

Abstract
Aims
Atherosclerotic development is exacerbated by two coupled pathophysiological phenomena in plaque-resident cells: modified lipid trafficking and inflammation. To address this therapeutic challenge, we designed and investigated the efficacy in vitro and ex vivo of a novel ‘composite’ nanotherapeutic formulation with dual activity, wherein the nanoparticle core comprises the antioxidant α-tocopherol and the shell is based on sugar-derived amphiphilic polymers that exhibit scavenger receptor binding and counteract atherogenesis.
Methods and results
Amphiphilic macromolecules were kinetically fabricated into serum-stable nanoparticles (NPs) using a core/shell configuration. The core of the NPs comprised either of a hydrophobe derived from mucic acid, M12, or the antioxidant α-tocopherol (α-T), while an amphiphile based on PEG-terminated M12 served as the shell. These composite NPs were then tested and validated for inhibition of oxidized lipid accumulation and inflammatory signalling in cultures of primary human macrophages, smooth muscle cells, and endothelial cells. Next, the NPs were evaluated for their athero-inflammatory effects in a novel ex vivo carotid plaque model and showed similar effects within human tissue. Incorporation of α-T into the hydrophobic core of the NPs caused a pronounced reduction in the inflammatory response, while maintaining high levels of anti-atherogenic efficacy.
Conclusions
Sugar-based amphiphilic macromolecules can be complexed with α-T to establish new anti-athero-inflammatory nanotherapeutics. These dual efficacy NPs effectively inhibited key features of atherosclerosis (modified lipid uptake and the formation of foam cells) while demonstrating reduction in inflammatory markers based on a disease-mimetic model of human atherosclerotic plaques.
Keywords: Atherosclerosis, Nanotechnology, Macrophages, Inflammation, Alpha-tocopherol
1. Introduction
Atherosclerosis, the build-up of lipid-laden plaques within arterial walls, is one of the primary triggers for cardiovascular disease (CVD). As the principal mortality factor for nearly 50% of deaths in western countries despite vast improvement in the understanding and therapeutic interventions, CVD remains a multifocal disease in need of novel management strategies.1 The key pathway for CVD pathogenesis involves the recruitment and deposition of high levels of circulating low-density lipoproteins (LDL) in the arterial wall, where they are modified to oxidized low-density lipoproteins (oxLDL).2 This initiates inflammatory cytokine secretion and adhesion molecule up-regulation, which signals enhanced recruitment of monocytes. Adherent monocytes undergo diapedesis and differentiation into macrophages, at which point they exhibit unregulated uptake of oxLDL via scavenger receptors (SRs), exacerbating inflammatory signalling, leading to plaque growth and narrowing of the artery.3,4 In the absence of inflammation, plaque development is severely restricted.5,6 SRs play a critical role in foam cell formation and plaque build-up, which can lead to increased susceptibility to the inflammatory phenotype.7–10 Thus, atherosclerosis embodies a self-perpetuating coupling of unregulated atherogenesis and inflammation.
While we have an understanding of the molecular changes in vascular wall biology that originate, sustain, and amplify the atherosclerotic process, the use of therapeutic agents to interfere with the uptake and/or processing of the LDL remains only modestly effective at local interventions. Statins are unable to address the localized oxidative damage and inflammation that accompany atherosclerosis.11 Antioxidant therapeutics could play a role in mitigating the pro-atherogenic effects of oxidized lipids and reactive oxygen species (ROS).12 However, clinical studies with orally administered antioxidants have not shown a clear benefit and many offer contradictory results, possibly due to the lack of specific localized delivery.13–15 A more lesion-directed, pharmacologic approach offers significant opportunity for reducing the negative sequelae of the atherosclerotic process.
Several synthetic approaches have been recently proposed to formulate lesion-targeted therapeutics for plaque stabilization.16,17 We have previously advanced sugar-derived amphiphilic macromolecules (AM), designed to competitively bind SRs, which have the potency to arrest lipid accumulation.18 The constituent AM unimers, designed around a charged sugar backbone that was modified with aliphatic chains and polyethylene glycol, were shown to have inherent affinity for macrophage SR.19,20 Additionally, it was shown that AMs could significantly reduce oxLDL uptake and foam cell formation.21,22
However, AMs exhibit an inherent limitation under physiological conditions, as they form micelles that have poor stability in vivo. To develop consistently efficacious AM formulations, we designed nanoparticles (NPs) fabricated from the bioactive AMs using a flash nanoprecipitation process (FNP).23 FNP utilizes the rapid, confined mixing of an amphiphile and hydrophobe dissolved in a water-miscible organic solvent with an excess of water, which results in rapid nucleation of the hydrophobe and stabilization into NPs by the encapsulating amphiphile.24–26 The hydrophobic core of the NP traps the amphiphile and prevents thermodynamic dissociation, minimizing partitioning into the lipid sinks that would reduce bioactivity. Packaging AMs into NPs also resulted in down-regulation of SR expression in primary human macrophages, reversing the atherogenic phenotype.27 When administered to an animal model of atherosclerosis, AM NPs demonstrated localization to both early-stage intimal fatty streaks and later stage lesions with minimal off-target organ accumulation.28
Despite the promise of AM-based NPs for treatment of lipid trafficking, addressing the inflammatory response of plaque-resident macrophages continues to pose a critical challenge.29 Because NPs bind strongly to macrophages, they provide a selective route to alter the inflammatory phenotype. We hypothesize that, given the intimate role of LDL oxidation and inflammation in the progression of atherosclerosis, composite nanoparticles that release antioxidant therapeutics could concomitantly attenuate the athero-inflammatory responses. α-Tocopherol (α-T), a potential cardioprotective antioxidant, was found to impart elevated resistance to oxidation of LDL and decreased plaque development.13,30 Intracellularly, α-T inhibits cyclooxygenase (COX) activity and expression of PGE2 in macrophages, resulting in lower peroxynitrite levels.31 Additionally, α-T has been shown to reduce adhesion molecule expression in endothelial cells.32 Thus, we hypothesized that incorporation of α-T into AM NPs could provide a mechanism for enhanced delivery to plaque-resident cells, where the antioxidant effect could be amplified and mitigate inflammation.
In this work, we fabricated AM NPs incorporating the antioxidant α-T and evaluated their athero-protective capabilities in both human primary cells (macrophages, smooth muscle and endothelial cells) and a novel ex vivo carotid plaque model that utilizes excised plaque specimens from human patients (Figure 1A). The human carotid plaque model developed in our lab provides a clinically relevant, three-dimensional multi-cellular tissue system for evaluating drug efficacy. By utilizing these different models, we sought to identify an athero-protective and anti-inflammatory therapeutic. Our hypothesis is that incorporating antioxidant compounds (α-T) into bioactive AM NPs would create innovative therapeutics with potential capabilities to counteract atherogenesis and attenuate the athero-inflammatory cycle.
Figure 1.
