

Abstract
The pharmacokinetic profile of gallocatechin-7-gallate (J10688) was studied in rats after intravenous administration. Male and female Sprague-Dawley (SD) rats received 1, 3, and 10 mg/kg (i.v.) of J10688 and plasma drug concentrations were determined by a high performance liquid chromatography-mass spectrometry (LC–MS) method. The pharmacokinetic software Data Analysis System (Version 3.0) was used to calculate the pharmacokinetic parameters. For different i.v. doses of J10688, the mean peak plasma concentration (C0) values ranged from 11.26 to 50.82 mg/L, and mean area under the concentration-time curve (AUC0–t) values ranged from 1.75 to 11.80 (mg·h/L). J10688 lacked dose-dependent pharmacokinetic properties within doses between 1 and 10 mg/kg, based on the power model. The method developed in this study was sensitive, precise, and stable. The pharmacokinetic properties of J10688 in SD rats were shown to have rapid distribution and clearance values. These pharmacokinetic results may contribute to an improved understanding of the pharmacological actions of J10688.
KEY WORDS: Gallocatechin-7-gallate, LC–MS, Pharmacokinetics, Dose proportionality, Non-compartment model
Graphical abstract
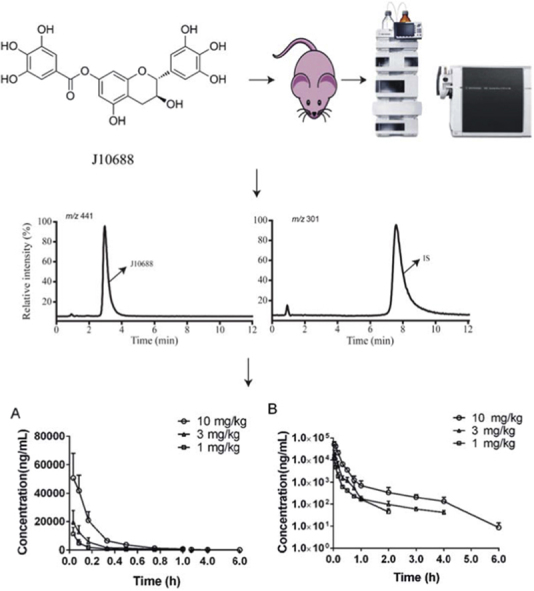
A simple, selective, and sensitive LC–MS method was developed and validated. Pharmacokinetic study of gallocatechin-7-gallate (J10688) in rats was carried out. The pharmacokinetic properties of J10688 in SD rats were characterized as quick distribution and clearance.
1. Introduction
Pithecellobium clypearia Benth., a member of Mimosaceae family, is a well-known and widely used traditional Chinese medicine1. It is mostly distributed in the South of China, such as Sichuan, Yunnan and Guangdong provinces. For many years, P. clypearia has been used in the treatment of respiratory tract diseases2, 3, 4. It is recorded in the Chinese Pharmacopoeia 2010 that, the aqueous extract of the leaves and twigs of P. clypearia has been used for the treatment of upper respiratory tract infections, laryngitis, pharyngitis, acute gastroenteritis, acute tonsillitis, and bacterial dysentery5, 6, 7. Until now, various flavonoids have been found in P. clypearia, most of which have anti-viral properties8. It has been confirmed that aqueous extracts from P. clypearia exhibit anti-influenza activity both in vitro and in vivo. The cytopathic effects induced by influenza virus in cells and pneumonia in mice were inhibited significantly by this aqueous extract9. (–)-Epigallocatechin-7-gallate isolated from P. clypearia exhibited a strong inhibition on the T lymphocytes proliferation induced by concanavalin A (ConA) with an IC50 of 4.4 μmol/L10.
