

Abstract
Hepatocarcinoma (HCC) is a highly prevalent cancer worldwide and its inflammatory background was established long ago. Recent studies have shown that innate immunity is closely related to the HCC carcinogenesis. An effective innate immunity response relies on the toll-like receptors (TLR) found in several different liver cells which, through different ligands and many signaling pathways can elicit, not only a pro-inflammatory but also an oncogenic or anti-oncogenic response. Our aim was to study the role of TLRs in the liver oncogenesis and as a consequence their value as potential therapeutic targets. We performed a systematic review of PubMed searching for original articles studying the relationship between HCC and TLRs until March 2015. TLR2 appears to be a fundamental stress-sensor as its absence reveals an augmented tendency to accumulate DNA-damages and to cell survival. However, pathways are still not fully understood as TLR2 up-regulation was also associated to enhanced tumorigenesis. TLR3 has a well-known protective role influencing crucial processes like angiogenesis, cell growth or proliferation. TLR4 works as an interesting epithelial-mesenchymal transition’s inducer and a promoter of cell survival probably inducing HCC carcinogenesis even though an anti-cancer role has already been observed. TLR9’s influence on carcinogenesis is also controversial and despite a potential anti-cancer capacity, a pro-tumorigenic role is more likely. Genetic polymorphisms in some TLRs have been found and its influence on the risk of HCC has been reported. As therapeutic targets, TLRs are already in use and have a great potential. In conclusion, TLRs have been shown to be an interesting influence on the HCC’s microenvironment, with TLR3 clearly determining an anti-tumour influence. TLR4 and TLR9 are considered to have a positive relationship with tumour development even though, in each of them anti-tumorigenic signals have been described. TLR2 presents a more ambiguous role, possibly depending on the stage of the inflammation-HCC axis.
Keywords: Hepatocarcinoma, Carcinogenesis, Toll-like receptor, Innate immunity, Chronic inflammation
Core tip: The importance of hepatocarcinoma (HCC) is undeniable in the current medical perspective. However, a lot still remains to be understood in this context. Therefore, this review aims to present the significance of innate immunity in HCC through toll-like receptors as they have already shown interesting effects on tumour’s microenvironment, influencing its progression or regression. As a result we also render some therapeutic usages of the established knowledge in this area.
INTRODUCTION
Liver cancer is one of the most common cancers worldwide, with hepatocarcinoma (HCC) being, by far, the most frequent type[1,2].
Due to its close contact with gut, via portal vein, liver faces a continuous exposure to gut-derived bacterial products, toxics and many other agents[3]. In the presence of such pathogens or irritants and associated molecules our body is able to respond in a manner that aims to prevent injury and combat infection. This protection system is called inflammation which, despite its tremendous defensive and antiviral/antibacterial importance in the short term, starts to become deleterious when prolonged or exaggerated - chronic inflammation - possibly leading to fibrosis, cirrhosis and, ultimately, HCC[4].
Therefore, the idea that hepatic carcinogenesis arouses from an inflammatory basis is not new. Several studies already focused on the development of HCC and possibilities like the c-Myc elevation or the deregulated SRY and SGF29 pathways have been proposed[5]. However, just in the last few years we have become aware of the critical role of innate immunity in chronic liver diseases, including HCC[6,7].
Toll-like receptors (TLRs) are a family of pattern-recognition receptors (PRRs) that can be activated by either pathogen-associated molecular patterns (PAMPs) or danger/damage-associated molecular patterns (DAMPs), with their own importance in eliciting innate immunity, regulation of inflammation and tissue regeneration. To date, 11 human TLRs have been identified[8]. In recent years, activation of several TLRs have been associated with viral hepatitis, steatohepatitis (alcoholic or non-alcoholic) and to the progression of the inflammation-fibrosis-HCC axis[9-11]. However, data is somewhat contradictory and no clear conclusions have been made.
In this line of thoughts, this review aims to present an overview of the expression of TLRs in the liver, its influence on the development of liver carcinogenesis as chronic inflammatory inducers or potential oncogenes as well as possible therapeutic targets.
RESEARCH
Specific criteria were defined in order to guide this systematic review. Firstly, a query to obtain the articles related to the theme on PubMed was built: [(Hepatocarcinoma) OR (Hepatocarcinogenesis) OR (hepatic cancer) OR (hepatocellular carcinoma) OR (liver cancer)] AND [(toll like receptors) OR (toll like receptor)]. With this query we intended to embrace a wide range of articles until March 2015, which then would be carefully selected.
A total amount of 277 articles were obtained through the referred search. After discarding the duplicates and adding 28 articles obtained through cross-referencing, 305 articles were available to be screened. The following inclusion criteria were used: (1) studies that were published until the end March 2015; (2) the article should be written in English; and (3) studies relevant to the theme (presenting original data). As exclusion criteria we defined: (1) studies considered by the authors as unrelated to the theme; and (2) non-original studies. These criteria were applied by reading the title and abstract resulting in 227 articles excluded. After this step, the remaining 78 studies were selected for full-text reading. On a second level of eligibility, 18 more studies were excluded and 60 studies were selected, analysed and included in this revision (Figure 1).
Figure 1.
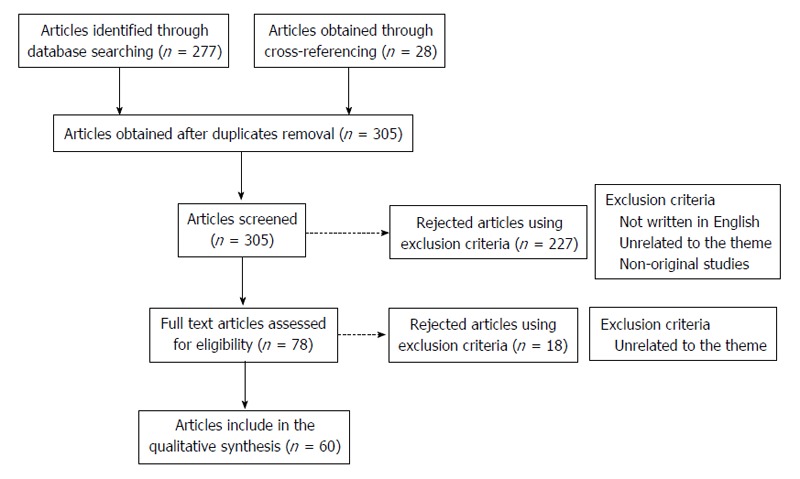
Methods’ flowchart. A total amount of 277 articles were obtained, on PubMed, through the query [(hepatocarcinoma) OR (hepatocarcinogenesis) OR (hepatic cancer) OR (hepatocellular carcinoma) OR (liver cancer)] AND [(toll like receptors) OR (toll like receptor)]. After discarding the duplicates and adding 28 articles obtained through cross-referencing, 305 articles were available to be screened. The following inclusion criteria were used: (1) studies that were published until the end March 2015; (2) the article should be written in English; and (3) studies relevant to the theme (presenting original data). As exclusion criteria we defined: (1) studies considered by the authors as unrelated to the theme; and (2) non-original studies. These criteria were applied by reading the title and abstract resulting in 227 articles excluded. After this step, the remaining 78 studies were selected for full-text reading. On a second level of eligibility, 18 more studies were excluded and 60 studies were selected, analysed and included in this revision.
Data about the defined topics were obtained from each article (Table 1) and the information was then summed up and organized in the present systematic review according to: The TLRs’ expression in each liver cell; separately role of TLR2, TLR3, TLR4 and TLR9 in inflammatory-driven hepatocarcinoma; known TLRs’ polymorphisms/genetic variations that influence the risk of hepatocarcinoma and lastly, TLRs modulators possibly used in hepatocarcinoma’s therapeutics.
Table 1.
Table of original studies
| Ref. | Year | Type of study | Methods | Limitations | Conclusions |
| Chew et al[1] | 2012 December | Experimental | Natural killer cell activation and cytotoxicity were assessed in vitro after treatment with the TLR3 ligand poly (I:C). The effect of TLR in a spontaneous liver tumor mouse model and a transplanted tumor mouse model were determined by Immunohistochemistry and PCR | The effect of poly (I:C) on tumor growth was only analyzed in a transplanted, nonorthotopic model of HCC. The effect of poly (I:C) on human NK cells was assessed only with cells from healthy donors. Not all HCC cell lines undergo apoptosis after TLR3 triggering and the reason is not known | TLR3 is an important modulator of HCC progression and is a potential target for novel immunotherapy |
| Mohamed et al[2] | 2015 March | Experimental | Tissue microarrays containing liver samples from patients with cirrhosis, viral hepatitis and HCC were examined for expression of TLR7 and TLR9. Proliferation of human HCC cell lines was studied following stimulation of TLR7 and TLR9 using agonists (imiquimod and CpG-ODN respectively) and inhibition with a specific antagonist (IRS-954) or chloroquine. The effect of these interventions was confirmed in a xenograft model and DEN/NMOR-induced model of HCC | Before translation to the clinical arena, it is important to further characterize the exact mechanisms through which TLR7 and TLR9 exert their actions and determine what effects their inhibition may have on the immune system | Inhibiting TLR7 and TLR9 with IRS-954 or chloroquine could potentially be used as a novel therapeutic approach for preventing HCC development and/or progression in susceptible patients |
| Dapito et al[3] | 2012 April | Experimental | TLR2-deficient mice, TLR4-deficient mice, TNFR1-/IL-1R1-double deficient and C57Bl/6 mice were used. HCC was induced by intraperitoneal injection of DEN. Gut-sterilization was done using a combination of ampicillin (1 g/L), neomycin (1 g/L), metronidazole (1 g/L) and vancomycin (500 mg/L) in drinking water. Samples from patients with features of alcoholic hepatitis were used. Liver biopsies were obtained from mice and from cadaveric donners or resection of liver metastases | Clinically feasible methods of targeting the intestinal microbiota or TLR4 need to be established. The quadruple combination of antibiotics employed is not suitable for long-term treatment due to known side effects in patients with advanced liver disease | Gut sterilization restricted to late stages of hepatocarcinogenesis reduced HCC, suggesting that the intestinal microbiota and TLR4 represent therapeutic targets for HCC prevention in advanced liver disease |
| Eiró et al[8] | 2014 July | Experimental | The expression levels of TLR3, TLR4 and TLR9 were analyzed from 30 patients with HCC and correlated with various clinicopathological findings and with overall survival | In the scoring system, after immunostaining analysis, when setting of the threshold for positive staining and the determination of the intensity different observers can set different thresholds and intensity levels | An association between TLR3, TLR4 and TLR9 expression and tumor aggressiveness and poor prognosis in HCC has been observed |
| Liu et al[12] | 2002 July | Experimental | Cultures of primary mouse hepatocytes were incubated with LPS to assess its effects on the global gene expression, hepatic transcription factors, and MAP kinase activation | Using hepatocytes' cell lines loses the capacity to observe the importance of a direct response to LPS by hepatocytes | NF-κB activation was reduced in TLR4-mutant or - null hepatocytes compared to control hepatocytes |
| Matsumura et al[13] | 2000 October | Experimental | PCR analysis of mice's hepatocytes and an murine hepatoma cell line Hepa 1-6 | Murine hepatoma cell line Hepa 1-6 may have reached an overquantitative level after stimulation | LPS and proin-flammatory cytokines differentially regulate gene expression of TLR2 and TLR4 in murine hepatocytes, which may lead to pathologic and host defense reactions in the liver |
| Thobe et al[14] | 2007 March | Experimental | Wester blotting and cytokine analysis in a cell culture. Evaluation of Kupfer cells response after a trauma-hemorrage procedure | Does not explain if the increase in MAPK-activity is due to TLRs' overexpression | Kupffer cell TLR signaling employs different MAPK pathways in eliciting cytokine and chemokine responses following trauma-hemorrhage |
| Knolle et al[15] | 1995 February | Experimental | Human Kupffer cells were isolated by collagenase perfusion followed by centrifugal elutriation and analyzed for cytokine secretion after 3 d in culture | Only IL-10 and IL-6 were analysed | The important role for IL-10 in the regulation of the local immune response in the liver sinusoid after Kupffer cells exposure to to lipopolysaccharide |
| Edwards et al[16] | 2003 April | Experimental | Splenocyte reparations were enriched for D11c+ and for Ly6C+ cells using magnetic selection. Four populations were routinely isolated and TLR's mRNA was amplified by PCR | To analyze the functional significance of TLR mRNA expression in DCs subsets it was only used ligands for TLR7 and TLR9 | mRNA for most TLRs is expressed at similar levels by murine splenic DC sub-types. TLR expression between plasmacytoid and non-plasmacytoid DC is not conserved between species |
| Sawaki et al[17] | 2007 March | Experimental | Total RNA was extracted, and mRNA for TLR1, 2, 3, 4, 5, 6, 7, 9 and b-actin was determined by reverse transcription-PCR. Nuclear localization of NF-κB was determined and cytokines and chemokines were measured by a commercially available kit | It was not evaluated precise roles of NK cell responses in vivo | Upon microbial infection, macrophages produce IL-12 that renders NK cells highly responsive to TLR agonists to produce IFN-γ and chemokines, which might in turn recruit and fully activate macrophages |
| Meyer-Bahlburg et al[18] | 2007 December | Experimental | It was compared the TLR response profile of germinal center after immunization vs naive mature B cell subsets, using real time PCR, ELISA and Western Blotting to evaluate MyD88 pathway | TLRs' role in B-cells immune response was only accessed in splenic B cells from MyD88 WT, Het, or KO, being studied only the MyD88-dependent pathway | B cell-intrinsic TLR signals are not required for antibody production or maintenance |
| Paik et al[19] | 2003 May | Experimental | LPS-associated signalling molecules in culture-activated HSCs and HSCs isolated from patients with hepatitis C virus-induced cirrhosis was evaluated by NF-κB-dependent luciferase reporter gene assays, electrophoretic mobility shift assays and in vitro kinase assays | It does not fully explain why only full activated HSCs respond to LPS. It was not evaluated the activation of TLR4 downstream molecules like MyD88 | Human activated HSCs utilize components of TLR4 signal transduction cascade to stimulate NF-ĸB and JNK and up-regulate chemokines and adhesion molecules |
| Wu et al[20] | 2010 March | Experimental | Isolated Kupffer cell and liver sinusoidal endothelial cells from wild-type C57BL/6 mice and examined their responses to TLR1 to TLR9 agonists. Characterization of cell surface protein expression was done by flow cytometry and quantification of mRNA was done by reverse transcription-polymerase chain reaction | The in vitro assay does not explore the organ-specific regulation of immune responses. For the identification of TLR-induced antiviral cytokine(s) only TLR3 and TLR4 were used | Non-parenchymal cells display a restricted TLR-mediated activation profile when compared with "classical" antigen-presenting cells which may, at least in part, explain their tolerogenic function in the liver |
| Huang et al[21] | 2012 July | Experimental | TLR expression in BLE-7402 cells was assayed by RT-PCR, real-time PCR and FCM. To investigate the function of TLR2 in hepatocarcinoma growth, BLE-7402 cells were transfected with recombinant plasmids expressing one TLR2 siRNA | Only the effect on tumour volume is evaluated after tumour implantation in nude mice | TLR2 knockdown inhibit proliferation of cultured hepatocarcinoma cells and decrease the secretion of cytokines |