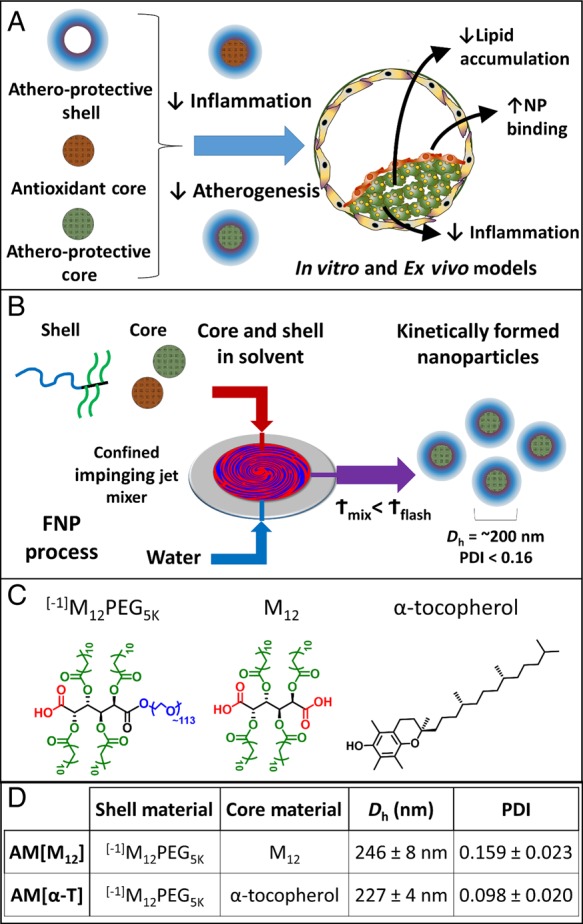
(A) AMs were fabricated into core/shell NPs with anti-atherogenic shells and either antioxidant or anti-atherogenic cores. (B) The flash nanoprecipitation process used to kinetically create serum-stable NPs from AMs. (C) Structures of the core and shell molecules. (D) AM NPs were characterized by DLS to identify hydrodynamic diameter (Dh) and PDI. The NP naming convention used is shell[core]. n = 3 for each NP formulation.
2. Materials and methods
2.1. Materials
All chemicals/materials were of molecular biology grade and purchased from Sigma-Aldrich (Milwaukee, WI, USA) or Fisher Scientific (Pittsburgh, PA, USA) unless otherwise noted. Deionized (DI) water with 18 MΩ cm resistivity was obtained using PicoPure 2 UV Plus (Hydro Service and Supplies, Durham, NC, USA). The following items were purchased from the indicated vendors: 1.077 g/cm3 Ficoll-Paque Premium from GE healthcare (Pittsburgh, PA, USA), RPMI 1640 from ATCC (Manassas, VA, USA), macrophage colony-stimulating factor (MCSF) from PeproTech (Rocky Hill, NJ, USA), FBS and AlexaFluor 680 carboxylic acid succinimidyl ester from Life Technologies (Grand Island, NY, USA), unlabelled oxLDL from Biomedical Technologies Inc. (Stoughton, MA, USA), 3,3′-dioctadecyloxacarbocyanine (DiO)-labelled oxLDL from Kalen Biomedical (Montgomery Village, MD, UDA), and human buffy coats from the Blood Center of New Jersey (East Orange, NJ, USA).
2.2. Nanoparticle fabrication
AMs were synthesized and fluorescently labelled as previously described.33,28 Kinetically assembled NPs were prepared via flash nanoprecipitation (Figure 1B).23,24,34 All fabrication equipment was sterilized in an autoclave, with 0.5 M NaOH + 1 M NaCl solution or 70% ethanol for 30 min and flushed with sterile DI H2O prior to use. Shell and core materials were dissolved in tetrahydrofuran (THF) and filtered with a 0.22 μm sterile nylon syringe filter. A confined impinging jet mixer was utilized to mix 500 µL of an aqueous stream with 500 µL of a THF stream containing 40 mg/mL shell (AM) and 20 mg/mL core solute. After mixing, the exit stream was immediately introduced into 4.5 mL of sterile DI H2O (H2O : THF volume ratio of 9 : 1) and subsequently dialyzed against sterile PBS. For all NPs, the AM [–1]M12PEG5K (hereafter referred to as AM, Figure 1C) was used as the NP shell. The core was composed of mucic acid acylated with lauroyl groups (M12) or α-T. Control NPs were created from polystyrene-block-poly(ethylene glycol) shells and polystyrene cores. To track NP fate and cellular association, selected experiments had 1.5 mol% of the shell amphiphiles labelled with Alexa Fluor 680. NP size and poly dispersity index (PDI) were determined by dynamic light scattering (DLS) using a Zetasizer Nano (Malvern).
2.3. Cell culture
Peripheral blood mononuclear cells (PBMCs) were isolated from human buffy coats and differentiated into macrophages using MCSF at 50 ng/mL in RPMI 1640, 1% penicillin/streptomycin, and 10% fetal bovine serum, as previously described.21 After differentiation, macrophages were trypsinized, scraped from flasks, and transferred into well plates at 50 000 cells/cm2. Treatments were administered after 24 h to allow for macrophage adherence. Human coronary artery endothelial cells (hCAECs) and human coronary artery smooth muscle cells (hCASMCs) were cultured according to the manufacturers protocol (Lonza, Walkersville, MD, USA).
2.4. oxLDL uptake
To measure AM NP anti-atherogenic efficacy at inhibiting oxLDL uptake, hMDMs, hCAECs, and hCASMCs were incubated with 5 µg/mL oxLDL (1 µg/mL DiO labelled, 4 µg/mL unlabelled) and 10−6 M or 10−5 M AM NPs in growth media for 24 h. Cells were removed from plates by vigorous pipetting in cold PBS with 2 mM EDTA, washed with PBS, centrifuged, and fixed in 1% paraformaldehyde. Uptake of fluorescently labelled oxLDL (DiO) was quantified using a Gallios flow cytometer (Beckman Coulter). Data for a minimum of 10 000 cells per sample was collected and quantified using the geometric mean fluorescence intensity (MFI) of oxLDL fluorescence associated with the hMDMs using FloJo software (Treestar). Results are the mean of three independent experiments with two technical replicates per experiment. Data are presented as % oxLDL uptake inhibition, which was calculated using the following formula:
For evaluation of oxLDL uptake by microscopy, hMDMs were washed and fixed with 4% paraformaldehyde and counterstained with Hoechst 33342 in a Labtek slide chamber. Cells were imaged on a Leica TCS SP2 confocal microscope using a ×63 oil immersion objective.
2.5. Foam cell formation
To measure foam cell formation, hMDMs were incubated with 50 µg/mL oxLDL and 10−5 M AM NPs in growth media for 24 h before fixation in 4% paraformaldehyde. Cells were washed with 60% isopropanol, stained with 3 mg/mL Oil Red O in 60% isopropanol, counterstained with Hoechst 33342, and imaged on a Nikon Eclipse TE2000S using a ×40 objective.
2.6. NP uptake
To measure cellular association of NPs, hMDMs, hCAECs, and hCASMCs were incubated with 10−6 M NPs with 1.5% shell amphiphile labelled with AF680 in growth media for 24 h. Cells were assessed using the same flow cytometry and microscopy procedure as described for oxLDL uptake.
2.7. NP inflammatory response
To measure the inflammatory response to AM NPs, hMDMs and hCAECs were incubated with 10−5 M AM NP in growth media for 24 h. RNA was isolated from hMDMs and hCAECs for quantitative gene expression studies, cell culture supernatant from hMDMs was collected for IL-8 cytokine quantification using ELISA, and hCAECs were collected and stained for VCAM.
