

Abstract
Human 5-lipoxygenase (5-LOX) is a well-validated drug target and its inhibitors are potential drugs for treating leukotriene-related disorders. Our previous work on structural optimization of the hit compound 2 from our in-house collection identified two lead compounds, 3a and 3b, exhibiting a potent inhibitory profile against 5-LOX with IC50 values less than 1 µmol/L in cell-based assays. Here, we further optimized these compounds to prepare a class of novel pyrazole derivatives by opening the fused-ring system. Several new compounds exhibited more potent inhibitory activity than the lead compounds against 5-LOX. In particular, compound 4e not only suppressed lipopolysaccharide-induced inflammation in brain inflammatory cells and protected neurons from oxidative toxicity, but also significantly decreased infarct damage in a mouse model of cerebral ischemia. Molecular docking analysis further confirmed the consistency of our theoretical results and experimental data. In conclusion, the excellent in vitro and in vivo inhibitory activities of these compounds against 5-LOX suggested that these novel chemical structures have a promising therapeutic potential to treat leukotriene-related disorders.
KEY WORDS: 5-Lipoxygenase, 5-LOX inhibitors, Pyrazole derivatives, Leukotrienes-related diseases, In vivo, Benzo-fused heterocycle, Ischemic incults, Brain inflammation
Graphical abstract
Previous work on structural optimization of the hit compound 2 from our in-house collection identified two lead compounds, 3a and 3b, exhibiting a potent inhibitory profile against 5-LOX with IC50 values less than 1 µmol/L in cell-based assays. Here, we further optimized these compounds to prepare a class of novel pyrazole derivatives by opening the fused-ring system. Their excellent in vitro and in vivo inhibitory activities of these compounds against 5-LOX suggested that these novel chemical structures have a promising therapeutic potential to treat leukotriene-related disorders.

1. Introduction
5-Lipoxygenase (5-LOX) is the key enzyme that metabolizes arachidonic acid (AA) into the bioactive leukotrienes (LTs), which are considered to be potent mediators of inflammatory responses1, 2. Accumulated evidence suggested that LTs play important roles in the development of allergic diseases such as asthma3, 4, 5, various inflammatory disorders such as rheumatoid arthritis and cardiovascular diseases5, 6. LTs also contribute to tumorigenesis including of the prostate, pancreatic cancer and leukemia7, 8. Moreover, 5-LOX expression and its enzymatic activity are increased after focal cerebral ischemia and 5-LOX plays an important role in the pathogenesis of cerebral ischemia9. Therefore, pharmacological interference with the 5-LOX pathway to down-regulate the formation of LTs is a promising therapeutic strategy for LT-related diseases.
In search of pharmacological agents that suppress the biosynthesis of LTs, different types of small molecular inhibitors of 5-LOX have been developed in the past two decades, such as redox inhibitors, iron-chelator agents and non-redox competitive inhibitors10, 11. To date, despite considerable efforts devoted to the development of efficient and safe drugs that target the 5-LOX pathway, only one 5-LOX inhibitor, Zileuton (1, Scheme 1), has been approved for asthma treatment12, although it still exhibits several drawbacks, including liver toxicity and unfavorable pharmacokinetic profile with a short half-life3. Therefore, there is an urgent need to develop novel drugs without the disadvantages of previous 5-LOX inhibitors10. In fact, considerable efforts have been directed recently toward the identification of higher efficacy 5-LOX inhibitors to reduce side effects through diverse medicinal chemistry approaches12, such as lead identification13, scaffold-hopping14, structure-based drug design15, dynamic modeling16, and structural optimization17.
Scheme 1.

Design of novel 1,5-disubstituted pyrazole-3-carboxamines by fused-ring cracking.
In our previous studies we identified a series of novel indole derivatives with potent inhibitory activities against 5-LOX by structural optimization for the hit compound 2, which originated from our in-house collection18. Among them, compounds 3a and 3b exhibited the most potent inhibitory profile with IC50 values of less than 1 µmol/L in the cell-based assay, and thus seem to be the promising lead compounds for treating LT-related diseases18. Here, we further optimized this class of compounds in an attempt to improve their druggability by splitting the indole ring to reconstruct the core scaffold and introducing a 5-LOX inhibitor pharmacophore (3,5-di-tert-butyl-4-hydroxyphenyl)19, 20, 21 to form substituted pyrazole-3-carboxamine derivatives (Scheme 1). Several optimized compounds displayed more potent inhibitory activities than the lead compounds 3a, 3b and the reference drug 1 against 5-LOX in peritoneal polymorphonuclear leukocytes (PMNLs), human whole blood (HWB), and enzymatic assays. Further investigations indicated that compound 4e not only effectively suppressed LPS-induced inflammation in brain inflammatory cells and protected neurons from oxidative toxicity in vitro, but also significantly decreased infarct volumes in a mouse model of cerebral ischemia.
2. Results and discussion
2.1. Chemistry
In the current studies, novel pyrazole-scaffold derivatives have been explored through a fused-ring cracking strategy with our previous lead compounds to further optimize the structural features and improve their in vitro and in vivo biological effects. As depicted in Scheme 1, compounds (4a–k, 5a–j and 6a–b, Table 1) were obtained by splitting the indole ring of the lead compounds (3a and 3b) to reconstruct the pyrazole scaffold and simultaneously introduce the 3,5-di-tert-butyl-4-hydroxyphenyl segment. Firstly, by introducing a 4-aminosulfonylphenyl group onto the 1-N position of the pyrazole ring, we obtained compounds 4a–k by introducing different R2 groups with various steric, electron-donating groups, electron-withdrawing groups and alkyl groups. Then compounds 5a–j were obtained by replacing the 4-aminosulfonylphenyl group of compounds 4a–j with a p-methylsulfonylphenyl group. Finally, we introduced a fluorine group to the R1 position to explore the biological influences against 5-LOX. As shown in Scheme 2, key intermediate 9 was synthesized by the nitration of the commercially available 2,6-di-tert-butylphenol (7) with aqueous HNO3 in hexane to give 2,6-di-tert-butyl-4-nitrophenol (8), which was then reduced with Sn/HCl under reflux conditions with a 70%–80% yield. Another key intermediate, 4-aminosulfonylphenylhydrazine hydrochloride (12), was prepared according to a similar method reported previously22, 23. Commercially available sulfanilamide (10) was converted into its diazonium salt by reaction of the amine with in situ generated nitrous acid. The diazonium salt 11 was subsequently reduced to hydrazine 12 with SnCl2 in acidic medium. The synthetic strategy for the target compounds are shown in Scheme 3. The important pyrazole intermediates 17a–k were synthesized starting from commercially available substituted actones, which were firstly reacted with diethyl oxalate in the presence of NaH to give β-diketone intermediates 14a–k, followed by reacting with 4-aminosulfonylphenylhydrazine hydrochloride (12) in ethanol under reflux condition for 12 h. Compounds 17a–k were also produced by a one-pot two-step process. Initially, Na was treated with absolute ethanol at room temperature until it was completely dissolved, substituted acetones (13a–k) and diethyl oxalate were then added, and the resulting mixture was refluxed for 15 h. After the reaction mixture was cooled to ambient temperature, equivalent HCl solution and the corresponding hydrazines were added to the crude reaction mixture, which was further heated to reflux for another 12 h to give the desired compounds. Subsequently, hydrolysis of compounds 17a–k gave the corresponding acids (20a–k), which then underwent a simple condensation reaction with the key intermediate 9 to give the target 1,5-disubstituted pyrazole-3-carboxamines 4a–k. Other desired compounds, 5a–j and 6a–b were also prepared using a similar process to 4a–k.
Table 1.