Our previous study has shown that three compounds in the ethyl acetate extract of P. clypearia exhibited influenza virus neuraminidase inhibitory activity and anti-inflammatory activity in vitro11, 12, 13. One of the potent compounds, gallocatechin-7-gallate (J10688) (Fig. 1), a novel polyphenol flavonoid with anti-influenza viral activity11, has been selected as the candidate compound for further research. In the previous study, we found that J10688 could inhibit influenza virus replication in human lung adenocarcinoma cell (A549), down-regulated the mRNA expression of influenza hemagglutinin gene, significantly down-regulated the influenza viral nucleoprotein and M2 ion channel expression, and also inhibited the secretion of inflammatory factors (data not published). To better understand the pharmacological actions of J10688, we presently performed pharmacokinetic research with this compound.
Figure 1.

The chemical structures of (A) J10688 and (B) quercetin (IS).
In this study, a simple, selective, and sensitive high performance liquid chromatography–mass spectrometry (LC–MS) method for the quantification of J10688 in rat plasma was developed, and the pharmacokinetic properties of this agent were studied after intravenous administration.
2. Materials and methods
2.1. Chemicals and reagents
J10688 had been successfully separated and identified in the previous work11. Samples purified and used presently had a purity of 99%. Quercetin (internal standard, IS) was purchased from the National Institutes for Food and Drug Control (Beijing, China).
Acetonitrile (LC–MS grade), methanol (HPLC grade) were obtained from J. T. Baker (Phillipsburg, NJ, USA), and formic acid (HPLC grade) bought from TEDIA Company (Fairfield, OH, USA). Pure water was purchased from Wahaha Company (Hangzhou, China). All the other chemical reagents were of analytical grade or higher level.
2.2. Instrumentation
The Agilent 1200 liquid chromatography-6100 mass spectrometer was employed for data acquisition and data post-processing. The system was equipped with G1311A quaternary gradient pump, G1379B vacuum degasser, G1329A auto-sampler, G1316A column oven and an ESI ion source.
2.3. Chromatographic conditions
The analytical column was Agilent Zorbax SD-C18 (100 mm×2.1 mm, 3.5 μm). The mobile phase was composed of acetonitrile-water (0.1% formic acid) (23:77, v/v). Injection volume was 20 μL, the total run time was 12 min and the flow rate was 0.3 mL/min. The column temperature was maintained at 35 °C.
The mass spectrometer was operated in the negative scan mode. The conditions of ESI source were as follows: drying gas flow was set at 10 L/min, drying gas temperature was 350 °C, nebulizer pressure was 35.0 psig and capillary voltage was 3000 V. The ESI was performed using nitrogen to assist nebulization. The typical compound parameters, fragmentor voltage and gain value, were set at 80 V and 1.5, respectively. The MS detector was operated in selective ion monitoring (SIM) mode using the quantification ions [M–H]– at m/z 441.0 for J10688 and m/z 301.0 for quercetin (IS).
2.4. Animals
Male and female Sprague-Dawley (SD) rats of 6–8 weeks old with weighing between 200 and 240 g were purchased from the Vital River Lab Animal Technology Co., Ltd. (Beijing, No. SCXK 2012-0001). All animals received care according to the Guide for the Care and Use of Laboratory Animals.
2.5. Preparation of stock solutions and standards
Primary stock solutions of J10688 and IS were prepared separately by dissolving analytes in methanol to obtain the concentration of 1 mg/mL and stored at −40 °C. Stock solutions were diluted to working solutions with methanol for use. Quality control (QC) samples were prepared by spiking corresponding standard working solutions with 135 μL of blank rat plasma at concentration of 50, 500 and 1500 ng/mL. To construct the plasma calibration curves, a series of working standard solutions at the concentrations of 10, 50, 100, 250, 500, 1000 and 2000 ng/mL J10688 were similarly prepared.
2.6. Preparation of plasma samples
Rat plasma (100 μL) and 10 μL of IS solution (quercetin, 500 ng/mL) were mixed, and 1000 μL ethyl acetate was added to extract the analytes from the plasma. After vortexing for 5 min, the mixture was centrifuged at 13,000 rpm for 10 min. Then the supernatant was transferred into another tube carefully and evaporated to dryness under a gentle flow of nitrogen gas. The residue was reconstituted in 50 μL 50% methanol–water solutions. After centrifugation, the supernatant was injected into LC–MS system for analysis.
