

ABSTRACT
Dengue virus is the most prevalent cause of arthropod-borne infection worldwide. Due to the limited coding capacity of the viral genome and the complexity of the viral life cycle, host cell proteins play essential roles throughout the course of viral infection. Host RNA-binding proteins mediate various aspects of virus replication through their physical interactions with viral RNA. Here we describe a technique designed to identify such interactions in the context of infected cells using UV cross-linking followed by antisense-mediated affinity purification and mass spectrometry. Using this approach, we identified interactions, several of them novel, between host proteins and dengue viral RNA in infected Huh7 cells. Most of these interactions were subsequently validated using RNA immunoprecipitation. Using small interfering RNA (siRNA)-mediated gene silencing, we showed that more than half of these host proteins are likely involved in regulating virus replication, demonstrating the utility of this method in identifying biologically relevant interactions that may not be identified using traditional in vitro approaches.
IMPORTANCE
Dengue virus is the most prevalent cause of arthropod-borne infection worldwide. Viral RNA molecules physically interact with cellular RNA-binding proteins (RBPs) throughout the course of infection; the identification of such interactions will lead to the elucidation of the molecular mechanisms of virus replication. Until now, the identification of host proteins bound to dengue viral RNA has been accomplished using in vitro strategies. Here, we used a method for the specific purification of dengue viral ribonucleoprotein (RNP) complexes from infected cells and subsequently identified the associated proteins by mass spectrometry. We then validated a functional role for the majority of these proteins in mediating efficient virus replication. This approach has broad relevance to virology and RNA biology, as it could theoretically be used to purify any viral RNP complex of interest.
INTRODUCTION
Dengue virus (DENV) is an arthropod-borne member of the family Flaviviridae. DENV is transmitted by the mosquito vectors Aedes aegypti and Aedes albopictus; nearly 40% of the world’s population is at risk of exposure to these vectors. Recent models estimate that nearly 400 million cases of dengue infection may occur annually worldwide, causing significant morbidity and negative economic impact (1). Importantly, there are currently no antivirals for treating DENV-associated disease or an effective vaccine available to prevent DENV infections. The discovery of strategies for preventing or treating DENV infection is critical and requires a detailed knowledge of the molecular mechanisms by which the virus replicates in both human and insect hosts.
The DENV genome is a single molecule of positive-sense RNA that is approximately 10.7 kb in length. The viral genomic RNA contains a 5′ m7GpppAm type 1 cap structure that mediates translation initiation, protection from cellular exonucleases, and masking from innate immune sensors (2). In contrast to the majority of cellular mRNAs, the DENV genome is not polyadenylated. The coding sequence of the genome is flanked by both 5′ and 3′ untranslated regions (UTRs) that contain many structural features critical for the efficient replication of the viral RNA (reviewed in reference 3). During the initiation of infection, incoming positive-sense genomes must be translated, and they then serve as templates for the synthesis of complementary negative strands. Negative-strand RNAs in turn serve as templates for the synthesis of additional positive-sense molecules, which are replicated, translated, and/or packaged into progeny virions.
In addition to full-length viral RNA (vRNA), another abundant RNA species present during flavivirus infection is the subgenomic flaviviral RNA (sfRNA). The sfRNA is generated from the degradation of full-length viral RNA, presumably by the cellular 5′-3′ exonuclease Xrn1, to produce a shorter (~400-nucleotide [nt]) RNA that is derived from the 3′ end of the viral genome (4–6). Generation of the sfRNA is essential for viral pathogenicity and functions at least in part to subvert the host cell interferon response by binding to cellular proteins (7–9).
Viral RNA molecules are exposed to and physically interact with cellular RNA-binding proteins (RBPs) throughout the course of infection. Interactions critical for the regulation of virus replication may occur at various stages of the viral life cycle, such as trafficking, translation, synthesis, and packaging of RNA genomes. As mentioned above, the sfRNA is also involved in critical interactions with host proteins. These interactions may impede or augment virus replication, and thus, they are of significant interest in elucidating the molecular interactions between the virus and its host cell. Until now, the identification of interactions involving DENV RNA has been accomplished predominantly using strategies involving the use of in vitro transcribed regions of vRNA (10–19). While these are powerful techniques these interactions may not necessarily reflect those that occur in a living cell in the context of infection.
Here, we identified interactions between DENV RNA and host cell proteins using a recently described in vivo UV cross-linking approach followed by antisense-mediated affinity purification of DENV ribonucleoprotein (RNP) complexes from infected cells (20). Proteins specifically purified with vRNA were then identified using mass spectrometry. Using a stringent set of selection criteria, we identified a list of twelve host proteins that bind DENV RNA in vivo. Of these, several have been previously reported to bind DENV UTRs in vitro (10–12). Seven of these interactions were independently validated in the context of infection using RNA immunoprecipitation (RIP). To assess the potential biological significance of these proteins during DENV replication, we used siRNA-mediated gene silencing followed by analysis of virus replication. We found that more than half of the host proteins found to bind DENV RNA in vivo appear to have an effect on virus replication. These results demonstrate that this approach can be used for the identification of biologically relevant interactions between DENV RNA and host proteins in the context of infected cells.