Structures and inhibitory activity against 5-LOX in rat peritoneal polymorphonuclear leukocytes (PMNLs). 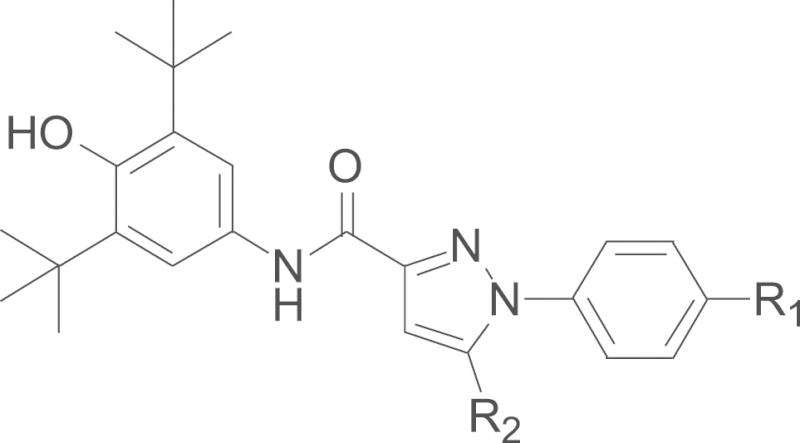
| Compd. | R1 | R2 | Inhibition at 5 μmol/L (%)a |
|---|---|---|---|
| 4a | SO2NH2 | 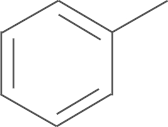 |
55.7 |
| 4b | SO2NH2 | 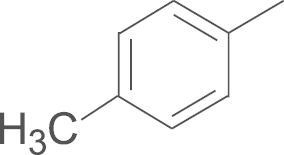 |
65.7 |
| 4c | SO2NH2 | 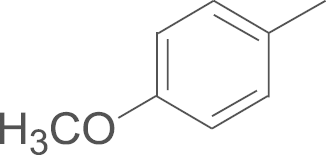 |
74.9 |
| 4d | SO2NH2 | 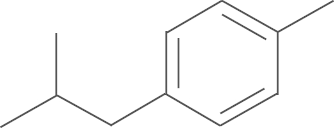 |
59.0 |
| 4e | SO2NH2 | 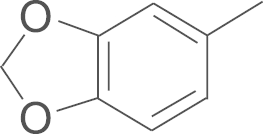 |
90.1 |
| 4f | SO2NH2 | 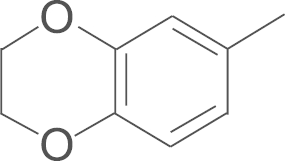 |
50.7 |
| 4g | SO2NH2 | 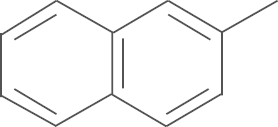 |
66.1 |
| 4h | SO2NH2 | 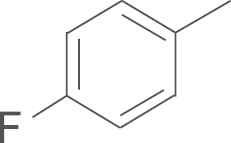 |
39.3 |
| 4i | SO2NH2 | 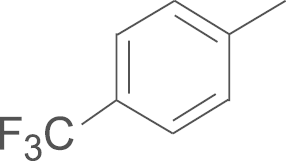 |
61.9 |
| 4j | SO2NH2 | 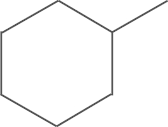 |
69.2 |
| 4k | SO2NH2 | 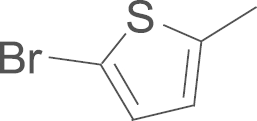 |
74.5 |
| 5a | SO2CH3 | 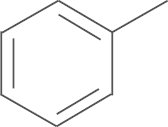 |
41.9 |
| 5b | SO2CH3 | 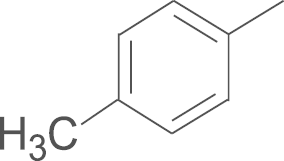 |
62.8 |
| 5c | SO2CH3 | 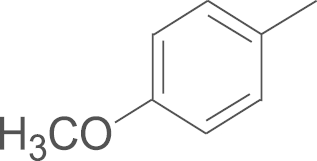 |
35.6 |
| 5d | SO2CH3 | 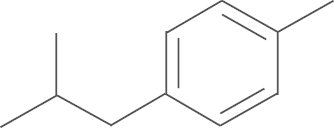 |
60.2 |
| 5e | SO2CH3 | 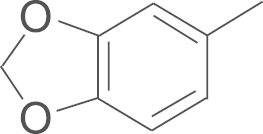 |
79.3 |
| 5f | SO2CH3 | 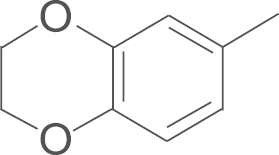 |
70.3 |
| 5g | SO2CH3 | 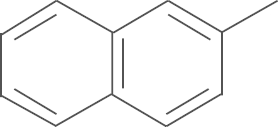 |
51.7 |
| 5i | SO2CH3 | 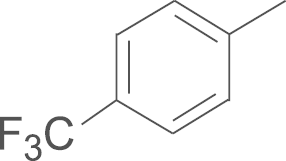 |
69.6 |
| 5j | SO2CH3 | 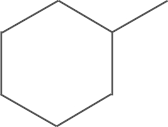 |
57.3 |
| 6a | F | 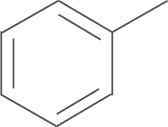 |
56.8 |
| 6b | F | 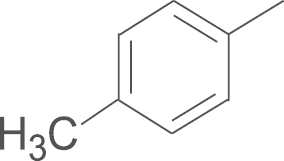 |
83.7 |
Values are means of three determinations and deviation from the mean is <10% of the mean value (Catalog No. 520111, Cayman Chemicals Inc.).
Scheme 2.

Synthetic routes of key intermediates 9 and 12. Reagent and conditions: (a) HNO3, H2O, hexane, 55 oC; (b) Sn/HCl, C2H5OH, reflux; (c) NaNO2, HCl, H2O; (d) SnCl2, HCl.
Scheme 3.

The synthetic strategy for the target compounds. Reagent and conditions: (a) diethyl oxalate, NaH, toluene, 45 oC, 2 h; (b) C2H5OH, reflux, 12 h; (c) (i) Na, C2H5OH, diethyl oxalate, 15 h, (ii) HCl, refluxing, 12 h; (d) LiOH, H2O, THF, rt, 12 h; (e) (i) SOCl2, CH2Cl2, reflux, 2 h, (ii) pyridine, CH2Cl2, 25 oC, 12 h.
2.2. Biological evaluations
All derivatives (4a–k, 5a–j, and 6a–b, Table 1) were evaluated using a cell-based assay for their inhibition of 5-LOX product synthesis at 5 μmol/L concentration in rat peritoneal polymorphonuclear leukocytes (PMNL) using zileuton, a potent 5-LOX inhibitor, as the reference drug18, 24. Nine compounds were selected to explore their concentration-response relationship in PMNLs, human whole blood (HWB) and 5-LOX enzymatic level assays. Compound 4e was further evaluated for its suppressive effects on lipopolysaccharide (LPS)-induced inflammation in brain inflammatory cells and protective effects against oxidative toxicity in a neuronal cell line. In addition, we investigated the efficacy of compound 4e for reducing brain infarction using a well-established mouse model of experimental stroke.
2.2.1. Structure–activity relationship of pyrazole-3-carboxamine derivatives
For the preliminary assay, the percent inhibition of 5-LOX formation of all compounds at 5 μmol/L in rat PMNLs was measured. The results are summarized in Table 1. The details of the bioassay procedures are described in the experimental section. As shown in Table 1, all compounds demonstrated good inhibitory activity against 5-LOX in rat PMNLs. The inhibition rates of 14 compounds were more than 60% at 5 μmol/L. Further studies showed that introducing substituents (electron-donating or electron-withdrawing groups), except the fluoro group, into the phenyl ring of R2 group increased the inhibitory activities against 5-LOX product synthesis when R1 was replaced by an aminosulfonyl group (4a–i). The introduction of a benzo[d] [1, 3] dioxol-5-yl group afforded the most potent inhibitory effects toward 5-LOX product synthesis, with 90.1% inhibition at 5 μmol/L. Additionally, introducing an alkyl group (cyclohexyl, 4j) and an aryl heterocyclic group (5-bromothiophen-2-yl, 4k) also improved inhibition potency against 5-LOX compared to the non-substituted compound 4a (the inhibition rate: 69.2% vs. 74.5%, respectively). In the second round of structural optimization, we replaced the 4-aminosulfonyl moiety on the phenyl ring of compounds 4a–k with a 4-methylsulfonyl moiety to give compounds 5a–j. Biological evaluation showed that the inhibition potency of most substituted derivatives was superior to that of non-substituted compound 5a. The compound substituted by a benzo-fused five-member heterocycle (5e) exhibited the most potent inhibitory activity against 5-LOX product synthesis in rat PMNLs with 79.3% inhibition at 5 μmol/L. Finally, we further explored the effects of a fluoro substituent in the R1 position on inhibitory potency of 5-LOX. The biological results indicated that introducing fluoro substituent to the pyrazole scaffold (6a and 6b) also improved the inhibitory activity.
To further characterize the potency of the compounds, nine were selected for further investigation in concentration-response studies. The results are summarized in Table 2. All selected compounds showed prominent inhibitory activities, with IC50 values ranging from 0.45 μmol/L to 1.07 μmol/L. Compounds 4c, 4e, 4g, 4k, 5e, 5f, 5i and 6b exhibited better inhibitory profiles (with IC50 values less than 1 μmol/L) compared with the reference drug zileuton. Meanwhile, the efficacies for inhibition of 5-LOX were also determined in the human whole blood (HWB) model. As shown in Table 2, the nine selected compounds showed good inhibitory activities against 5-LOX product synthesis. Compounds with benzo-fused five-member heterocycles (4e and 5e) exhibited the most potent inhibitory activity against 5-LOX, and these results were consistent with the results obtained from the PMNL model.
Table 2.
Determination of IC50 values of the selected compounds in PMNL, HWB and in the enzymatic assay.
| Compd. | IC50/μmol/L |
||
|---|---|---|---|
| PMNL | HWB | Enzymatic assaya | |
| 4c | 0.45 | 4.87 | 0.136±0.015 |
| 4e | 0.69 | 1.08 | 0.133±0.014 |
| 4g | 0.75 | 2.11 | 0.165±0.017 |
| 4j | 1.07 | 33.9 | −b |
| 4k | 0.90 | 5.83 | 0.159±0.016 |
| 5e | 0.64 | 0.94 | 0.169±0.016 |
| 5f | 0.81 | 4.65 | 0.273±0.021 |
| 5i | 0.80 | 1.13 | 0.127±0.029 |
| 6b | 0.82 | 10.2 | 0.327±0.022 |
| Zileuton | 1.19 | 4.83 | 0.206 |
HWB, human whole blood; PMNL, rat peritoneal polymorphonuclear leukocytes.
Each concentration was tested in triplicate. The IC50 was calculated with the sigmoidal dose-response (variable slope) fitting equation in GraphPad Prism.
Not determined.
To assess the inhibitory potency directly against 5-LOX, we used a cell-free assay to examine the inhibitory effects of eight compounds on the enzymatic activity of human 5-LOX. As shown in Table 2, most of the compounds exhibited much higher 5-LOX inhibition than the reference drug zileuton. Compounds 4e, 4g, 5e and 5i not only displayed excellent inhibitory activities against 5-LOX in rat PMNL and HWB models, but also effectively inhibited the enzymatic activity of human 5-LOX in the cell-free assay. It appears that introducing benzo-fused five-member heterocyle (4e and 5e) is favorable for the inhibitory activity against 5-LOX. These results indicated that these compounds were highly effective 5-LOX inhibitors, which may possess potential for the treatment of LT-related diseases. Therefore, compound 4e was selected to further evaluate its protective effects against ischemic insults using in vitro and in vivo approaches.
2.2.2. Compound 4e suppresses LPS-induced inflammation in vitro
Nitric oxide (NO) served as a proinflammatory marker and its production was induced upon LPS stimulation in the BV2 cells. Thus, we used a mouse microglia cell line BV2 to evaluate the suppressive effects of compound 4e on LPS-induced inflammation. NO production of microglia was generally evaluated by measuring nitrite released into the media. As shown in Fig. 1A, LPS at 200 ng/mL significantly induced the production of NO in BV2 cells, as indicated by significantly higher levels of nitrite in BV2 cells exposed to LPS vs. those exposed to saline. LPS-induced generation of NO was remarkably attenuated by the addition of 4e at 10, 50 or 100 μmol/L, while at the tested concentrations 4e did not reduce the viabilities of BV2 cells regardless of the presence or absence of LPS (MTT results, Fig. 1B). The results suggest that 4e was effective in suppressing LPS-induced inflammation in brain inflammatory cells.
Figure 1.

Compound 4e inhibited LPS-induced inflammation in BV2 cells. (A) LPS-induced production of NO compared to saline: compound 4e at 10, 50 and 100 μmol/L significantly attenuated NO production in cells treated with LPS; (B) cell viability in MTT assay: compound 4e at the tested concentrations did not exhibit toxicity to BV2 cells regardless of the presence or absence of LPS. **P<0.01 compared with the cells only treated with LPS (n = 3).