2.8. Gene expression
RNA was extracted by RNeasy Plus Mini Kit with Qiashredder columns (Qiagen) following the kit instructions. RNA was reverse transcribed to cDNA with High Capacity cDNA Kit (Life Technologies) using a RapidCycler thermal cycler (Idaho Technology). Real-Time PCR was performed on a Lightcycler 480 (Roche) with Fast SYBR Green Master Mix (Life Technologies) for 40 cycles and a melting curve. Fold change was analysed using the ΔΔCt method with endogenous controls (ACTB and GAPDH) relative to the basal condition. Primers were designed by Harvard Primer Bank or Primer-BLAST and synthesized by Integrated DNA Technology; sequences are available in Supplementary materials.
2.9. Cytokine secretion
Sandwich enzyme-linked immunosorbant assays (ELISAs) were used to quantify IL-8 protein secretion from hMDMs. ELISA kits were purchased from Biolegend and the protocol followed accordingly.
2.10. VCAM expression
To measure AM NP effects on VCAM expression, hCAECs were incubated with 10−5 M AM NPs and/or 10 ng/mL TNFα in growth media for 24 h. Cells were removed from plates by vigorous pipetting in cold PBS with 2 mM EDTA, washed with PBS, and centrifuged. Cells were incubated with blocking buffer (PBS containing 0.5% BSA, 0.1% sodium azide, and 1% normal goat serum) for 30 min. Following blocking, they were incubated with labelled antibodies for human PE VCAM1 (Biolegend) for 1 h at 4°C. After the incubation, they were washed with PBS, fixed in 1% paraformaldehyde, and analysed using the same flow cytometry procedure as described for oxLDL uptake.
2.11. Human carotid plaque treatment
Plaque specimens were excised from human carotid arteries in accordance with IRB-approved protocols for de-identified specimens. The plaques were washed with RPMI 1640, sectioned into uniform 2–5 mm cylindrical sections, and incubated in media (RPMI 1640, 1% penicillin/streptomycin, and 10% fetal bovine serum) at 35°C and 5% CO2 for 24 h. The ex vivo carotid artery sections were then treated with fluorescent AM NPs (1 × 10−5 M) and 5 µg/mL DiO oxLDL for 24 h (Figure 2A).
Figure 2.
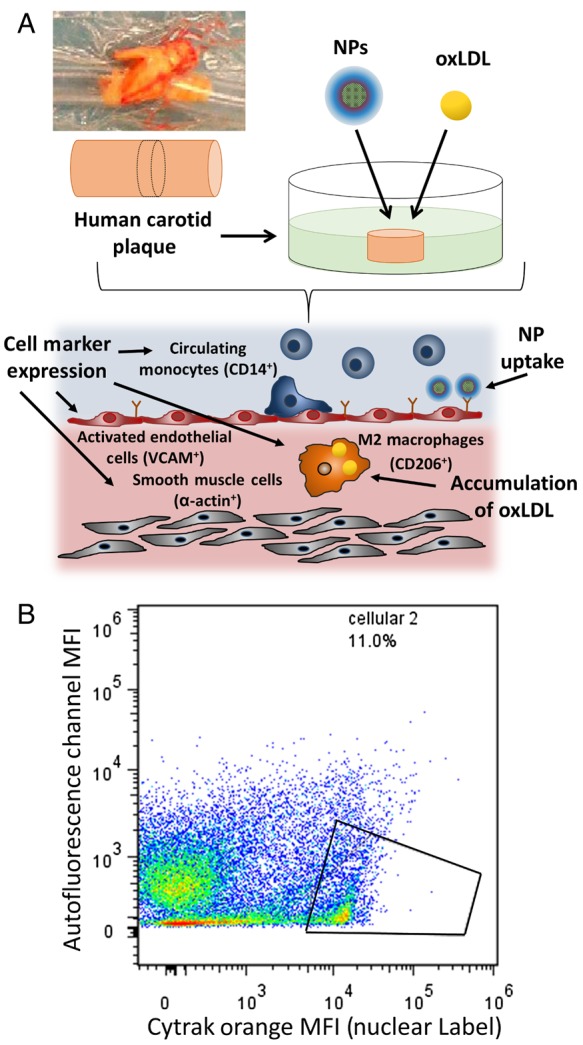
(A) Plaque specimens were excised from human carotid arteries, sectioned into uniform 2–5 mm cylindrical sections, and treated with fluorescent AM NPs (1 × 10−5 M) and 5 µg/mL DiO oxLDL. Cell subsets within the plaque were evaluated for NP and oxLDL uptake, including CD206+ (surface protein of M2 phenotype), VCAM+ (surface protein of inflamed endothelial cells), CD14+ (surface protein of monocytes and macrophages), and α-actin+ (surface and intracellular protein of smooth muscle cells). (B) To ensure proper evaluation via flow cytometry of the cellular population within the plaque, Cytrak orange was used as a nuclear label, and only populations positive for this marker were used in the data analysis.
2.12. Ethics statement
Plaque tissue specimens were taken in accordance with approved protocols that conform to the principles outlined in the Declaration of Helsinki (Rutgers University IRB protocol #E12–132 and Robert Wood Johnson Hospital IRB Protocol #0220080081). Patients were given informed consent, and tissue specimens were de-identified prior to inclusion in the study.
2.13. Cellular analysis of plaques
Following treatment, tissues were homogenized (Tissue Tearor) and then passed through a 40 µm cell strainer and washed with PBS. Solids were allowed to settle for 2 min and the cell suspension removed. Cells were incubated with blocking buffer (PBS containing 0.5% BSA, 0.1% sodium azide, and 1% normal goat serum) for 30 min. Following blocking they were incubated with labelled antibodies for human Pacific Blue CD14 (Biolegend), PE-Cy7 CD206 (MMR) (Biolegend), APC CD106 (VCAM1) (Biolegend) and Cy3 α-smooth muscle actin (Sigma-Aldrich) for 1 h at 4°C. After the incubation they were washed twice with PBS, incubated with CyTRAK Orange (eBioscience) for 30 min, fixed in 400 µL of 1% paraformaldehyde, and then analysed for CyTRAK orange positive cellular events using a Gallios flow cytometer (Beckman Coulter). Samples were quantified with FloJo (Treestar) by gating on the cellular events (cells positive for CyTRAK orange) as seen in Figure 2B. Data points for cellular events were then gated on for different surface markers (CD14, CD206, VCAM1, or α-actin) to identify different cell types within the plaque tissue. From these cell subsets, oxLDL and/or NP uptake was evaluated by quantifying the MFI of that component. Results are the mean of three independent experiments. NP uptake data are presented as fold change in NP MFI (MFI), which was calculated using the following formula:
OxLDL uptake data are presented as % oxLDL uptake inhibition, which was calculated using the following formula:
2.14. Histologic analysis of plaques
Plaques were sectioned to examine morphology and binding of AM NPs via microscopy. Plaques were fixed in formalin and prepared for cryosectioning by immersion in 30% sucrose. Tissue was embedded in OCT media (Tissue Tek), frozen, and sectioned into 10 µm serial sections on a cryostat (Leica). NP accumulation was evaluated on a Leica TCS SP2 confocal microscope using a ×63 oil immersion objective following counterstaining with ProLong Gold with DAPI (Life Technologies). To visualize areas of lipid deposits, sections were washed in 60% isopropyl alcohol before staining with 3 mg/mL Oil Red O in 60% isopropyl alcohol, counterstaining with Mayers haematoxylin, and coverslipped with Prolong Gold. To determine levels of inflammation in tissue, sections were incubated with rabbit polyclonal anti-COX-2 (Abcam, ab15191) or rabbit control IgG (Pro-Sci). Sections were delipidized in xylene (8 min) and a decreasing series of alcohol (2 min each) for before steam antigen retrieval and neutralization with 3% H2O2, followed by washing and blocking with 100% goat serum with streptavidin (Vector Labs) for 2 h at room temperature. Sections were incubated with primary antibodies (1 μg/mL COX-2) in blocking buffer with biotin (Vector Labs) overnight at 4°C and then washed before secondary incubation with Vectastain Elite anti-rabbit IgG (Vector Labs). Staining was visualized with DAB peroxidase (Vector Labs) and counterstained with Mayers haematoxylin for 1 min. Sections were mounted on coverslips with permount and imaged with an Olympus VS120.