| Kim et al[22] | 2009 January | Experimental | LLC cells were implanted in mice. Metastasis enhancing factors were identified on a QSTAR XL qQTOF mass spectrometer. Gene and protein expression were monitored by Q-PCR and immunoblot analysis. Tumors were analyzed by immunohistochemistry and indirect immunofluorescence | It does not explain if the interaction between versican and TLR2 is direct or depends on a versican’s ligand | By activating TLR2:TLR6 complexes and inducing TNF-α secretion by myeloid cells, versican strongly enhances lewis lung carcinoma metastatic growth |
| Lin et al[24] | 2013 January | Experimental | A DEN injection was done in TLR2-/- and WT mice. Than they were sham-treated or treated with interferon-gamma. TUNEL, heterochromatin and SA b-gal staining were performed | The mechanism by which TLR2 signaling participates in the regulation of cellular senescence to maintain growth arrest and promote programmed cell death remains inconclusive | Loss of immune networks may play a role in the failure of initiating and maintaining cellular senescence and autophagy flux in the TLR2-mutant liver tissue |
| Lin et al[25] | 2013 October | Experimental | WT mice were pre-treated with anti-TLR2 antibody and a subset of TLR2-/- mice were pre-treatment with NAC (antioxidant) or physiological saline. Both were submitted to DEN. Histology was submitted to western blotting, ROS assay, immunohistochemistry and immunofluorescence | It does not report any results about the effects on non-parenchymal cells like Kuppfer cells. It does not reveal interactions that regulate the signal from TLR2 activation to suppression of oxidant and ER stressors in HCC | A TLR2 activity defends against hepatocarcinogenesis through diminishing the accumulation of ROS and alleviating ER stress and unfold protein response |
| Li et al[26] | 2015 March | Experimental | WT and Tlr2-/- mice were used. Flow cytometry, Histopathological analysis and Immunofluorescence, Western blot and ELISA were performed. MDSC induction in vitro and functional T cell suppression assay and knockdown of IL-18 and caspase-8 in hepatocytes with quantitative PCR were also done | The exact role of IL-18 in MDSC generation is still unknown. It does not reveal the levels of TLR2 that determine the possible use of IL-18 as a therapeutic target | TLR2 deficiency accelerates IL-18-mediated immunosuppression during liver carcinogenesis, providing new insights into immune control that may assist the design of effective immunotherapies to treat HCC |
| Soares et al[27] | 2012 October | Analytic - cross sectional | It was used samples from patients with hepatitis, cirrhosis and hepatocarcinoma. mRNA isolation and quantification of TLR2, TLR4, NF-κB, TNF-α and COX-2 were performed. Immunohistochemical evaluation of TLR2 and TLR4 was also done | Most patients included in the reference group have evidence of NAFLD and it was demonstrated that NAFLD is associated with increased hepatic TLR2 and TLR4-mRNA expression. the hepatitis, cirrhosis and hepatocarcinoma groups included both patients with HBV infection or HCV infection. Included only patients with virus-induced chronic hepatitis. The method used for quantification of protein expression was semi-quantitative | Increased expression of TLR2 and TLR4 in hepatitis and cirrhosis and maintained expression in hepatocarcinoma. Up-regulation of TLR2, TLR4 and their pro-inflammatory mediators is associated with virus-induced hepatic IFC sequence |
| Dolado et al[31] | 2007 February | Experimental | WT and p38a-/- were used. Growth in soft agar was evaluated. Intracellular ROS levels were determined, immunoblot Analysis was performed. To induce p38 MAPK activation, cells were treated with H2O2, sorbitol and cisplatin | The tumorigenesis enhanced by ROS is not evaluated on hepatocarcinoma | Oxidative stress sensing plays a key role in the inhibition of tumor initiation by p38alpha |
| Kang et al[32] | 2011 November | Experimental | For transposon-mediated intra-hepatic gene transfer mice received a transposon- to transposase encoding vector (30 mg total DNA). DNA was administered by hydrodynamic tail vein injection. Immunohistochemical analyses were performed | It was not investigated if factors secreted from pre-malignant senescent hepatocytes also contribute to the oncogenic transformation of neighbouring cells | Indicates that senescence surveillance represents an important extrinsic component of the senescence anti-tumour barrier, and illustrates how the cellular senescence program is involved in tumour immune surveillance |
| Ogata et al[34] | 2006 December | Experimental | Electron microscopic analysis was performed using neuroblastoma SK-N-SH cells exposed to ER stressors. GFP-LC3 fluorescence was used to monitor autophagy in cells transiently transfected with an expression vector for GFP-LC3. Then was performed an Amino acid uptake assay and autophagosome formation was evaluated | A signalling pathway other than the IRE1-JNK pathway may also play important roles in the activation of autophagy signalling after ER stress. The detailed signalling pathway for activation of the autophagy induced by ER stress is still unknown | Disturbance of autophagy rendered cells vulnerable to ER stress, suggesting that autophagy plays important roles in cell survival after ER stress |
| Pikarsky et al[36] | 2004 September | Experimental | The possibility that NF-κB activation is involved in Mdr2-knockout hepatocarcinogenesis was investigated by RelA (p65) immunostaining. Hystological analysis was performed. To study the relationship between the | It does not explain how the inflammatory process in Mdr2-knockout mice is maintained in the double mutants as it is independent of hepatocyte NF-κB activity | NF-κB is essential for promoting inflammation-associated cancer, and is therefore a potential target for cancer prevention in chronic inflammatory diseases |
| TNF-α-producing cells and NF-κB activation in the hepatocytes, liver sections were stained for both TNF-α and p65 | |||||
| Gong et al[37] | 2013 September | Experimental | BALB/c mice were used and inoculated with H22 hepatocarcinoma cells into the hind thigh muscle. They were treated with TLR2/4 ligands, HSP70 and HMGB1. The main tumor nodules were measured and satellite tumor nodes counted. To downregulate HMGB1, RAGE or Beclin-1 in tumor cells, cells were transduced with short interfering RNA | It does not explain the mechanisms responsible by the NF-κB's phosphorylation in the first 30 min. It was observed only one of the pathways responsible for the involvement of HMGB1/RAGE in the NF-κB signaling | Activation of NF-κB was indispensable for the effect of HSP70. HSP70 induced a positive feedback loop involving Beclin-1/HMGB1 production, causing re-phosphorylation of NF-κB |
| Shi et al[38] | 2014 October | Experimental | Human hepatocellular carcinoma cell lines were used. Into the cell lines were transfected small-interfeering-RNAs and at 48 h after transfection, the TLR2-siRNA-transfected group, scramble control group, and blank group were treated with recombinant-HMGB1. Evaluation included real time PCR, Western blot, MTT assay, Transwell assay and Flow cytometry assay | It does not explore the signaling pathway that regulates NF-κB through TLR2 inhibition or stimulation with recombinant-HMGB1 | TLR2-siRNA could effectively inhibit the growth, migration, invasion, and expression of NF-κB/P65, and HMGB1 promoted HCC progression via TLR2 |
| Wu et al[39] | 2012 April | Experimental | It was used mice and HCC cell lines. Eukaryotic expression vectors psTLR2 and psTLR4 were created. An adhesion assay, a tumor cell proliferation assay, a flow cytometric analysis, an apoptosis analysis, an analysis of gene expression by RT-PCR and a western blot analysis were performed | More than one signaling pathways activated by HSPA1A might be required for the survival of tumor cells. The effect of eHSPA1A was only evaluated in one cell line. Injection of HSPA1A suppressed tumor growth in early stage of tumor development, but promoted tumor growth in later stage | Extracellular HSPA1A functions as endogenous ligand for TLR2 and TLR4 to facilitate tumor growth |
| Yoneda et al[41] | 2008 November | Experimental | HCC cell lines and 74 HCC samples were used. Poly (I:C), cycloheximide and actinomycin were included in the study. Profiling analysis of TLRs recognized by viral components, flow cytometric analysis, immunohistochemical staining, Detection of TLR3 by immunofluorescence, Detection of cell viability and apoptosis assays, Detection of apoptosis-related proteins by immunoblotting, NF-κB activity assays and measurement of IFN-β were also performed | Further evaluation of the possible roles and the type of regulation associated with TLR3 needs to be undertaken | Intracellular TLR3 signalling is involved in cell death, while in contrast, the cell surface TLR3 signalling is responsible for activation of NF-κB |
| Zorde-Khvalevsky et al[42] | 2009 July | Experimental | It was used TLR3-WT mice and TLR3-/- mice. Partial hepatectomy was done followed by immuonhistochemistry stainings, plasma aminotransferase activity assay, measurements of serum cytokine levels, semi-quantitative reverse-transcription polymerase chain reaction, Western blotting, caspase-8 immunopurification and injection with poly (I:C) or saline solution | It is not explained what happens to the levels of ALT in mice's serum before the 10-h time point following 70% PHx. Cytokine evaluation only includes IL-6 and IL-22 | TLR3 plays an inhibitory role in the priming of liver regeneration, thus reinforcing the role of the innate immune system in balancing tissue regeneration |
| Khvalevsky et al[44] | 2007 April | Experimental | Various cell lines and plasmids pTLR7, pTLR8, and pTLR9, carrying the respective human TLR gene, were used. Transfection | The role of TLR3 signaling in normal hepatocytes requires further investigation in vivo. It is not specified the degree of | Preferential induction of the apoptotic pathway over the cytokine induction pathway by TLR3 signaling in |
| assays, RNA quantification, immuno-staining and flow cytometry, were performed | NF-κB activation obtained from the overexpression of TLR3 nor the degree of this overexpression that is needed | hepatocellular carcinoma cells with potential implications for therapeutic strategies | |||
| Chen et al[45] | 2012 July | Experimental | The human HCC cell line HepG2.2.15 was used. After treating HepG2.2.15 with BM-06 or poly (I:C), NF-κB activity was checked by dual luciferase reporter gene kit. Then it was performed a nuclear and cytoplasmic extraction, Western blot analysis, a cell proliferation assay, cell invasion assays and flow-cytometry was used to determine the apoptotic rate | The role of TLR3 in the antiviral defense against HBV was not analyzed according to differences in the type of viruses, the type of cells that are infected, the viral load, its model of infection (endoplasmic cytoplasmic), and stage of infection | BM-06 inhibited the proliferation, invasion and secretion of HBV, and induced apoptosis in HepG2.2.15 cells. In addition, the antitumor effects of BM-06 were superior to poly (I:C) |
| Guo et al[46] | 2012 February | Experimental | Cell cultures were used and submitted to BM-06 and poly (I:C) treatment. RNA isolation and one-step quantitative real-time PCR were performed. Analysis included detection of TLR3 by immunocytochemistry, luciferase reporter assays, Endothelial cell tube formation assay, rat aortic ring assay, annexin V/PI for cell apoptotic analysis and Cell migration assays | It does not evaluate the molecular mechanisms after TLR3 stimulation that lead to modulation of endothelial tube-forming activity of HUVECs and vascular sprouting or enhanced apoptosis | TLR3 agonists not only affect tumor microenvironment by suppressing angiogenesis but also directly induce tumor cell apoptosis and inhibit tumor cell migration |
| Bergé et al[47] | 2010 December | Experimental | It was injected transgenic mice developing HCC with either control siRNAs or siRNA targeting neuropilin-1. The study used antibodies (goat anti-TLR3 and rabbit anti-tubulin antibody), Western Blotting, and Immunofluorescence Analysis. Real-time RT-PCR, ELISA, MTT assay and three-dimensional collagen assay were also performed | It is not known why INF-γ does not inhibit cells' functions in the in vitro study despite the high levels in HCC. In vivo evaluation was not performed | Synthetic siRNAs inhibit target-independently HCC growth and angiogenesis through the activation of the innate interferon response and by directly inhibiting endothelial cell function |
| Xu et al[48] | 2013 October | Experimental | Thirty rats were used, all 30 were fed with 2-acetylaminofluorene to establish the HCC model. Two animal groups were treated, respectively, with the drug candidate (BM-06) and poly (I:C). It was performed a H and E staining, an Immunohistochemical staining, a Western blot analysis | It does not explore the pathway though which BM-06 and poly (I:C) are capable of inducing cell death. It is not evaluated TLR3's downstream molecules to explain the signalling pathway responsible for these results | Treatment with BM-06, showed a decrease in tumor growth and cell proliferation, and an increase in apoptosis compared with that in a phosphate-buffered saline control group |
| Wang et al[49] | 2013 August | Experimental | Fifty-three HCC and ten normal liver specimens were analyzed by immunohistochemistry, and three cell lines were used for in vitro studies. Lipopolysaccharide was used to activate TLR4 signaling. Cell survival, proliferation and invasion were examined | Only a specific amount of LPS has shown to have an effect on the mRNA expression of IL-6, EGFR and HB-EGF. Opposing to HL-7702 cell line, PLC/PRF/5, with a moderate level of TLR4 expression, was not affected by inhibiting p38 | Indicate that TLR4 signaling in cancer cells promotes cell survival and proliferation in HCC |
| Liu et al[50] | 2015 March | Experimental | Two HCC cell lines and a splenic vein metastasis of the nude mouse model were used. A total of 88 clinical samples from HCC patients were used. A fluorescence activated cell sorting system and flow cytometry analysis were performed. Nude mouse splenic vein metastasis assay, immunohistochemistry analysis, real-time quantitative PCR, Western blot analysis, immunofluorescence and cell apoptosis assay were also done | More pathological specimens should be enrolled to verify the tendencies of association between TLR4 expression and malignant characteristics of HCC found in this study. A particular signaling pathway involved in the relationship between TLR4 expression and stem cell features remains elusive | There is a relationship between TLR4 expression and CSC's features, TLR4 may act as a CSC marker, prompting tumor invasion and migration, which contributes to the poor prognosis of HCC |
| Li et al[52] | 2014 October | Experimental | A HCC cell line was used where a Scratch assay was performed. Invasion assay, Western blot analysis, quantitative real-time reverse transcription PCR and siRNA knockdown of TLR4 gene expression were also done | It does not reveal the time needed for induction of epithelial-mesenchymal transition after LPS stimulus. Does not explore influence of LPS on TLR2 | TLR4/JNK/MAPK signaling is required for LPS-induced EMT, tumor cell invasion and metastasis, which provide molecular insights for LPS-related pathogenesis and a basis for developing new strategies against metastasis in HCC |
| Jing et al[53] | 2012 August | Experimental | Four HCC cell lines and a splenic vein metastasis of the nude mouse model were used and stable TLR4-expressed and knocked-down cell lines were generated. 106 clinical samples from HCC patients were also used. Quantitative real-time PCR, Western-blot analysis, Immunofluorescence, FACS Analysis and IHC analysis were performed | HCC development is a multifactorial and complicated process, which has a close association with various risk factors. Many gene alterations and cytokines also could induce EMT. HCC cells with low expression or even a lack of TLR4 are not susceptible to LPS, they might perform EMT induced by other TLR4-independent mechanisms | TLR4 signaling is required for LPS-induced EMT, tumor cell invasion and metastasis, which provide molecular insights for LPS-related pathogenesis and a basis for developing new strategies against metastasis in HCC |