2.2.3. Neuroprotective effects of 4e against glutamate-induced oxidative toxicity
Glutamate toxicity is a critical contributor to the pathogenesis of multiple neurological diseases, including ischemic stroke. In HT22 cells which lack functional glutamate receptors, glutamate-induced neurotoxicity has been exclusively attributed to oxidative stress25. Therefore, we assessed the neuroprotective effects of compound 4e against glutamate-induced oxidative toxicity in HT22 cells with MTT assay. The viability of HT22 was decreased significantly at 24 h after glutamate exposure. The novel compound 4e at concentrations between 1–100 μmol/L remarkably reduced glutamate-induced HT22 cell death (Fig. 2), suggesting that compound 4e was effective in protecting neurons from oxidative toxicity.
Figure 2.

Compound 4e protected HT22 neuronal cells against glutamate oxidative toxicity at 1, 10, 50 and 100 μmol/L. **P<0.01 compared with the glutamate-treated cells (n = 3). Glu: glutamate.
2.2.4. Protective effects against ischemic damage in vivo
As a major enzyme responsible for leukotriene biosynthesis and with an induced expression pattern following cerebral ischemia, 5-LOX plays an important role in the pathogenesis of cerebral ischemia1. The above in vitro results demonstrated that compound 4e not only effectively suppressed LPS-induced inflammation in brain inflammatory cells, but also directly protected neurons from oxidative toxicity. Here, we further investigated the protective effects of compound 4e against infarct damage in the in vivo mouse middle cerebral artery occlusion (MCAO) model. As shown in Fig. 3, compared with vehicle treatment, post-ischemic intraperitoneal (IP) administration of compound 4e significantly decreased cortical, striatal and hemispheric infarct volumes after 24 h in the MCAO model.
Figure 3.

Compound 4e reduced infarct volumes at 24 h after reperfusion in the mouse MCAO model. Compared to vehicle treatment, post-ischemic administration of 4e significantly decreased cortical, striatal and hemispheric infarct volumes. (A) representative images of 5-tripheyltetrazolium chloride (TTC) staining (white: infracted area; red: viable tissue); (B) the bar graph of TTC histology. **P<0.01 and *P<0.05 compared with vehicle-treated mice (n = 7).
2.2.5. Molecular modeling
To better rationalize the structure-activity relationships (SARs) for the pyrazole derivatives, molecular docking studies were performed for compound 4e. Here the computations aimed to elucidate the structural requisites responsible for binding at the 5-LOX active sites. As shown in Fig. 4, compound 4e fits into the binding site with the 3,5-di-tert-butyl-4-hydroxyphenyl head approaching the Fe2+ ion, and the pyrazole ring inserting into a narrow groove formed by Gln363 and Ala603. The central carboxamide group bridging between these two moieties forms a hydrogen bond via its oxygen atom to Gln363. This binding pattern was followed by all the pyrazole derivatives examined here, which explains the overall effectiveness of this series of compounds against 5-LOX. In addition to this common binding feature, key interactions were established by the reference compound within the active site. Specifically, the 1,3-benzodioxol ring neatly binds into a sterically restricted hydrophobic cavity and is involved in a π–π stacking with Phe359. A detailed schematic depiction of the interaction of 4e within the active site of 5-LOX is shown in Fig. 5, from which we observed that the hydrophobic contacts involve Phe359, Ala561, ILe564, Leu601, Ala603 and Val604. A less favorable occupation of the cavity formed by these residues could contribute significantly to the potency. As shown in Table 1, decreasing the size of R2 led to a similar activity order of compounds 4e>4c>4b>4a, while more sterically hindering groups than the 1,3-benzodioxol ring (i.e., compounds 4d, 4f and 4g) also exhibited reduced activities. Furthermore, the p-sulfonamide-phenyl branch is exposed to solvent interactions and located adjacent to a polar region formed by Gln303, Thr570, His360 and His600. This group establishes an extra key hydrogen bond via its amino group with the backbone carbonyl oxygen of Pro569, which explains why replacement of the sulfonamide substituent with a methylsulfonyl group (compound 5e) resulted in a loss of the inhibitory activity against 5-LOX. Taken together, the consistency between the results of our investigations into the binding mode from theoretical results and the experimental data further support the rationality of our structure optimization from the lead compound, which will pave the way for the discovery of more effective inhibitors of 5-LOX.
Figure 4.

Putative binding mode of 4e in the 5-LOX structure, where the catalytic pocket of 5-LOX is depicted as a gray surface and the key interacting amino acids are shown. The image was generated using PyMOL, version 1.3.
Figure 5.

A schematic depiction of detailed interaction of 4e within the active site of 5-LOX, where distinctive colors and shapes are used to describe the different types of interactions. The diagram was generated using Maestro 9.1.
3. Conclusions
A new class of pyrazole derivatives was designed and synthesized by structural optimization of a lead compound, which originated from an in-house compound collection, and were evaluated as 5-LOX inhibitors in PMNLs, HWB and cell-free enzymatic assays. These pyrazole derivatives exhibited good to excellent inhibitory activities against 5-LOX in the PMNLs assay. Nine compounds were selected for concentration–response studies and displayed prominent inhibitory activities. Moreover, the nine compounds also displayed excellent biological activities in the HWB assay. Among of them, compounds with a benzo-fused heterocycle (4e and 5e) exhibited the most potent inhibitory activities against 5-LOX. In the cell-free assay, several exhibited much higher 5-LOX inhibitory potency than the reference drug zileuton. In view of the excellent in vitro 5-LOX inhibitory potency, compound 4e was evaluated for its ability to protect against ischemic insults. The results suggested that 4e not only effectively suppressed LPS-induced inflammation in brain inflammatory cells and protected neurons from oxidative toxicity, but also significantly decreased cortical, striatal and hemispheric infarct volumes after 24 h in the mouse MCAO model. Our binding model also demonstrated the consistency of our results between the analysis of the binding model from theoretical study and the experimental data, which further supported the rationale of our structural optimization. In summary, on the basis of the excellent inhibitory activities against 5-LOX in vitro and in vivo, these novel chemical structures have a promising therapeutic potential for LTs-related disorders.
4. Experimental section
4.1. Chemistry
The reagents (chemicals) were purchased from Shanghai Chemical Reagent Company, Lancaster Company, and Acros Company, and used without further purification. The analytical thin-layer chromatography (TLC) was HSGF 254 (0.15–0.2 mm thickness, Yantai Huiyou Company, China). Yields were not optimized. Melting points (m.p.) were measured in capillary tube on a SGW X-4 melting point apparatus without correction. Nuclear magnetic resonance (NMR) spectra were performed on a Brucker AMX-400 NMR (IS as TMS). Chemical shifts were reported in parts per million (ppm, δ) downfield from tetramethylsilane. Proton coupling patterns were described as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), and broad (br). Low- and high-resolution mass spectra (LRMS and HRMS) were given with electric (EI) produced by Finnigan MAT-95 mass spectrometer.
4.1.1. General procedure for the synthesis of 3,5-di-tert-butyl-4-hydroxyaniline hydrochloride (9)
2,6-Di-tert-butyl-4-nitrophenol (8) was prepared by the previously reported procedure26. 2,6-Di-tert-butylphenol (10 g) was dissolved in 40 mL of hexane with heating to 50 °C. Aqueous nitric acid solution (30%, 17 mL, w/w) was added dropwise to the hexane solution over 30 min. The reaction temperature was maintained between 50 °C and 55 °C overnight. The yellow precipitate was filtered and washed three times with hexane (10 mL×3) followed by water (10 mL). The resulting yellow solid was dried under vacuum. Yield: 95%; m.p. 152–155 °C26 (m.p. 155–156 °C). 1H NMR (CDCl3) δ 8.14 (s, 2 H), 5.94 (s, 1 H), 1.49 (s, 18 H). The 1H NMR spectrum is consistent with that of the reference reported26.
Compound 8 (1.0 mmol) was dissolved in 20 mL of ethanol and 2.0 mmol of Sn and 4 mL of concentrated hydrochloric acid were added into the ethanol solution. The resulting reaction mixture was heated to reflux for 2 h27. After filtering the mixture, most solvent was evaporated under reduced pressure and the residue was cooled in refrigerator. The white precipitate, 3,5-di-tert-butyl-4-hydroxyaniline hydrochloride (6), was obtained by filtering, washing three times with cooled-ethanol, and dried in vacuum. Yield: 90%; m.p. 199–201 °C. 1H NMR (CDCl3) δ 6.61 (s, 2 H), 1.43 (s, 18 H); EI–MS m/z 221 (M+).
4.1.2. General procedure for the synthesis of (4-aminosulfonyl)phenylhydrazine hydrochloride (12)
The key intermediate (4-aminosulfonyl)phenylhydrazine hydrochloride (12) could prepared according to the reported method2, 3. To a solution of sulfanilamide (6.9 g, 40.0 mmol, 1.0 eq) in conc. HCl (30 mL) was added a solution of sodium nitrate (3.1 g, 44.0 mmol, 1.1 eq) in 12.0 mL of water over 15 min in an ice-water bath. The resulting mixture was rapidly added to a cooled (0 °C) solution of tin (II) chloride dihydrate (27.1 g, 120.0 mmol, 3.0 eq) in conc. HCl (30 mL), followed by a stirring for 1 h, then overnight at ambient temperature. The precipitate was collected by filtration and successively washed with cool water and Et2O to give the pure compound, which was used directly without further purification.
