

Abstract
Cancer stem cells (CSCs) are undifferentiated cancer cells with a high tumorigenic activity, the ability to undergo self‐renewal, and a multilineage differentiation potential. Cancer stem cells are responsible for the development of tumor cell heterogeneity, a key feature for resistance to anticancer treatments including conventional chemotherapy, radiation therapy, and molecularly targeted therapy. Furthermore, minimal residual disease, the major cause of cancer recurrence and metastasis, is enriched in CSCs. Cancer stem cells also possess the property of “robustness”, which encompasses several characteristics including a slow cell cycle, the ability to detoxify or mediate the efflux of cytotoxic agents, resistance to oxidative stress, and a rapid response to DNA damage, all of which contribute to the development of therapeutic resistance. The identification of mechanisms underlying such characteristics and the development of novel approaches to target them will be required for the therapeutic elimination of CSCs and the complete eradication of tumors. In this review, we focus on two prospective therapeutic approaches that target CSCs with the aim of disrupting their quiescence or redox defense capability.
Keywords: CD44, Fbw7, intratumoral heterogeneity, niche, plasticity
Cancer Stem Cells and Intratumoral Heterogeneity
Heterogeneity of tumor tissue is highly associated with failure of conventional anticancer therapies. Tumors have been found to be composed of genetically distinct clones of cancer cells that arise in the face of selection pressure imposed by the tumor microenvironment. Even genetically homogeneous tumor cells show different patterns of gene expression.1, 2 Intratumoral heterogeneity is thus generated by a combination of genetic and functional diversities and is consequently highly complex.
Normal tissues are constructed from heterogeneous cell types that are derived from tissue stem cells and they develop in a hierarchical manner.3 Such heterogeneity is determined by differential gene expression, which is itself under precise and programmed epigenetic control.4 Recent studies have suggested that tumors also show cellular hierarchy, with a subpopulation of cancer cells having a tumorigenic potential much greater than that of other cancer cells. This highly tumorigenic subpopulation of cells at the top of the hierarchy comprises CSCs and gives rise to progenitors and cells at various levels of differentiation along various lineages in a manner similar to that of normal tissue stem cells (Fig. 1a). Before the CSC theory became widely accepted, tumor heterogeneity was thought to result predominantly from the stochastic accumulation of genetic mutations.3 Both genetic and epigenetic mechanisms are now thought to contribute to tumor heterogeneity in a parallel manner.
Figure 1.
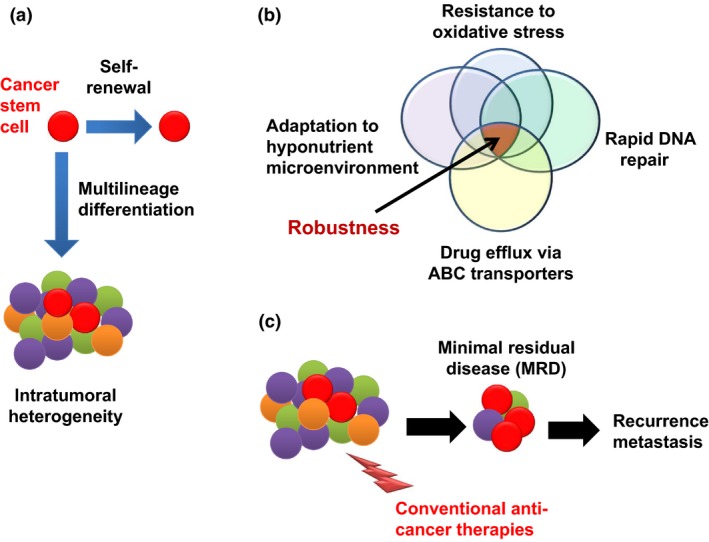
Biological characteristics of cancer stem cells. Cancer stem cells possess both self‐renewal ability and multilineage differentiation potential, leading to the composition of intratumoral heterogeneity (a). Cancer stem cells possess the property of “robustness,” which is established by a combination of various phenotypes (b). Cancer stem cells are more resistant to various therapeutic interventions, leading to the generation of minimal residual disease (MRD) that is mainly composed by CSCs, and MRD is a major cause of recurrence and metastasis (c). ABC, ATP‐binding cassette.
Human CSCs were first identified in AML as the CD34+/CD38− cell subpopulation by transplantation into immunodeficient mice.5, 6 Since then, several approaches have been adopted to distinguish CSCs from other cancer cells, including surface marker characterization, sphere formation assays,7, 8 analysis of persistent tumorigenic potential after serial transplantation,9 and side‐population detection.10 Flow cytometric analysis of surface markers has been applied to the detection of breast CSCs that are enriched within a cell subpopulation that is CD44high/CD24low/aldehyde dehydrogenase‐1high.10 It is important to bear in mind, however, that no markers have been identified to date that are expressed only in CSCs.7, 11 CD133 (also known as prominin 1, or PROM1) has long been used to identify CSCs,12 but cancer cells negative for this glycoprotein have also been shown to possess tumorigenic potential, which questions the legitimacy of CD133 as a bona fide CSC marker.13 Thus, the functional characterization of cell subpopulations defined by putative CSC markers is thus crucial for CSC research.