| Xu et al[54] | 2014 October | Experimental | HCC and adjacent tissues were obtained from 84 patients. HCC cell lines were used and a PLV-PTPRO-GFP plasmid was constructed. Real-time PCR, immunofluorescence, Western blot analysis and cell proliferation assay were performed | It does not specify the doses of NF-κB specific inhibitor needed to result in a decreasing of PTPRO's levels in HuH7 cells stimulated with LPS | The effect of PTPRO on TLR4 signaling is dependent on NF-κB pathway, suggesting an interesting PTPRO/TLR4/NF-κB signaling feedback loop in HCC carcinogenesis and progression |
| Wang et al[55] | 2015 January | Experimental | It was used LPS-induced human hepatocellular carcinoma cell lines. Cell viability was assessed using the MTT assay. Double staining for annexin V-FITC and propidium iodide was performed. Inflammatory mediators were evaluated through a specific ELISA kit. Immunoprecipitation and Western blot analysis were also used | Only one type of cell line is used to observe the effect of CXC-195. It does not reveal the level (high or low) of TLR4 expression. It does not explore the influence of LPS in TLR2 | Treatment with CXC195 could attenuate the TLR4-mediated proliferation and inflammatory response in LPS-induced HepG2 cells |
| Yu et al[56] | 2010 October | Experimental | Rats and mice were used, including TLR4-deficient mice. Immunohistochemical analysis and bone marrow transplantation were performed | It does not explore the effect of modulating gut flora. It does not evaluate the effect of different LPS' levels | Sustained LPS accumulation represents a pathological mediator of inflammation-associated HCC and manipulation of the gut flora to prevent pathogenic bacterial translocation |
| Lin et al[58] | 2012 September | Experimental | It was used wild-type and TLR4-deficient mice. A flow cytometry analysis and Isolation and Culture of CD4+ cells were performed | TLR4 knockout showed decreased liver injury induced by Con A, contrarily to what was expected. It is needed to determine whether the regimen with antiendotoxin effects will prove beneficial in preventing or delaying T cell-mediated hepatitis and hepatitis-induced HCC | Gut-derived LPS and TLR4 play important positive roles in Con A-induced hepatitis and modulation of the gut microbiota may represent a new avenue for therapeutic intervention |
| Chen et al[60] | 2013 July | Experimental | It used HCV Tg mouse models and patients with HCC functional cDNA. Then, functional cDNA screening for oncogenes was performed. In vitro and in vivo oncogenic activities were evaluated. It was also done a liver TIC engraftment via splenic injection | The degree of attenuation of TLR4 expression in TICs by Nanog, implying a feedback loop is not shown. Besides this, the underlying mechanisms are not known | TLR4/NANOG oncogenic pathway is linked to suppression of cytostatic TGF-β signaling and could potentially serve as a therapeutic target for HCV-related HCC |
| French et al[63] | 2013 August | Experimental | Liver biopsies from patients diagnosed with alcoholic hepatitis, with or without cirrhosis were selected. Double Immunohistochemistry was performed | The antibody stain was only against TLR4 | The Mallory-Denk-bodies forming cells expressed two additional progenitor cell markers. These markers were CD49f and TLR4 |
| Machida et al[64] | 2014 November | Experimental | An immunostaining of liver tumor sections from alcohol-fed Ns5a mice was performed along with TLR4 silencing with lentiviral short-hairpin RNA | LPS-independent mechanisms of TLR4 activation in TICs remain to be elucidated. The oncogenic role of TLR4 is explored only around the synergism alcohol-HCV | TLR4-dependent mechanisms of TIC generation actually contribute to or at least promote the initiation of HCC |
| Yan et al[65] | 2012 June | Experimental | Human HCC liver samples and mice were used. Stable HMGB1-expressing cells and HMGB1 knockdown cells were established. immunoblotting analysis, RNA Interference by short interfering RNA, enzyme-linked immunosorbent assay, confocal microscopy exam, caspase-1 colorimetric assay, cell migration and invasion assays and metastatic potential exam were all performed | Mechanisms by which caspase-1 affects tumor cancer progression remain incompletely understood | In hypoxic HCC cells, HMGB1 activates TLR4- and RAGE-signalling pathways to induce caspase-1 activation which, in turn, promote cancer invasion and metastasis |
| Xu et al[67] | 2008 February | Analytic - cross sectional | 52 patients were studied. The protein and mRNA levels of TLR7 and TLR9 were evaluated using real-time PCR, Western blot analysis, and flow cytometry. We also detected the serum viral load of HBV in the patients and analyzed the correlation between HBV-DNA copies and the TLR expression | The statistical analysis indicated no difference in the TLR9 levels among the HCC and LC groups. If the sample size was enlarged, the results may be different. The expression of TLR7 was not different among the groups of patients, suggesting that TLR7 has no correlation with HCC | There are downregulations of TLR7 expression and TLR9 mRNA in PBMC of HBV-infected patients, but an increased TLR9 expression at the protein level |
| Tanaka et al[68] | 2010 October | Experimental | HCC cell lines and 42 HCC tissues were used. The type C CpG oligonucleotide was used as TLR9 ligand. Flow cytometric analysis, Immunohistochemical staining, Cell proliferation assay, Immunoblotting, NF-κB activity assays and expression analysis of IRF-7, RNA extraction and oligonucleotide microarray and Microarray data analysis were all performed | Despite being present both intracellular or extracellular TLR9's intracellular function is not observed with TLR9 ligands and its function is not known | Functional cell surface expression of TLR9 in human HCC may play an important role in tumorigenesis and cancer progression |
| Liu et al[69] | 2015 February | Experimental | C57BL6 mice were injected with Hepa1-6 cancer cells. TLR9 and HMGB1 were inhibited using shRNA or direct antagonists. HuH7 and Hepa1-6 cancer cells were investigated in vitro to determine how the interaction of HMGB1 and mtDNA activates TLR9 signaling pathways | The contribution of TLR9 to cancer pathophysiology remains incompletely understood. The regulation of TLR9 signaling and the physiological ligands which may induce TLR9 mediated tumor growth remain poorly characterized | Reveals a novel mechanism by which the interactions of HMGB1 and mtDNA activate TLR9 signaling during hypoxia to induce tumor growth |
| Zhang et al[70] | 2014 December | Experimental | It was used HCC cell lines to where was transfected CpG oligodeoxynucleotide and poly (I:C). Proliferation analyses, Detection of apoptosis with an Apoptosis Detection Kit, quantitative real-time PCR analysis, Western blot analysis and Fluorescence microscopy were also performed | The precise molecular interactions that likely occur between CpG ODNs and poly (I:C) to block poly (I:C) entry, remain to be established. Poly (I:C) may be influenced by many molecules in the microenvironment | When combining poly (I:C) and CpG ODN for cancer therapy, these agents should be used in an alternating rather than simultaneous manner to avoid the blocking effect of phosphorothioate-modified TLR9 ligands |
| Zhang et al[71] | 2014 April | Experimental | Human hepatoma cell lines were used. Cells were transfected with CpG ODNs or small interfering RNAs targeting TLR9. Reverse transcriptase polymerase chain reaction assay, Proliferation measurements, cell cycle analysis, detection of apoptosis, quantitative real-time PCR analysis, Western blot analysis were all performed. An in vivo study was also done | Apoptosis induced by ODN M362 Ctrl and ODN M362 occurred independently of TLR9 stimulation. TLR9- and MyD88-independent mechanisms in ODN-stimulated immune cells, including B lymphocytes and neutrophils may exist | Phosphorothioate-modified TLR9 agonist ODN M362, and its control, elicit antitumor activity in HCC cells and may serve as a novel therapeutic target for HCC therapy |
| Bubici et al[74] | 2004 December | Perspective | Induction of FHC and Mn-SOD represents an additional, indirect means by which NF-κB controls proapoptotic JNK signaling | ||
| Liu et al[75] | 2009 April | Experimental | Cell cultures were used. Immunocytochemistry stain for TLR9, a Cell proliferation assay, reverse transcriptase PCR for TLR9 and real-time reverse transcriptase PCR for DNMT-1 and Bcl-2, NF-κB activation measurement and Cellular apoptosis analysis were all performed | L-02 cells were used to allow in vitro studies but cells may behave differently in vivo. Future in vivo models are needed | Identified a possible novel mechanism that indicates how CpG DNA of HBV DNA may contribute to the malignant transformation of benign liver cells |
| Nischalke et al[76] | 2012 March | Analytic - cross sectional | A total of 197 patients with HCV-associated HCC, 192 HCV-infected patients without HCC and 347 healthy controls were included. HCV antibodies were detected for diagnosis. Determination of TLR2-196 to -174 del/ins polymorphism was performed by LightCycler real-time PCR. In vitro induction of TLR2 expression and IL-8 was performed | Analysis of the functional role of TLR2-196 to -174 del/ins alleles with respect to TLR2 expression was based on in vitro stimulation studies but it is not known if an in vivo analysis would have the same results | TLR2-196 to -174 del allele to affect HCV viral loads and to increase the risk for HCC in HCV genotype 1-infected patients |
| Junjie et al[77] | 2012 February | Single center-based case-control | SNaPshot method was used to genotype sequence variants of TLR2 and TLR9 in 211 patients with HCC and 232 subjects as controls | Despite the SNP rs3804099 and rs3804100 were out of HWE (P = 0.01-0.02), they were retained in the analyses | TLR2 rs3804099 C/T and rs3804100 C/T polymorphisms were closely associated with HCC. In addition, the haplotypes composed of these two TLR2 synonymous SNPs have stronger effects on the susceptibility of HCC |
| Jiang et al[79] | 2014 December | Single center-based case-control study | 426 HCC subjects and 438 cancer-free control subjects were used. SNP genotyping was performed. A Vector was constructed and luciferase reporter assays were done. TLR4 mRNA levels were evaluated and Western blotting was done | The hypothesis that the overexpression of TLR4 induced by the rs1057317 polymorphism miRNA-disrupting function may influence the development of hepatocellular carcinoma is possible but still not proved. More studies in this area are needed | The risk of hepatocellular carcinoma was associated with a functional variant at miR-34a binding site in TLR4 gene. miR-34a/TLR4 axis may play an important role in the development of HCC |
| Minmin et al[80] | 2011 April | Analytic-case-control | A systematic genetic analysis of sequence variants of TLR4 by evaluating ten single-nucleotide polymorphisms was performed from 216 hepatocellular carcinoma cases and 228 controls | The contribution of the SNPs in TLR4 to HCC is modest. More studies are needed to validate this finding in independent populations and to understand the mechanism by which TLR4 sequence variants affect the pathological role of TLR4 in the signaling pathways that control carcinogenesis | The risk of hepatocellular carcinoma was associated with TLR4 sequence variation. TLR4 single nucleotide polymorphisms may play an important protective role in the development of hepatocellular carcinoma |
| Kawamoto et al[82] | 2008 April | Experimental | Mouse cells were used together with plasmids containing TLRs. Cells were submitted to LPS and TAK-242. Nitrite and TNF-α were measured. Reporter gene assay for ligand-dependent signaling by TLRs, Reporter gene assay for ligand-independent signaling by TLR4, CD4-TLR or adaptors and Western blot analysis were performed | Human studies are needed as the interacting affinity of TAK-242 with TLR4 may be affected by a subtle difference in the amino acid sequences of TIR between humans and mice | TAK-242 selectively suppresses TLR4-signaling mediated by the intracellular domain |
| Matsunaga et al[83] | 2011 January | Experimental | 293 cells of human embryonic kidney and murine resident peritoneal macrophages were used. They were subited to TAK-242 and LPS. Vectors for FLAG-TLR4 and FLAG-TLR2 were cloned. Measurement of nitrite and | To fully understand the physical basis whereby TAK-242 disturbs signaling complex formation and intracellular signal transduction, a crystal structure analysis of the TLR4-TAK-242 complex is needed | TAK-242 binds selectively to TLR4 and subsequently disrupts the interaction of TLR4 with adaptor molecules, thereby inhibiting TLR4 signal transduction and its downstream |
| cytokine concentrations in culture supernatants, radiolabeling of the cells, immunoprecipitation, Western blot analysis and autoradiography, reporter gene assay and in vitro IL-1 receptor-associated kinase-1 kinase assay were all performed | signaling events | ||||
| Xu et al[84] | 2013 November | Experimental | Four dsRNAs were designed and synthesized. The expression of proteins was compared. The migration, proliferation and apoptosis of HepG2.2.15 cells were evaluated in presence of BM-06, sorafenib alone or in combination of both. The similar treatments were also applied in an SD rat primary HCC model | Since synthetic siRNAs must be transfected into the target cells through a vector, such as Lipofectamine™ 2000 reagent, they always exhibit cytotoxicity, which may limit their use in clinic | dsRNA alone was capable of inhibiting the proliferation of HepG2.2.15 cells and tumor growth of orthotopic HCC SD rats, but the effect of combination of dsRNA with sorafenib was more prominent |
| Behm et al[85] | 2014 December | Experimental | Rabbits were randomised to receive RFA, CpG B, their combination or no therapy, further tested by rechallenging a separate group with intravenously injected VX2 tumour cells after 120 d. Animals were assessed for survival, tumour size and spread, and tumour and immune related histological markers after 120 d. Peripheral blood mononuclear cells were tested for tumour-specific T cell activation and cytotoxicity. Immune modulatory cytokines were measured in serum | Lack of antibody reagents for the VX2-tumour model in rabbits. It was not possible to elucidate in depth histopathological changes | The combination of TLR9 stimulation with RFA resulted in a potentiated antitumour T cell response and cytotoxicity in the VX2 tumour model. Only this combination prevented subsequent tumour spread and resulted in a significantly improved survival |
TLR: Toll-like receptor; PCR: Polymerase chain reaction; HCC: Hepatocarcinoma; LPS: Lipopolysaccharides; TNF-α: Tumour necrosis factor α; DEN: Diethylnitrosamine; NAC: N-acetyl cysteine; MAPK: Mitogen-activated protein kinase; NF-κB: Nuclear transcription factor kappa B; ER: Endoplasmic reticulum; MDSC: Myeloid-derived suppressor cells; NAFLD: Non-alcoholic fatty liver disease; JNK: Junamino-terminal kinase; HMGB1: High mobility group box 1; INF-γ: Interferon gamma; HBV: Hepatitis B virus; HCV: Hepatitis C virus; HSCs: Hepatic stellate cells; ODN: Oligodeoxynucleotides; NMOR: N-nitrosomorpholine; TNFR1: Tumor necrosis factor receptor 1; IL: Interleukine; DC: Dendritic cells; NK: Natural killer; HSCs: Hematopoietic stem cells; TUNEL: Terminal deoxynucleotidyl transferase dUTP nick end labeling; SA b-gal: Senescence-associated beta-galactosidase; ELISA: Enzyme-linked Immunosorbent Assay; COX: Ciclo-oxigenase; IFC: Inflammation-fibrosis-carcinoma; ROS: Reactive oxigen species; HMGB1: High mobility group box 1; RAGE: Receptor for advanced glycation endproducts; HSP: Heat shock protein; HUVECs: Human umbilical vein endothelial cells; CSC: Colony stem cells; EMT: Epithelial-mesenchymal transition; TICs: Tumor-initiating cells; IRF: Interferon regulatory transcription factor; SNPs: Single nucleotide polymorphisms; HWE: Hardy-Weinberg equilibrium.
