO→DKO chimera group compared to the Het→DKO and DKO→Het chimeras demonstrated an absolute requirement for DKO radioresistant cells in the development of all the Unc93b1 clinical manifestations. Because the DKO→Unc93 TKO mice completely recapitulated the phenotype of the DKO→DKO chimeras, the host component is most likely to be due to inflammation driven by one or more cytosolic sensors, and not an endosomal cytosolic sensor. Both STING TKO and Unc93 TKO mice failed to develop the hypercellularity we observed in the long bone marrow, and therefore we propose that at a minimum, bone marrow hypocellularity depends on a STING triggered host cell, perhaps located in the hematopoietic niche of the marrow, and a TLR responsive hematopoietic cell. In both BM hypercellularity and splenomegaly, the relevant TLR is expected to be an RNA sensor (TLR7, TLR8, TLR3 or perhaps even TLR13), since endogenous DNA is not sufficiently degraded in DKO mice to activate TLR9 (19, 25). The BM infiltrate includes a high percentage of neutrophils and a similar accumulation of neutrophils in the BM of pristane-injected mice has been shown to be TLR7 dependent. These observations may be related to the activated neutrophils recently identified in the marrow of SLE patients (32).
In summary, the combined data from the Het/DKO and Unc93 TKO/DKO chimeras demonstrate a critical interplay between cytosolic and endosomal sensors in the development of all the clinical manifestations of DKO mice and suggest that cytosolic sensors may play a more general role in promoting SLE and related autoimmune disorders. Cytosolic DNA sensors appear to be particularly responsive to inherent cell stress such as mitochondrial depolarization and the subsequent release of mitochondrial DNA into the cytosol (33, 34), or to the excessive accumulation of damaged DNA resulting from defective autophagosome formation (35). We previously demonstrated a role for the inflammasome-associated DNA sensor AIM2 in the development of arthritis in DKO mice. Whether RNA-sensing cytosolic sensors such as MDA-5 or RIG-I also play a role remains to be determined. From a broader perspective, cytosolic sensors may play an active role in detecting environmental insults that can trigger the onset and/or perpetuation of systemic autoimmune or autoinflammatory conditions. A better understanding of the in vivo networks that promote the distinct features of systemic autoimmune diseases will provide a platform for the design of therapeutics that address the unmet needs of patient populations.
Acknowledgments
Grant Support: This work was supported by National Institutes of Health grants AR050256 (A.M.R.), AI093752 and AI083713 (K.A.F.), AR067394 (E.M.G.), T32 AI 095213 (R.B.), and by grants from the Lupus Research Institute (A.M.R.), and the Musculoskeletal Center of Excellence Grants Program, University of Massachusetts (E.M.G.) and Alliance for Lupus Research Grant (S.S.).
The authors would like to thank Dr. S. Nagata for providing the DNase II deficient mice, Dr. B. Beutler for providing the Unc93b13d/3d mice, Drs. G. Barber for generating and D. Stetson for providing the STING-deficient mice, Dr. R. Gerstein for providing the GFP Tg mice, and T. Robidoux for technical assistance.
References
- 1.Sharma S, Fitzgerald KA. Innate immune sensing of DNA. PLoS Pathog. 2011;7:e1001310. doi: 10.1371/journal.ppat.1001310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhat N, Fitzgerald KA. Recognition of cytosolic DNA by cGAS and other STING-dependent sensors. Eur J Immunol. 2014;44:634–640. doi: 10.1002/eji.201344127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xiao TS, Fitzgerald KA. The cGAS-STING pathway for DNA sensing. Mol Cell. 2013;51:135–139. doi: 10.1016/j.molcel.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg MC, Goudin N, Fremond ML, Nitschke P, Molina TJ, Blanche S, Picard C, Rice GI, Crow YJ, Manel N, Fischer A, Bader-Meunier B, Rieux-Laucat F. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J Clin Invest. 2014;124:5516–5520. doi: 10.1172/JCI79100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Montealegre Sanchez GA, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS, Lee CC, DiMattia MA, Cowen EW, Gonzalez B, Palmer I, DiGiovanna JJ, Biancotto A, Kim H, Tsai WL, Trier AM, Huang Y, Stone DL, Hill S, Kim HJ, St Hilaire C, Gurprasad S, Plass N, Chapelle D, Horkayne-Szakaly I, Foell D, Barysenka A, Candotti F, Holland SM, Hughes JD, Mehmet H, Issekutz AC, Raffeld M, McElwee J, Fontana JR, Minniti CP, Moir S, Kastner DL, Gadina M, Steven AC, Wingfield PT, Brooks SR, Rosenzweig SD, Fleisher TA, Deng Z, Boehm M, Paller AS, Goldbach-Mansky R. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371:507–518. doi: 10.1056/NEJMoa1312625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crow YJ, Rehwinkel J. Aicardi-Goutieres syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Hum Mol Genet. 2009;18:R130–136. doi: 10.1093/hmg/ddp293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Namjou B, Kothari PH, Kelly JA, Glenn SB, Ojwang JO, Adler A, Alarcon-Riquelme ME, Gallant CJ, Boackle SA, Criswell LA, Kimberly RP, Brown E, Edberg J, Stevens AM, Jacob CO, Tsao BP, Gilkeson GS, Kamen DL, Merrill JT, Petri M, Goldman RR, Vila LM, Anaya JM, Niewold TB, Martin J, Pons-Estel BA, Sabio JM, Callejas JL, Vyse TJ, Bae SC, Perrino FW, Freedman BI, Scofield RH, Moser KL, Gaffney PM, James JA, Langefeld CD, Kaufman KM, Harley JB, Atkinson JP. Evaluation of the TREX1 gene in a large multi-ancestral lupus cohort. Genes Immun. 2011;12:270–279. doi: 10.1038/gene.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, de Silva U, Bailey SL, Witte T, Vyse TJ, Kere J, Pfeiffer C, Harvey S, Wong A, Koskenmies S, Hummel O, Rohde K, Schmidt RE, Dominiczak AF, Gahr M, Hollis T, Perrino FW, Lieberman J, Hubner N. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. 2007;39:1065–1067. doi: 10.1038/ng2091. [DOI] [PubMed] [Google Scholar]
- 9.Ramantani G, Kohlhase J, Hertzberg C, Innes AM, Engel K, Hunger S, Borozdin W, Mah JK, Ungerath K, Walkenhorst H, Richardt HH, Buckard J, Bevot A, Siegel C, von Stulpnagel C, Ikonomidou C, Thomas K, Proud V, Niemann F, Wieczorek D, Hausler M, Niggemann P, Baltaci V, Conrad K, Lebon P, Lee-Kirsch MA. Expanding the phenotypic spectrum of lupus erythematosus in Aicardi-Goutieres syndrome. Arthritis Rheum. 2010;62:1469–1477. doi: 10.1002/art.27367. [DOI] [PubMed] [Google Scholar]
- 10.Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, Corry PC, Cowan FM, Frints SG, Klepper J, Livingston JH, Lynch SA, Massey RF, Meritet JF, Michaud JL, Ponsot G, Voit T, Lebon P, Bonthron DT, Jackson AP, Barnes DE, Lindahl T. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet. 2006;38:917–920. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- 11.Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, Fuller JC, Jackson RM, Lamb T, Briggs TA, Ali M, Gornall H, Couthard LR, Aeby A, Attard-Montalto SP, Bertini E, Bodemer C, Brockmann K, Brueton LA, Corry PC, Desguerre I, Fazzi E, Cazorla AG, Gener B, Hamel BC, Heiberg A, Hunter M, van der Knaap MS, Kumar R, Lagae L, Landrieu PG, Lourenco CM, Marom D, McDermott MF, van der Merwe W, Orcesi S, Prendiville JS, Rasmussen M, Shalev SA, Soler DM, Shinawi M, Spiegel R, Tan TY, Vanderver A, Wakeling EL, Wassmer E, Whittaker E, Lebon P, Stetson DB, Bonthron DT, Crow YJ. Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet. 2009;41:829–832. doi: 10.1038/ng.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, Jenkinson EM, Bacino CA, Battini R, Bertini E, Brogan PA, Brueton LA, Carpanelli M, De Laet C, de Lonlay P, del Toro M, Desguerre I, Fazzi E, Garcia-Cazorla A, Heiberg A, Kawaguchi M, Kumar R, Lin JP, Lourenco CM, Male AM, Marques W, Jr, Mignot C, Olivieri I, Orcesi S, Prabhakar P, Rasmussen M, Robinson RA, Rozenberg F, Schmidt JL, Steindl K, Tan TY, van der Merwe WG, Vanderver A, Vassallo G, Wakeling EL, Wassmer E, Whittaker E, Livingston JH, Lebon P, Suzuki T, McLaughlin PJ, Keegan LP, O’Connell MA, Lovell SC, Crow YJ. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet. 2012;44:1243–1248. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rice GI, Reijns MA, Coffin SR, Forte GM, Anderson BH, Szynkiewicz M, Gornall H, Gent D, Leitch A, Botella MP, Fazzi E, Gener B, Lagae L, Olivieri I, Orcesi S, Swoboda KJ, Perrino FW, Jackson AP, Crow YJ. Synonymous mutations in RNASEH2A create cryptic splice sites impairing RNase H2 enzyme function in Aicardi-Goutieres syndrome. Hum Mutat. 2013;34:1066–1070. doi: 10.1002/humu.22336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134:587–598. doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stetson DB. Endogenous retroelements and autoimmune disease. Curr Opin Immunol. 2012;24:692–697. doi: 10.1016/j.coi.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kimura-Kataoka K, Yasuda T, Fujihara J, Toga T, Ono R, Otsuka Y, Ueki M, Iida R, Sano R, Nakajima T, Kominato Y, Kato H, Takeshita H. Genetic and expression analysis of SNPs in the human deoxyribonuclease II: SNPs in the promoter region reduce its in vivo activity through decreased promoter activity. Electrophoresis. 2012;33:2852–2858. doi: 10.1002/elps.201200260. [DOI] [PubMed] [Google Scholar]