Cancer stem cells possess the property of “robustness”, which refers to several biological characteristics including resistance to redox stress,14, 15 the capacity to carry out rapid repair of damaged DNA,16, 17 the ability to adapt to a hyperinflammatory or hyponutritious microenvironment,18, 19 plasticity in the transition to transit‐amplifying cells,20 metabolic reprogramming,15, 21, 22 and an enhanced ability to expel anticancer drugs through ATP‐binding cassette transporters23 (Fig. 1b). Such characteristics of CSCs are responsible for the formation of MRD,24, 25 corresponding to clinically undetectable lesions enriched in CSCs that remain after therapy and that later give rise to progression of cancer as a relapse or distant metastasis (Fig. 1c). Indeed, the chemotherapy‐induced formation of MRD has been shown to be accompanied by CSC enrichment in diverse mechanisms. The number of CSCs in mouse pancreatic ductal adenocarcinoma increased as a result of metabolic reprogramming after transient ablation of oncogenic mutant KRAS (G12D).26 In another instance, keratin14‐positive bladder CSCs in the dormant state were induced to proliferate on exposure to prostaglandin E2 released from non‐CSC cancer cells undergoing apoptosis in response to anticancer agents.27 It was also reported that cell subpopulations positive for CSC markers increased after chemotherapy for both liver cancer and osteosarcoma occurring simultaneously in a patient with Li–Fraumeni syndrome.28 Dynamic changes in CSCs after chemotherapy have thus attracted much attention as predictors of therapeutic efficacy and prognosis.
The Niche, a Favorable Microenvironment for CSCs to Maintain their Stemness
Normal tissue stem cells are located within or adjacent to a microenvironment, known as the “niche,” that is favorable for the maintenance of their stemness. Niches are composed of various cell types as well as ECM, cytokines, and growth factors released by the niche cells. For instance, Paneth cells located in intestinal crypts and melanocyte stem cells located in the bulge area of hair follicles form niches for normal intestinal stem cells and hair follicle stem cells, respectively.29, 30 Cancer stem cells have also been shown to possess niches whose components include endothelial cells, osteoblasts, and ECM molecules composed of osteopontin and hyaluronic acid.31 In addition, cancer‐associated fibroblasts, tumor‐associated macrophages, undifferentiated mesenchymal stem cells, and immune cells in the tumor stroma serve as niches for CSCs by providing growth factors such as transforming growth factor‐β, epidermal growth factor, and hepatocyte growth factor as well as pro‐inflammatory cytokines such as tumor necrosis factor‐α and various interleukins including IL‐1β and IL‐6.32, 33 The inflammatory microenvironment is beneficial for cancer cells in that it results in activation of the NF‐κB signaling pathway.34 The cytokine network not only promotes tumor development but also maintains CSC characteristics that underlie tumor metastasis and recurrence.
Accumulating evidence thus supports the importance of a cellular niche for maintenance of the stem cell pool.29, 30, 35, 36 Lineage tracing has suggested that Paneth cells are required for the support not only of Lgr5‐expressing normal stem cells in the intestine but also of APC‐mutant adenoma‐initiating cells.35 Furthermore, melanocytic stem cells localized to the secretory portion of sweat glands in the volar skin are responsible for the development of acral melanoma associated with amplification of the driver oncogene CCND1, which encodes cyclin D1.36 These findings suggest that some CSCs are derived from normal tissue stem cells and share their niche.
Glioma stem cells reside in contact with endothelial cells, in what is referred to as a perivascular niche.37 On the other hand, a hypoxic or perinecrotic microenvironment has been found to be advantageous for the survival and proliferation of other types of CSCs.38, 39 Both HIF1 and HIF2α are activated under hypoxic conditions and promote the stem‐like properties of cancer cells. Furthermore, HIF2α was recently shown to contribute in a cooperative manner with the intracellular domain of CD44 generated by γ‐secretase to the acquisition of radioresistance by glioma stem cells in a perivascular niche rich in osteopontin.40
The concept of the niche is important in terms of the “seed and soil” theory of tumor dissemination proposed by Paget, which stipulates that the distant metastasis of cancer cells is dependent on the site of the primary tumor.41 Such organ‐selective tumor metastasis is thought to be established by homing of CSCs present among CTCs to a premetastatic niche (Fig. 2). For instance, progressive prostate cancer frequently metastasizes to bone in association with the production by the cancer cells of parathyroid hormone‐related protein. This protein influences bone remodeling, which may promote the homing of prostate cancer cells to bone marrow and their occupation of the osteoblastic niches for HSCs.42, 43 Furthermore, CXCL12, an osteoblast‐secreted cytokine, interacts with its receptor CXCR4, located on the prostate cancer cell surface, leading to homing of the cancer cells to the bone more efficiently than do HSCs,42 suggesting that the axis might promote CSC survival and proliferation. Indeed, neutralizing antibodies to CXCR4 were found to be effective for prevention of prostate cancer metastasis to the premetastatic niche.44
Figure 2.
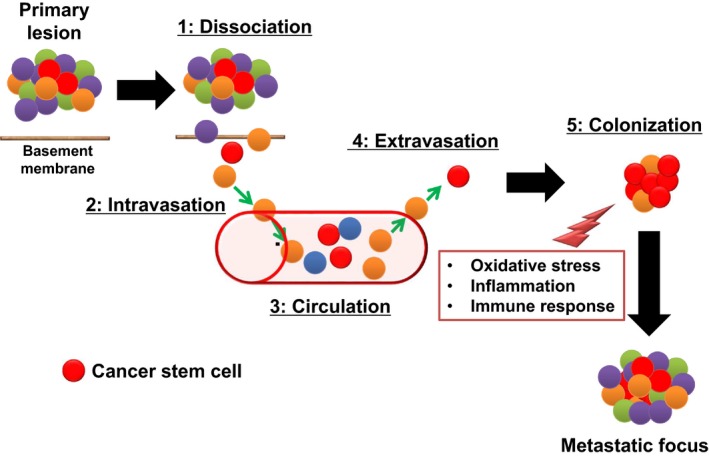
Steps in the development of distant metastasis based on cancer stem cell theory. Distant metastasis of tumor cells occurs by way of several distinct steps: dissociation of cancer cells from the primary tumor after they have undergone epithelial–mesenchymal transition (1) is followed by their intravasation (2), circulation in the blood as CTCs (3), extravasation (4), and homing to the premetastatic niche and colonization of the metastatic site (5). Organ‐selective tumor metastasis may depend on whether the premetastatic niche is a favorable microenvironment for circulating cancer stem cells.