4.1.3. General procedures for the synthesis of intermediates 17a–k, 18a–j and 19a–b as described for ethyl 1-(4-aminosulfonylphenyl)-5-phenyl-1H-pyrazole-3-carboxylate (17a)
To a stirred solution of NaH (2 mmol) in 10 mL of toluene was added acetophenone (1 mmol) in 10 mL of toluene. The mixture was heated to 45–50 °C, followed by the addition of diethyl oxalate (2 mmol) in 5 mL of toluene, and the resulting mixture was stirred at refluxing for 2 h. The solvent was removed under reduced pressure and the residue was extracted with EtOAc (20 mL×3), the organic layer was washed by 20 mL of aqueous sodium bicarbonate solution and brine, dried with anhydrous MgSO4, and filtered and concentrated under reduced pressure to obtain the crude product, which was further purified by flash chromatography on silica gel (hexane/EtOAc=4:1) to afford the desired products. The resulting intermediate 14a (1 mmol) was added to a solution of 1-(4-(aminosulfonyl)phenyl)hydrazine (12, 1 mmol) in 20 mL of ethanol, and heated to reflux overnight. After cooling the reaction mixture to ambient temperature, the solvent was removed under reduced pressure. Water (20 mL) was added to the residue, extracted with EtOAc (20 mL×3), the resulting organic layer was washed by brine, dried with anhydrous MgSO4, filtered and concentrated under reduced pressure to obtain the crude product, which was further purified by flash chromatography on silica gel (hexane/EtOAc=2:1) to afford the target products 14a as a yellow solid.
Alternatively, a one-pot, two-step process was also carried out to synthesize the target 14a. To a solution of sodium ethoxide, prepared by dissolving Na (1 mmol) in 20 mL of anhydrous ethanol, was added acetophenone (1 mmol). After about 30 min, diethyl oxalate (1 mmol) was added. The resulting mixture was heated to reflux for 15 h and then cooled to the room temperature. During this time the sodium salt of the intermediate β-diketonate condensation product precipitated as a thick yellow paste. HCl (1.2 mmol) and the corresponding hydrazines (1 mmol) were added and the mixture was stirred for another 12 h. Most of solvent was removed under reduced pressure and the residue was cooled to precipitate a thick yellow solid, which was filtered off and dried in vacuum to obtain the title compound 14a. m.p. 182–186 °C. 1H NMR (CDCl3) δ 1.43 (t, 3H), 4.46 (q, 2H), 5.11 (br, 2H), 7.04 (s, 1H), 7.20 (m, 2 H), 7.36 (m, 3H), 7.46 (d, 2H), 7.87 (d, 2H); LRMS (EI) m/z 371 (M+).
4.1.4. General procedure for the preparations of compounds 4a–k, 5a–j, 6a–b are described as that for N-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-(4-aminosulfonyl-phenyl)-5-phenyl-1H-pyrazole-3-carboxamide (4a)
A solution of ethyl 1-(4-aminosulfonylphenyl)-5-phenyl-1H-pyrazole-3-carboxylate (17a, 1 mmol) and potassium hydroxide (2 mmol) in water (30 mL) was heated to reflux for 0.5 h, and then allowed to cool to room temperature. After acidification with aqueous hydrochloric acid (2 mol/L), the mixture was extracted with EtOAc (20 mL×3) and the organic layer was washed by brine, dried with anhydrous MgSO4, filtered and concentrated under reduced pressure to afford the crude product 20a as a yellow solid, which was directly used without further purification.
To a solution of the intermediate 17a (1 mmol) in 15 mL of CH2Cl2 was added 2 mL of SOCl2 and the resulting mixture was heated to reflux for 3 h. After evaporation of the solvent under reduced pressure, 15 mL of CH2Cl2 was added, followed by the addition of 3,5-di-tert-butyl-4-hydroxyaniline hydrochloride (1 mmol) and 5 mL of pyridine and the resulting mixture was stirred overnight at room temperature. The solvent was evaporated under reduced pressure and the residue was dissolved in 20 mL of H2O, extracted by CH2Cl2 (20 mL×3), the organic layer was washed by aqueous hydrochloric acid, saturated NaHCO3, and brine, and dried with anhydrous MgSO4. The solvent was evaporated under reduced pressure and the residue was purified by column chromatography to obtain the target compounds 4a as a pale yellow solid. m.p. 233–235 °C; 1H NMR (500 MHz, DMSO-d6) δ 9.91 (s, 1H), 7.88 (d, J=8.6Hz, 2H), 7.64 (s, 2H), 7.59 (d, J=8.6 Hz, 2H), 7.50 (s, 2H), 7.45–7.41 (m, 3H), 7.36–7.31 (m, 2H), 7.14 (s, 1H), 6.81 (s, 1H), 1.40 (s, 18H); 13C NMR (125 MHz, DMSO-d6) δ 159.53, 150.57, 148.63, 145.07, 144.04, 142.06, 140.14, 131.60, 129.53, 129.34, 129.23, 127.15, 126.34, 117.96, 109.08, 35.24, 30.87; LRMS (EI) m/z 546 (M+); HRMS (EI) m/z Calcd. for C30H34N4O4S (M+) 546.2301, Found: 546.2312.
4.1.5. N-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-(4-aminosulfonylphenyl)-5-p-tolyl-1H-pyrazole-3-carboxamide (4b)
This compound was prepared from 4′-methylacetophenone by using a procedure similar to that described for the preparation of 4a. A pale yellow solid, mp 184–187 °C; 1H NMR (500 MHz, DMSO-d6) δ 9.89 (s, 1H), 7.89 (d, J=8.5 Hz, 2 H), 7.67–7.56 (m, 4H), 7.50 (s, 2H), 7.28–7.19 (m, 4H), 7.09 (s, 1H), 6.81 (d, J=2.9 Hz, 1H), 2.33 (s, 3H), 1.40 (s, 18H); 13C NMR (125 MHz, DMSO-d6) δ 159.56, 150.55, 148.59, 145.13, 143.98, 142.14, 140.13, 139.15, 131.61, 129.91, 129.10, 127.15, 126.64, 126.32, 117.94, 108.79, 35.24, 30.87, 21.29; LRMS (EI) m/z 560 (M+); HRMS (EI) m/z Calcd. C31H36N4O4S (M+) 560.2457, Found 560.2441.
4.1.6. N-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-(4-aminosulfonylphenyl)-5-p-meth- oxyphenyl-1H-pyrazole-3-carboxamide (4c)
This compound was prepared from 4′-methoxyacetophenone by using a procedure similar to that described for the preparation of 4a. A white solid, mp 219–221 °C; 1H NMR (500 MHz, DMSO-d6) δ 9.87 (s, 1H), 7.89 (d, J=8.6 Hz, 2H), 7.63 (s, 2H), 7.59 (d, J=8.6 Hz, 2H), 7.50 (s, 2H), 7.26 (d, J=8.7 Hz, 2H), 7.06 (s, 1 H), 6.99 (d, J=8.8 Hz, 2H), 6.81 (s, 1H), 3.78 (s, 3H), 1.39 (s, 18H); 13C NMR (125 MHz, DMSO-d6) δ 160.18, 159.61, 150.55, 148.53, 144.98, 143.91, 142.20, 140.13, 131.62, 130.64, 127.15, 126.28, 117.94, 114.80, 108.56, 55.74, 35.24, 30.87; LRMS (EI) m/z 576 (M+); HRMS (EI) m/z Calcd. C31H36N4O5S (M+) 576.2406, Found 576.2410.
4.1.7. N-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-(4-aminosulfonylphenyl)-5-p-iso-butylphenyl-1H-pyrazole-3-carboxamide (4d)
This compound was prepared from 4′-isobutylacetophenone by using a procedure similar to that described for the preparation of 4a. A yellow solid, mp 155–158 °C; 1H NMR (500 MHz, DMSO-d6) δ 9.89 (s, 1 H), 7.90–7.86 (m, 2H), 7.64 (s, 2 H), 7.61–7.57 (m, 2H), 7.51 (s, 2 H), 7.29–7.17 (m, 4H), 7.12 (d, J=9.6 Hz, 1H), 6.81 (s, 1H), 2.54–2.45 (m, 3H), 1.40 (s, 18H), 0.87 (d, J=6.6 Hz, 6H); 13C NMR (125 MHz, DMSO-d6) δ 159.57, 150.56, 148.61, 145.20, 143.95, 142.78, 142.16, 140.13, 131.61, 129.88, 128.98, 127.11, 126.95, 126.21, 117.94, 108.85, 44.67, 35.24, 30.87, 29.93, 22.69; LRMS (EI) m/z 602 (M+); HRMS (EI) m/z Calcd. C34H42N4O4S (M+) 602.2927, Found 602.2932.
4.1.8. N-(3,5-di-tert-butyl-4-hydroxyphenyl)-5-(benzo [1, 3] dioxol-5-yl)-1-(4-amino-sulfonylphenyl)-1H-pyrazole-3-carboxamide (4e)
This compound was prepared from 3,4-methylenedioxyacetophenone by using a procedure similar to that described for the preparation of 4a. A white solid, mp 180–183 oC; 1H NMR (500 MHz, DMSO-d6 δ 9.88 (s, 1H), 7.90 (d, J=8.6 Hz, 2H), 7.68–7.59 (m, 4H), 7.51 (s, 2H), 7.06 (s, 1H), 6.96 (d, J=8.0 Hz, 1H), 6.92 (s, 1H), 6.84–6.75 (m, 2H), 6.08 (s, 2H), 1.40 (s, 18H); 13C NMR (125 MHz, DMSO-d6) δ 159.56, 150.55, 148.47, 148.39, 147.97, 144.73, 143.92, 142.08, 140.12, 131.52, 127.18, 126.25, 123.53, 123.11, 117.95, 109.54, 109.15, 108.90, 102.04, 35.23, 30.86; LRMS (EI) m/z 590 (M+); HRMS (EI) m/z Calcd. C31H34N4O6S (M+) 590.2199, Found 590.2207.
4.1.9. N-(3,5-di-tert-butyl-4-hydroxyphenyl)-5-(2,3-dihydrobenzo [1, 4] dioxin-7-yl)-1-(4-aminosulfonylphenyl)-1H-pyrazole-3-carboxamide (4f)
This compound was prepared from 1,4-benzodioxan-6-yl methyl ketone by using a procedure similar to that described for the preparation of 4a. A white solid, mp 276–278 oC; 1H NMR (500 MHz, DMSO-d6) δ 9.88 (s, 1H), 7.90 (d, J=8.5 Hz, 2H), 7.65–7.59 (m, 4H), 7.52 (s, 2H), 7.05 (s, 1 H), 6.92–6.85 (m, 2H), 6.82 (s, 1H), 6.72 (dd, J=8.3, 1.9 Hz, 1H), 4.26 (dd, J=9.8, 4.6 Hz, 4H), 1.39 (s, 18H); 13C NMR (125 MHz, DMSO-d6) δ 159.57, 150.54, 148.49, 144.64, 144.60, 143.97, 143.85, 142.17, 140.12, 131.59, 127.14, 126.31, 122.44, 122.32, 117.96, 117.94, 117.87, 108.71, 64.65, 64.51, 35.23, 30.86; LRMS (EI) m/z 604 (M+); HRMS (EI) m/z Calcd C32H36N4O6S (M+) 604.2356, Found 604.2373.