2.15. Statistical analysis
Results are presented as mean ± S.E.M. and data evaluated by one-way ANOVA and post hoc Tukey's test for comparisons between multiple conditions. A P-value of 0.05 or less was considered statistically significant.
3. Results
3.1. NP fabrication
NPs were fabricated via flash nanoprecipitation, using the varied hydrophobic cores M12 and α-T. AM NPs had an average Dh of 230 nm and exhibit narrow size distributions (Figure 1D). Non-biologically active control NPs based on polystyrene were fabricated as a control for in vitro studies (see Supplementary material online, Figure S1).
3.2. NP inhibition of oxLDL uptake
The ability to block oxLDL uptake was used as the key measure of anti-atherogenic efficacy of NPs. Figure 3A shows the uptake inhibition potential of the NP formulations in all three key cell types, hMDMs, hCAECs, and hCASMCs. Variation in the core material of the NPs did not significantly affect their ability to reduce oxLDL uptake at 10−5M, as both AM NP formulations were able to inhibit ∼85% of oxLDL uptake, even in the presence of 10% serum. However, when administered at one order of magnitude lower concentration of 10−6M NP, the α-T core NPs had a significant reduction in oxLDL inhibition potential. Control NPs minimally inhibited oxLDL uptake at both concentrations and elicited similar oxLDL uptake levels as cells treated only with oxLDL (see Supplementary material online, Figure S2A).
Figure 3.

The ability of NPs to inhibit oxLDL uptake (A) and be internalized (B) was examined in smooth muscle cells (SMCs), macrophages (MΦ), and endothelial cells (EC). (A) NPs with M12 cores were most effective at preventing oxLDL uptake while AM[α-T] lost efficacy at lower concentrations. (B) NP uptake in SMCM12 core NPs had significantly higher levels of cellular internalization relative to α-T core NPs. Macrophages had approximately three-fold higher NP uptake relative to endothelial and smooth muscle cells. (C) Representative images of macrophages after treatment with oxLDL (green) and AF680-labelled NPs (red). Nuclei were counterstained with Hoechst 33342 (blue). hMDMs were also stained with Oil Red O to indicate transition to the foam cell phenotype. For oxLDL and NP uptake, hMDMs, HCAECs, and HCASMCs were co-incubated with 5 μg/mL oxLDL and 10−5 M or 10−6M NPs for 24 h. For foam cell, hMDMs were co-incubated with 50 μg/mL oxLDL and 10−5M NPs for 24 h and then stained with Oil Red O. n = 3, all conditions had P < 0.001 from the control (no AM, oxLDL only for oxLDL uptake, and basal cells for NP uptake). Treatments with the same letter are not statistically significant from one another.
3.3. NP cellular association
Cellular internalization of NPs by cultured hMDMs, hCAECs, and hCASMCs is shown in Figure 3B. AM NPs with M12 in the core displayed high levels of association with hMDMs and were readily internalized at 10−5M concentration. In contrast, hCAECs and hCASMCs had significantly lower internalization of M12 core NPs relative to macrophages (approximately three-fold less). α-T core NPs were internalized to a lesser extent by all three types of cultured cells. Control NPs were minimally internalized (see Supplementary material online, Figure S2B).
3.4. Gene regulation of macrophages and endothelial cells after treatment with AM NPs
The inflammatory response was evaluated following treatment with the NPs in hMDMs and hCAECs by assessing gene expression, cytokine secretion, and adhesion molecule expression. qRT–PCR was used to measure expression changes in hMDMs treated with NPs for an array of genes that modulate inflammation (IL-1β, IL-6, IL-8, IL-10, TNFα, NFκB1, MCP-1, MMP9), oxLDL uptake (CD36, MSR1), and lipid trafficking (NR1H3, PPARγ, ABCA1). The quantitative fold change in mRNA levels is shown in Figure 4A and B.
Figure 4.

The ability of NPs to modulate the inflammatory and atherogenic phenotype was examined by qRT–PCR gene expression in macrophages (A) and endothelial cells (B), IL-8 cytokine secretion by macrophages (C), and VCAM expression by endothelial cells (D). (A) M12 core NPs increased inflammatory cytokine expression while down-regulating scavenger receptor expression in macrophages. Data are presented as fold change relative to untreated cells. Positive numbers (green) represent gene up-regulation and negative (red) represent gene down-regulation. (B) M12 core NPs up-regulated inflammatory cytokine and adhesion molecule expression. (C) AM[M12] NPs amplified IL-8 secretion while α-T core NPs maintained basal levels. (D) Both NP formulations did not increase VCAM expression when added to resting endothelial cells and lowered VCAM expression on EC that had been exposed to TNFα. For these experiments, hMDMs and HCAECs were incubated with 10−5M NP for 24 h. TNFα was added to HCAECs prior to (pre-inflamed) or during (co-inflamed) NP treatment to induce external inflammation relative to resting (quiescent) cells. n = 3, conditions with the same letter are not statistically significant from one another and the asterisk (*) indicates statistical significance (P < 0.05) from the control (basal cells).
NPs with M12 in the core up-regulated pro-inflammatory gene expression, notably IL-1β, IL-6 and IL-8. M12 cores also had the effect of down-regulating scavenger receptor expression for CD36 and MSR1. α-T core NPs elicited basal, low levels of inflammatory gene expression. Control NPs did not affect gene regulation (see Supplementary material online, Figure S2D).
3.5. IL-8 secretion by human macrophages after treatment with AM NPs
ELISAs were used to quantitatively measure cytokine secretion by hMDMs after treatment with AM NPs. Given its role in monocyte recruitment, IL-8 was chosen as a representative protein as it was consistently highly up-regulated.5 The secretion profile of the inflammatory cytokine, IL-8, following differential treatment of hMDMs with the NP conditions is graphed in Figure 4C. M12 core NPs significantly increased secretion of IL-8, while α-T core NPs did not induce an increase in secretion relative to basal cells. Control NPs did not induce IL-8 secretion (see Supplementary material online, Figure S2C).