2.7. Method validation
According to the Industrial Guidelines for Bioanalytical Method Validation from the US FDA the current LC-MS method was validated in biological samples of specificity, linearity, the lower limit of quantification (LLOQ), precision, accuracy, matrix effect, extraction recovery and stability.
2.7.1. Specificity
The specificity of the established method was evaluated by comparing the chromatograms of blank plasma from no less than six individual rats with the plasma sample 20 min after intravenous injection of J10688 and a blank rat plasma sample spiked with J10688 and IS. The purpose was to ensure that there is no endogenous interference at the retention times of J10688 and IS.
2.7.2. Linearity and sensitivity
The calibration standards of J10688 were prepared by spiking blank rat plasma with the standard solution to the concentrations ranged 10–2000 ng/mL. Calibration curves were established by plotting the peak area ratio of J10688 to IS (Y-axis) versus the nominal concentration of J10688 (X-axis), and fitted with by least square weighted linear regression. The lower limit of quantification (LLOQ) was defined as the lowest concentration of which the detected response reaches 10-fold of signal-to-noise ratio (10 S/N).
2.7.3. Accuracy and precision
The precision of the method was defined as the relative standard deviation (RSD) calculated from replicate measurements of QCs. Intra-day and inter-day precision were determined by measuring samples at three concentrations with five QC samples per level (50, 250 and 1500 ng/mL). The accuracy of the method was characterized as the RE (%), which was the mean of the replicate measurements of QCs from the theoretical values. It was acceptable when the RE (%) and RSD (%) values were less than 15%.
| (1) |
2.7.4. Extraction recovery and matrix effect
Recovery was calculated by comparing the peak area obtained from an extracted sample with that obtained from unextracted standard solution prepared with the same solvent.
The matrix effects were determined by comparing the peak areas obtained from samples where the extracted matrix was spiked with standard solutions to those of the pure samples prepared in mobile phase containing equivalent amounts of the analyte at three concentrations.
2.7.5. Stability
The stability of J10688 in rat plasma was evaluated at three levels of QC samples. Short-term stability was tested after QC samples had been kept at 25 °C for 4 h. And in the long-term stability, the QC samples were stored at −40 °C for four weeks before detection. QC samples were also analyzed following three freeze/thaw cycles. In each freeze/thaw cycle, the samples were frozen for 24 h at −20 °C and thawed to ambient temperature. At last, the processed samples were stored in the autosampler for 24 h before analyzed. Samples were considered to be stable if the deviation from the nominal concentration was within ±15.0%.
2.8. Pharmacokinetic study
J10688 was dissolved in 60% hydroxypropyl-β-cyclodextrin prepared with normal saline, vortexed until the solution became transparent. According to the drug non-clinical pharmacokinetic guidelines14, three dose groups (1, 3 and 10 mg/kg) had been set up in this study. The high dose group (10 mg/kg) is the maximum tolerated dose, and the low-dose group (1 mg/kg) is the minimum effective dose. The middle dose group (3 mg/kg) was the logarithmic midpoint between 10 and 1 mg/kg. After intravenous administration of J10688, the blood samples were collected into heparinized tubes by retro-orbital puncture at 0.033, 0.083, 0.167, 0.333, 0.5, 0.75, 1, 2, 4 and 6 h postinjection. After centrifuged at 5000 rpm for 10 min, 100 μL plasma were achieved. These samples were stored at −80 °C before analysis.
2.9. Parameters calculation
The software DAS 3.0 pharmacokinetic program (Chinese Pharmacology Society) was used to calculate the pharmacokinetic parameters and a non-compartment model analysis was employed.