4.1.10. N-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-(4-aminosulfonylphenyl)-5-naphth-2-yl-1H-pyrazole-3-carboxamide (4g)
This compound was prepared from 2′-acetonaphthone by using a procedure similar to that described for the preparation of 4a. A yellow solid, mp 176–177 oC; 1H NMR (500 MHz, DMSO-d6) δ 9.95 (s, 1H), 8.05 (s, 1H), 7.99–7.85 (m, 5H), 7.69–7.56 (m, 6H), 7.49 (s, 2H), 7.32 (dd, J=8.5, 1.5 Hz, 1H), 7.26 (s, 1H), 6.82 (s, 1H), 1.41 (s, 18H); 13C NMR (125 MHz, DMSO-d6) δ 159.54, 150.55, 148.73, 145.05, 144.01, 142.10, 140.16, 133.12, 133.08, 131.62, 128.79, 128.74, 128.73, 128.16, 127.67, 127.40, 127.21, 126.96, 126.42, 126.23, 117.97, 109.47, 35.25, 30.88; LRMS (EI) m/z 596 (M+); HRMS (EI) m/z Calcd. C34H36N4O4S (M+) 596.2457, Found 596.2467.
4.1.11. N-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-(4-aminosulfonylphenyl)-5-p-fluorophenyl-1H-pyrazole-3-carboxamide (4h)
This compound was prepared from 4′-fluoroacetophenone by using a procedure similar to that described for the preparation of 4a. A pale yellow solid, mp 234–236 oC; 1H NMR (500 MHz, DMSO-d6) δ 9.91 (s, 1H), 7.89 (d, J=8.6 Hz, 2H), 7.63 (s, 2H), 7.59 (d, J=8.6 Hz, 2H), 7.50 (s, 2H), 7.42–7.36 (m, 2H), 7.29 (t, J=8.9 Hz, 2H), 7.14 (s, 1H), 6.81 (s, 1H), 1.40 (s, 18H); 13C NMR (125 MHz, DMSO-d6) δ 162.82 (d, J=247.0 Hz), 159.49, 150.57, 148.59, 144.06, 141.90, 140.13, 131.62 (d, J=8.9 Hz), 127.19, 126.35, 126.06, 126.03, 117.96, 116.40 (d, J=21.8 Hz), 109.20, 35.12, 30.87; LRMS (EI) m/z 564 (M+); HRMS (EI) m/z Calcd. C30H33FN4O4S (M+) 564.2207, Found 564.2203.
4.1.12. N-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-(4-aminosulfonylphenyl)-5-p-trifluoromethylphenyl-1H-pyrazole-3-carboxamide (4i)
This compound was prepared from 4′-trifluoroacetophenone by using a procedure similar to that described for the preparation of 4a. A white solid, mp 186–189 oC; 1H NMR (500 MHz, DMSO-d6) δ 9.93 (s, 1H), 8.03 (d, J=8.7 Hz, 2H), 7.67 (d, J=8.6 Hz, 2H), 7.64 (s, 2H), 7.44–7.37 (m, 2H), 7.29 (t, J=8.8 Hz, 2H), 7.16 (s, 1H), 6.82 (s, 1H), 3.28 (s, 2H), 1.40 (s, 18H); 13C NMR (125 MHz, DMSO-d6) δ 163.84, 161.88, 159.43, 150.59, 148.81, 144.19, 143.33, 140.59, 140.14, 131.66 (d, J=8.6 Hz), 131.56, 128.66, 126.49, 125.95, 117.94, 116.45 (d, J=21.8 Hz), 109.44, 35.23, 30.86; LRMS (EI) m/z 614 (M+); HRMS (EI) m/z Calcd. C31H33F3N4O4S (M+) 614.2175, Found 614.2180.
4.1.13. N-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-(4-aminosulfonylphenyl)-5-cyclo-hexyl-1H-pyrazole-3-carboxamide (4j)
This compound was prepared from cyclohexyl methyl ketone by using a procedure similar to that described for the preparation of 4a. A white solid, mp 273–275 oC; 1H NMR (500 MHz, DMSO-d6) δ 9.74 (s, 1H), 8.02 (d, J=8.5 Hz, 2H), 7.82 (d, J=8.5 Hz, 2H), 7.57 (d, J=18.2 Hz, 4H), 6.79 (d, J=14.1 Hz, 2H), 2.75 (t, J=11.7 Hz, 1 H), 1.84 (d, J=12.4 Hz, 2H), 1.71 (s, 2H), 1.64 (s, 1H), 1.38 (s, 20H), 1.22 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 159.86, 151.51, 150.46, 148.15, 144.46, 142.02, 140.08, 131.63, 127.41, 126.68, 117.94, 105.08, 35.22, 35.04, 33.06, 30.86, 26.12, 25.74; LRMS (EI) m/z 552 (M+); HRMS (EI) m/z Calcd. C30H40N4O4S (M+) 552.2770, Found 552.2758.
4.1.14. N-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-(4-aminosulfonylphenyl)-5-(5-bromothiophen-2-yl)-1H-pyrazole-3-carboxamide (4k)
This compound was prepared from 2-Acetyl-5-bromothiophene by using a procedure similar to that described for the preparation of 4a. A white solid, mp 224–225 oC; 1H NMR (500 MHz, DMSO-d6) δ 9.93 (s, 1H), 7.98 (d, J=8.6 Hz, 2H), 7.76 (d, J=8.6 Hz, 2H), 7.62 (s, 2H), 7.57 (s, 2H), 7.27 (d, J=3.9 Hz, 1H), 7.26 (s, 1H), 7.05 (d, J=3.9 Hz, 1H), 6.81 (s, 1H), 1.39 (s, 18H); 13C NMR (125 MHz, DMSO-d6) δ 159.18, 150.59, 148.58, 145.18, 141.38, 140.11, 137.93, 131.67, 131.52, 131.27, 130.16, 127.52, 127.34, 117.99, 114.30, 109.02, 35.23, 30.86; LRMS (EI) m/z 630 (M+); HRMS (EI) m/z Calcd. C28H31BrN4O4S2 (M+) 630.0970, Found 630.0982.
4.1.15. N-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-(4-methylsulfonylphenyl)-5-phenyl-1H-pyrazole-3-carboxamide (5a)
This compound was prepared from acetophenone by using a procedure similar to that described for the preparation of 4a. A yellow solid, mp 165–167 oC; 1H NMR (500 MHz, DMSO-d6) δ 9.93 (s, 1H), 8.02 (d, J=8.7 Hz, 2H), 7.67 (d, J=8.7 Hz, 2H), 7.64 (s, 2 H), 7.47–7.41 (m, 3H), 7.38–7.33 (m, 2H), 7.15 (s, 1H), 6.82 (s, 1H), 3.28 (s, 3H), 1.40 (s, 18H); 13C NMR (125 MHz, DMSO-d6) δ 159.47, 150.58, 148.85, 145.17, 143.48, 140.57, 140.14, 131.58, 129.63, 129.42, 129.39, 129.26, 128.61, 126.46, 117.94, 109.33, 43.70, 35.12, 30.86; LRMS (EI) m/z 545 (M+); HRMS (EI) m/z Calcd. C31H35N3O4S (M+) 545.2348, Found 545.2350.
4.1.16. N-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-(4-methylsulfonylphenyl)-5-p-tolyl-1H-pyrazole-3-carboxamide (5b)
This compound was prepared from 4′-methylacetophenone by using a procedure similar to that described for the preparation of 4a. A yellow solid, mp 205–207 oC; 1H NMR (500 MHz, DMSO-d6) δ 9.91 (s, 1H), 8.02 (d, J=8.6 Hz, 2H), 7.67 (d, J=8.6 Hz, 2H), 7.64 (s, 2H), 7.26–7.20 (m, 4H), 7.10 (s, 1H), 6.81 (s, 1H), 3.29 (s, 3H), 2.33 (s, 3H), 1.40 (s, 18H); 13C NMR (125 MHz, DMSO-d6) δ 159.47, 150.58, 148.91, 145.23, 143.57, 140.52, 140.14, 139.24, 131.68, 129.97, 129.12, 128.62, 126.55, 126.44, 117.93, 109.06, 43.83, 35.24, 30.87, 21.30; LRMS (EI) m/z 559 (M+); HRMS (EI) m/z Calcd. C32H37N3O4S (M+) 559.2505, Found 559.2493.
4.1.17. N-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-(4-methylsulfonylphenyl)-5-p-methoxyphenyl-1H-pyrazole-3-carboxamide (5c)
This compound was prepared from 4′-methoxyacetophenone by using a procedure similar to that described for the preparation of 4a. A yellow solid, mp 187–189 oC; 1H NMR (500 MHz, DMSO-d6) δ 9.90 (s, 1H), 8.02 (d, J=8.6 Hz, 2H), 7.67 (d, J=8.6 Hz, 2H), 7.64 (s, 2H), 7.27 (d, J=8.7 Hz, 2H), 7.07 (s, 1H), 6.99 (d, J=8.7 Hz, 2H), 6.81 (s, 1H), 3.78 (s, 3H), 3.28 (s, 3H), 1.40 (s, 18H); 13C NMR (125 MHz, DMSO-d6) δ 160.24, 159.55, 150.57, 148.77, 145.09, 143.63, 140.43, 140.14, 131.60, 130.67, 128.62, 126.41, 121.60, 117.93, 114.85, 108.81, 55.73, 43.84, 35.24, 30.87; LRMS (EI) m/z 575 (M+); HRMS (EI) m/z Calcd. C32H37N3O5S (M+) 575.2454, Found 575.2447.