3.6. Endothelial cell VCAM expression after treatment with AM NPs
VCAM expression was used as a marker of endothelial inflammation and represents a necessary step in the recruitment of macrophages to a growing lesion. NPs with α-T cores lowered VCAM expression in resting cells relative to the basal cell control. Both M12 and α-T core NPs lowered VCAM expression for cells pre-treated and co-treated with TNFα, an inflammatory agent that significantly increases adhesion molecule expression.
3.7. Interactions of AM NPs and oxLDL with cellular sub-cultures of human carotid plaques
The clinically relevant human carotid plaque model was utilized to further evaluate the ability of AM NPs to interact with the plaque components. The ability of AM NPs of varying core solutes to enter the plaque and be internalized by plaque-resident cells was examined with flow cytometry and confocal microscopy. NPs were found to penetrate thoroughly into the plaque as seen by the overall ∼2.5-fold increase in cell-associated NP fluorescence (Figure 5A). This infiltration is also seen in a representative cross-section of the plaque, in which AM NP presence is seen throughout the section (Figure 5B). The ability of NPs to be internalized was found to be independent of the core hydrophobe, as both NP formulations had similar overall cellular accumulation. Cell subsets within the plaque were evaluated for NP uptake and correlated with the expression of CD206 (surface marker of M2 macrophage phenotype), VCAM (surface marker of inflamed endothelial cells), CD14 (surface marker of monocytes and macrophages), and α-SMC actin (surface and intracellular marker of smooth muscle cells) (Figure 5A). Minimal NP uptake was observed in CD14+ cells, which is to be expected as monocytes exhibit minimal phagocytosis. However, significant NP uptake was observed in CD206+, VCAM+, as well as α-actin+ cells with the α-T core NPs showing stronger cell internalization and association. This may be related to the presence of SRs on these cell types as NPs were previously shown to demonstrate the ability to bind to SRs.27,35
Figure 5.
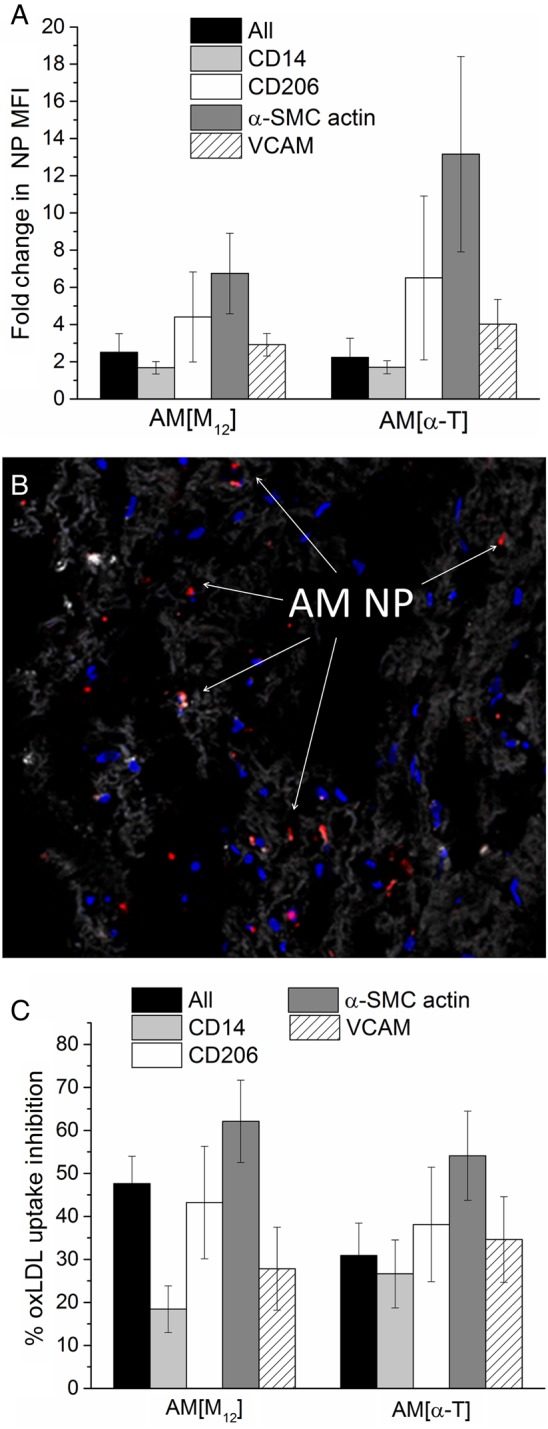
(A) Evaluation of the entire cellular population of the plaque and specific subsets within the cellular population (CD14, C206, α-SMC actin, or VCAM) for NP uptake. AM NPs readily associate with plaque-resident cells, exhibiting the strongest uptake within CD206+, α-actin+, and VCAM+ populations. (B) Representative cross-section images of plaque showing NP co-localization with plaque-resident cells. (C) Evaluation of the entire cellular population of the plaque and specific subsets within the cellular population (CD14, C206, α-SMC actin, or VCAM) for inhibition of oxLDL uptake. All cellular population subsets exhibited inhibition of oxLDL uptake following treatment with AM NPs, with the CD206+ and α-actin+ population showing the greatest reduction in uptake. n = 10 plaque samples and error bars represent the S.E.M. All conditions shown are P < 0.05 from the control (non-treated plaque).
The ability of NPs to prevent oxLDL uptake within each cell subset was examined following oxLDL administration to the plaque cultures (Figure 5C). The NPs best inhibited oxLDL uptake in CD206+ and α-actin+ cells, which may again be, related to their increased expression of the SRs MSR1, and CD36, on the cell surface.
3.8. Influence of AM NPs on cell markers and lipid uptake of human carotid plaque specimens
NPs reduced the inflammatory phenotype of cells resident to the human carotid plaque specimens (Figure 6A). Both M12 and α-T core NPs reduced CD206 and α-SMC actin expression on cells within the human plaques. Qualitatively, the α-T core NPs had a more pronounced effect on the reduction of the surface expression of these proteins on plaque cells, particularly CD206 and α-actin. This may be related to the anti-inflammatory nature of α-T, rescuing cells from the athero-inflammatory phenotype. When these plaques were sectioned and stained for evaluation of lipid accumulation and inflammatory signalling (Figure 6B), similar anti-athero-inflammatory effects were observed as both AM NPs reduced lipid deposits (Oil Red O) and inflammatory marker expression (COX-2).
Figure 6.

(A) Cellular composition of plaque sections. AM NPs are able to reduce the percentages of cells positive for CD206 and α-actin markers in human carotid plaques. n = 10 plaque samples. (B) Plaque cross-sections were stained to show lipid deposits (Oil Red O) and inflammation (COX-2). Both AM NPs reduced amounts of lipid deposits and severity of inflammation.
4. Discussion
Molecular interventions to block lipid uptake and the ensuing inflammatory phenotype are among the possible strategies for the inhibition of plaque development in atherosclerosis.17,36–39 While AM micelles have been demonstrated to inhibit oxLDL uptake in vitro, they are not ideal for in vivo applications as micelles often exhibit relatively short half-lives due to the natural thermodynamic dissolution that results from hydrophobic partitioning.16 Using flash nanoprecipitation, we have created serum-stable anti-atherogenic NPs from AMs, which can further enhance the therapeutic utility of AMs in vivo. The NP arrangement is able to stabilize the AMs with a hydrophobic core, thereby potentially extending the circulation half-life of AMs. In this work, a second generation of NPs was envisioned to enhance anti-atherogenic potential of these serum-stable particles by reducing inflammation and oxidized lipid uptake in primary human cells as well as a clinically relevant human carotid plaque model.