The process by which circulating tumor cells return to the primary tumor is termed “tumor self‐seeding”.45 Indeed, CTCs circulate in the bloodstream in the whole body and show dissemination, seeding not only distant sites but also their primary tumor site.46 Circulating tumor cells can colonize their own tumors of origin because the primary lesion provides a familiar microenvironment and factors for CTCs. Such cancer cell dynamics implies the importance of the niche as a foundation for expansion of the primary tumor as well as establishment of distant metastases. Recent findings show that self‐seeding is mediated by “aggressive” CTCs that highly express MMP 1, collagenase‐1, and the actin cytoskeleton component fascin‐1.47 Therefore, it is possible that a fraction of CTCs alter the microenvironment to make it more suitable for CSCs to form tumors in both the primary lesion and distant organs.
Molecular Mechanisms Underlying Plasticity of CSCs
Originally, CSCs were defined as a static entity and thought to stably locate at the top of the hierarchy among tumor cells. However, it is becoming more acceptable that CSCs undergo dynamic and reversible changes depending on the surrounding microenvironment. This is referred to as the dynamic stemness model.48 Epigenetic changes induced by various factors including chronic inflammation, excessive redox stress, and hypoxic stimuli enhance the plasticity of the transition between CSCs and non‐CSCs.49 Indeed, the dual nature of CSCs makes them possible to show hyperadaptation to the tumor microenvironment. This plasticity of CSCs is thought to hinder the identification of CSCs clinically in vivo.50, 51
Importantly, a treatment‐induced transient decrease in the extent of cancer heterogeneity has been found to reflect enrichment of CSCs. Such CSC enrichment is caused by not only selection of therapeutic resistant CSCs but also induction of CSC properties in non‐CSCs. For example, long‐term treatment with vemurafenib, a drug targeted to the V600E active mutant form of the protein kinase BRAF, upregulates expression in melanoma of the histone demethylase Jumonji AT‐rich interactive domain 1B protein, an enzyme that is highly expressed in slow‐cycling melanoma cells with stem‐like properties.52 Cancer stem cells are generally slow‐cycling or dormant under unfavorable conditions, which is why melanoma CSCs exposed to vemurafenib lose their addiction to oncogenic BRAF(V600E)‐mediated signaling and are responsible for the development of adaptive resistance of the tumor.52 Cross‐talk between signaling pathways and reversible epigenetic changes are thus thought to give rise to adaptive resistance to exogenous stress or anticancer treatment. It is therefore expected that combination therapies with two or more molecularly targeted drugs will be required for eradication of cancer.
Furthermore, it has recently been reported that relatively differentiated progenitor cells outside of the intestinal crypt niche are responsible for intestinal tumorigenesis through the activation of Wnt or NF‐κB signal transduction.53, 54 Given that Paneth cells correspond to the “cellular niche” for Lgr5‐positive intestinal stem cells which have been shown to form adenoma by cell‐lineage tracing analysis.35 Remarkably, aberrant expression of the bone morphogenetic antagonist leads to altering cell fate determination by promoting the dedifferentiation of Lgr5‐negative progenitor cells.53 It is also notable that the constitutive activation of Wnt or the NF‐κB signaling pathway leads to emergence of CSCs derived from non‐stem cells in normal tissue.54 These reports strongly suggest that the origin of CSCs is not necessarily a normal tissue stem cell.
Novel CSC‐Targeted Therapies: Targeting of CSC Quiescence
The dormant status of CSCs has long been thought to reduce their susceptibility to chemotherapy. Mitotic inhibitors such as paclitaxel and vincristine preferentially kill proliferating cancer cells during M phase of the cell cycle. Antimetabolite drugs such as 5‐fluorouracil, 6‐mercaptopurine, and methotrexate damage cancer cells during S phase. Topoisomerase inhibitors such as irinotecan (CPT‐11) and etoposide (VP‐16) interfere with the separation of DNA strands during DNA replication and transcription.55 These agents, however, exhibit anticancer effects only when tumor cells are under proliferative conditions. By striking contrast, CSCs in the quiescent state (G0 phase of the cell cycle) are thus refractory to such conventional anticancer drugs whose action is dependent on operation of the cell cycle.
The cell cycle status of CSCs is determined by many factors. Endogenous cyclin‐dependent kinase inhibitors and Fbw7 can contribute to the cell cycle delay or arrest manifest in CSCs.56 Fbw7 is a component of E3 ubiquitin ligase and promotes the ubiquitin–proteasome‐dependent degradation of several proto‐oncogene products such as c‐Myc, cyclin E, Notch, and JUN.57 Thus, Fbw7 inactivation triggers “awakening” of quiescent CSCs in the niche (Fig. 3). A recent study uncovered an important role for the cyclin‐dependent kinase inhibitor p57, which is expressed at a high level in HSCs, in the maintenance of quiescence in these cells. Ablation of p57 in HSCs was thus found to induce the aberrant proliferation of these cells in bone marrow and consequent HSC exhaustion.58
Figure 3.