TLR EXPRESSION IN HEPATIC CELL POPULATION
The liver is a very special organ when it comes to dealing with pathogens. Due to its vascular links, contact with gut-derived bacteria is constant and, therefore, mechanisms not only to defend the organism from these pathogens but also to tolerate them, had to be developed[8]. In this duality, TLRs play an interesting role as it is known that, in a healthy liver, mRNA levels of TLRs like TLR1, 2, 4, 6, 7, 8, 9 and 10 are decreased when compared to other organs[9].
In the liver, hepatocytes represent 60%-80% of the total cell population. Here, it can be found mRNA from all TLRs; however, only a response from TLR2 and TLR4, to their ligands, can be obtained[12]. Interestingly, only the response of TLR2 is up-regulated under inflammatory conditions[13].
Besides hepatocytes, it is possible to find, in the liver, non-parenchymal cells which consist of Kupffer cells (KCs), dendritic cells (DCs), Lymphocytes, hepatic stellate cells (HSCs) and liver sinusoidal endothelial cells (LSECs).
KCs not only express lipopolysaccharide (LPS)-responsive TLR4 but also TLR2, TLR3 and TLR9 that respond to their ligands[14]. These cells develop an inflammatory response to high levels of LPS but produce an anti-inflammatory cytokine (IL-10) in response to continuous low levels of LPS, known as LPS tolerance[15]. DCs represent a small population (< 1%). In humans, the plasmocytoid DCs subset expresses TLR1, TLR7 and TLR9 while other subsets carry all TLRs with the exception of TLR9[16]. When it comes to Lymphocytes population and TLRs relationship it is important to notice that differences can be found from one subpopulation to another. Natural killer (NK) cells contain TLR1, TLR2, TLR3, TLR4, TLR6, TLR7, TLR8 and TLR9[1,17] but T cells are only activated through TLR2 while B cells rely on TLR2, TLR4, TLR7 and TLR9[18]. HSCs, also in small proportion (< 1%), when activated are able to express TLR4 responsive to LPS which, in turn, enables inflammatory cytokines’ secretion[19]. LSECs express mRNA from TLR1 to TLR9 despite not being able to respond to TLR5 ligands[20].
IMPORTANCE OF TLRS IN INFLAMMATORY-INDUCED HCC
HCC has long been considered a chronic-inflammation driven cancer independently of the possible risk factors; virus induced hepatitis, smoke, alcohol or metabolic diseases. Despite that, the liver is an organ with several mechanisms readily available to defend against carcinogenesis. Among these, we found PRRs, with special attention to TLRs which were already shown to exhibit different roles in the regulation of tumorigenesis and tumour progression. To date several works have documented its influence not only in specific types of cancer - breast, ovarian, prostate and lung - but also in processes directly linked with cancer - resistance to apoptosis, increased invasiveness and metastasis. This is the reflection of their actions on metalloproteases and integrins, tumour cell immune escape, among others[21-23]. However, the cellular and molecular effectors mediating the interplay between TLRs and HCC are still largely unknown.
TLR2
When the TLR2 signal is triggered, the downstream cascade initiates through a “Myd88 dependent pathway” with the activation of the apoptosis signal regulating kinase 1 (ASK1)/p38 mitogen-activated protein kinase (p38 MAPK)/nuclear factor kappa B (NF-κB), or through a “Myd88 independent manner/toll/interleukin-1 receptor domain-containing adaptor protein inducing interferon beta (TRIF) dependent” with the extracellular signal-regulated kinase (ERK)/Junamino-terminal kinase (JNK) and PI3K/Akt pathways[24]. Besides this, TLR2 signal is also involved in processes like autophagy and senescence in response to oxidative stress and DAMPS release[25].
Diethylnitrosamine (DEN) is a chemical carcinogen capable of inducing HCC through accumulation of reactive oxygen species (ROS) and endoplasmatic reticulum (ER) stress. It was found that when a TLR2-deficient (TLR2-/-) mouse was submitted to DEN treatment the ROS and ER stress were abundantly accumulated, even though less apoptosis was observed[25].
In fact, Lin et al[24] demonstrated, in two separated works, that both TLR2-/- and wild-type (WT) mice developed HCC after being submitted to a DEN-treatment. However, the TLR2-/- mice revealed earlier tumours (every TLR2-/- mouse developed HCC at 6 mo after DEN treatment vs only 68% WTs)[24] that were, not only significantly increased in number and in volume, but also less differentiated[24,25]. This increase reached the 3 fold (20.1% ± 4.5% vs 6.4% ± 1.0%, P < 0.01) in the tumour area and 5 fold in visible tumour nodules (29.1% ± 2.8% vs 5.5% ± 0.9%, P < 0.001)[24]. Meanwhile, in the WT mice, is possible to attenuate HCC development if a TLR2 agonist is used[26]. Ultimately, TLR2-/- mice had shorter mean survival times with HCC than WTs[24]. Moreover, similar scenery was observed when a WT was pre-treated with an anti-TLR2 antibody[25]. Indeed, when observing liver samples from patients in different stages of liver diseases it is notorious that, in patients with HCC, not only the mRNA levels of TLR2 are lower but also TLR2 immunohistochemical expression grade and intensity are reduced, when compared to patients with hepatitis or cirrhosis[27].
A ROS-generation reaction in cytochrome p450 2E1 is responsible for DEN metabolism. In spite of not finding any significant difference in cytochrome activity, TLR2-/- mice still revealed enhanced accumulation of ROS in their liver tissue[24].
Generation of ROS results in oxidative stress, which is often the source of DNA mutation or a direct link with chronic inflammation[28-30]. The ASK1/p38 MAPK/NF-κB pathway is one of the major sensors for ROS accumulation contributing to induced senescence cell death when risk of mutation is present[31]. However, in TLR2-/- mice submitted to DEN treatment, it is possible to assist to an attenuation of this major pathway[25] together with a suppression of biomarkers of autophagy-associated cell death and cellular senescence, like β-galactosidase[24]. Moreover, unlike the WT, TLR2-/- mice fail not only, to induce other important channels to premature cellular senescence like the p16-pRb/p21 pathway[24], but also to activate DNA damage repair mechanisms[32].
Furthermore, ER-stress is augmented after DEN-treatment in TLR2-/- mice as a result of ROS accumulation[25]. This leads to an enhanced unfold protein response (UPR) and activation of UPR-JNK pathway[33], necessary for autophagy activation under ER-stress which, paradoxically, plays a dominant pro-survival role[34]. Lin et al[25] noticed that, in livers from TLR2-/- mice there was an increased JNK activity.
Overall this data indicates that in the absence of TLR2, a down-regulation of common ROS neutralizing mechanisms, due to supressed activation of ASK1/p38 MAPK/NF-κB, results in HCC cells containing higher ROS and DNA damages that, because of an up-regulated UPR-JNK pathway, have more chances to survive.
However, other pathways relating to TLR2 and hepatocarcinogenesis exist. Li et al[26] focus their work on IL-18, which was found to be fundamental to carcinogenesis in TLR2-/- mice. In these mice, HCC developing after DEN treatment was capable of inducing IL-18 up-regulation in a caspase-8-dependent manner, therefore contributing to promotion of angiogenesis and suppression of NK cell arm of tumour immunosurveillance[26].
Another perspective is related to the High mobility group box 1 (HMGB1), a nuclear protein released from dead/dying cells or even from cancer cells. It has the ability to bind to TLR2 and, with that, successfully activate NF-κB[35] which, in turn, can have an important role as a tumour promoter in inflammation-associated cancer[36]. Up-regulation of HMGB1 in an HCC cell line can result in increased matrix metalloprotease 9 and satellite tumour nodules in the liver, while blocking it supresses tumour growth[37]. A recombinant HMGB1 (rHMGB1) was used by Shi et al[38] in order to simulate TLR2 activation in an HCC cell line. Interestingly, rHMGB1 not only reduced cell apoptosis but also accelerated the tumour’s growth and enhanced the ability of migration and invasion. Additionally, rHMGB1 activity significantly declined when HCC cells were pre-treated with a TLR2 inhibitor[38].
Similarly to HMGB1, HSPA1A - a member of the HSP70 family - is also a TLR2’s ligand released by the tumour’s necrotic cells. With a resembling pathway based on up-regulation of NF-κB, HSPA1A is capable of promoting the proliferation and survival of tumour cells[39].
TLR2 clearly represents an important modulator of cells’ response to stress situations. It has influence in mechanisms like autophagy, apoptosis or even DNA damage repair, possibly contributing to a protective role against HCC. However, it is, also, important to notice that these pathways may not be, already, clearly understood as studies reveal that TLR2’s ligands like HMGB1 and HSPA1A, can result in tumour enhancement (Figure 2).
Figure 2.
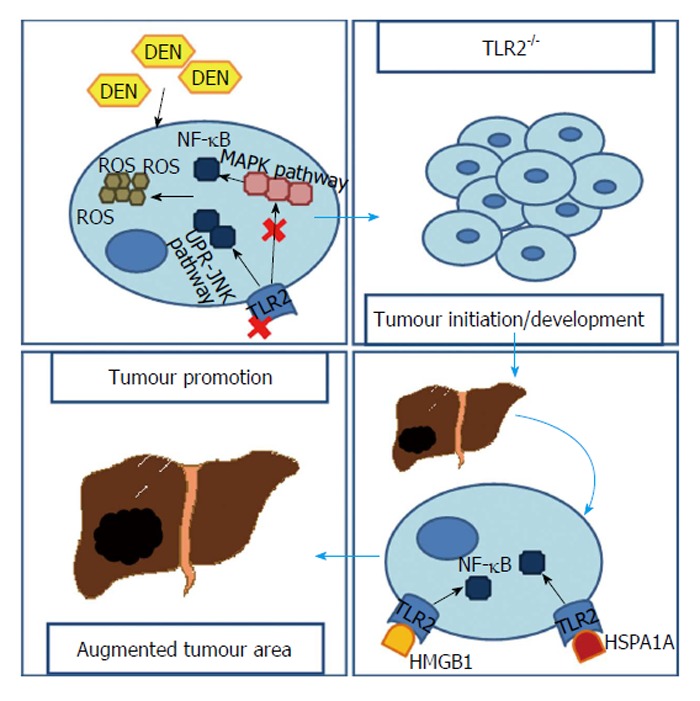
Toll-like receptor 2’s signalling pathways contributing to hepatocarcinoma. In the absence of TLR2, cells are incapable of responding to an increasing ROS when submitted to DEN. This is the result of an absence of MAPK/NF-κB pathway and an up-regulation of the UPR-JNK pathway. Consequently, cells containing higher ROS and DNA damages have more chances to survive and, HCC develops. In a second stage, where HCC is already established, HMGB1 and HSPA1A released by tumour’s dying cells, through TLR2 stimulation, lead to an NF-κB up-regulation which, in this contest, seems to contribute to tumour’s growth. HCC: Hepatocarcinoma; TLR2: Toll-like receptor 2; ROS: Reactive oxigen species; DEN: Diethylnitrosamine; MAPK: Mitogen-activated protein kinase; NF-κB: Nuclear transcription factor kappa B; UPR: Unfold protein response; JNK: Junamino-terminal kinase; HMGB1: High mobility group box 1; HSPA1A: Heat shock protein A1A.