4.1.18. N-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-(4-methylsulfonylphenyl)-5-p-iso-butylphenyl-1H-pyrazole-3-carboxamide (5d)
This compound was prepared from 4′-isobutylacetophenone by using a procedure similar to that described for the preparation of 4a. A yellow solid, mp 238–240 oC; 1H NMR (500 MHz, DMSO-d6) δ 9.91 (s, 1H), 8.01 (d, J=8.6 Hz, 2H), 7.66 (d, J=8.6 Hz, 2H), 7.64 (s, 2H), 7.24 (q, J=8.2 Hz, 4H), 7.12 (s, 1H), 6.81 (s, 1H), 3.28 (s, 3H), 2.48 (d, J=7.2 Hz, 2H), 1.92–1.80 (m, 1H), 1.40 (s, 18H), 0.87 (s, 3H), 0.86 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 159.47, 150.58, 148.87, 145.32, 143.59, 142.79, 140.49, 140.14, 131.59, 129.93, 129.01, 128.57, 126.85, 126.38, 117.92, 109.09, 44.75, 43.83, 35.24, 30.87, 29.93, 22.62; LRMS (EI) m/z 601 (M+); HRMS (EI) m/z Calcd. C35H43N3O4S (M+) 601.2974, Found 601.2965.
4.1.19. N-(3,5-di-tert-butyl-4-hydroxyphenyl)-5-(benzo [1, 3] dioxol-5-yl)-1-(4-methylsulfonylphenyl)-1H-pyrazole-3-carboxamide (5e)
This compound was prepared from 3,4-methylenedioxyacetophenone by using a procedure similar to that described for the preparation of 1a. A pale yellow solid, mp 181–183 oC; 1H NMR (500 MHz, DMSO-d6) δ 9.90 (s, 1H), 8.03 (d, J=8.7 Hz, 2H), 7.69 (d, J=8.7 Hz, 2H), 7.63 (s, 2H), 7.07 (s, 1H), 6.99–6.92 (m, 2H), 6.81 (s, 1 H), 6.79 (dd, J=8.0, 1.7 Hz, 1H), 6.08 (s, 2H), 3.29 (s, 3H), 1.40 (s, 18H); 13C NMR (125 MHz, DMSO-d6) δ 159.50, 150.57, 148.70, 148.47, 148.04, 144.94, 143.51, 140.44, 140.14, 131.58, 128.59, 126.37, 123.59, 123.03, 117.94, 109.59, 109.19, 109.16, 102.07, 43.82, 35.24, 30.86; LRMS (EI) m/z 589 (M+); HRMS (EI) m/z Calcd. C32H35N3O6S (M+) 589.2247, Found 589.2235.
4.1.20. N-(3,5-di-tert-butyl-4-hydroxyphenyl)-5-(2,3-dihydrobenzo [1, 4] dioxin-7-yl)-1-(4-methylsulfonylphenyl)-1H-pyrazole-3-carboxamide (5f)
This compound was prepared from 1,4-benzodioxan-6-yl methyl ketone by using a procedure similar to that described for the preparation of 4a. A yellow solid, mp 153–156 oC; 1H NMR (500 MHz, DMSO-d6) δ 9.89 (s, 1H), 8.03 (d, J=8.6 Hz, 2H), 7.69 (d, J=8.6 Hz, 2H), 7.63 (s, 2H), 7.06 (s, 1H), 6.91 (d, J=2.1 Hz, 1H), 6.89 (d, J=8.4 Hz, 1H), 6.81 (s, 1H), 6.72 (dd, J=8.3, 2.1 Hz, 1H), 4.27 (dd, J=10.1, 5.0 Hz, 4H), 3.29 (s, 3H), 1.39 (s, 18H); 13C NMR (125 MHz, DMSO-d6) δ 159.52, 150.57, 148.72, 144.77, 144.66, 143.92, 143.60, 140.48, 140.14, 131.59, 128.60, 126.44, 122.37, 118.02, 117.93, 108.98, 64.66, 64.52, 43.84, 35.24, 30.86; LRMS (EI) m/z 603 (M+); HRMS (EI) m/z Calcd. C33H37N3O6S (M+) 603.2403, Found 603.2411.
4.1.21. N-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-(4-methylsulfonylphenyl)-5-naphth-2-yl-1H-pyrazole-3-carboxamide (5g)
This compound was prepared from 2′-acetonaphthone by using a procedure similar to that described for the preparation of 4a. A white solid, mp 241–243 oC; 1H NMR (500 MHz, DMSO-d6) δ 9.98 (s, 1H), 8.06 (s, 1H), 8.01 (d, J=8.6 Hz, 2H), 7.98–7.89 (m, 3H), 7.72 (d, J=8.6 Hz, 2H), 7.67 (s, 2 H), 7.62–7.55 (m, 2H), 7.34 (dd, J=8.5, 1.3 Hz, 1H), 7.28 (s, 1H), 6.84 (s, 1H), 3.28 (s, 3H), 1.42 (s, 18H); 13C NMR (125 MHz, DMSO-d6) δ 159.48, 150.61, 148.96, 145.15, 143.52, 140.53, 140.17, 133.14, 131.60, 128.88, 128.77, 128.74, 128.68, 128.18, 127.71, 127.42, 126.87, 126.43, 126.36, 117.96, 109.74, 43.83, 35.25, 30.88; LRMS (EI) m/z 595 (M+); HRMS (EI) m/z Calcd. C35H37N3O4S (M+) 595.2505, Found 595.2500.
4.1.22. N-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-(4-methylsulfonylphenyl)-5-p-tri-fluoromethylphenyl-1H-pyrazole-3-carboxamide (5i)
This compound was prepared from 2′-trifluoronaphthone by using a procedure similar to that described for the preparation of 4a. A pale yellow solid, mp 146–149 oC; 1H NMR (500 MHz, DMSO-d6) δ 9.97 (s, 1H), 8.05 (d, J=8.4 Hz, 2H), 7.81 (d, J=8.3 Hz, 2H), 7.71 (d, J=8.4 Hz, 2H), 7.64 (s, 2H), 7.58 (d, J=8.1 Hz, 2H), 7.29 (s, 1H), 6.83 (s, 1H), 3.29 (s, 3H), 1.40 (s, 18H); 13C NMR (125 MHz, DMSO-d6) δ 159.30, 150.63, 149.01, 143.64, 143.18, 140.85, 140.15, 133.46, 131.54, 130.07, 128.80, 126.65, 126.29, 126.27, 117.97, 110.23, 43.83, 35.24, 30.86; LRMS (EI) m/z 613 (M+); HRMS (EI) m/z Calcd. C32H34F3N3O4S (M+) 613.2222, Found 613.2221.
4.1.23. N-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-(4-methylsulfonylphenyl)-5-cyclo-hexyl-1H-pyrazole-3-carboxamide (5j)
This compound was prepared from cyclohexyl methyl ketone by using a procedure similar to that described for the preparation of 4a. A white solid, mp 183–185 oC; 1H NMR (500 MHz, DMSO-d6) δ 9.76 (s, 1H), 8.14 (d, J=8.6 Hz, 2H), 7.90 (d, J=8.5 Hz, 2H), 7.59 (s, 2H), 6.83 (s, 1H), 6.78 (s, 1H), 3.33 (s, 3H), 2.78 (t, J=11.6 Hz, 1H), 1.89–1.59 (m, 5H), 1.48–1.16 (m, 23H); 13C NMR (125 MHz, DMSO-d6) δ 159.80, 151.67, 150.48, 148.40, 143.47, 141.00, 140.09, 131.58, 128.92, 126.83, 117.93, 105.34, 43.88, 35.22, 35.03, 33.13, 30.86, 26.09, 25.69; LRMS (EI) m/z 551 (M+); HRMS (EI) m/z Calcd. C31H41N3O4S (M+) 551.2818, Found 551.2820.
4.1.24. N-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-(4-fluorophenyl)-5-phenyl-1H-pyrazole-3-carboxamide (6a)
This compound was prepared from acetophenone by using a procedure similar to that described for the preparation of 4a. A white solid, mp 232–234 oC; 1H NMR (500 MHz, DMSO-d6) δ 9.88 (s, 1H), 7.64 (s, 2H), 7.49–7.43 (m, 2H), 7.42–7.37 (m, 3H), 7.35–7.28 (m, 4H), 7.11 (s, 1H), 6.80 (s, 1H), 1.40 (s, 18H); 13C NMR (125 MHz, DMSO-d6) δ 161.98 (d, J=254.5 Hz), 159.68, 150.50, 148.05, 145.03, 140.09, 136.24, 131.67, 129.60, 129.29, 129.19, 129.11, 128.60 (d, J=8.9 Hz), 117.96, 116.56 (d, J=23.1 Hz), 108.30, 35.24, 30.87; LRMS (EI) m/z 485 (M+); HRMS (EI) m/z Calcd. C30H32FN3O2 (M+) 485.2479, Found 485.2473.
4.1.25. N-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-(4-fluorophenyl)-5-p-tolyl-1H-pyrazole-3-carboxamide (6b)
This compound was prepared from 4′-methylacetophenone by using a procedure similar to that described for the preparation of 4a. A white solid, mp 225–227 oC; 1H NMR (500 MHz, CDCl3) δ 8.63 (s, 1H), 7.56 (s, 2H), 7.40–7.33 (m, 2H), 7.18–7.09 (m, 7H), 5.11 (s, 1H), 2.38 (s, 3H), 1.49 (s, 18H); 13C NMR (125 MHz, CDCl3) δ 162.09 (d, J=249.1 Hz), 159.42, 150.56, 147.61, 145.54, 138.96, 136.51, 135.82, 129.87, 129.43, 128.66, 127.41 (d, J=8.6 Hz), 126.52, 117.49, 116.14 (d, J=23.0 Hz), 107.97, 34.54, 30.26, 21.30; LRMS (EI) m/z 499 (M+); HRMS (EI) m/z Calcd. C31H34FN3O2 (M+) 499.2635, Found 499.2633.