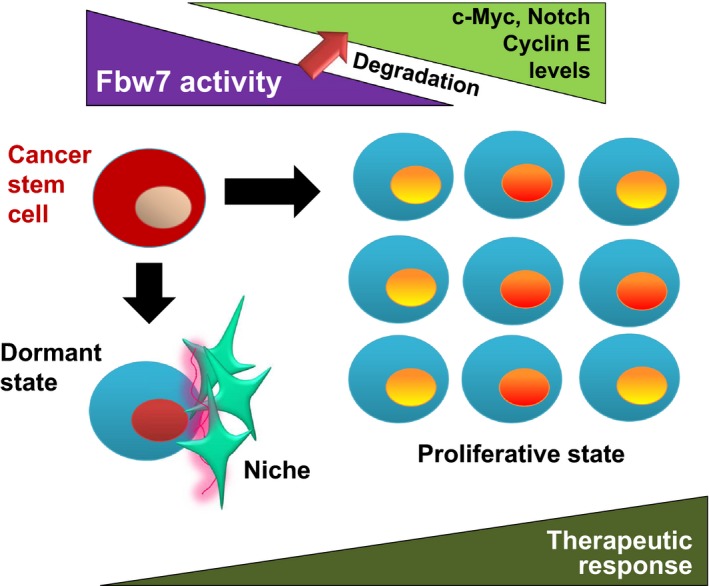
Mechanism by which cancer stem cells become quiescent. F‐box and WD40 repeat domain‐containing protein 7 (Fbw7), a subunit of an SCF‐type ubiquitin ligase, negatively regulates cell cycle progression by promoting the ubiquitin‐dependent proteasomal degradation of c‐Myc, Notch, and cyclin E. For instance, leukemia stem cells that lack Fbw7 activity become much more proliferative than hematopoietic stem cells as a result of the accumulation of such driver molecules of the cell cycle. Conversely, upregulation of Fbw7 induces cell cycle arrest, leading to the dormant state of cancer stem cells.
The IGF family of proteins has also been implicated in acquired or adaptive resistance of CSCs to conventional anticancer therapies. Repeated irradiation enhances the self‐renewal potential of glioma stem cells by increasing IGF1 secretion and upregulating expression of the IGF type 1 receptor. Consequent chronic receptor activation results in inhibition of the phosphatidylinositol 3‐kinase‐Akt signaling pathway, which in turn activates the transcription factor FoxO3a, leading to a slowing of the cell cycle. Acute irradiation of the slow‐cycling CSCs, however, induces rapid activation of IGF1–Akt survival signaling and thereby further promotes radioprotection.59 Chemotherapy was also found to induce excess IGF2 expression that paradoxically leads to a dormant growth state in osteosarcoma cells maintained in the absence of serum and thereby promotes survival and confers resistance to various treatments.60 Together, these findings suggest that blockade of aberrant IGF signaling is a potential novel therapeutic strategy to selectively attack quiescent CSCs of glioma and osteosarcoma.
Fbw7 has also attracted much attention as a potential novel target for CSC elimination. This protein is a subunit of a ubiquitin ligase responsible for the degradation of the proto‐oncoprotein c‐Myc.61, 62 In addition to the stabilization of c‐Myc, Fbw7 shows antimetastatic function through regulation of the NOTCH1–CCL2 axis in tumor stroma.63
Many patients with CML treated with imatinib, a drug targeted to the oncogenic fusion protein produced by the Philadelphia chromosome,64 eventually develop acquired resistance to this tyrosine kinase inhibitor. Chronic myeloid leukemia stem cells that are resistant to this agent as a result of their quiescence, and which are responsible for MRD, express Fbw7 at a high level, which results in the degradation of c‐Myc by the ubiquitin–proteasome system and cell cycle arrest. Ablation of Fbw7 in imatinib‐resistant CML cells was found to markedly enhance the anticancer effect of this drug in mice, with the loss of Fbw7 expression resulting in molecular stabilization of c‐Myc and consequent induction of cell proliferation (Fig. 3).65 Intriguingly, heterogeneity of Fbw7 expression has been detected at the invasive front of solid tumors such as gastric and breast cancer, which show a collective cell migration pattern.66 This heterogeneity might reflect coexistence of dormant and proliferative cancer cells at the invasive front.
The “locked‐out” therapeutic strategy combining Fbw7 inhibition with conventional anticancer agents to lock CSCs out of G0 dormant phase is thus potentially effective for overcoming the low susceptibility of CSCs to anticancer drugs, but its possible side‐effects will need to be fully investigated before its clinical implementation. The inhibition of Fbw7 and consequent upregulation of c‐Myc might promote tumor cell proliferation before the combined modality therapy is able to eliminate CSCs. By contrast, “locked‐in” therapy might be expected to prevent further tumor growth as well as relapse due to MRD if the proliferative potential of CSCs remains inactivated for the lifetime of the patient.
Novel CSC‐Targeted Therapies: Targeting of CSC Resistance to Oxidative Stress
The adhesion molecule CD44, which binds to osteopontin and hyaluronic acid,67 has recently been identified as a CSC marker.68, 69 Alternative splicing of the CD44 gene results in the generation of various CD44 isoforms, which are classified as either CD44 standard or CD44v isoforms according to the absence or presence of sequences encoded by variant exons.70 The isoforms CD44v8–10 and CD44v6 have been shown to enhance the metastatic potential of colon cancer and melanoma cells, respectively.71, 72 CD44v6 interacts with c‐Met, a receptor tyrosine kinase that binds hepatocyte growth factor, and thereby increases the potential of melanoma cells to migrate to the brain.72 Epithelial splicing regulatory protein 1 (ESRP1), an RNA binding protein, as well as heterochromatin protein 1γ, an epigenetic modulator, contribute to the alternative splicing of CD44 pre‐mRNA.73, 74 Three‐dimensional culture experiments have revealed that both normal and cancer cells change the splicing pattern of CD44 to upregulate CD44v expression during the formation and maintenance of organoids or spheroids in ECM,75, 76 suggesting that expression of variant forms is associated with epithelial organization.
We have shown that CD44v including sequences encoded by variant exons 8, 9, and 10 (CD44v8–10) interacts with and stabilizes the protein xCT at the cell membrane. This latter protein, together with CD98 heavy chain, forms an antiporter known as system Xc(−) that exchanges intracellular glutamate for extracellular cystine.77 Cysteine as well as glycine and glutamate are essential substrates for synthesis of GSH. CD44v8–10 thus promotes GSH synthesis by increasing the import of cystine and thereby increasing the intracellular concentration of cysteine.14 The elimination of ROS by GSH inhibits the activation of p38 MAPK signaling78 and thereby prevents ROS‐induced senescence, apoptosis, or differentiation of cancer cells. The CD44v8–10–xCT–GSH axis thus protects CSCs from redox stress (Fig. 4).