Taken altogether this data suggests that TLR2 activation may slow down initiation and development of HCC (anti-oncogenic potential) in the earlier phases of HCC carcinogenesis. However, at later stages its activation may influence the progression of inflammation and fibrosis (pro-oncogenic potential). Therefore, new studies are required in order to understand the exactly pathways through which this receptor is able to work and to conclude if its role in HCC carcinogenesis is different or not depending on the stage of the Inflammation-fibrosis-carcinoma axis.
TLR3
Several studies have already shown that TLR3 is expressed in many cancer cells such as colonic adenocarcinoma, lung cancer, breast cancer and melanoma. In HCC it was found that 17-fold longer median survival accompanied patients with higher intratumoral TLR3 expression[40]. However, Yoneda et al[41] observed that 52.7% of the HCC tissues and 34.8% of the HCC metastasis studied expressed TLR3. Furthermore, the receptor was not only present in the cytoplasm, but also in the membrane, particularly in the exterior, suggesting a cell surface recognition mechanism for TLR3 agonists[41].
Other works have already implied that the TRIF-dependent pathway of TLR3 signalling could have a special contribution to a tumours response. In fact, this adaptor molecule can promote either an inflammatory or an apoptotic response. The first one pending on NF-κB, the second in caspase-8 activation and interferon-γ (INF-γ) release[42]. Experiments showed that using synthetic TLR3 agonists resulted in a rise in NF-κB. In fact, as it was seen with TLR2 signalling, NF-κB is normally associated with augmented tumour necrosis factor α (TNF-α) responsible for cells’ growth and proliferation[43]. However, here, an NF-κB rise is responsible for affecting the tumour microenvironment and driving HCC and endothelial cells to apoptosis[44], accompanied by a significantly decreased tumour invasiveness and angiogenesis/vascular endothelial growth factor (VEGF) levels[45-47]. Thus, it seems that, whether NF-κB promotes or inhibits hepatocarcinogenesis depends on the presence of inflammation and the degree of NF-κB inhibition/promotion[3].
Moreover, INF-γ - a potent inhibitor of endothelial cell proliferation/angiogenesis - and caspase-8/caspase-3 - inhibitors of hepatocytes proliferation - were found to be significantly increased in HCC cell lines pre-treated with TLR3 agonists[48].
However, it is important to notice that, when stimulated through polyinosinicpolycytidylic acid, the surface TLR3 is only able to induce apoptosis if a protein synthesis inhibitor or a RNA synthesis inhibitor are present[41]. This might indicate that, in an HCC cell line, endogenous suppressors of TLR3-mediated apoptosis are present. Curiously, stimulation of intracellular TLR3, even without protein or RNA synthesis’ inhibitors, was able to elicit cell apoptosis in a tumour necrosis factor-related apoptosis-inducing ligand-dependent manner that synergistically accompanies a down-regulation of anti-apoptotic proteins[41].
Notably, despite overall tumour growth could be reduced through TLR3 activation (from a 3-fold increase, when no TLR3’s stimulus is present, to an only 1.9-fold increase after TLR3’s agonists being used), the number of tumour nodules increases even after eliciting TLR3 signalling, leading to the conclusion that it does not affects the incidence but limits their growth[47].
Interestingly, it appears that in HCC carcinogenesis TLR3 is a TLR that works as a protector against cancer. This is possible through molecules, downstream to TLR3, such as caspases, INF-γ or NF-κB, influencing crucial processes like angiogenesis, cell growth or proliferation (Figure 3).
Figure 3.
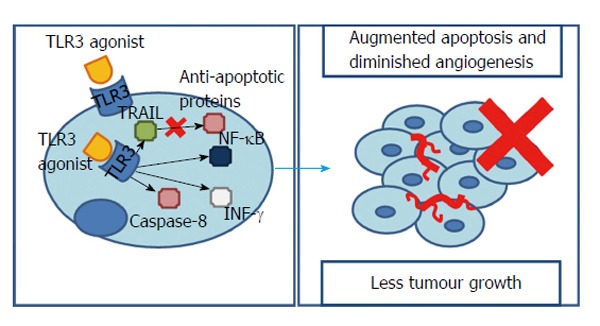
How toll-like receptor 3 stimulus works against hepatocarcinoma. Stimulation of intracellular TLR3 is able to elicit cell apoptosis in a TRAIL-dependent manner that synergistically accompanies a down-regulation of anti-apoptotic proteins. Additionally, TLR3 stimulus can promote either an inflammatory or an apoptotic response. The first one pending on NF-κB, the second in caspase-8 activation and INF-γ release. As a result, TLR3 works as a protector against cancer which stimulation results in diminished tumour growth. TLR3: Toll-like receptor 3; TRAIL: Tumour necrosis factor-related apoptosis-inducing ligand; INF-γ: Interferon gamma; NF-κB: Nuclear transcription factor kappa B.
TLR4
It is known that, despite being present in multiple liver cells, TLR4 expression is relatively low in this organ[49]. However, following liver damage and inflammation it is possible to assist to an up-regulation of this receptor[50]. Emerging evidence associates TLR4 to several types of tumours, enlightening its role in carcinogenesis, metastasis and cancer progression[51]. Observation of human’s livers detected a high expression of TLR4 in cancer cells of HCC patients[49].
Bacterial LPS is capable of initiating TLR4 signalling and subsequently activating NF-κB and MAPK signalling pathways - p38, ERK, JNK. In fact, in a HCC cell line incubated with bacterial LPS both TLR4 expression[52-54] and MAPK signalling pathways are significantly augmented[52]. However, it was found that, in contrast with a normal hepatocytes cell line, in a HCC cell line, the cellular growth was augmented and the cytotoxicity induced by LPS was decreased and dependent on TLR4 expression (higher expression is equal to less cytotoxicity)[49]. Additionally, these effects are reduced after inhibiting TLR4 signalling[55].
The explanation of these results is based on two perspectives. One based on the fact that, in TLR4-overexpressing cells, ERK and JNK’s activity is promoted[49] contributing to cell survival and proliferation. Nonetheless, loss of TLR4 results in a substantial decrease in proliferating hepatocytes as well as in a reduced duration of JNK and ERK mitogenic signals[56]. This pro-survival effect, when facing LPS, can also be blocked by down-regulating this TLR4-downstream molecules - ERK and JNK[49].
A second and slightly opposing situation relies on p38 - capable of inducing cell cycle arrest and apoptosis - and NF-κB - capable of stimulating pro-inflammatory cytokines (IL-1, -6, -10, TNF-α)[57] - that were inhibited by LPS, in a TLR4-overexpressing HCC cell line, allowing cell proliferation[49]. In fact, after stimulating TLR4, either blocking[49] or augmenting[39,55] NF-κB have been reported to promote tumour’s survival. Once more we face an ambiguity in interpreting NF-κB values. However, in this situation, the explanation can rely on the degree of the stimuli/block and the underlying inflammation[3].
Consequently, we are able to conclude that increased expression of TLR4 may protect HCC cells from LPS-induced cytotoxicity and promote cell HCC survival and proliferation.
This pro-tumorigenic effect of TLR4 is confirmed by the fact that, in TLR4-/- mice subjected to DEN, tumour incidence is 25% lower and diameters are smaller accompanied by less inflammation, proliferation as well as enhanced apoptosis[56]. Moreover, using antibiotics to reduce the LPS levels results in diminished activation of T helper 1 cells[58] and consequently less liver damage, and lower cell proliferation in tumour mass[56].
However, Xu et al[54] presented a different vision when reported increased expression of protein tyrosine phosphatase receptor type O (PTPRO) in TLR4-overexpressing HCC cell lines after LPS treatment. Here, cell proliferation was inhibited and apoptosis was augmented as a result of the tumour suppressor capability of PTPRO[54]. To that end, it was found that, contrarily to the effects on LPS-induced cytotoxicity, TLR4-overexpression might also have a protective role through PTPRO and thus, worth being subjected to new studies.
Li et al[52] also observed that, with TLR4 over-expression, came a gradual disappearance of epithelial cell markers and increased mesenchymal ones, suggesting an epithelial-mesenchymal transition (EMT). This EMT is considered to be the molecular basis of tumour cell infiltration and metastasis[59] and can, actually, be induced by two possible pathways related to TLR4 and LPS stimulus. On one hand, the TLR4 - MAPK/JNK pathway, confirmed by the fact that, blocking directly MAPK/JNK or indirectly through TLR4, lead to inhibition of LPS-induced EMT[52]. On the other hand, Snail, a transcription factor handled by NF-κB and a major inducer of EMT[53].
For this reason, LPS, via activation of TLR4 signalling pathway and consequently MAPK/JNK pathway activation or NF-κB up-regulation, can significantly induce EMT.
This EMT phenotype is conveyed by cancer stem cells[60] which, in turn, are thought to be involved in processes like formation and progression of cancer, being, inclusively, responsible for chemotherapy resistance, metastasis and postoperative recurrence[61,62]. Recent studies revealed that TLR4 positive cells exhibit a series of stem cells characteristics[50,60]. These cells not only display a higher invasive ability, when compared to TLR4 negatives, but also express many stem cell markers (CD133 increase 85% when TLR4 is overexpressed[60]) as well as a stronger colony forming ability and increased chemotherapy/apoptosis resistance[50].
In agreement with these results, Chen et al[60] proposed that TLR4 could work as a proto-oncogene which aberrant expression/activation leads to induction of pluripotency genes and genesis of tumour-initiating stem-like cells (TICs). This process is possible through activation of a TLR4/NANOG pathway[60,62-64] and consequent inhibition of the transforming growth factor β (TGF-β)[60,62,64].
NANOG is per se a core transcription factor found in pluripotent stem cells[62]. TGF-β is an effective proliferation inhibitor and an apoptosis promoter that, when down-regulated, is able to initiate tumorigenesis via stemness gene induction in an epithelial tissue such as liver[60]. In fact, knockdown of TLR4 attenuated the induction of stem cell genes as well as DNA synthesis of TICs in 50% to 80% and blocking NANOG, results in a tumour growth reduction of 60% to 75%[60].
However, some cancer cells grow efficiently in vitro without addition of LPS but this growth is still reduced by TLR4 knockdown, suggesting LPS-independent mechanisms of TLR4 activation in these cells[64]. One possibility includes non-LPS ligands influencing tumorigenesis through TLR4 signalling.
Yan et al[65] observed that hypoxia was also responsible for TLR4 up-regulation in an HCC cell line. Hypoxia is a hallmark of several solid tumours, including HCC, and an important factor in tumour progression[66]. A possible explanation of this relationship may involve hypoxia-induced HMGB1 release, capable of activating TLR4 signalling and consequently augment caspase-1. This one is, in turn, related with maturation of pro-inflammatory cytokines and consequent tumorigenesis and tumour progression. After TLR4 blockage, caspase-1 expression diminished significantly[65]. Additionally, caspase-1 blocking was capable of decreasing HCC cell invasiveness[65]. This suggests that hypoxia-induced caspase-1 activation, as well as caspase-1-mediated tumour progression, can depend on TLR4 signalling.
In spite of several evidences attributing a pro-tumorigenic role to TLR4, the pathways to that end are many and still not fully understood. Diminished apoptotic-response to LPS, EMT-induction or caspase-1 up-regulation through TLR4 were already proposed but, opposing effects mediated by tumour suppressors like PTPRO were also found (Figure 4). According to these results new studies are suggested to clarify not only how each work, but also how they are related. However, contrarily to TLR2 most data suggests that TLR4 activation not only has an important role in inflammation and fibrosis but also in HCC initiation and progression.
Figure 4.
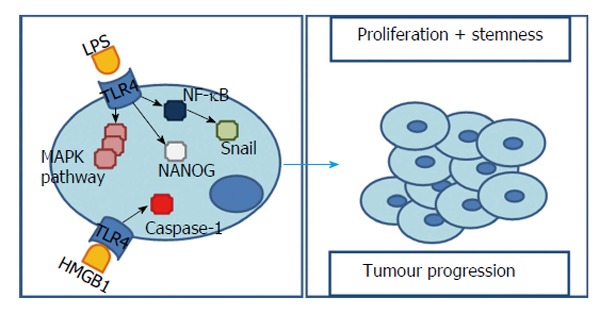
Toll-like receptor 4’s signalling pathways influencing hepatocarcinogenesis. Bacterial LPS is capable of initiate TLR4 signalling and subsequently activate NF-κB and MAPK signalling pathways. In one hand, in a HCC cell line incubated with bacterial LPS both TLR4 expression and MAPK signalling pathways are significantly augmented, contributing to cell survival and proliferation. On the other hand, TLR4 over-expression contributes to an EMT through MAPKs pathway and Snail. Additionally, NANOG induces TICs’ formation. Both are considered to be the molecular basis of tumour cell infiltration and metastasis. HMGB1’s stimulation of TLR4 with caspase-1 activation is related with maturation of pro-inflammatory cytokines and consequent tumorigenesis and tumour progression. TLR4: Toll-like receptor 4; LPS: Lipopolysaccharides; HCC: Hepatocarcinoma; MAPK: Mitogen-activated protein kinase; EMT: Epithelial-mesenchymal transition; TICs: Tumour-initiating stem-like cells; NF-κB: Nuclear transcription factor kappa B; HMGB1: High mobility group box 1.