- 17.Ahn J, Gutman D, Saijo S, Barber GN. STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci U S A. 2012;109:19386–19391. doi: 10.1073/pnas.1215006109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baum R, Sharma S, Carpenter S, Li QZ, Busto P, Fitzgerald KA, Marshak-Rothstein A, Gravallese EM. Cutting edge: AIM2 and endosomal TLRs differentially regulate arthritis and autoantibody production in DNase II-deficient mice. J Immunol. 2015;194:873–877. doi: 10.4049/jimmunol.1402573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pawaria S, Moody K, Busto P, Nundel K, Choi CH, Ghayur T, Marshak-Rothstein A. Cutting Edge: DNase II deficiency prevents activation of autoreactive B cells by double-stranded DNA endogenous ligands. J Immunol. 2015;194:1403–1407. doi: 10.4049/jimmunol.1402893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gall A, Treuting P, Elkon KB, Loo YM, Gale M, Jr, Barber GN, Stetson DB. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity. 2012;36:120–131. doi: 10.1016/j.immuni.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawane K, Tanaka H, Kitahara Y, Shimaoka S, Nagata S. Cytokine-dependent but acquired immunity-independent arthritis caused by DNA escaped from degradation. Proc Natl Acad Sci U S A. 2010;107:19432–19437. doi: 10.1073/pnas.1010603107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tabeta K, Hoebe K, Janssen EM, Du X, Georgel P, Crozat K, Mudd S, Mann N, Sovath S, Goode J, Shamel L, Herskovits AA, Portnoy DA, Cooke M, Tarantino LM, Wiltshire T, Steinberg BE, Grinstein S, Beutler B. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol. 2006;7:156–164. doi: 10.1038/ni1297. [DOI] [PubMed] [Google Scholar]
- 23.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pettit AR, Ji H, von Stechow D, Muller R, Goldring SR, Choi Y, Benoist C, Gravallese EM. TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am J Pathol. 2001;159:1689–1699. doi: 10.1016/S0002-9440(10)63016-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chan MP, Onji M, Fukui R, Kawane K, Shibata T, Saitoh S, Ohto U, Shimizu T, Barber GN, Miyake K. DNase II-dependent DNA digestion is required for DNA sensing by TLR9. Nat Commun. 2015;6:5853. doi: 10.1038/ncomms6853. [DOI] [PubMed] [Google Scholar]
- 26.Green NM, Laws A, Kiefer K, Busconi L, Kim YM, Brinkmann MM, Trail EH, Yasuda K, Christensen SR, Shlomchik MJ, Vogel S, Connor JH, Ploegh H, Eilat D, Rifkin IR, van Seventer JM, Marshak-Rothstein A. Murine B cell response to TLR7 ligands depends on an IFN-beta feedback loop. J Immunol. 2009;183:1569–1576. doi: 10.4049/jimmunol.0803899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turner JD, Filer A. The role of the synovial fibroblast in rheumatoid arthritis pathogenesis. Curr Opin Rheumatol. 2015;27:175–182. doi: 10.1097/BOR.0000000000000148. [DOI] [PubMed] [Google Scholar]
- 29.Raza K, Falciani F, Curnow SJ, Ross EJ, Lee CY, Akbar AN, Lord JM, Gordon C, Buckley CD, Salmon M. Early rheumatoid arthritis is characterized by a distinct and transient synovial fluid cytokine profile of T cell and stromal cell origin. Arthritis Res Ther. 2005;7:R784–795. doi: 10.1186/ar1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buckley CD. Why does chronic inflammation persist: An unexpected role for fibroblasts. Immunol Lett. 2011;138:12–14. doi: 10.1016/j.imlet.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lichanska AM, Browne CM, Henkel GW, Murphy KM, Ostrowski MC, McKercher SR, Maki RA, Hume DA. Differentiation of the mononuclear phagocyte system during mouse embryogenesis: the role of transcription factor PU.1. Blood. 1999;94:127–138. [PubMed] [Google Scholar]
- 32.Zhuang H, Han S, Xu Y, Li Y, Wang H, Yang LJ, Reeves WH. Toll-like receptor 7-stimulated tumor necrosis factor alpha causes bone marrow damage in systemic lupus erythematosus. Arthritis Rheumatol. 2014;66:140–151. doi: 10.1002/art.38189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rongvaux A, Jackson R, Harman CC, Li T, West AP, de Zoete MR, Wu Y, Yordy B, Lakhani SA, Kuan CY, Taniguchi T, Shadel GS, Chen ZJ, Iwasaki A, Flavell RA. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell. 2014;159:1563–1577. doi: 10.1016/j.cell.2014.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, Kaech SM, Smiley JR, Means RE, Iwasaki A, Shadel GS. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520:553–557. doi: 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lan YY, Londono D, Bouley R, Rooney MS, Hacohen N. Dnase2a deficiency uncovers lysosomal clearance of damaged nuclear DNA via autophagy. Cell Rep. 2014;9:180–192. doi: 10.1016/j.celrep.2014.08.074. [DOI] [PMC free article] [PubMed] [Google Scholar]