Figure 4.
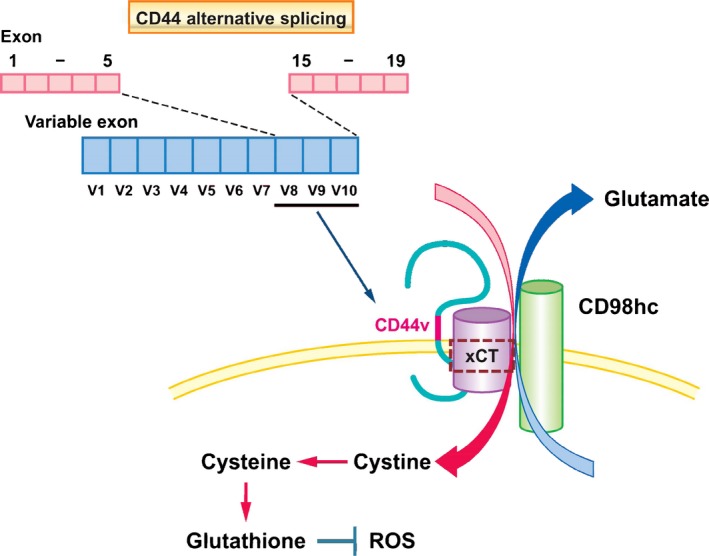
Function of CD44 variant isoform (CD44v) in promoting resistance to oxidative stress. Alternative splicing of the CD44 gene results in the generation of multiple protein isoforms. CD44v8–10 is overexpressed in epithelial cancer stem cells, and their colocalization with the xCT subunit of system Xc(−), a glutamate/cystine antiporter, promotes the uptake of cystine and the consequent synthesis of the antioxidant glutathione, which reduces reactive oxygen species (ROS).
Regulation of oxidative stress is thought to be important not only for therapeutic resistance but also for the metastatic potential of cancer. Highly metastatic 4T1 mouse breast cancer cells include a subpopulation positive for CD44v8–10, and ESRP1‐depleted 4T1 cells form significantly fewer nodules and smaller metastatic foci in lungs after their injection into mammary fat pads compared with control 4T1 cells. Furthermore, microarray analysis revealed that ESRP1‐positive cancer cells were more undifferentiated than ESRP1‐negative cells, consistent with the notion that CD44v8–10‐positive cancer cells have characteristics of CSCs, serving as the cell of origin for metastatic lesions in this model.79 Differential expression of ESRP1 may thus determine CSC robustness by affecting resistance to oxidative stress.
The expression of ESRP1 is regulated by epigenetic factors. Trimethylation of lysine 4 residue on histone H3 (H3K4me3) is recognized in the promoter region of ESRP1 gene of CD44v8–10‐positive 4T1 cells. In contrast, H3K27 residues in the promoter region of this gene are trimethylated in CD44v8–10‐negative 4T1 cells.79 Those 4T1 cells positive for ESRP1‐induced CD44v8–10 expression also have greater tumorigenic potential compared with those negative for such expression. Indeed, clinical data suggest that ESRP1 is a marker for poor prognosis in breast cancer.79 Epigenetic regulation of ESRP1 expression is therefore a potential therapeutic target. However, given that ESRP1 regulates the alternative splicing of p120 and fibroblast growth factor receptor genes in addition to that of the CD44 gene,79, 80 the altered regulation of alternative splicing by ESRP1 might also give rise to unwanted side‐effects.
Sulfasalazine, a drug given for the treatment of rheumatoid arthritis or inflammatory bowel disease,81 inhibits the transport activity of xCT. Given that targeting of xCT with sulfasalazine increased the sensitivity of CD44v8–10‐positive cancer cells to ROS,14 we are currently carrying out clinical trials using this drug for patients with advanced gastric adenocarcinomas and non‐small‐cell lung cancer without driver gene mutations. Furthermore, the combination of sulfasalazine and auranofin, both disease‐modifying antirheumatic drugs, were reported to inhibit cysteine uptake by xCT transporter and compensatory‐activated Nrf2‐dependent anti‐ROS machinery.82
Of note, CSCs of head and neck squamous cell carcinoma that had survived treatment with an epidermal growth factor receptor‐targeted drug, cetuximab, were found to be sensitive to the induction of death by sulfasalazine. The cetuximab‐resistant cancer cells tended to be undifferentiated and CD44v8–10‐positive, whereas sulfasalazine‐resistant cancer cells were relatively differentiated and CD44v8–10‐negative.83 Combination therapy with both drugs might therefore prove effective for the elimination of heterogeneous tumor tissue.
In addition to being more susceptible to redox stress, CD44v‐negative cancer cells tend to show a higher level of ROS‐induced signaling by the Wnt/β‐catenin pathway compared with CD44v‐positive cancer cells. Furthermore, there is an inverse relation between the expression of CD44v8–10, which is a CSC marker, and that of c‐Myc, which is a target of canonical Wnt signaling.66 This negative relation between CD44v8–10 expression and ROS‐induced canonical Wnt signaling might support the survival and proliferation of CSCs under the evolutionary selection pressure of oxidative stress in the tumor microenvironment.66 Collectively, these various observations suggest that CD44v is not only a promising diagnostic or prognostic marker but also a therapeutic target for various types of cancer. Given that CD44v expression reflects the cellular heterogeneity of tumor tissue, novel therapeutic strategies targeted to CD44v would be expected to reduce both tumor heterogeneity and resistance to conventional anticancer therapies, and thereby to limit relapse and distant metastasis.