3. Results and discussion
3.1. LC–MS optimization
The chromatographic conditions were optimized through several trials using various columns and mobile phases to achieve good chromatographic separation of J10688 and IS. In this section of work, the HPLC–DAD was employed for optimizing the chromatographic condition. The chromatograms under the wavelengths of 280 nm and 256 nm (Supplementary Fig. 1) were selected to monitor the drug and internal standard, respectively. To make the LC condition transferable to the LC–MS method, the identical HPLC instrument was employed. Three different types and sizes of C18 columns in our laboratory, Eclipse Plus C18 (100 mm×2.1 mm, 3.5 μm), Eclipse XDB C18 (150 mm×4.6 mm, 5 μm) and ZORBAX SB C18 (100 mm×2.1 mm, 3.5 μm) were selected as the candidate columns. Different acetonitrile ratios in mobile phase ranged 20%–40% were also investigated. As shown in Supplementary Figs. 2–4, the ZORBAX SB C18 (100 mm×2.1 mm, 3.5 μm) yielded optimal column efficiency and peak shape for the compounds. The drug and the internal standard could be separated ideally in all three columns under the acetonitrile ratios between 20% and 30%. Further, we intended to set retention time of the drug peak longer than 2.0 min in order to avoid interference from these polar endogenous metabolites which might cause interfering ion suppression or enhancement to the drug ion. Finally, the acetonitrile–water (0.1% formic acid) (23:77, v/v) was set as the mobile phase composition.
To achieve the higher signal response, both positive and negative scan modes were investigated. The results showed that the target molecule could give better ion intensity in the negative mode than in the positive mode. Based on the chemical structures showed in Fig. 1, we supposed that this flavane tends to dissociate and gives several neutral losses due to its galloyl and hydroxyl groups in the positive mode after protonation. In contrast, this flavane maintained stability after deprotonation in negative mode due to the existence of the conjugated system and electron-donating groups. In this condition, IS could be ionized efficiently as well. In full-scan mass spectra, J10688 and IS predominantly formed [M–H]– ions at m/z 441.0 and m/z 301.0, respectively (Fig. 2).
Figure 2.

Full scan mass spectra of (A) J10688 with the m/z 441.0 [M–H]– and (B) IS with the m/z 301.0 [M–H]–.
3.2. Method validation
3.2.1. Specificity
The specificity of the method was evaluated by comparing the chromatograms of blank plasma, plasma sample spiked with J10688 and IS, and a real rat plasma sample obtained 20 min after intravenous administration of J10688 at 10 mg/kg (Fig. 3). No endogenous or other interfering peaks were observed from plasma at the retention time of J10688 and IS, indicating that the method was specific.
Figure 3.

Typical chromatograms of J10688 and IS. (A) Blank rat plasma sample; (B) blank rat plasma sample spiked with J10688 (10 ng/mL) and IS (50 ng/mL); (C) rat plasma sample at 60 min after intravenous injection of 10 mg/kg J10688 spiked with IS (50 ng/mL).
3.2.2. Linearity and LLOQ
Each calibration curve was constructed by plotting the peak area ratios (y) of the analyte (J10688) to the IS versus the nominal concentration (x) of the analyte (10–2000 ng/mL for J10688). The calibration curve showed good linearity over the plasma concentration with correlation coefficients (r) ranging from 0.9993 to 0.9999. The LLOQ, defined as the lowest concentration detected, was 10 ng/mL with an RSD of 2.07% for J10688 in rat plasma. Accuracy was within ±20%, precision was ≤20%. These results confirmed that the method was sensitive enough for the pharmacokinetic study of J10688 in rats.
3.2.3. Accuracy and precision
The intra- and inter-day precision and accuracy were calculated by analyzing three concentration levels of QC samples. The results are exhibited in Table 1. The intra- and inter-day precision for J10688 was less than 7.98%, and the accuracy for J10688 was within 100.15% and 107.27%, respectively. These results indicated that the method was precise and accurate for determination of J10688 and IS in rat plasma.
Table 1.
Intra- and inter-day precision and accuracy of J10688 in the rat plasma.
| Spiked conc. (ng/mL) | Measured concentration (ng/mL)a | RE (%) | RSD (%) | |
|---|---|---|---|---|
| Intra-day | 50 | 53.64±1.39 | 107.27 | 2.59 |
| 250 | 251.67±5.26 | 100.67 | 2.09 | |
| 1500 | 1502.19±16.49 | 100.15 | 1.10 | |
| Inter-day | 50 | 53.21±4.24 | 106.42 | 7.98 |
| 250 | 251.78±9.37 | 100.71 | 3.72 | |
| 1500 | 1505.57±50.34 | 100.37 | 3.34 |
Data are expressed as mean±SD, n =5.