TLR9
A possible relationship between TLR9 and carcinogenesis came to light when its high expression levels of TLR9 were found in samples of lung and breast cancer cell lines[67]. HCC cells exhibit a broad repertoire of TLRs, also including TLR9. This receptor plays a crucial role in cell survival as it recognises several bacterial and viral components, including unmethylated CpG-DNA. Different works revealed that there is an augmented TLR9 positivity in human HCC cells[8,68,69] with Eiró et al[8] showing a TLR9’s prevalence of 60% (in a population of 30 cases) and Tanaka et al[68] reaching the 85.7% (in a population of 42 cases) in their works with human samples of HCC. Moreover, in the later, 7 of 8 cases of HCC metastasis presented TLR9 positivity[68]. Additionally, it was found that, in both HCC cell line or HCC human samples, TLR9 was present not only in the cytoplasm but also on cells’ membrane[68]. However, is important to notice that, possibly, only the stimulation of membrane receptors could result in increased cell viability as transfecting a TLR9 agonist, CpG-oligodesoxynucleotide (CpG-ODN), which stimulates intracellular TLR9 receptors, may not affect proliferation and survival[68]. The explanation for this tumour-promoter role of TLR9 comes from the fact that, after TLR9 stimulation, a HCC cell line is able to, not only up-regulate apoptosis inhibitors such as survivin, Bcl-xL, XIAP and cFLIP, but also, to closely modulate oncogenic genes with a major contribution in tumorigenesis and cancer progression[68] (Figure 5).
Figure 5.
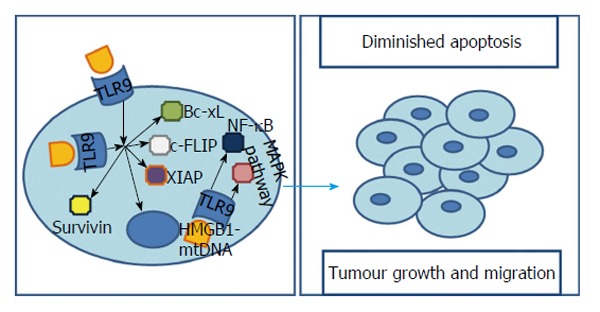
Toll-like receptor 9’s signalling pathways influencing hepatocarcinoma. TLR9 is present not only in the cytoplasm but also on cells’ membrane. However, it is still controversial whether cytoplasmatic stimulation results in increased cell viability. Independently, membrane receptors’ stimulation results, not only, in up-regulation of apoptosis inhibitors such as survivin, Bcl-xL, XIAP and c-FLIP, but also, in a modulation of oncogenic genes with a major contribution in tumorigenesis and cancer progression. Additionally, a cytoplasmatic HMGB1-mtDNA interaction was proved to be capable of activating TLR9 and MAPK pathway as well as NF-κB leading to augmented survival, growth, proliferation, differentiation and migration of cancer cells. TLR9: Toll-like receptor 9; XIAP: X-linked inhibitor of apoptosis protein; c-FLIP: Cellular FLICE-Like inhibitory protein; MAPK: Mitogen-activated protein kinase; NF-κB: Nuclear transcription factor kappa B; HMGB1: High mobility group box 1.
Although, this data is not that linear, and, somehow, different from what Zhang et al[70,71] stated in their studies. Here, transfecting a TLR9 agonist into a HCC cell line lead to a marked increase in IFN-α, IFN-β, TNF-α, IL-6 and IL-8 without activating NF-αB. As a result a cell-proliferation’s inhibition rate was increased approximately 50% and apoptosis was augmented[70,71].
The contradictory findings about the influence on tumour’s environment of intracellular TLR9 agonists could be explained by the fact that the phosphorothioate-modified backbone of CpG-ODN are able to form a complex with or cause conformational changes in other compounds, like Poly (I:C) that, normally would result in enhanced apoptosis but, when together with CpG-ODN, are unable to act[70]. Moreover is important to look at the protocols used, as CpG-ODN induces HCC cell apoptosis in a dose-dependent manner, at concentrations below 0.5 μg/mL. In contrast, high concentrations of this agonist (e.g., 5 μg/mL) had no effect on HCC cells[71].
Additionally, this pathway from TLR9 signalling to carcinogenesis is supported by HMGB1. We have already seen that HMGB1 and hypoxia could influence tumorigenesis through different TLRs. Interestingly, they are, also, both involved with TLR9. It was seen that along with TLR9 overexpression, hypoxic cancer cells accumulate structurally and functionally abnormal mitochondria, which release mitochondrial DNA (mt-DNA) to the cytosol, and induce translocation of HMGB1 from nucleus to cytoplasm[69]. The role of HMGB1 as a promoter of invasion, metastasis and angiogenesis when its location is extracellular is not new[72]. However, Liu et al[69] revealed that, on top of this, an cytoplasmatic HMGB1-mtDNA interaction is required for complete activation of TLR9 signalling cascade and therefore essential for HCC cells to proliferate under hypoxic conditions. The underlying mechanism in this pro-tumorigenic pathway lies in MAPKs activation - fundamental in growth, proliferation, differentiation and migration[73] - and also in NF-κB signalling - capable of suppressing apoptosis in response to stress[74] - after the interaction between HMGB1/mtDNA and TLR9[69,75] (Figure 5).
TLRS GENETIC POLYMORPHISMS AND VARIANTS AND HCC SUSCEPTIBILITY
Several authors have already focused their studies on the relationship between TLRs’ genetics and carcinogenesis, approaching different cancers such as non-Hodgkin lymphoma, endometrial cancer, cervical cancer, non-cardiac gastric cancer, among others.
Genetic studies on the TLR2 gene have shown a number of polymorphisms capable of interfering with host defenses and disease progression[76]. In fact, it was already seen that inherited variation in TLR2 influence the risk of HCC. Genetic TLR2 analysis revealed that two single nucleotide polymorphisms (SNP), rs3804099 and rs3804100, had a significantly different distribution between HCC patients and the healthy controls[77]. Interestingly, in what is concerned to these SNPs, Junjie et al[77] suggested that, TLR2 gene variation could play an important protective role in HCC as the heterozygous genotype comprise lesser HCC risk (OR from 0.331 to 0.759, P < 0.001) when compared to wild-type homozygous genotype. In fact, individuals carrying the TT haplotype had a significantly decreased risk of HCC [odds ratio (OR) = 0.524, 95%CI: 0.394-0.697, P < 0.001]. Contrarily, the CC haplotype had greater risk (OR = 2.743, 95%CI: 1.915-3.930, P < 0.001). Unfortunately, the authors do not reveal the real influence of the referred SNPs on the TLR2’s activity and more studies are suggested to clarify this information.
Moreover, the frequency of a -196 to -174 deletion allele was, also, significantly higher in HCC patients than in healthy controls (22.5% vs 15.3%) and HCV-infected patients without HCC (22.5% vs 15.6%)[76]. Nischalke et al[76] observations indicate that the -196 to -174 deletion allele possibly augment the risk of HCV-induced HCC, probably as a result of diminished TLR2 signalling and thus increased viral loads. This -196 to -174 deletion not only had greater viral loads than -196 to -174 ins/ins but also, contribute to a 3-fold increase in HCC risk relatively to this -196 to -174 ins/ins when both are compared to healthy controls or a 1.5 fold increase when both are compared to hepatitis C patients without HCC[76].
Researchers have already studied the possible presence of polymorphisms in the area of TLR3. It was found that, at least in the chinese population, a +1234CT polymorphism is present which might contribute to increased susceptibility to HCC (specially 1234CT and TT genotypes)[78]. The presence of this SNP is responsible for a markedly diminished TLR3 function, which may result in up-regulated vasculature remodelling and tumour growth and, in that way, contributing to HCC[78].
The TLR4 is probably the more extensively studied TLR and therefore, not an exception when it comes to having polymorphisms or variants capable of influence carcinogenesis. Growing evidence has shown that TLR4 polymorphisms are related to chronic inflammation and inflammatory-related cancer. As a matter of fact, a polymorphism in microRNA-34a binding site in TLR4 (rs1057317) was significantly associated with higher HCC risk, especially in HBsAg (+) patients and in the AA homozygous genotypes[79]. MicroRNA-34a is capable of inducing apoptosis, G1 arrest and senescence explaining why its down-regulation may be associated with malignancy. However, there are not only polymorphisms related to augmented risk. Indeed, some mutations of TLR4 gene - four SNPs in 5’-UTR (rs10759930, rs2737190, rs10116253 and rs1927914) and one intron polymorphism (rs1927911) - may allow a two-fold decrease in HCC risk, especially in heterozygous genotypes when compared to wild-type homozygous[80]. The justification can rely on the fact that 5’-UTR is involved in regulation of proteins concerned with growth and differentiation in normal tissues and these SNPs may, therefore, exert regulator effects in these proteins[80]. Therefore, according to the TLR4’s polymorphism observed, an augmented or diminished risk of HCC is possible, even though its magnitude is small.
TLRS AS THERAPEUTIC TARGETS FOR HCC
So far we have seen that different TLRs could work as specific modulators of HCC. Therefore it is logical to think that its use as therapeutic targets could open the door to new promising strategies in the fighting against HCC. In fact, the modulation of TLRs’ signalling, by targeting either the TLRs or their adaptors or downstream signalling molecules, is not new and they have already proved to be useful in ovarian, colorectal or head and neck cancer[81].
TLR4 modulators seem to be important chemotherapy adjuvants that enhance chemotherapy efficacy and prolong survival[81]. TAK-242 is a TLR4 ligand capable of selectively supress both ligand-dependent and independent signalling via the intracellular domain of TLR4, disrupting the TRAM and TIRAP interactions with TLR4[82]. This small molecule is, therefore, able to down-regulate NF-κB and consequently diminish inflammatory mediators such as nitric oxide, TNF-α, IL-1, -6 and with that, reduce the proliferation/invasion activity induced by LPS in the liver cancer cell lines[57,82,83]. Furthermore, TAK-242 might also show an efficacy against inflammation mediated by excessive expression of TLR4, what, in fact, has been shown to happen in HCC[82]. Independently, new studies are still required for better evaluating effects, doses and other characteristics of TAK-242.
To date, we still lack an effective systemic curative therapy for advanced cases of HCC and, in most cases the only alternative is palliative treatment.
Even though, sorafenib, a multi-kinase inhibitor, represents an important chemotherapeutic drug in the treatment of this type of cancer. Xu et al[84] found that a combination of sorafenib with a TLR3-synergist (BM-06) results in a superior inhibition of tumour growth in HCC cell lines or rat models when compared to the two different agents alone. Their results were based on a significantly reduced proliferative capacity, invasion ability, tumour volume and an increased apoptotic rate[84]. Therefore, BM-06 emerges as a possible adjuvant agent in the therapeutic against HCC.
In the TLR9 domain several studies were already conducted. It was reported that using TLR9 antagonists like chloroquine could be useful in several autoimmune diseases[2]. In fact, a markedly reduced proliferation is seen in a HCC cell line when TLR9 is inhibited by chloroquine[2]. This antimalaric agent works as a direct TLR9 antagonist, being proposed that its activity on HCC cells may be brought about via its effects on the protein kinase AKT, tumour-associated angiogenesis factor VEGF as well as NF-κB. Moreover, the same tumour growth restriction, followed by smaller volume and reduction in tumour’s markers of aggressiveness was seen when this treatment was used in HCC cell lines intrahepatic implanted in mice[2].
However, as it was said, sometimes, the only option is the palliation and radiofrequency ablation (RFA) which has already established an important role in this setting. Behm et al[85] successfully demonstrated that, in a rabbit model, TLR9 agonists could work together with RFA in an anti-tumour response through a strong cytotoxic immune response mediated by increased tumour-specific lymphocytes. In fact, it was not only a good predictor of containment of tumour growth and spread but also of prolonged survival[85].
Moreover, some studies focus on the use of TLRs as vaccine’s adjuvants against HCV or HBV mediated hepatocarcinogenesis. There are also some proposals for using TLR4’s antagonists in patients with septic shock[86]. Besides this, the use of TLR4’s antagonists is being investigated in the prevention of alcoholic liver injury and Non Alcoholic Steato-Hepatitis[9].
Despite the good results, when it comes to using TLRs as a novel HCC therapeutic it still has a long run before every mechanism is understood. TLRs’ signalling pathways are too many and effects remain controversial but a lot is to be expected from these innate immunity receptors.
CONCLUSION
HCC occupies the third place when it comes to mortality in cancer[1]. Several factors are known to contribute to the carcinogenic process including viral hepatitis, alcohol, auto-immune or metabolic diseases, among others. The link between all these factors is inflammation or, more precisely, chronic inflammation.
However, despite all this malignant potential or this knowledge around the inflammatory causality, most of the pathways of carcinogenesis are still unknown or, at least, not entirely known.
TLRs’ role in the tumour formation is part of a more recent concept that involves innate immunity but, despite all the advances, a lot is still waiting to be studied. The reasons for this lack of information include the range of responses that can be obtained from a single TLR signalling. Humans dispose of 11 TLRs capable of initiating a signal cascade from only five molecular adaptors. Consequently, some can be used by more than one receptor. Moreover, each of this activated adaptor molecules, depending on the initial TLR, elicit a response based on the production of several effectors, pro-inflammatory or anti-inflammatory cytokines, interferons and many others with countless results.
To understand the role of TLRs we must remember that HCC comes from an inflammatory background where a complex and progressive process appears with fibrosis and cirrhosis until the last stage, HCC, is reached. This review tried to show that, the path taken can be closely influenced by innate immunity/TLRs (Figure 6). TLR2 was shown to be an important stress manager so that, in its absence, an attenuated ASK1/p38 MAPK/NF-κB pathway and an increased JNK activity result in a larger and less differentiated HCC. Contrarily, TLR2’s stimulation through HMGB1 and HSPA1A also indicates a tumour-promoter role. TLR3 may be responsible for driving HCC and endothelial cells to apoptosis and decreasing invasiveness and angiogenesis by mediating NF-κB, caspase-8 and INF-γ up-regulation. TLR4 is closely related to LPS cytotoxic, which is diminished in TLR4-overexpressing HCC due to promoted ERK’s and JNK’s activity and limited NF-κB and p38 activation. Moreover, this receptor is tightly involved in EMT and progression of cancer based on non-LPS ligands like NANOG and HMGB1. TLR9 activity is different whether the membrane or the intracellular receptor is activated. The first promotes HCC through apoptosis inhibitors and oncogenic genes. The second augments apoptosis by increasing Interferons and interleukines. Consequently, initial findings attribute a pro-tumorigenic role to TLR4 and TLR9 and a protective capacity to TLR3. When it comes to TLR2, the available data suggests that its influence may go both ways (pro-tumorigenic and protective) depending on the liver’s stage in the inflammation-cirhosis-carcinoma axis. However this is not that simple or linear as a closer look easily reveals studies with interesting but opposing conclusions from the ones before.