Conclusions
The development of molecularly targeted drugs to destroy CSCs has been pursued as a “silver bullet” for eradication of cancer composed of heterogeneous cell populations. However, such strategies would not be expected to be successful if they do not take into account the roles of stromal cells such as cancer‐associated fibroblasts and tumor‐associated macrophages as well as reversible transitions between CSCs and non‐CSCs. Furthermore, inhibition of a single signal transduction pathway to which CSCs are addicted eventually becomes ineffective as a result of the activation of alternative survival pathways, and consequently induces the paradoxical enrichment of CSCs in MRD after chemotherapy. This phenomenon of adaptive resistance highlights the importance of simultaneous blockade of multiple signaling pathways. Therapeutic approaches to overcome the robustness of CSCs and their incorporation into regimens that simultaneously target both CSCs and non‐CSCs are urgently required.
Disclosure Statement
H.S. received research grants from Daiichi Sankyo Inc. and Eisai Co. Ltd. G.J.Y. declares no conflict of interest.
Abbreviations
- CD44v
CD44 variant
- CML
chronic myeloid leukemia
- CSC
cancer stem cell
- CTC
circulating tumor cell
- CXCR4
C‐X‐C chemokine receptor type 4
- ECM
extracellular matrix
- ESRP1
epithelial splicing regulatory protein 1
- Fbw7
F‐box and WD40 repeat domain‐containing protein 7
- GSH
glutathione
- HIF
hypoxia‐inducible factor
- HSC
hematopoietic stem cell
- IGF
insulin‐like growth factor
- Lgr5
leucine‐rich repeat‐containing G‐protein coupled receptor 5
- MRD
minimal residual disease
- NF‐κB
nuclear factor‐κB
- ROS
reactive oxygen species
Acknowledgments
We thank Dr. Oltea Sampetrean for critical reading of this manuscript. This work was supported by a grant from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (to H.S.).
Cancer Sci 107 (2016) 5–11
Funding Information
Ministry of Education, Culture, Sports, Science, and Technology of Japan.
References
- 1. Fisher R, Pusztai L, Swanton C. Cancer heterogeneity: implications for targeted therapeutics. Br J Cancer 2013; 108: 479–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gerlinger M, Rowan AJ, Horswell S et al Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012; 366: 883–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer 2012; 12: 133–43. [DOI] [PubMed] [Google Scholar]
- 4. Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem‐like states, and drug resistance. Mol Cell 2014; 54: 716–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lapidot T, Sirard C, Vormoor J et al A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994; 367: 645–8. [DOI] [PubMed] [Google Scholar]
- 6. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997; 3: 730–7. [DOI] [PubMed] [Google Scholar]
- 7. Jiang X, Gwye Y, Russell D et al CD133 expression in chemo‐resistant Ewing sarcoma cells. BMC Cancer 2010; 10: 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kanwar SS, Yu Y, Nautiyal J, Patel BB, Majumdar AP. The Wnt/beta‐catenin pathway regulates growth and maintenance of colonospheres. Mol Cancer 2010; 9: 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sugihara E, Shimizu T, Kojima K et al Ink4a and Arf are crucial factors in the determination of the cell of origin and the therapeutic sensitivity of Myc‐induced mouse lymphoid tumor. Oncogene 2012; 31: 2849–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Charafe‐Jauffret E, Ginestier C, Birnbaum D. Breast cancer stem cells: tools and models to rely on. BMC Cancer 2009; 9: 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rocco A, Liguori E, Pirozzi G et al CD133 and CD44 cell surface markers do not identify cancer stem cells in primary human gastric tumors. J Cell Physiol 2012; 227: 2686–93. [DOI] [PubMed] [Google Scholar]
- 12. Sanai N, Alvarez‐Buylla A, Berger MS. Neural stem cells and the origin of gliomas. N Engl J Med 2005; 353: 811–22. [DOI] [PubMed] [Google Scholar]
- 13. LaBarge MA, Bissell MJ. Is CD133 a marker of metastatic colon cancer stem cells? J Clin Investig 2008; 118: 2021–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ishimoto T, Nagano O, Yae T et al CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(−) and thereby promotes tumor growth. Cancer Cell 2011; 19: 387–400. [DOI] [PubMed] [Google Scholar]
- 15. Wu Z, Wei D, Gao W et al TPO‐induced metabolic reprogramming drives liver metastasis of colorectal cancer CD110 + tumor‐initiating cells. Cell Stem Cell 2015; 17: 47–59. [DOI] [PubMed] [Google Scholar]
- 16. Maugeri‐Sacca M, Bartucci M, De Maria R. DNA damage repair pathways in cancer stem cells. Mol Cancer Ther 2012; 11: 1627–36. [DOI] [PubMed] [Google Scholar]
- 17. Skvortsov S, Debbage P, Lukas P, Skvortsova I. Crosstalk between DNA repair and cancer stem cell (CSC) associated intracellular pathways. Semin Cancer Biol 2015; 31: 36–42. [DOI] [PubMed] [Google Scholar]
- 18. Ishimoto T, Oshima H, Oshima M et al CD44 + slow‐cycling tumor cell expansion is triggered by cooperative actions of Wnt and prostaglandin E2 in gastric tumorigenesis. Cancer Sci 2010; 101: 673–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yoshida GJ, Saya H. EpCAM expression in the prostate cancer makes the difference in the response to growth factors. Biochem Biophys Res Commun 2014; 443: 239–45. [DOI] [PubMed] [Google Scholar]
- 20. Weinberg R, Fisher DE, Rich J. Dynamic and transient cancer stem cells nurture melanoma. Nat Med 2010; 16: 758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shen YA, Wang CY, Hsieh YT, Chen YJ, Wei YH. Metabolic reprogramming orchestrates cancer stem cell properties in nasopharyngeal carcinoma. Cell Cycle 2015; 14: 86–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Saga I, Shibao S, Okubo J et al Integrated analysis identifies different metabolic signatures for tumor‐initiating cells in a murine glioblastoma model. Neuro‐oncology 2014; 16: 1048–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dean M. ABC transporters, drug resistance, and cancer stem cells. J Mammary Gland Biol Neoplasia 2009; 14: 3–9. [DOI] [PubMed] [Google Scholar]