The development of non-inflammatory AM NPs is critical to their success as a viable therapeutic. FNP remains an attractive way of synthesizing NPs due to the consistent size of NPs generated and a unimodal size distribution. AM NPs created with flash nanoprecipitation have a larger size (100–250 nm) than AM micelles which have an average hydrodynamic diameter (Dh) of 22 nm. As size is a critical factor for cellular internalization, being able to generate consistently sized materials with a narrow size distribution is important for process scale up and clinical studies.40 Using flash nanoprecipitation, NP size can be predictably controlled by altering the strength of hydrophobic interactions between the core and shell molecules. Increasingly, hydrophobic core molecules lead to smaller NPs due to increased nucleation sites as supersaturation is enhanced. It can also lead to more stable NPs, with individual unimers having slower release kinetics from the particle into other matrix or cellular sinks. The larger size of nanoparticles relative to micelles protects them from renal filtration, which has a size cut-off of ∼8 nm, enabling longer circulation times.41 However, the NP formulation may also modulate an immune response from the macrophages, making inflammatory responses a critical aspect to consider in the design and development of nanoscale therapeutics.40,42 One potential consideration in the design of AM NPs is the inherent pro-inflammatory nature of nanosized materials.43 Two imaging nanosystems exploited this selective association/internalization by monocytes/macrophages for the visualization of plaques using PET and MRI.44,45 Nanoparticles, in particular, can stimulate inflammatory pathways that could exacerbate the signalling cascade that leads to atherogenesis.46 Ensuring that NPs do not cause cellular or systemic inflammatory responses is a major consideration in the design of implantable or injectable nanosystems.47
In this work, NPs were fabricated with different core materials using a consistent amphiphilic shell to evaluate both the anti-atherogenic and anti-inflammatory capabilities of the NPs. Polystyrene-based amphiphilic NPs were used as a control system for primary cell work and were found to be non-bioactive, consistent with previous studies.27,28 Examining the inhibition of oxLDL uptake (anti-atherogenic bioactivity) demonstrated that the core composition of the NP is critical for efficacy. NPs with α-T cores showed a significant diminution of oxLDL inhibition bioactivity when tested at the lower concentration (10−6 M), while NPs containing M12 in the core were effective at inhibiting ∼80% of oxLDL uptake at both concentrations. The M12 solute was originally chosen for incorporation into the NP core as it corresponds to the hydrophobic portion of the bioactive AM, [−1]M12P5, and had increased miscibility when formulated in this way. It is hypothesized that the M12 hydrophobic core contributes to more complete blockage of the SRs, thus inhibiting oxLDL uptake to a greater extent than the α-T core NPs, which do not demonstrate comparable efficacy at the lower concentration.27 However, when evaluated at the higher concentration with the human carotid plaque specimens, both cores containing NPs resulted in equivalent levels of oxLDL uptake inhibition. Furthermore, enhanced inhibition of oxLDL uptake was observed in the CD206+ and α-SMC actin+ cell types, which may be related to their increased expression of SRs.48,49 Previous studies have shown that AM NPs bind MSR1 and inhibit oxLDL uptake.23 These results in this clinically relevant model suggest that the α-T NPs are likely to be efficacious in vivo as long as their local concentration is optimally maintained.
The cellular association of AM NPs with macrophages appears to be strongly correlated with oxLDL uptake inhibition. NPs with M12 cores had high levels of internalization by hMDMs, whereas α-T core NPs showed lower levels of internalization. This is consistent with previous work which suggests that the M12 core promotes SR binding and internalization by hMDMs, thus lowering SR expression, further inhibiting oxLDL uptake.27 Cell types likely to express SRs are also those that up-regulate other atherogenic markers (VCAM+, CD206+, and α-actin+) in the human carotid plaque specimens, which exhibit the strongest uptake of the AM NPs. The scavenger receptor profiles notwithstanding, the analysed cell populations indicate that the α-T core NPs bind most strongly to these three cell subtypes. The α-T in the core may facilitate cellular uptake via different receptor interactions than typically observed with AMs.50 Despite the PEG shell surrounding the bioactive AM NPs, all of the different cell types examined in this work elicited significant NP uptake, consistent with previous studies.23,27 Thus, specific cellular delivery of other therapeutic drugs that have limited targeting capability (lipid efflux enhancers, antioxidants, or anti-inflammatory agents) to cells with high levels of lipid accumulation could be achieved by incorporation into AM NPs, with the goal of further reducing the cellular atherogenic phenotype.
Recent work has clarified the phenotypic distribution of plaque macrophages with plaque progression.29,51 Alternatively activated macrophages (M2) are primarily responsible for oxidized lipid uptake and early-stage growth of the plaque when cholesterol homeostasis is lost due to dyslipidaemia.49 However, inflammatory signalling is pivotal for exacerbation of late-stage disease and increased risk of plaque rupture, when cells in the necrotic core shift to a M1 phenotype.39,52 Reducing this early-stage lipid uptake in conjunction with lowering inflammatory signalling or preventing further exacerbation of it remains a therapeutic challenge. In this work, the first-generation AM NPs utilizing M12 for the hydrophobic core were found to induce an elevation in the extent of the inflammatory phenotype in human macrophages. Cytokines such as IL-1β, IL-6, and IL-8, which are classical markers of macrophage activation, were up-regulated as was secretion of IL-8, indicating a potential transition from the alternative M2 to a more M1-like activated macrophage phenotype.53 In the context of a disease lesion signalling, this could then lead to further monocyte recruitment and macrophage differentiation. The high charge density of the M12 hydrophobic NP core may be the stimulus for this inflammatory gene up-regulation and subsequent cytokine secretion. Interestingly, the SRs responsible for oxLDL uptake, CD36 and MSR1, were down-regulated by the M12 core NPs, also indicating a potential shift to the M1 macrophage phenotype. This is consistent with previous work that revealed the AM NP's ability to down-regulate surface expression of SRs on hMDMs.27 The α-T core NPs demonstrated a different profile in their ability to reduce inflammatory gene expression back to basal levels and minimize secretion of IL-8. However, these NPs have a limited ability to down-regulate scavenger receptor expression. On the other hand, the human plaque model revealed that the α-T core NPs retained the potential to down-regulate surface expression of CD206 and α-actin, key cell markers of M2 macrophages and smooth muscle cells.49,54 These findings suggest a unique dual property profile of the α-T core NPs, namely, strong anti-inflammatory and anti-lipid uptake outcomes, which may also have an effect on reducing SMC proliferation and stenosis. While the M12 core may contribute to inflammation, it also plays a key role in scavenger receptor down-regulation, thought to prevent oxLDL uptake. Thus, a combination particle with smaller fraction of M12 and larger fraction of α-T, or M12 with a more potent anti-inflammatory agent can be envisioned. Additionally, the ability to modulate macrophage phenotype from M2 to M1 and vice versa is a promising avenue that merits further study.