Figure 6.
Toll-like receptors influence on the pathway to hepatocarcinoma. Several factors are known to contribute to the carcinogenic process including viral hepatitis, alcohol, auto-immune or metabolic diseases, among others and the link between all this factors is chronic inflammation which, in turn, is an important hepatocarcinoma’s percursor. Moreover, innate immunity represents an important player in this equation with TLRs such as 4 and 9 having, mainly, a positive contribution to hepatocarcinogenesis and TLR3, essentially, a negative/protective one. TLR2 still presents an ambiguous role, possibly depending on liver’s stage in the inflammation-cirrhosis-carcinoma axis to exert its pro-tumorigenic or anti-tumorigenic capacity. TLR: Toll-like receptor.
Independently of this lack of knowledge, one thing is certain; TLRs can have a determining influence on the cancer’s progression. Therefore, the usage of TLRs as therapeutic targets has already been established, especially as adjuvants to other agents currently in use. However, the possibilities are many and with a deeper insight over the mechanisms involved new ways of dealing with HCC are expected to emerge.
In conclusion, we cannot say that TLRs came to facilitate our understanding of HCC mechanisms. Instead they came to open the door to a new reality and, with that, to possible new approaches, perhaps in a closer future than we might know.
Footnotes
Conflict-of-interest statement: All the authors certify that they have no conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: July 11, 2015
First decision: August 16, 2015
Article in press: December 8, 2015
P- Reviewer: Dang SS, Ding MX, Sugimura H S- Editor: Kong JX L- Editor: A E- Editor: Liu SQ
References
- 1.Chew V, Tow C, Huang C, Bard-Chapeau E, Copeland NG, Jenkins NA, Weber A, Lim KH, Toh HC, Heikenwalder M, et al. Toll-like receptor 3 expressing tumor parenchyma and infiltrating natural killer cells in hepatocellular carcinoma patients. J Natl Cancer Inst. 2012;104:1796–1807. doi: 10.1093/jnci/djs436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mohamed FE, Al-Jehani RM, Minogue SS, Andreola F, Winstanley A, Olde Damink SW, Habtesion A, Malagó M, Davies N, Luong TV, et al. Effect of toll-like receptor 7 and 9 targeted therapy to prevent the development of hepatocellular carcinoma. Liver Int. 2015;35:1063–1076. doi: 10.1111/liv.12626. [DOI] [PubMed] [Google Scholar]
- 3.Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, Mederacke I, Caviglia JM, Khiabanian H, Adeyemi A, Bataller R, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell. 2012;21:504–516. doi: 10.1016/j.ccr.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kipanyula MJ, Seke Etet PF, Vecchio L, Farahna M, Nukenine EN, Nwabo Kamdje AH. Signaling pathways bridging microbial-triggered inflammation and cancer. Cell Signal. 2013;25:403–416. doi: 10.1016/j.cellsig.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 5.Kurabe N, Murakami S, Tashiro F. SGF29 and Sry pathway in hepatocarcinogenesis. World J Biol Chem. 2015;6:139–147. doi: 10.4331/wjbc.v6.i3.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakamoto N, Kanai T. Role of toll-like receptors in immune activation and tolerance in the liver. Front Immunol. 2014;5:221. doi: 10.3389/fimmu.2014.00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soares JB, Pimentel-Nunes P, Roncon-Albuquerque R, Leite-Moreira A. The role of lipopolysaccharide/toll-like receptor 4 signaling in chronic liver diseases. Hepatol Int. 2010;4:659–672. doi: 10.1007/s12072-010-9219-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eiró N, Altadill A, Juárez LM, Rodríguez M, González LO, Atienza S, Bermúdez S, Fernandez-Garcia B, Fresno-Forcelledo MF, Rodrigo L, et al. Toll-like receptors 3, 4 and 9 in hepatocellular carcinoma: Relationship with clinicopathological characteristics and prognosis. Hepatol Res. 2014;44:769–778. doi: 10.1111/hepr.12180. [DOI] [PubMed] [Google Scholar]
- 9.Mencin A, Kluwe J, Schwabe RF. Toll-like receptors as targets in chronic liver diseases. Gut. 2009;58:704–720. doi: 10.1136/gut.2008.156307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roh YS, Seki E. Toll-like receptors in alcoholic liver disease, non-alcoholic steatohepatitis and carcinogenesis. J Gastroenterol Hepatol. 2013;28 Suppl 1:38–42. doi: 10.1111/jgh.12019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aravalli RN. Role of innate immunity in the development of hepatocellular carcinoma. World J Gastroenterol. 2013;19:7500–7514. doi: 10.3748/wjg.v19.i43.7500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu S, Gallo DJ, Green AM, Williams DL, Gong X, Shapiro RA, Gambotto AA, Humphris EL, Vodovotz Y, Billiar TR. Role of toll-like receptors in changes in gene expression and NF-kappa B activation in mouse hepatocytes stimulated with lipopolysaccharide. Infect Immun. 2002;70:3433–3442. doi: 10.1128/IAI.70.7.3433-3442.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsumura T, Ito A, Takii T, Hayashi H, Onozaki K. Endotoxin and cytokine regulation of toll-like receptor (TLR) 2 and TLR4 gene expression in murine liver and hepatocytes. J Interferon Cytokine Res. 2000;20:915–921. doi: 10.1089/10799900050163299. [DOI] [PubMed] [Google Scholar]
- 14.Thobe BM, Frink M, Hildebrand F, Schwacha MG, Hubbard WJ, Choudhry MA, Chaudry IH. The role of MAPK in Kupffer cell toll-like receptor (TLR) 2-, TLR4-, and TLR9-mediated signaling following trauma-hemorrhage. J Cell Physiol. 2007;210:667–675. doi: 10.1002/jcp.20860. [DOI] [PubMed] [Google Scholar]
- 15.Knolle P, Schlaak J, Uhrig A, Kempf P, Meyer zum Büschenfelde KH, Gerken G. Human Kupffer cells secrete IL-10 in response to lipopolysaccharide (LPS) challenge. J Hepatol. 1995;22:226–229. doi: 10.1016/0168-8278(95)80433-1. [DOI] [PubMed] [Google Scholar]
- 16.Edwards AD, Diebold SS, Slack EM, Tomizawa H, Hemmi H, Kaisho T, Akira S, Reis e Sousa C. Toll-like receptor expression in murine DC subsets: lack of TLR7 expression by CD8 alpha+ DC correlates with unresponsiveness to imidazoquinolines. Eur J Immunol. 2003;33:827–833. doi: 10.1002/eji.200323797. [DOI] [PubMed] [Google Scholar]
- 17.Sawaki J, Tsutsui H, Hayashi N, Yasuda K, Akira S, Tanizawa T, Nakanishi K. Type 1 cytokine/chemokine production by mouse NK cells following activation of their TLR/MyD88-mediated pathways. Int Immunol. 2007;19:311–320. doi: 10.1093/intimm/dxl148. [DOI] [PubMed] [Google Scholar]
- 18.Meyer-Bahlburg A, Khim S, Rawlings DJ. B cell intrinsic TLR signals amplify but are not required for humoral immunity. J Exp Med. 2007;204:3095–3101. doi: 10.1084/jem.20071250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paik YH, Schwabe RF, Bataller R, Russo MP, Jobin C, Brenner DA. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology. 2003;37:1043–1055. doi: 10.1053/jhep.2003.50182. [DOI] [PubMed] [Google Scholar]
- 20.Wu J, Meng Z, Jiang M, Zhang E, Trippler M, Broering R, Bucchi A, Krux F, Dittmer U, Yang D, et al. Toll-like receptor-induced innate immune responses in non-parenchymal liver cells are cell type-specific. Immunology. 2010;129:363–374. doi: 10.1111/j.1365-2567.2009.03179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang Y, Cai B, Xu M, Qiu Z, Tao Y, Zhang Y, Wang J, Xu Y, Zhou Y, Yang J, et al. Gene silencing of Toll-like receptor 2 inhibits proliferation of human liver cancer cells and secretion of inflammatory cytokines. PLoS One. 2012;7:e38890. doi: 10.1371/journal.pone.0038890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim S, Takahashi H, Lin WW, Descargues P, Grivennikov S, Kim Y, Luo JL, Karin M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature. 2009;457:102–106. doi: 10.1038/nature07623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang W, Xu GL, Jia WD, Ma JL, Li JS, Ge YS, Ren WH, Yu JH, Liu WB. Ligation of TLR2 by versican: a link between inflammation and metastasis. Arch Med Res. 2009;40:321–323. doi: 10.1016/j.arcmed.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 24.Lin H, Yan J, Wang Z, Hua F, Yu J, Sun W, Li K, Liu H, Yang H, Lv Q, et al. Loss of immunity-supported senescence enhances susceptibility to hepatocellular carcinogenesis and progression in Toll-like receptor 2-deficient mice. Hepatology. 2013;57:171–182. doi: 10.1002/hep.25991. [DOI] [PubMed] [Google Scholar]
- 25.Lin H, Liu XB, Yu JJ, Hua F, Hu ZW. Antioxidant N-acetylcysteine attenuates hepatocarcinogenesis by inhibiting ROS/ER stress in TLR2 deficient mouse. PLoS One. 2013;8:e74130. doi: 10.1371/journal.pone.0074130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li S, Sun R, Chen Y, Wei H, Tian Z. TLR2 limits development of hepatocellular carcinoma by reducing IL18-mediated immunosuppression. Cancer Res. 2015;75:986–995. doi: 10.1158/0008-5472.CAN-14-2371. [DOI] [PubMed] [Google Scholar]
- 27.Soares JB, Pimentel-Nunes P, Afonso L, Rolanda C, Lopes P, Roncon-Albuquerque R, Gonçalves N, Boal-Carvalho I, Pardal F, Lopes S, et al. Increased hepatic expression of TLR2 and TLR4 in the hepatic inflammation-fibrosis-carcinoma sequence. Innate Immun. 2012;18:700–708. doi: 10.1177/1753425912436762. [DOI] [PubMed] [Google Scholar]
- 28.Marra M, Sordelli IM, Lombardi A, Lamberti M, Tarantino L, Giudice A, Stiuso P, Abbruzzese A, Sperlongano R, Accardo M, et al. Molecular targets and oxidative stress biomarkers in hepatocellular carcinoma: an overview. J Transl Med. 2011;9:171. doi: 10.1186/1479-5876-9-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol. 2011;54:795–809. doi: 10.1016/j.jhep.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin H, Hua F, Hu ZW. Autophagic flux, supported by toll-like receptor 2 activity, defends against the carcinogenesis of hepatocellular carcinoma. Autophagy. 2012;8:1859–1861. doi: 10.4161/auto.22094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dolado I, Swat A, Ajenjo N, De Vita G, Cuadrado A, Nebreda AR. p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell. 2007;11:191–205. doi: 10.1016/j.ccr.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 32.Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–551. doi: 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- 33.Clarke R, Cook KL, Hu R, Facey CO, Tavassoly I, Schwartz JL, Baumann WT, Tyson JJ, Xuan J, Wang Y, et al. Endoplasmic reticulum stress, the unfolded protein response, autophagy, and the integrated regulation of breast cancer cell fate. Cancer Res. 2012;72:1321–1331. doi: 10.1158/0008-5472.CAN-11-3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Beijnum JR, Buurman WA, Griffioen AW. Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1) Angiogenesis. 2008;11:91–99. doi: 10.1007/s10456-008-9093-5. [DOI] [PubMed] [Google Scholar]
- 36.Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 37.Gong W, Wang ZY, Chen GX, Liu YQ, Gu XY, Liu WW. Invasion potential of H22 hepatocarcinoma cells is increased by HMGB1-induced tumor NF-κB signaling via initiation of HSP70. Oncol Rep. 2013;30:1249–1256. doi: 10.3892/or.2013.2595. [DOI] [PubMed] [Google Scholar]
- 38.Shi W, Su L, Li Q, Sun L, Lv J, Li J, Cheng B. Suppression of toll-like receptor 2 expression inhibits the bioactivity of human hepatocellular carcinoma. Tumour Biol. 2014;35:9627–9637. doi: 10.1007/s13277-014-2268-3. [DOI] [PubMed] [Google Scholar]
- 39.Wu FH, Yuan Y, Li D, Liao SJ, Yan B, Wei JJ, Zhou YH, Zhu JH, Zhang GM, Feng ZH. Extracellular HSPA1A promotes the growth of hepatocarcinoma by augmenting tumor cell proliferation and apoptosis-resistance. Cancer Lett. 2012;317:157–164. doi: 10.1016/j.canlet.2011.11.020. [DOI] [PubMed] [Google Scholar]
- 40.Chew V, Abastado JP. Immunomodulation of the tumor microenvironment by Toll-like receptor-3 (TLR3) ligands. Oncoimmunology. 2013;2:e23493. doi: 10.4161/onci.23493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoneda K, Sugimoto K, Shiraki K, Tanaka J, Beppu T, Fuke H, Yamamoto N, Masuya M, Horie R, Uchida K, et al. Dual topology of functional Toll-like receptor 3 expression in human hepatocellular carcinoma: differential signaling mechanisms of TLR3-induced NF-kappaB activation and apoptosis. Int J Oncol. 2008;33:929–936. [PubMed] [Google Scholar]