- 24. Meads MB, Gatenby RA, Dalton WS. Environment‐mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer 2009; 9: 665–74. [DOI] [PubMed] [Google Scholar]
- 25. Creighton CJ, Li X, Landis M et al Residual breast cancers after conventional therapy display mesenchymal as well as tumor‐initiating features. Proc Natl Acad Sci USA 2009; 106: 13820–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Viale A, Pettazzoni P, Lyssiotis CA et al Oncogene ablation‐resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014; 514: 628–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kurtova AV, Xiao J, Mo Q et al Blocking PGE‐induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 2015; 517: 209–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yoshida GJ, Fuchimoto Y, Osumi T et al Li–Fraumeni syndrome with simultaneous osteosarcoma and liver cancer: increased expression of a CD44 variant isoform after chemotherapy. BMC Cancer 2012; 12: 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sato T, van Es JH, Snippert HJ et al Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 2011; 469: 415–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rabbani P, Takeo M, Chou W et al Coordinated activation of Wnt in epithelial and melanocyte stem cells initiates pigmented hair regeneration. Cell 2011; 145: 941–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guerrouahen BS, Al‐Hijji I, Tabrizi AR. Osteoblastic and vascular endothelial niches, their control on normal hematopoietic stem cells, and their consequences on the development of leukemia. Stem Cells Int 2011; 2011: 375857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. De Veirman K, Rao L, De Bruyne E et al Cancer associated fibroblasts and tumor growth: focus on multiple myeloma. Cancers 2014; 6: 1363–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jinushi M, Baghdadi M, Chiba S, Yoshiyama H. Regulation of cancer stem cell activities by tumor‐associated macrophages. Am J Cancer Res 2012; 2: 529–39. [PMC free article] [PubMed] [Google Scholar]
- 34. Hoesel B, Schmid JA. The complexity of NF‐kappaB signaling in inflammation and cancer. Mol Cancer 2013; 12: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schepers AG, Snippert HJ, Stange DE et al Lineage tracing reveals Lgr5 + stem cell activity in mouse intestinal adenomas. Science 2012; 337: 730–5. [DOI] [PubMed] [Google Scholar]
- 36. Okamoto N, Aoto T, Uhara H et al A melanocyte–melanoma precursor niche in sweat glands of volar skin. Pigment Cell Melanoma Res 2014; 27: 1039–50. [DOI] [PubMed] [Google Scholar]
- 37. Calabrese C, Poppleton H, Kocak M et al A perivascular niche for brain tumor stem cells. Cancer Cell 2007; 11: 69–82. [DOI] [PubMed] [Google Scholar]
- 38. Keith B, Simon MC. Hypoxia‐inducible factors, stem cells, and cancer. Cell 2007; 129: 465–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liang D, Ma Y, Liu J et al The hypoxic microenvironment upgrades stem‐like properties of ovarian cancer cells. BMC Cancer 2012; 12: 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pietras A, Katz AM, Ekstrom EJ et al Osteopontin‐CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell 2014; 14: 357–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ribatti D, Mangialardi G, Vacca A. Stephen Paget and the ‘seed and soil’ theory of metastatic dissemination. Clin Exp Med 2006; 6: 145–9. [DOI] [PubMed] [Google Scholar]
- 42. Shiozawa Y, Pedersen EA, Havens AM et al Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Investig 2011; 121: 1298–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yoneda T, Hiraga T. Crosstalk between cancer cells and bone microenvironment in bone metastasis. Biochem Biophys Res Commun 2005; 328: 679–87. [DOI] [PubMed] [Google Scholar]
- 44. Sun YX, Schneider A, Jung Y et al Skeletal localization and neutralization of the SDF‐1(CXCL12)/CXCR4 axis blocks prostate cancer metastasis and growth in osseous sites in vivo. J Bone Miner Res 2005; 20: 318–29. [DOI] [PubMed] [Google Scholar]
- 45. Norton L, Massague J. Is cancer a disease of self‐seeding? Nat Med 2006; 12: 875–8. [DOI] [PubMed] [Google Scholar]
- 46. Comen E, Norton L, Massague J. Clinical implications of cancer self‐seeding. Nat Rev Clin Oncol 2011; 8: 369–77. [DOI] [PubMed] [Google Scholar]
- 47. Kim MY, Oskarsson T, Acharyya S et al Tumor self‐seeding by circulating cancer cells. Cell 2009; 139: 1315–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Islam F, Qiao B, Smith RA, Gopalan V, Lam AK. Cancer stem cell: fundamental experimental pathological concepts and updates. Exp Mol Pathol 2015; 98: 184–91. [DOI] [PubMed] [Google Scholar]
- 49. Csermely P, Hodsagi J, Korcsmaros T et al Cancer stem cells display extremely large evolvability: alternating plastic and rigid networks as a potential mechanism: network models, novel therapeutic target strategies, and the contributions of hypoxia, inflammation and cellular senescence. Semin Cancer Biol 2015; 30: 42–51. [DOI] [PubMed] [Google Scholar]
- 50. Harris JF, Best MW. Dynamic heterogeneity: metastatic variants to liver are generated spontaneously in mouse embryonal carcinoma cells. Clin Exp Metastasis 1988; 6: 451–62. [DOI] [PubMed] [Google Scholar]