To more effectively treat atherosclerosis, anti-atherogenic therapeutics must be able to not only counteract lipid accumulation but also able to minimize inflammation. To address this therapeutic challenge, we created a second-generation nanotherapeutic formulation, incorporating the antioxidant α-T into the core with the anti-atherogenic [−1]M12PEG5k in the shell. These AM NPs inhibited key features of atherosclerosis (modified lipid uptake and the formation of foam cells) while minimizing inflammation in the most advanced ex vivo models available today. These studies suggest that by integrating anti-inflammatory molecules with bioactive AM, innovative anti-atherosclerotic nanotherapeutics can be realized.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
The authors acknowledge the following financial support: National Heart, Lung and Blood Institute at the National Institute of Health (R01HL107913, R21HL93753—P.V.M., K.E.U.), the Coulter Foundation for Biomedical Engineering Translational Research Award (P.V.M.), and National Institute of Health T32 training programs and fellowships (EB005583—A.W.Y., T32GM008339—D.R.L.).
Acknowledgements
The authors thank Douglas Matijakovich, Rebecca Chmielowski, Allison Faig, and Ricky Li for technical help.
Conflict of interest: none declared.
References
- 1.Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Stafford R, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J. Executive Summary: Heart Disease and Stroke Statistics—2010 Update: a report from the American Heart Association. Circulation 2010;121:948–954. [DOI] [PubMed] [Google Scholar]
- 2.Berliner JA, Heinecke JW. The role of oxidized lipoproteins in atherogenesis. Free Radic Biol Med 1996;20:707–727. [DOI] [PubMed] [Google Scholar]
- 3.Boullier A, Bird DA, Chang M-K, Dennis EA, Friedman P, Gillotte-Taylor K, Hörkkö S, Palinski W, Quehenberger O, Shaw P, Steinberg D, Terpstra V, Witztum JL. Scavenger receptors, oxidized LDL, and atherosclerosis. Ann NY Acad Sci 2001;947:214–223. [DOI] [PubMed] [Google Scholar]
- 4.Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med 1999;340:115–126. [DOI] [PubMed] [Google Scholar]
- 5.Boisvert WA, Santiago R, Curtiss LK, Terkeltaub RA. A leukocyte homologue of the IL-8 receptor CXCR-2 mediates the accumulation of macrophages in atherosclerotic lesions of LDL receptor-deficient mice. J Clin Invest 1998;101:353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gu L, Okada Y, Clinton SK, Gerard C, Sukhova GK, Libby P, Rollins BJ. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol Cell 1998;2:275–281. [DOI] [PubMed] [Google Scholar]
- 7.Febbraio M, Podrez EA, Smith JD, Hajjar DP, Hazen SL, Hoff HF, Sharma K, Silverstein RL. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest 2000;105:1049–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whitman SC, Rateri DL, Szilvassy SJ, Cornicelli JA, Daugherty A. Macrophage-specific expression of class A scavenger receptors in LDL receptor(-/-) mice decreases atherosclerosis and changes spleen morphology. J Lipid Res 2002;43:1201–1208. [PubMed] [Google Scholar]
- 9.van Tits LJH, Stienstra R, van Lent PL, Netea MG, Joosten LAB, Stalenhoef AFH. Oxidized LDL enhances pro-inflammatory responses of alternatively activated M2 macrophages: a crucial role for Krüppel-like factor 2. Atherosclerosis 2011;214:345–349. [DOI] [PubMed] [Google Scholar]
- 10.Robbesyn F, Salvayre R, Negre-Salvayre A. Dual role of oxidized LDL on the NF-kappaB signaling pathway. Free Radic Res 2004;38:541–551. [DOI] [PubMed] [Google Scholar]
- 11.Gotto AM. Antioxidants, statins, and atherosclerosis. J Am Coll Cardiol 2003;41:1205–1210. [DOI] [PubMed] [Google Scholar]
- 12.Niki E. Antioxidants and atherosclerosis. Biochem Soc Trans 2004;32:156–159. [DOI] [PubMed] [Google Scholar]
- 13.Saremi A, Arora R. Vitamin E and cardiovascular disease. Am J Ther 2010;17:e56–e65. [DOI] [PubMed] [Google Scholar]
- 14.Steinberg D, Witztum JL. Is the oxidative modification hypothesis relevant to human atherosclerosis?: Do the antioxidant trials conducted to date refute the hypothesis? Circulation 2002;105:2107–2111. [DOI] [PubMed] [Google Scholar]
- 15.Kissner R, Nauser T, Bugnon P, Lye PG, Koppenol WH. Formation and properties of peroxynitrite as studied by laser flash photolysis, high-pressure stopped-flow technique, and pulse radiolysis. Chem Res Toxicol 1997;10:1285–1292. [DOI] [PubMed] [Google Scholar]
- 16.Peters D, Kastantin M, Kotamraju VR, Karmali PP, Gujraty K, Tirrell M, Ruoslahti E. Targeting atherosclerosis by using modular, multifunctional micelles. Proc Natl Acad Sci 2009;106:9815–9819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lewis DR, Kamisoglu K, York AW, Moghe PV. Polymer-based therapeutics: nanoassemblies and nanoparticles for management of atherosclerosis. Wiley Interdiscip Rev Nanomed Nanobiotechnol 2011;3:400–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chnari E, Nikitczuk JS, Wang J, Uhrich KE, Moghe PV. Engineered polymeric nanoparticles for receptor-targeted blockage of oxidized low density lipoprotein uptake and atherogenesis in macrophages. Biomacromolecules 2006;7:1796–1805. [DOI] [PubMed] [Google Scholar]
- 19.Plourde NM, Kortagere S, Welsh W, Moghe PV. Structure−activity relations of nanolipoblockers with the atherogenic domain of human macrophage scavenger receptor A. Biomacromolecules 2009;10:1381–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hehir S, Plourde NM, Gu L, Poree DE, Welsh WJ, Moghe PV, Uhrich KE. Carbohydrate composition of amphiphilic macromolecules influences physicochemical properties and binding to atherogenic scavenger receptor A. Acta Biomater 2012;8:3956–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lewis DR, Kholodovych V, Tomasini MD, Abdelhamid D, Petersen LK, Welsh WJ, Uhrich KE, Moghe PV. In silico design of anti-atherogenic biomaterials. Biomaterials 2013;34:7950–7959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iverson NM, Sparks SM, Demirdirek B, Uhrich KE, Moghe PV. Controllable inhibition of cellular uptake of oxidized low-density lipoprotein: structure-function relationships for nanoscale amphiphilic polymers. Acta Biomater 2010;6:3081–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.York AW, Zablocki KR, Lewis DR, Gu L, Uhrich KE, Prud'homme RK, Moghe PV. Kinetically assembled nanoparticles of bioactive macromolecules exhibit enhanced stability and cell-targeted biological efficacy. Adv Mater 2012;24:733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson BK, Prud'homme RK. Flash nanoprecipitation of organic actives and block copolymers using a confined impinging jets mixer. Aust J Chem 2003;56:1021–1024. [Google Scholar]