3.2.4. Extraction recovery and matrix effect
The extraction recovery and matrix effect were evaluated by determining QC samples at 50, 250 and 1500 ng/mL with five replicates. Data are shown in Table 2. The extraction recoveries of J10688 at three concentration levels ranged from 99.65% to 102.91% and the IS value was 72.19%. The matrix effect values for the analyte J10688 were 99.13%, 96.42% and 97.12% for the three QC concentration samples, respectively. These data demonstrate that acetonitrile is an appropriate and feasible medium for J10688 and IS extraction. No significant ion suppression or enhancement from plasma matrix was observed under the present experimental conditions.
Table 2.
Recovery and matrix effect of J10688 and IS in rat plasma (n =5).
| Analyte | Spiked conc. (ng/mL) | Recovery (%) | Matrix effect (%) |
|---|---|---|---|
| J10688 | 50 | 102.91 | 99.13 |
| 250 | 102.44 | 96.42 | |
| 1500 | 99.65 | 97.12 | |
| IS | 500 | 72.19 | 91.94 |
3.2.5. Stability
To test the possible effect of all experienced conditions for samples. QC samples of three concentrations (50, 250 and 1500 ng/mL) were stored under different conditions, including sample handling, processing and analysis. The results were presented in Table 3, indicating that the samples were stable for 4 h at 25 °C and 24 h in the autosampler (25 °C), and no significant degradation occurred after three freeze-thaw cycles and long term storage (–40 °C for four weeks). The RSD (%) values were less than 6.75% for all QC samples, which demonstrated the reproducibility of this method for J10688.
Table 3.
Stability of J10688 in rat plasma.
| Condition | Spiked conc. (ng/mL) | Measured conc. (ng/mL)a | RSD (%) | RE (%) |
|---|---|---|---|---|
| Three freeze/thaw cycles | 50 | 50.04±0.54 | 1.07 | 100.08 |
| 250 | 252.75±11.50 | 4.55 | 101.10 | |
| 1500 | 1481.97±29.58 | 2.00 | 98.80 | |
| Short-term (25 °C for 4 h) | 50 | 51.29±0.43 | 0.84 | 102.58 |
| 250 | 257.05±3.52 | 1.37 | 102.82 | |
| 1500 | 1489±28.88 | 1.94 | 99.27 | |
| Autosampler for 24 h (25 °C) | 50 | 46.7±2.87 | 6.15 | 93.40 |
| 250 | 253.02±5.09 | 2.01 | 101.21 | |
| 1500 | 1520.68±24.80 | 1.63 | 101.33 | |
| Long-term (−40 °C for four weeks) | 50 | 50.16±3.38 | 6.75 | 100.32 |
| 250 | 246.43±13.53 | 5.49 | 98.57 | |
| 1500 | 1469.50±53.41 | 3.63 | 97.97 |
Data are expressed as mean±SD, n =5.
3.3. Pharmacokinetic studies
The presently developed LC–MS method was successfully applied to the pharmacokinetic study of J10688 in rats. Plasma drug concentration-time curves of J10688 were obtained after three different i.v. doses. The results are displayed in Fig. 4. The main pharmacokinetic parameters of J10688 were calculated by a non-compartment model analysis (Table 4). After injection of J10688, it was rapidly distributed in the blood. Time for half-life elimination (t1/2) was 1.32, 1.62 and 0.53 h, respectively, indicating that J10688 was relatively quickly eliminated from plasma with a short elimination half-life. The maximum plasma drug concentration (C0) of J10688 ranged from 11.26–50.83 g/L, and values of AUC0–t were 11806.62±1651.11, 3678.648±1235 and 1752.29±538.72 μg·h/L, respectively.
Figure 4.

Mean plasma concentration-time curves of J10688 in rats. (A) Mean plasma concentration-time curves of J10688 (i.v., n =6); (B) semi-logarithmic axis mean plasma concentration-time curves of J10688 (i.v., n =6).
Table 4.