- 51. Roesch A, Fukunaga‐Kalabis M, Schmidt EC et al A temporarily distinct subpopulation of slow‐cycling melanoma cells is required for continuous tumor growth. Cell 2010; 141: 583–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Roesch A, Vultur A, Bogeski I et al Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow‐cycling JARID1B(high) cells. Cancer Cell 2013; 23: 811–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Davis H, Irshad S, Bansal M et al Aberrant epithelial GREM1 expression initiates colonic tumorigenesis from cells outside the stem cell niche. Nat Med 2015; 21: 62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schwitalla S, Fingerle AA, Cammareri P et al Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem‐cell‐like properties. Cell 2013; 152: 25–38. [DOI] [PubMed] [Google Scholar]
- 55. Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol 2010; 17: 421–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cicenas J, Valius M. The CDK inhibitors in cancer research and therapy. J Cancer Res Clin Oncol 2011; 137: 1409–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer 2008; 8: 83–93. [DOI] [PubMed] [Google Scholar]
- 58. Matsumoto A, Takeishi S, Kanie T et al p57 is required for quiescence and maintenance of adult hematopoietic stem cells. Cell Stem Cell 2011; 9: 262–71. [DOI] [PubMed] [Google Scholar]
- 59. Osuka S, Sampetrean O, Shimizu T et al IGF1 receptor signaling regulates adaptive radioprotection in glioma stem cells. Stem Cells 2013; 31: 627–40. [DOI] [PubMed] [Google Scholar]
- 60. Shimizu T, Sugihara E, Yamaguchi‐Iwai S et al IGF2 preserves osteosarcoma cell survival by creating an autophagic state of dormancy that protects cells against chemotherapeutic stress. Cancer Res 2014; 74: 6531–41. [DOI] [PubMed] [Google Scholar]
- 61. Yada M, Hatakeyama S, Kamura T et al Phosphorylation‐dependent degradation of c‐Myc is mediated by the F‐box protein Fbw7. EMBO J 2004; 23: 2116–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cheng Y, Li G. Role of the ubiquitin ligase Fbw7 in cancer progression. Cancer Metastasis Rev 2012; 31: 75–87. [DOI] [PubMed] [Google Scholar]
- 63. Yumimoto K, Akiyoshi S, Ueo H et al F‐box protein FBXW7 inhibits cancer metastasis in a non‐cell‐autonomous manner. J Clin Investig 2015; 125: 621–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schiffer CA. BCR‐ABL tyrosine kinase inhibitors for chronic myelogenous leukemia. N Engl J Med 2007; 357: 258–65. [DOI] [PubMed] [Google Scholar]
- 65. Takeishi S, Matsumoto A, Onoyama I, Naka K, Hirao A, Nakayama KI. Ablation of Fbxw7 eliminates leukemia‐initiating cells by preventing quiescence. Cancer Cell 2013; 23: 347–61. [DOI] [PubMed] [Google Scholar]
- 66. Yoshida GJ, Saya H. Inversed relationship between CD44 variant and c‐Myc due to oxidative stress‐induced canonical Wnt activation. Biochem Biophys Res Commun 2014; 443: 622–7. [DOI] [PubMed] [Google Scholar]
- 67. Goodison S, Urquidi V, Tarin D. CD44 cell adhesion molecules. Mol Pathol 1999; 52: 189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Prince ME, Sivanandan R, Kaczorowski A et al Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci USA 2007; 104: 973–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res 2005; 65: 10946–51. [DOI] [PubMed] [Google Scholar]
- 70. Lynch KW. Consequences of regulated pre‐mRNA splicing in the immune system. Nat Rev Immunol 2004; 4: 931–40. [DOI] [PubMed] [Google Scholar]
- 71. Tanabe KK, Ellis LM, Saya H. Expression of CD44R1 adhesion molecule in colon carcinomas and metastases. Lancet 1993; 341: 725–6. [DOI] [PubMed] [Google Scholar]
- 72. Marzese DM, Liu M, Huynh JL et al Brain metastasis is predetermined in early stages of cutaneous melanoma by CD44v6 expression through epigenetic regulation of the spliceosome. Pigment Cell Melanoma Res 2015; 28: 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Horiguchi K, Sakamoto K, Koinuma D et al TGF‐beta drives epithelial–mesenchymal transition through deltaEF1‐mediated downregulation of ESRP. Oncogene 2012; 31: 3190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Saint‐Andre V, Batsche E, Rachez C, Muchardt C. Histone H3 lysine 9 trimethylation and HP1gamma favor inclusion of alternative exons. Nat Struct Mol Biol 2011; 18: 337–44. [DOI] [PubMed] [Google Scholar]
- 75. Olsson E, Honeth G, Bendahl PO et al CD44 isoforms are heterogeneously expressed in breast cancer and correlate with tumor subtypes and cancer stem cell markers. BMC Cancer 2011; 11: 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yoshida GJ, Saya H, Zouboulis CC. Three‐dimensional culture of sebaceous gland cells revealing the role of prostaglandin E2‐induced activation of canonical Wnt signaling. Biochem Biophys Res Commun 2013; 438: 640–6. [DOI] [PubMed] [Google Scholar]
- 77. Bassi MT, Gasol E, Manzoni M et al Identification and characterisation of human xCT that co‐expresses, with 4F2 heavy chain, the amino acid transport activity system xc. Pflugers Arch 2001; 442: 286–96. [DOI] [PubMed] [Google Scholar]
- 78. Ito K, Hirao A, Arai F et al Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med 2006; 12: 446–51. [DOI] [PubMed] [Google Scholar]
- 79. Yae T, Tsuchihashi K, Ishimoto T et al Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat Commun 2012; 3: 883. [DOI] [PubMed] [Google Scholar]
- 80. Tavanez JP, Valcarcel J. A splicing mastermind for EMT. EMBO J 2010; 29: 3217–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Plosker GL, Croom KF. Sulfasalazine: a review of its use in the management of rheumatoid arthritis. Drugs 2005; 65: 1825–49. [DOI] [PubMed] [Google Scholar]
- 82. Harris IS, Treloar AE, Inoue S et al Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015; 27: 211–22. [DOI] [PubMed] [Google Scholar]
- 83. Yoshikawa M, Tsuchihashi K, Ishimoto T et al xCT inhibition depletes CD44v‐expressing tumor cells that are resistant to EGFR‐targeted therapy in head and neck squamous cell carcinoma. Cancer Res 2013; 73: 1855–66. [DOI] [PubMed] [Google Scholar]