- 42.Zorde-Khvalevsky E, Abramovitch R, Barash H, Spivak-Pohis I, Rivkin L, Rachmilewitz J, Galun E, Giladi H. Toll-like receptor 3 signaling attenuates liver regeneration. Hepatology. 2009;50:198–206. doi: 10.1002/hep.22973. [DOI] [PubMed] [Google Scholar]
- 43.Maeda S. NF-κB, JNK, and TLR Signaling Pathways in Hepatocarcinogenesis. Gastroenterol Res Pract. 2010;2010:367694. doi: 10.1155/2010/367694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khvalevsky E, Rivkin L, Rachmilewitz J, Galun E, Giladi H. TLR3 signaling in a hepatoma cell line is skewed towards apoptosis. J Cell Biochem. 2007;100:1301–1312. doi: 10.1002/jcb.21119. [DOI] [PubMed] [Google Scholar]
- 45.Chen L, Xu YY, Zhou JM, Wu YY, E Q, Zhu YY. TLR3 dsRNA agonist inhibits growth and invasion of HepG2.2.15 HCC cells. Oncol Rep. 2012;28:200–206. doi: 10.3892/or.2012.1791. [DOI] [PubMed] [Google Scholar]
- 46.Guo Z, Chen L, Zhu Y, Zhang Y, He S, Qin J, Tang X, Zhou J, Wei Y. Double-stranded RNA-induced TLR3 activation inhibits angiogenesis and triggers apoptosis of human hepatocellular carcinoma cells. Oncol Rep. 2012;27:396–402. doi: 10.3892/or.2011.1538. [DOI] [PubMed] [Google Scholar]
- 47.Bergé M, Bonnin P, Sulpice E, Vilar J, Allanic D, Silvestre JS, Lévy BI, Tucker GC, Tobelem G, Merkulova-Rainon T. Small interfering RNAs induce target-independent inhibition of tumor growth and vasculature remodeling in a mouse model of hepatocellular carcinoma. Am J Pathol. 2010;177:3192–3201. doi: 10.2353/ajpath.2010.100157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu YY, Chen L, Zhou JM, Wu YY, Zhu YY. Inhibitory effect of dsRNA TLR3 agonist in a rat hepatocellular carcinoma model. Mol Med Rep. 2013;8:1037–1042. doi: 10.3892/mmr.2013.1646. [DOI] [PubMed] [Google Scholar]
- 49.Wang L, Zhu R, Huang Z, Li H, Zhu H. Lipopolysaccharide-induced toll-like receptor 4 signaling in cancer cells promotes cell survival and proliferation in hepatocellular carcinoma. Dig Dis Sci. 2013;58:2223–2236. doi: 10.1007/s10620-013-2745-3. [DOI] [PubMed] [Google Scholar]
- 50.Liu WT, Jing YY, Yu GF, Han ZP, Yu DD, Fan QM, Ye F, Li R, Gao L, Zhao QD, et al. Toll like receptor 4 facilitates invasion and migration as a cancer stem cell marker in hepatocellular carcinoma. Cancer Lett. 2015;358:136–143. doi: 10.1016/j.canlet.2014.12.019. [DOI] [PubMed] [Google Scholar]
- 51.Chen R, Alvero AB, Silasi DA, Steffensen KD, Mor G. Cancers take their Toll--the function and regulation of Toll-like receptors in cancer cells. Oncogene. 2008;27:225–233. doi: 10.1038/sj.onc.1210907. [DOI] [PubMed] [Google Scholar]
- 52.Li H, Li Y, Liu D, Liu J. LPS promotes epithelial-mesenchymal transition and activation of TLR4/JNK signaling. Tumour Biol. 2014;35:10429–10435. doi: 10.1007/s13277-014-2347-5. [DOI] [PubMed] [Google Scholar]
- 53.Jing YY, Han ZP, Sun K, Zhang SS, Hou J, Liu Y, Li R, Gao L, Zhao X, Zhao QD, et al. Toll-like receptor 4 signaling promotes epithelial-mesenchymal transition in human hepatocellular carcinoma induced by lipopolysaccharide. BMC Med. 2012;10:98. doi: 10.1186/1741-7015-10-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu D, Wang X, Yan S, Yin Y, Hou J, Wang X, Sun B. Interaction of PTPRO and TLR4 signaling in hepatocellular carcinoma. Tumour Biol. 2014;35:10267–10273. doi: 10.1007/s13277-014-2302-5. [DOI] [PubMed] [Google Scholar]
- 55.Wang Y, Tu Q, Yan W, Xiao D, Zeng Z, Ouyang Y, Huang L, Cai J, Zeng X, Chen YJ, et al. CXC195 suppresses proliferation and inflammatory response in LPS-induced human hepatocellular carcinoma cells via regulating TLR4-MyD88-TAK1-mediated NF-κB and MAPK pathway. Biochem Biophys Res Commun. 2015;456:373–379. doi: 10.1016/j.bbrc.2014.11.090. [DOI] [PubMed] [Google Scholar]
- 56.Yu LX, Yan HX, Liu Q, Yang W, Wu HP, Dong W, Tang L, Lin Y, He YQ, Zou SS, et al. Endotoxin accumulation prevents carcinogen-induced apoptosis and promotes liver tumorigenesis in rodents. Hepatology. 2010;52:1322–1333. doi: 10.1002/hep.23845. [DOI] [PubMed] [Google Scholar]
- 57.Yu P, Cheng X, Du Y, Huang L, Dong R. TAK-242 can be the potential agents for preventing invasion and metastasis of hepatocellular carcinoma. Med Hypotheses. 2013;81:653–655. doi: 10.1016/j.mehy.2013.06.034. [DOI] [PubMed] [Google Scholar]
- 58.Lin Y, Yu LX, Yan HX, Yang W, Tang L, Zhang HL, Liu Q, Zou SS, He YQ, Wang C, et al. Gut-derived lipopolysaccharide promotes T-cell-mediated hepatitis in mice through Toll-like receptor 4. Cancer Prev Res (Phila) 2012;5:1090–1102. doi: 10.1158/1940-6207.CAPR-11-0364. [DOI] [PubMed] [Google Scholar]
- 59.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 60.Chen CL, Tsukamoto H, Liu JC, Kashiwabara C, Feldman D, Sher L, Dooley S, French SW, Mishra L, Petrovic L, et al. Reciprocal regulation by TLR4 and TGF-β in tumor-initiating stem-like cells. J Clin Invest. 2013;123:2832–2849. doi: 10.1172/JCI65859. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 61.Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012;21:283–296. doi: 10.1016/j.ccr.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Machida K, Chen CL, Liu JC, Kashiwabara C, Feldman D, French SW, Sher L, Hyeongnam JJ, Tsukamoto H. Cancer stem cells generated by alcohol, diabetes, and hepatitis C virus. J Gastroenterol Hepatol. 2012;27 Suppl 2:19–22. doi: 10.1111/j.1440-1746.2011.07010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.French SW, Vitocruz E, French BA. Balloon liver cells forming Mallory-Denk-bodies are progenitor cells. Exp Mol Pathol. 2013;95:117–120. doi: 10.1016/j.yexmp.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Machida K, Feldman DE, Tsukamoto H. TLR4-dependent tumor-initiating stem cell-like cells (TICs) in alcohol-associated hepatocellular carcinogenesis. Adv Exp Med Biol. 2015;815:131–144. doi: 10.1007/978-3-319-09614-8_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yan W, Chang Y, Liang X, Cardinal JS, Huang H, Thorne SH, Monga SP, Geller DA, Lotze MT, Tsung A. High-mobility group box 1 activates caspase-1 and promotes hepatocellular carcinoma invasiveness and metastases. Hepatology. 2012;55:1863–1875. doi: 10.1002/hep.25572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Finger EC, Giaccia AJ. Hypoxia, inflammation, and the tumor microenvironment in metastatic disease. Cancer Metastasis Rev. 2010;29:285–293. doi: 10.1007/s10555-010-9224-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xu N, Yao HP, Sun Z, Chen Z. Toll-like receptor 7 and 9 expression in peripheral blood mononuclear cells from patients with chronic hepatitis B and related hepatocellular carcinoma. Acta Pharmacol Sin. 2008;29:239–244. doi: 10.1111/j.1745-7254.2008.00711.x. [DOI] [PubMed] [Google Scholar]
- 68.Tanaka J, Sugimoto K, Shiraki K, Tameda M, Kusagawa S, Nojiri K, Beppu T, Yoneda K, Yamamoto N, Uchida K, et al. Functional cell surface expression of toll-like receptor 9 promotes cell proliferation and survival in human hepatocellular carcinomas. Int J Oncol. 2010;37:805–814. [PubMed] [Google Scholar]
- 69.Liu Y, Yan W, Tohme S, Chen M, Fu Y, Tian D, Lotze M, Tang D, Tsung A. Hypoxia induced HMGB1 and mitochondrial DNA interactions mediate tumor growth in hepatocellular carcinoma through Toll-like receptor 9. J Hepatol. 2015;63:114–121. doi: 10.1016/j.jhep.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang Y, Lin A, Sui Q, Zhang C, Tian Z, Zhang J. Phosphorothioate modification of the TLR9 ligand CpG ODN inhibits poly(I: C)-induced apoptosis of hepatocellular carcinoma by entry blockade. Cancer Lett. 2014;355:76–84. doi: 10.1016/j.canlet.2014.09.013. [DOI] [PubMed] [Google Scholar]
- 71.Zhang Y, Lin A, Zhang C, Tian Z, Zhang J. Phosphorothioate-modified CpG oligodeoxynucleotide (CpG ODN) induces apoptosis of human hepatocellular carcinoma cells independent of TLR9. Cancer Immunol Immunother. 2014;63:357–367. doi: 10.1007/s00262-014-1518-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kang R, Zhang Q, Zeh HJ, Lotze MT, Tang D. HMGB1 in cancer: good, bad, or both? Clin Cancer Res. 2013;19:4046–4057. doi: 10.1158/1078-0432.CCR-13-0495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–3290. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 74.Bubici C, Papa S, Pham CG, Zazzeroni F, Franzoso G. NF-kappaB and JNK: an intricate affair. Cell Cycle. 2004;3:1524–1529. doi: 10.4161/cc.3.12.1321. [DOI] [PubMed] [Google Scholar]
- 75.Liu X, Xu Q, Chen W, Cao H, Zheng R, Li G. Hepatitis B virus DNA-induced carcinogenesis of human normal liver cells by virtue of nonmethylated CpG DNA. Oncol Rep. 2009;21:941–947. doi: 10.3892/or_00000307. [DOI] [PubMed] [Google Scholar]
- 76.Nischalke HD, Coenen M, Berger C, Aldenhoff K, Müller T, Berg T, Krämer B, Körner C, Odenthal M, Schulze F, et al. The toll-like receptor 2 (TLR2) -196 to -174 del/ins polymorphism affects viral loads and susceptibility to hepatocellular carcinoma in chronic hepatitis C. Int J Cancer. 2012;130:1470–1475. doi: 10.1002/ijc.26143. [DOI] [PubMed] [Google Scholar]
- 77.Junjie X, Songyao J, Minmin S, Yanyan S, Baiyong S, Xiaxing D, Jiabin J, Xi Z, Hao C. The association between Toll-like receptor 2 single-nucleotide polymorphisms and hepatocellular carcinoma susceptibility. BMC Cancer. 2012;12:57. doi: 10.1186/1471-2407-12-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li G, Zheng Z. Toll-like receptor 3 genetic variants and susceptibility to hepatocellular carcinoma and HBV-related hepatocellular carcinoma. Tumour Biol. 2013;34:1589–1594. doi: 10.1007/s13277-013-0689-z. [DOI] [PubMed] [Google Scholar]
- 79.Jiang ZC, Tang XM, Zhao YR, Zheng L. A functional variant at miR-34a binding site in toll-like receptor 4 gene alters susceptibility to hepatocellular carcinoma in a Chinese Han population. Tumour Biol. 2014;35:12345–12352. doi: 10.1007/s13277-014-2547-z. [DOI] [PubMed] [Google Scholar]
- 80.Minmin S, Xiaoqian X, Hao C, Baiyong S, Xiaxing D, Junjie X, Xi Z, Jianquan Z, Songyao J. Single nucleotide polymorphisms of Toll-like receptor 4 decrease the risk of development of hepatocellular carcinoma. PLoS One. 2011;6:e19466. doi: 10.1371/journal.pone.0019466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Garay RP, Viens P, Bauer J, Normier G, Bardou M, Jeannin JF, Chiavaroli C. Cancer relapse under chemotherapy: why TLR2/4 receptor agonists can help. Eur J Pharmacol. 2007;563:1–17. doi: 10.1016/j.ejphar.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 82.Kawamoto T, Ii M, Kitazaki T, Iizawa Y, Kimura H. TAK-242 selectively suppresses Toll-like receptor 4-signaling mediated by the intracellular domain. Eur J Pharmacol. 2008;584:40–48. doi: 10.1016/j.ejphar.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 83.Matsunaga N, Tsuchimori N, Matsumoto T, Ii M. TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol Pharmacol. 2011;79:34–41. doi: 10.1124/mol.110.068064. [DOI] [PubMed] [Google Scholar]
- 84.Xu YY, Chen L, Wang GL, Zhou JM, Zhang YX, Wei YZ, Zhu YY, Qin J. A synthetic dsRNA, as a TLR3 pathwaysynergist, combined with sorafenib suppresses HCC in vitro and in vivo. BMC Cancer. 2013;13:527. doi: 10.1186/1471-2407-13-527. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 85.Behm B, Di Fazio P, Michl P, Neureiter D, Kemmerling R, Hahn EG, Strobel D, Gress T, Schuppan D, Wissniowski TT. Additive antitumour response to the rabbit VX2 hepatoma by combined radio frequency ablation and toll like receptor 9 stimulation. Gut. 2016;65:134–143. doi: 10.1136/gutjnl-2014-308286. [DOI] [PubMed] [Google Scholar]
- 86.Chen Y, Sun R. Toll-like receptors in acute liver injury and regeneration. Int Immunopharmacol. 2011;11:1433–1441. doi: 10.1016/j.intimp.2011.04.023. [DOI] [PubMed] [Google Scholar]