Pharmacokinetic parameters of J10688 after intravenous administration in rat.
| Parameter | Unit | J10688 (mg/kg)a |
||
|---|---|---|---|---|
| 10 | 3 | 1 | ||
| AUC0–t | μg·h/L | 11,806.62±1651.11 | 3678.648±1235 | 1752.29±538.72 |
| AUC0–∞ | μg·h/L | 11,929.63±1536.63 | 3773.395±1223.312 | 1788.694±533.7 |
| MRT0–t | h | 0.366±0.122 | 0.283±0.083 | 0.217±0.062 |
| MRT0–∞ | h | 0.452±0.159 | 0.495±0.339 | 0.275±0.094 |
| t1/2 | h | 1.316±0.884 | 1.619±1.268 | 0.526±0.123 |
| Vd | L/kg | 1.669±1.19 | 2.128±2.151 | 0.457±0.161 |
| CL | L·kg/h | 0.85±0.112 | 0.864±0.265 | 0.607±0.195 |
| C0 | μg/L | 50,827.117±17151.46 | 19,528.1±8253.442 | 11,264.783±4684.666 |
Data are expressed as mean±SD, n =6.
Dose proportionality was assessed using the power model in a manner analogous to a published method15, 16. Dose correlation results showed that, in doses ranging from 1 to 10 mg/kg, the slopes (β1) of ln(AUC0–t), ln(AUC0–∞) and ln(C0) to ln(dose) were 0.8456, 0.8407 and 0.6752,respectively (Table 5). According to the power model17, 18, 90% confidence intervals were 0.7204–0.9708 for AUC0–t, 0.7206–0.9608 for AUC0–∞, and 0.5083–0.8422 for C0, which were out of the range 0.9031–1.0969 and 0.8451–1.155. The relationships between ln (Exposure) and ln (Dose) are shown in Fig. 5. The regression analysis indicated less perfect linearity of the kinetics of J10688 after intravenous administration.
Table 5.
Assessment of dose proportionality of J10688 based on power model.
| Parameter | Acceptance range | β1 | 90% Confidence interval |
|---|---|---|---|
| AUC0–t | 0.9031–1.0969 | 0.8456 | 0.7204–0.9708 |
| AUC0–∞ | 0.9031–1.0969 | 0.8407 | 0.7206–0.9608 |
| C0 | 0.8451–1.155 | 0.6752 | 0.5083–0.8422 |
β1: the slopes of lnAUC0–t, lnAUC0–∞ and lnC0 to lnDose.
Figure 5.

Relationship between lnPK and lnDose. (A): AUC0–t; (B): AUC0–∞; (C): C0. The blank dots are the individual observed values; the full lines are the fitted values calculated by the power model and the dotted lines are the 90% confidence interval.
J10688 showed relatively quick elimination rates in rat after tail vein injection. The drug disappeared completely 6 h after the high dose (10 mg/kg) after 2 h after the low (1 mg/kg) dose. The pharmacokinetic parameters showed that the exposure level (characterized with AUC) correlated with certain doses, and the dose of 10 mg/kg J10688 demonstrated non-linearity with pharmacokinetics parameters. It is likely that this high dose exceeds maximum capacity of a drug metabolic or transporter mechanism. When specific transporters or the metabolic enzymes become saturated due to the high drug concentration level, biochemical elimination processes like active transport in kidney tubules or phase I/II metabolism slow down19, 20, 21. Unlike the case for first-order kinetic processes, the pharmacokinetic parameters of J10688 (half-life and clearance) are not constant, but varied with different doses. Consistent with this conclusion, the AUC and C0 values were not proportional to dose. All of these patterns indicate the existence of non-linear pharmacokinetic processes for J10688.
4. Conclusions
This study is the first to evaluate the pharmacokinetics of gallocatechin-7-gallate, an active anti-influenza virus candidate isolated from P. clypearia. A sensitive, rapid and reliable LC–MS method was developed to analyze J10688 in rat plasma. The established method was successfully applied to the pharmacokinetic study of J10688 in SD rats following i.v. administration of three doses. These results will provide more information about the pharmacokinetic profiles of J10688 for its subsequent preclinical studies and guide the pharmacokinetic study of structurally similar compounds.