4.2. Biological evaluation
4.2.1. In vitro inhibition assays for 5-LOX product synthesis in PMNL model
All derivatives (4a–k, 5a–j, and 6a–b, Table 1) were evaluated by the cell-based assay for their inhibitory activities against 5-LOX product synthesis in rat peritoneal leukocytes2, 3 using zileuton as the control drug. Mixed peritoneal leukocytes, including PMNLs and mononuclear leukocytes, were obtained from SD rats receiving IP injection of 20 mL of 0.2% glycogen solution (10K154, Sigma-Aldrich Co.). Briefly, PMNLs were harvested with peritoneal lavage using 10 mL of Hanks balanced salt solution (HBSS). The resulting fluid was centrifuged at 2000 r/min for 10 min. The cells were resuspended with 5 mL of ice-cold distilled water to disrupt blood red cells. After 1 min, 5 mL of 1.8% saline was added. After centrifugation at 2000 r/min for 5 min, the cells were washed twice with HBSS. PMNLs) were counted by wright-Giemsa stain (PMNLs>80%). The cell densities were adjusted using HBSS to the concentration of 5×106 cells/mL. Aliquots (0.5 mL) of the cell suspensions were preincubated at 37 °C for 10 min, followed by the addition of l-cysteine (10 mmol/L), indomethacin (1 mg/L), and tested compounds or vehicle (0.1% DMSO) in 96-well plates (Costar). Indomethacin was added to prevent ex vivo formation of eicosanoids, which have the potential to interfere with the assay. After 30 min, the cells were further incubated with the calcium ionophore A23187 (5 μmol/L) at 37 °C for 30 min. The mixtures were centrifuged at 14,000 r/min at 4 °C for 5 min and the supernatants were collected and stored at −70 oC until analysis. The amounts of LTB4 in the medium was determined spectrophotometrically at 412 nm using commercial enzyme immunoassay kits (Cayman Chemical Company, Ann Arbor, USA).
4.2.2. In vitro inhibition assays for 5-LOX product synthesis in HWB model
Nine compounds were also evaluated for their inhibitory potency against 5-LOX product synthesis in HWB challenged with Ca2+-ionophore A23187, using zileuton as the reference drug. Fresh blood from a healthy volunteer was collected into test tubes with heparin. Tested compounds were added to aliquots (0.5 mL) of the whole blood and incubated for 15 min at 37 °C. After addition of the Ca2+ ionophore A23187, the blood was incubated at 37 °C for another 15 min. The mixtures were centrifuged at 12,000 r/min at 4 °C for 15 min and 400 μL of methanol was added to the supernatant fractions. After further vortexing and incubation, the supernatants were collected and stored at −70 oC until analysis. The amounts of LTB4 in the medium was determined spectrophotometrically at 412 nm using commercial enzyme immunoassay kits (Lot No. 143772, Cayman Chemical Company, Ann Arbor, USA).
4.2.3. Evaluation of 5-LOX inhibitory activity in a cell-free assay
The enzyme assay is based on the oxidation of the substrate H2DCFDA to the highly fluorescent product 2′,7′-dichloro-fluorescein (DCF)28. Nonspecific ester cleavage of the acetate groups of H2DCFDA was carried out prior to the oxidation by incubation of 20 μmol/L H2DCFDA with 3.34 mU/μL 5-LOX in the assay buffer (50 mmol/L Tris, 2 mmol/L CaCl2, pH 7.5) (Reagent A). Specifically, 40 μL of reagent A was added into the well of a 96-well plate (Costar Catalog No. 3915). The plate was shaken for 1 min and allowed to stand for 5 min. 1 μL of various concentrations of inhibitors were then added to the wells. After incubation for 10 min, the reaction was initiated by the addition of 40 μL of Reagent B (10 μmol/L AA, 20 μmol/L ATP) with shaking for 1 min. The reaction was stopped by the addition of 80 μL pure acetonitrile after 20 min. The fluorescence at 520 nm was measured with the excitation light of 485 nm on POLARstar OPTIMA microplate reader (BMG LabTech, Offenburg, Germany). Zileuton was used as the positive control.
4.2.4. Quantification of LPS-induced generation of the proinflammatory mediator nitric oxide in microglial BV2 cells
To assess the suppressive effects of compound 4e on LPS-induced inflammation in brain-resident inflammatory cells, we used a mouse microglia cell line BV2. BV2 cells were cultured in Dulbecco׳s modified Eagle׳s Medium (DMEM, Invitrogen Corporation, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS, Gibco Laboratories, Grand Island, NY, USA) and 100 μg/mL penicillin-streptomycin at 37 °C with 5% CO2. Cells were trypsinized, seeded onto 96-well plates at the density of 2×104 cells per well per 100 μL and exposed to compound 4e in the presence or absence of LPS (200 ng/mL) at 24 h after seeding. Cell Supernatants were harvested at 24 h after exposure for the measurement of the stable end product of NO, nitrite (NO−), using Griess reagents as described previously29. Briefly, supernatants (50 μL) were mixed with 50 μL Griess reagent, which reacted with nitrite to form a compound spectrophotometrically detectable at 570 nm. Furthermore, the effects of compound 4e on cell viability was assessed with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)30. Briefly, cells were incubated in medium containing of MTT (0.5 mg/mL) at 37 °C with 5% CO2 for 2 h. Then, medium was aspirated and cells were lysed with 100 µL DMSO to dissolve dark blue crystals of formazan, a MTT metabolite formed only in live cells but not in dead cells. The absorbance was measured at 570 nm using the Microplate Reader (Infinite M200 PRO, Tecan, Switzerland). Relative cell viabilities were calculated and presented as percentages of controls without glutamate treatment.
4.2.5. Assessment of neuroprotective effects of 4e against glutamate-induced oxidative toxicity in the hippocampal neuronal cell line HT22
We assessed the neuroprotective effects of the compound 4e against glutamate-induced oxidative toxicity in HT22 cells, as previously described31. Briefly, mouse hippocampal HT22 cells were cultured in Dulbecco׳s modified Eagle׳s Medium (DMEM, Invitrogen Corporation, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS, Gibco Laboratories, Grand Island, NY, USA) and 100 μg/mL penicillin-streptomycin at 37 °C with 5% CO2. The medium was changed every two days. For the glutamate toxicity assay, cells were trypsinized and seeded onto 24-well plates at the density of 2×104 cells per well. After a 24 h incubation the cells were exposed to glutamate at final concentration of 5 mmol/L in the presence or absence of compound 4e. At 24 h after glutamate addition cell viabilities were measured with MTT4 as described in earlier. Relative cell viabilities were calculated and presented as a percentage of control without glutamate treatment.
4.2.6. Assessment of protective effects of 4e against ischemic damage in a mouse middle cerebral artery occlusion model
Animal experiments were approved by the Ethical Review Panel of Soochow University. Adult male CD-1 mice weighing 30±2 g were used in the study. Experimental stroke was induced in mice by middle cerebral artery occlusion (MCAO) via the intraluminal suture technique, as described32. Briefly, mice were anesthetized with isoflurane and body temperature was maintained by warming pads during surgery33. A skin incision was made to expose the right external and common carotid arteries. After the common carotid artery was tightened, a silicon-coated nylon suture with a heat-blunted tip (diameter 0.22±0.02 mm) was inserted into the right internal carotid artery via the external carotid artery. The suture was secured and the surgical site was closed when the tip of the suture reached the origin of the middle cerebral artery. After 60 min of occlusion, the suture was withdrawn to allow for reperfusion. Vascular occlusion (<25% of baseline) and reperfusion (>75% of baseline) were verified with laser Doppler flowmetry (PeriFlux System 5000, Perimed Inc, Stockholm, Sweden) by affixing a laser probe to the mouse skull to monitor cortical perfusion. At 3 h after reperfusion, mice showing obvious neurological deficits were randomly divided into two groups to receive: 1) intraperitoneal (IP) injection of the compound 4e at a dose of 50 mg/kg; or 2) an equal volume of vehicle (10 μL DMSO suspended in 490 μL corn oil). At 24 h after MCAO, mice were decapitated. Brains were harvested and cut into 2-mm-thick coronal sections. Slices were stained in a 1.2% solution of 2,3,5-triphenyltetrazolium chloride (TTC, Sigma, St Louis, MO, USA) and then photographed. Unstained (infarcted) and stained (uninfarcted) areas were analyzed with digital image analysis software (SigmaScan Pro, Jandel, San Rafael, CA, USA) and integrated across all slices, as previously described33.
4.3. Molecular modeling
4.3.1. Preparing the protein structure
The X-ray crystal structure of arachidonic acid bound to 5-LOX was retrieved from the Protein Data Bank (PDB ID: 3V99). The solvent molecules were deleted, and the remaining protein structure was prepared using the Protein Preparation Wizard module in the Maestro program (version 9.1. Schrödinger, LLC, New York, NY; 2010). In brief, the hydrogen atoms were properly added to the complexes, bond corrections were applied to the cocrystallized ligand, the hydrogen bond networks and flip orientations/tautomeric states of Gln, Asn, and His residues were optimized to maximize hydrogen bond formation, and an exhaustive sampling was performed with regard to hydrogen bond assignment. Finally, a restrained minimization on the ligand–protein complexes was performed with the OPLS_2005 force field and the default value for rmsd of 0.30 Å for non-hydrogen atoms was used.
4.3.2. Ligand preparation
The three-dimensional (3D) coordinates of the synthesized compounds were generated with LigPrep (version 2.4. Schrödinger, LLC, New York, NY; 2010), and their protonation states were determined at a target pH 7.0±2.0 with Epik (version 2.1. Schro–dinger, LLC, New York, NY; 2010). The resulting structures were used as the starting point for the followed docking simulations.
4.3.3. Induced fit docking
The Glide (version 5.6. Schrödinger, LLC, New York, NY; 2010) grid was defined as an enclosing box centered at the native ligand arachidonic acid, with the box size 18 Å×18 Å×18 Å. The IFD protocol was carried out in three consecutive steps. Firstly, the ligand was docked into a flexible receptor model, with manually chosen rotatable residues: Tyr181, Phe177, Thr427. Next, the Prime (version 2.2. Schrödinger, LLC, New York, NY; 2010.) program was used to generate the induced-fit protein–ligand complex with default parameters. Finally, the extra precision (XP) Glide docking method was used to dock the compound of interest flexibly into the arachidonic acid binding site of 5-LOX. To verify the docking protocol, a redocking validation was firstly performed and the top-scored (Gscore) binding pose of the native ligand (arachidonic acid) is close to its crystal structure conformation, with a small RMSD value of 1.2 Å. Accordingly, the IFD protocol was used for the followed binding mode analysis of our active compounds.
Acknowledgments
We gratefully acknowledge financial support from the National Natural Science Foundation of China (Grants Nos. 91229204 and 81220108025), Major Project of Chinese National Programs for Fundamental Research and Development (No. 2015CB910304), National High Technology Research and Development Program of China (No. 2012AA020302), National Basic Research Program of China (No. 2012CB518005), National S&T Major Projects (Nos. 2012ZX09103101-072, 2014ZX09507002-001, and 2013ZX09507-001).
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
Contributor Information
Dongxiang Liu, Email: dxl@mail.shcnc.ac.cn.
Jian Cheng, Email: chengjian@suda.edu.cn.