- 25.Kumar V, Wang L, Riebe M, Tung H-H, Prud'homme RK. Formulation and stability of itraconazole and odanacatib nanoparticles: governing physical parameters. Mol Pharm 2009;6:1118–1124. [DOI] [PubMed] [Google Scholar]
- 26.Gindy ME, Panagiotopoulos AZ, Prud'homme RK. Composite block copolymer stabilized nanoparticles: simultaneous encapsulation of organic actives and inorganic nanostructures. Langmuir 2007;24:83–90. [DOI] [PubMed] [Google Scholar]
- 27.Petersen LK, York AW, Lewis DR, Ahuja S, Uhrich KE, Prud'homme RK, Moghe PV. Amphiphilic nanoparticles repress macrophage atherogenesis: novel core/shell designs for scavenger receptor targeting and down-regulation. Mol Pharm 2014;11:2815–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewis DR, Petersen LK, York AW, Zablocki KR, Joseph LB, Kholodovych V, Prud'homme RK, Uhrich KE, Moghe PV. Sugar-based amphiphilic nanoparticles arrest atherosclerosis in vivo. Proc Natl Acad Sci 2015;112:2693–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chinetti-Gbaguidi G, Colin S, Staels B. Macrophage subsets in atherosclerosis. Nat Rev Cardiol 2015;12:10–17. [DOI] [PubMed] [Google Scholar]
- 30.Esterbauer H, Dieber-Rotheneder M, Striegl G, Waeg G. Role of vitamin E in preventing the oxidation of low-density lipoprotein. Am J Clin Nutr 1991;53:314S–321S. [DOI] [PubMed] [Google Scholar]
- 31.Beharka AA, Wu D, Serafini M, Meydani SN. Mechanism of vitamin E inhibition of cyclooxygenase activity in macrophages from old mice: role of peroxynitrite. Free Radic Biol Med 2002;32:503–511. [DOI] [PubMed] [Google Scholar]
- 32.Koga T, Kwan P, Zubik L, Ameho C, Smith D, Meydani M. Vitamin E supplementation suppresses macrophage accumulation and endothelial cell expression of adhesion molecules in the aorta of hypercholesterolemic rabbits. Atherosclerosis 2004;176:265–272. [DOI] [PubMed] [Google Scholar]
- 33.Tian L, Yam L, Zhou N, Tat H, Uhrich KE. Amphiphilic scorpion-like macromolecules:design, synthesis, and characterization. Macromolecules 2004;37:538–543. [Google Scholar]
- 34.Liu Y, Cheng C, Liu Y, Prud'homme RK, Fox RO. Mixing in a multi-inlet vortex mixer (MIVM) for flash nano-precipitation. Chem Eng Sci 2008;63:2829–2842. [Google Scholar]
- 35.Tomasini MD, Zablocki K, Petersen LK, Moghe PV, Tomassone MS. Coarse grained molecular dynamics of engineered macromolecules for the inhibition of oxidized low-density lipoprotein uptake by macrophage scavenger receptors. Biomacromolecules 2013;14:2499–2509. [DOI] [PubMed] [Google Scholar]
- 36.Iverson N, Plourde N, Chnari E, Nackman GB, Moghe PV. Convergence of nanotechnology and cardiovascular medicine. Biodrugs 2008;22:1–10. [DOI] [PubMed] [Google Scholar]
- 37.Mantovani A, Garlanda C, Locati M. Macrophage diversity and polarization in atherosclerosis: a question of balance. Arterioscler Thromb Vasc Biol 2009;29:1419–1423. [DOI] [PubMed] [Google Scholar]
- 38.Mantovani A, Sica A, Locati M. New vistas on macrophage differentiation and activation. Eur J Immunol 2007;37:14–16. [DOI] [PubMed] [Google Scholar]
- 39.Saha P, Modarai B, Humphries J, Mattock K, Waltham M, Burnand KG, Smith A. The monocyte/macrophage as a therapeutic target in atherosclerosis. Curr Opin Pharmacol 2009;9:109–118. [DOI] [PubMed] [Google Scholar]
- 40.Clift MJD, Rothen-Rutishauser B, Brown DM, Duffin R, Donaldson K, Proudfoot L, Guy K, Stone V. The impact of different nanoparticle surface chemistry and size on uptake and toxicity in a murine macrophage cell line. Toxicol Appl Pharmacol 2008;232:418–427. [DOI] [PubMed] [Google Scholar]
- 41.Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Itty Ipe B, Bawendi MG, Frangioni JV. Renal clearance of quantum dots. Nat Biotechnol 2007;25:1165–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brown DM, Wilson MR, MacNee W, Stone V, Donaldson K. Size-dependent proinflammatory effects of ultrafine polystyrene particles: a role for surface area and oxidative stress in the enhanced activity of ultrafines. Toxicol Appl Pharmacol 2001;175:191–199. [DOI] [PubMed] [Google Scholar]
- 43.Stevenson R, Hueber AJ, Hutton A, McInnes IB, Graham D. Nanoparticles and Inflammation. Sci World J 2011;11:1300–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Metz S, Bonaterra G, Rudelius M, Settles M, Rummeny E, Daldrup-Link H. Capacity of human monocytes to phagocytose approved iron oxide MR contrast agents in vitro. Eur Radiol 2004;14:1851–1858. [DOI] [PubMed] [Google Scholar]
- 45.Nahrendorf M, Zhang H, Hembrador S, Panizzi P, Sosnovik DE, Aikawa E, Libby P, Swirski FK, Weissleder R. Nanoparticle PET-CT imaging of macrophages in inflammatory atherosclerosis. Circulation 2008;117:379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tedgui A, Mallat Z. Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol Rev 2006;86:515–581. [DOI] [PubMed] [Google Scholar]
- 47.Monteiro-Riviere NA, Tran CL. Nanotoxicology: Characterization, Dosing and Health Effects. New York: Informa Healthcare; 2007. [Google Scholar]
- 48.Boyle JJ. Macrophage activation in atherosclerosis: pathogenesis and pharmacology of plaque rupture. Curr Vasc Pharmacol 2005;3:63–68. [DOI] [PubMed] [Google Scholar]
- 49.Waldo SW, Li Y, Buono C, Zhao B, Billings EM, Chang J, Kruth HS. Heterogeneity of human macrophages in culture and in atherosclerotic plaques. Am J Pathol 2008;172:1112–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Traber MG, Kayden HJ. Vitamin E is delivered to cells via the high affinity receptor for low-density lipoprotein. Am J Clin Nutr 1984;40:747–751. [DOI] [PubMed] [Google Scholar]
- 51.Chinetti-Gbaguidi G, Staels B. Macrophage polarization in metabolic disorders: functions and regulation. Curr Opin Lipidol 2011;22:365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khallou-Laschet J, Varthaman A, Fornasa G, Compain C, Gaston A-T, Clement M, Dussiot M, Levillain O, Graff-Dubois S, Nicoletti A, Caligiuri G. Macrophage plasticity in experimental atherosclerosis. PLoS One 2010;5:e8852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gordon S. Alternative activation of macrophages. Nat Rev Immunol 2003;3:23–35. [DOI] [PubMed] [Google Scholar]
- 54.Auge N, Garcia V, Maupas-Schwalm F, Levade T, Salvayre R, Negre-Salvayre A. Oxidized LDL-induced smooth muscle cell proliferation involves the EGF receptor/PI-3 kinase/Akt and the sphingolipid signaling pathways. Arterioscler Thromb Vasc Biol 2002;22:1990–1995. [DOI] [PubMed] [Google Scholar]