Acknowledgments
This work was supported by Beijing Natural Science Foundation (7152103), the National Great Science and Technology Projects (2014ZX09507003-002 and 2012ZX09301002-2013HXW-11) and the International Collaboration Project (2011DFR31240).
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.apsb.2015.10.001.
Contributor Information
Ailin Liu, Email: liuailin@imm.ac.cn.
Guanhua Du, Email: dugh@imm.ac.cn.
Appendix A. Supplementary material
Supplementary material: Fig. 1 The ultraviolet spectra of (A) J10688 and (B) internal standard (IS).
Supplementary material: Fig. 2 The chromatograms of different acetonitrile ratios in mobile phase ranged 20–40% using Eclipse Plus C18 column (100 mm×2.1 mm, 3.5 μm), 0.3 mL/min, 2 μL. Sample: the blank plasma spiked with the drug and IS.
Supplementary material: Fig. 3 The chromatograms of different acetonitrile ratios in mobile phase ranged 25–40% using Eclipse XDB C18 column (150 mm×4.6 mm, 5 μm) 1.0 mL/min, 10 μL. Sample: the blank plasma spiked with the drug and IS.
Supplementary material: Fig. 4 The chromatograms of different acetonitrile ratios in mobile phase ranged 25–30% using ZORBAX SB C18 column (100 mm×2.1 mm, 3.5 μm) 0.3 mL/min, 2 μL. Sample: the blank plasma spiked with the drug and IS.
References
- 1.Li Y., Ooi L.S., Wang H., But P.P., Ooi V.E. Antiviral activities of medicinal herbs traditionally used in southern mainland China. Phytother Res. 2004;18:718–722. doi: 10.1002/ptr.1518. [DOI] [PubMed] [Google Scholar]
- 2.Li Y., Leung K.T., Yao F., Ooi L.S., Ooi V.E. Antiviral flavans from the leaves of Pithecellobium clypearia. J Nat Prod. 2006;69:833–835. doi: 10.1021/np050498o. [DOI] [PubMed] [Google Scholar]
- 3.Xie C.Y., Lin L.W. Study on the chemical constituents of Pithecellobium clypearia. J Chin Med Mater. 2011;34:1060–1062. [PubMed] [Google Scholar]
- 4.Li Y.L., LK Su M.X., Leung K.T., Chen Y.W., Zhang Y.W. Studies on antiviral constituents in stems and leaves of Pithecellibium clypearia. China J Chin Mater Med. 2006;31:397–400. [PubMed] [Google Scholar]
- 5.Su M.X., Tang Z.Y., Huang W.H., Li Y.L., Cen Y.Z. Studies on the chemical comstituents of Pithecellobium clypearia. J Chin Med Mater. 2009;32:705–707. [PubMed] [Google Scholar]
- 6.Bao L., Yao X., Xu J., Guo X., Liu H., Kurihara H. Effects of Pithecellobium clypearia Benth extract and its main components on inflammation and allergy. Fitoterapia. 2009;80:349–353. doi: 10.1016/j.fitote.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 7.Li L., Peng Y., Hu C., Liu C., Liu Q., Li L., Wu J., Song S. Antioxidant activity of chemical constituents isolated from Pithecellobium clypearia. Rec Nat Prod. 2015;9:284–291. [Google Scholar]
- 8.Kujumgiev A., Tsvetkova I., Serkedjieva Y., Bankova V., Christov R., Popov S. Antibacterial, antifungal and antiviral activity of propolis of different geographic origin. J Ethnopharmacol. 1999;64:235–240. doi: 10.1016/s0378-8741(98)00131-7. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Z.X., Huang J.Z., Li P.B. Experimental study on anti-viral activity of the water extract from Pitecollobium clypearia Benth. China TropMed. 2008;1:30–34. [Google Scholar]
- 10.Guo X.Y., Wang N.L., Bo L., Li Y.H., Xu Q., Yao X.S. Chemical constituents from Pithecellobium clypearia and their effects on T lymphocytes proliferation. J Chin Pharm Sci. 2007;16:208–213. [Google Scholar]