Hong Liu, Email: hliu@mail.shcnc.ac.cn.
References
- 1.Shang E.C., Liu Y., Wu Y.R., Zhu W., He C., Lai L.H. Development of 3,5-dinitrobenzoate-based 5-lipoxygenase inhibitors. Bioorg Med Chem. 2014;22:2396–2402. doi: 10.1016/j.bmc.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 2.Peters-Golden M., Henderson W.R. Leukotrienes. N Engl J Med. 2007;357:1841–1854. doi: 10.1056/NEJMra071371. [DOI] [PubMed] [Google Scholar]
- 3.Steinhilber D., Hofmann B. Recent advances in the search for novel 5-lipoxygenase inhibitors. Basic Clin Pharmacol Toxicol. 2014;114:70–77. doi: 10.1111/bcpt.12114. [DOI] [PubMed] [Google Scholar]
- 4.Montuschi P. Leukotrienes, antileukotrienes and asthma. Mini Rev Med Chem. 2008;8:647–656. doi: 10.2174/138955708784567395. [DOI] [PubMed] [Google Scholar]
- 5.Hofmann B., Steinhilber D. 5-Lipoxygenase inhibitors: a review of recent patents (2010–2012) Expert Opin Ther Pat. 2013;23:895–909. doi: 10.1517/13543776.2013.791678. [DOI] [PubMed] [Google Scholar]
- 6.Werz O., Steinhilber D. Therapeutic options for 5-lipoxygenase inhibitors. Pharmacol Ther. 2006;112:701–718. doi: 10.1016/j.pharmthera.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 7.Chen Y.Y., Hu Y.G., Zhang H.J., Peng C., Li S.G. Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat Genet. 2009;41:783–792. doi: 10.1038/ng.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang D.Z., DuBois R.N. Eicosanoids and cancer. Nat Rev Cancer. 2010;10:181–193. doi: 10.1038/nrc2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou Y., Wei E.Q., Fang S.H., Chu L.S., Wang M.L., Zhang W.P. Spatio-temporal properties of 5-lipoxygenase expression and activation in the brain after focal cerebral ischemia in rats. Life Sci. 2006;79:1645–1656. doi: 10.1016/j.lfs.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 10.Landwehr J., George S., Karg E.M., Poeckel D., Steinhilber D., Troschuetz R. Design and synthesis of novel 2-amino-5-hydroxyindole derivatives that inhibit human 5-lipoxygenase. J Med Chem. 2006;49:4327–4332. doi: 10.1021/jm050801i. [DOI] [PubMed] [Google Scholar]
- 11.Charlier C., Hénichart J.P., Durant F., Wouters J. Structural insights into human 5-lipoxygenase inhibition: combined ligand-based and target-based approach. J Med Chem. 2006;49:186–195. doi: 10.1021/jm050870x. [DOI] [PubMed] [Google Scholar]
- 12.Pergola C., Gaboriaud-Kolar N., Jestädt N., König S., Kritsanida M., Schaible A.M. Indirubin core structure of glycogen synthase kinase-3 inhibitors as novel chemotype for intervention with 5-lipoxygenase. J Med Chem. 2014;57:3715–3723. doi: 10.1021/jm401740w. [DOI] [PubMed] [Google Scholar]
- 13.Werz O., Greiner C., Koeberle A., Hoernig C., George S., Popescu L. Novel and potent inhibitors of 5-lipoxygenase product synthesis based on the structure of pirinixic acid. J Med Chem. 2008;51:5449–5453. doi: 10.1021/jm800588x. [DOI] [PubMed] [Google Scholar]
- 14.Hofmann B., Franke L., Proschak E., Tanrikulu Y., Schneider P., Steinhilber D. Scaffold-hopping cascade yields potent inhibitors of 5-lipoxygenase. ChemMedChem. 2008;3:1535–1538. doi: 10.1002/cmdc.200800153. [DOI] [PubMed] [Google Scholar]
- 15.Reddy N.P., Chandramohan R.T., Aparoy P., Achari C., Sridhar P.R., Reddanna P. Structure based drug design, synthesis and evaluation of 4-(benzyloxy)-1-phenylbut-2-yn-1-ol derivatives as 5-lipoxygenase inhibitors. Eur J Med Chem. 2012;47:351–359. doi: 10.1016/j.ejmech.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 16.Wu Y.R., He C., Gao Y., He S., Liu Y., Lai L.H. Dynamic modeling of human 5-lipoxygenase-inhibitor interactions helps to discover novel inhibitors. J Med Chem. 2012;55:2597–2605. doi: 10.1021/jm201497k. [DOI] [PubMed] [Google Scholar]
- 17.Karg E.M., Luderer S., Pergola C., Bühring U., Rossi A., Northoff H. Structural optimization and biological evaluation of 2-substituted 5-hydroxyindole-3-carboxylates as potent inhibitors of human 5-lipoxygenase. J Med Chem. 2009;52:3474–3483. doi: 10.1021/jm900212y. [DOI] [PubMed] [Google Scholar]
- 18.Zheng M.F., Zheng M.Y., Ye D.J., Deng Y.M., Qiu S.F., Luo X.M. Indole derivatives as potent inhibitors of 5-lipoxygenase: design, synthesis, biological evaluation, and molecular modeling. Bioorg Med Chem Lett. 2007;17:2414–2420. doi: 10.1016/j.bmcl.2007.02.038. [DOI] [PubMed] [Google Scholar]
- 19.Kramer J.B., Capiris T., Sircar J.C., Connor D.T., Bornemeier D.A., Dyer R.D. Hydroxylamine analogs of 2,6 di-t-butylphenols: dual inhibitors of cyclooxygenase and 5-lipoxygenase or selective 5-lipoxygenase inhibitors. Bioorg Med Chem. 1995;3:403–410. doi: 10.1016/0968-0896(95)00027-e. [DOI] [PubMed] [Google Scholar]
- 20.Ruiz J., Pérez C., Pouplana R. QSAR study of dual cyclooxygenase and 5-lipoxygenase inhibitors 2,6-di-tert-butylphenol derivatives. Bioorg Med Chem. 2003;11:4207–4216. doi: 10.1016/s0968-0896(03)00449-8. [DOI] [PubMed] [Google Scholar]
- 21.Cuadro A.M., Valenciano J., Vaquero J.J., Alvarez-Builla J., Sunkel C., de Casa-Juana M.F. Synthesis and biological evaluation of 2,6-di-tert-butylphenol hydrazones as 5-lipoxygenase inhibitors. Bioorg Med Chem. 1998;6:173–180. doi: 10.1016/s0968-0896(97)10018-9. [DOI] [PubMed] [Google Scholar]
- 22.Hu J.T., Chen S., Sun Y.H., Yang J., Rao Y. Synthesis of tri- and tetrasubstituted pyrazoles via Ru(II) catalysis: intramolecular aerobic oxidative C–N coupling. Org Lett. 2012;14:5030–5033. doi: 10.1021/ol3022353. [DOI] [PubMed] [Google Scholar]
- 23.Winum J.Y., Dogné J.M., Casini A., de Leval X., Montero J.L., Scozzafava A. Carbonic anhydrase inhibitors: synthesis and inhibition of cytosolic/membrane-associated carbonic anhydrase isozymes I, II, and IX with sulfonamides incorporating hydrazino moieties. J Med Chem. 2005;48:2121–2125. doi: 10.1021/jm0494826. [DOI] [PubMed] [Google Scholar]
- 24.Robinson B.S., Rathjen D.A., Trout N.A., Easton C.J., Ferrante A. Inhibition of neutrophil leukotriene B4 production by a novel synthetic N-3 polyunsaturated fatty acid analogue, β-oxa 21:3n-3. J Immunol. 2003;171:4773–4779. doi: 10.4049/jimmunol.171.9.4773. [DOI] [PubMed] [Google Scholar]
- 25.van Leyen K., Arai K., Jin G., Kenyon V., Gerstner B., Rosenberg P.A. Novel lipoxygenase inhibitors as neuroprotective reagents. J Neurosci Res. 2008;86:904–909. doi: 10.1002/jnr.21543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ellington J.C., Arnett E.M. Kinetics and thermodynamics of phenolate silylation and alkylation. J Am Chem Soc. 1988;110:7778–7785. [Google Scholar]
- 27.Zhou Y., Li J., Liu H., Zhao L.X., Jiang H.L. An unusual de-nitro reduction of 2-substituted-4-nitroquinolines. Tetrahedron Lett. 2006;47:8511–8514. [Google Scholar]
- 28.Pufahl R.A., Kasten T.P., Hills R., Gierse J.K., Reitz B.A., Weinberg R.A. Development of a fluorescence-based enzyme assay of human 5-lipoxygenase. Anal Biochem. 2007;364:204–212. doi: 10.1016/j.ab.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 29.Kolb J.P., Paul-Eugene N., Damais C., Yamaoka K., Drapier J.C., Dugas B. Interleukin-4 stimulates cGMP production by IFN-γ-activated human monocytes. Involvement of the nitric oxide synthase pathway. J Biol Chem. 1994;269:9811–9816. [PubMed] [Google Scholar]
- 30.Kimura Y., Dargusch R., Schubert D., Kimura H. Hydrogen sulfide protects HT22 neuronal cells from oxidative stress. Antioxid Redox Signal. 2006;8:661–670. doi: 10.1089/ars.2006.8.661. [DOI] [PubMed] [Google Scholar]
- 31.Fukui M., Song J.H., Choi J., Choi H.J., Zhu B.T. Mechanism of glutamate-induced neurotoxicity in HT22 mouse hippocampal cells. Eur J Pharmacol. 2009;617:1–11. doi: 10.1016/j.ejphar.2009.06.059. [DOI] [PubMed] [Google Scholar]
- 32.Cheng J., Uchida M., Zhang W.R., Grafe M.R., Herson P.S., Hurn P.D. Role of salt-induced kinase 1 in androgen neuroprotection against cerebral ischemia. J Cereb Blood Flow Metab. 2011;31:339–350. doi: 10.1038/jcbfm.2010.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kitano H., Young J.M., Cheng J., Wang L., Hurn P.D., Murphy SJ. Gender-specific response to isoflurane preconditioning in focal cerebral ischemia. J Cereb Blood Flow Metab. 2007;27:1377–1386. doi: 10.1038/sj.jcbfm.9600444. [DOI] [PMC free article] [PubMed] [Google Scholar]