- 11.Kang J., Liu C., Wang H., Li B., Li C., Chen R. Studies on the bioactive flavonoids isolated from Pithecellobium clypearia Benth. Molecules. 2014;19:4479–4490. doi: 10.3390/molecules19044479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma S.G., Hu Y.C., Yu S.S., Zhang Y., Chen X.G., Liu J. Cytotoxic triterpenoid saponins acylated with monoterpenic acid from Pithecellobium lucidum. J Nat Prod. 2008;71:41–46. doi: 10.1021/np0703794. [DOI] [PubMed] [Google Scholar]
- 13.Maldini M., Montoro P., Hamed A.I., Mahalel U.A., Oleszek W., Stochmal A. Strong antioxidant phenolics from Acacia nilotica: profiling by ESI-MS and qualitative-quantitative determination by LC–ESI-MS. J Pharm Biomed Anal. 2011;56:228–239. doi: 10.1016/j.jpba.2011.05.019. [DOI] [PubMed] [Google Scholar]
- 14.Gordi T., Huong D.X., Hai T.N., Nieu N.T., Ashton M. Artemisinin pharmacokinetics and efficacy in uncomplicated-malaria patients treated with two different dosage regimens. Antimicrob AgentsChemother. 2002;46:1026–1031. doi: 10.1128/AAC.46.4.1026-1031.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith B.P., Vandenhende F.R., DeSante K.A., Farid N.A., Welch P.A., Callaghan J.T. Confidence interval criteria for assessment of dose proportionality. Pharm Res. 2000;17:1278–1283. doi: 10.1023/a:1026451721686. [DOI] [PubMed] [Google Scholar]
- 16.Song J., Zhang W., Sun J., Xu X., Zhang X., Zhang L. Pharmacokinetic study of salvianolic acid D after oral and intravenous administration in rats. Acta Pharm Sin B. 2015;5:246–253. doi: 10.1016/j.apsb.2015.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hummel J., McKendrick S., Brindley C., French R. Exploratory assessment of dose proportionality: review of current approaches and proposal for a practical criterion. PharmStat. 2009;8:38–49. doi: 10.1002/pst.326. [DOI] [PubMed] [Google Scholar]
- 18.Gordi T., Huong D.X., Hai T.N., Nieu N.T., Ashton M. Artemisinin pharmacokinetics and efficacy in uncomplicated-malaria patients treated with two different dosage regimens. Antimicrob Agents Chemother. 2002;46:1026–1031. doi: 10.1128/AAC.46.4.1026-1031.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jambhekar S.S., Breen P.J. Pharmaceutical Press; London: 2009. Basic pharmacokinetics. [Google Scholar]
- 20.Boroujerdi M. McGraw-Hill Medical Pub.; New York: 2011. Pharmacokinetics: principles and applications. [Google Scholar]
- 21.Velez de Mendizabal N., Jackson K., Eastwood B., Swanson S., Bender D.M., Lowe S. A population PK model for citalopram and its major metabolite, N-desmethyl citalopram, in rats. J Pharmacokinet Pharmacodyn. 2015;42:721–733. doi: 10.1007/s10928-015-9448-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material: Fig. 1 The ultraviolet spectra of (A) J10688 and (B) internal standard (IS).
Supplementary material: Fig. 2 The chromatograms of different acetonitrile ratios in mobile phase ranged 20–40% using Eclipse Plus C18 column (100 mm×2.1 mm, 3.5 μm), 0.3 mL/min, 2 μL. Sample: the blank plasma spiked with the drug and IS.
Supplementary material: Fig. 3 The chromatograms of different acetonitrile ratios in mobile phase ranged 25–40% using Eclipse XDB C18 column (150 mm×4.6 mm, 5 μm) 1.0 mL/min, 10 μL. Sample: the blank plasma spiked with the drug and IS.
Supplementary material: Fig. 4 The chromatograms of different acetonitrile ratios in mobile phase ranged 25–30% using ZORBAX SB C18 column (100 mm×2.1 mm, 3.5 μm) 0.3 mL/min, 2 μL. Sample: the blank plasma spiked with the drug and IS.