RESULTS
Identification of host proteins associated with DENV RNA in vivo.
We recently described an approach employing antisense-mediated affinity purification of DENV RNPs from infected cells to identify host RBPs that bind directly to vRNA in vivo (Fig. 1) (20). This method relies on the use of in vivo UV cross-linking and was developed through modification of similar strategies used to identify RBPs associated with cellular polyadenylated RNA (21). Huh7 cells were infected with dengue virus type 2 strain New Guinea C (DENV NGC) at a multiplicity of infection (MOI) of 1 for 30 h. Infections were carried out for 30 h to allow initiation of a subsequent round of infection, enabling us to capture interactions that may occur during all phases of the viral life cycle. Infected cell cultures were exposed to 254 nm UV to induce covalent cross-links between protein and RNA that are in direct contact (22). Cells were lysed under denaturing conditions that prevent the maintenance or formation of the vast majority of noncovalent associations between protein and protein or between protein and RNA (21). DENV RNP complexes were recovered by affinity purification using biotinylated antisense DNA oligonucleotides complementary to the vRNA (Table 1). Bound proteins were liberated from the RNA by treatment with RNase and identified by mass spectrometry. Lysates from mock-infected cells were processed in parallel as a negative control to aid in the elimination of nonspecific background during the evaluation of mass spectrometry data. Data for all samples from three independent experiments, including the numbers of unique peptides and spectra for each of the 233 proteins identified, are shown in Table S1 in the supplemental material. Experiment 1 was a small-scale experiment using ~2 × 108 cells as starting material for the affinity purification. Therefore, the total number of spectra in experiment 1 is approximately 5-fold less than in full-scale experiments 2 and 3, where ~1 × 109 cells were used.
FIG 1 .

Schematic of affinity purification technique. Cells were infected with dengue virus at an MOI of 1. Mock-infected cells served as a negative control. RNA-protein cross-links were induced 30 h postinfection by exposing the cells to 254 nm UV. Cells were lysed under denaturing conditions and incubated with biotinylated antisense DNA oligonucleotides. RNA-protein complexes were captured on streptavidin-coated magnetic beads. Proteins were liberated from the RNA by RNase treatment and identified by mass spectrometry.
TABLE 1 .
Antisense oligonucleotides used for affinity purificationa
| Sequence (5′ to 3′) | Nucleotide position |
|---|---|
| TAATTGTTCCCCATCTC | 313-297 |
| TCAAAACATTAATGGCTTTTG | 340-320 |
| TGTTTCTATCAGTTCAAAAT | 1080-1061 |
| CCTTGTGTTGGGCAGCGAGA | 1169-1150 |
| TTTCCTTGCCATGTTTTCCTG | 1420-1400 |
| ACCAAATTCTCTTCTTTCTCTTTCAAT | 3458-3432 |
| TCTTTCTTCATCCATGATTGGTGCATTG | 5532-5505 |
| ATTAGGTCCACAAAGGTTTTCCTTGCTTCT | 6131-6102 |
| TTTGTTACCATTTCTTCATTTTT | 9233-9211 |
| TTGTTGCTGCGATTTGTAA | 10539-10521 |
Nucleotide positions are based on dengue virus type 2 strain New Guinea C (GenBank accession number M29095.1). Affinity purification was performed using 250 pmol of an equimolar oligonucleotide mix per batch of 8 × 107 cells. Each sequence had a biotin-TEG tag at the 5′ end.
Selection criteria were applied to these data to assemble a high-confidence candidate list of RBPs that interact with vRNA. First, only those protein IDs that were represented by a minimum of two high-confidence peptides per experiment were considered. Each protein was then evaluated based on the number of unique spectra per sample. Proteins were considered to be high-confidence candidate interactors with vRNA if the sum of the unique spectra from all experiments was enriched ≥2-fold in the infected samples relative to the mock-infected samples. Finally, the first two selection criteria had to be met in at least two of three independent experiments. Based on these stringent selection criteria, we derived a list of twelve candidate host proteins that may be associated with DENV RNA in vivo (Table 2).
TABLE 2 .
Proteins found to be associated with DENV RNA by antisense-mediated affinity purification of dengue viral ribonucleoprotein complexes followed by mass spectrometrya
| Protein | Gene ID | No. of spectra |
Ratio (DENV:mock) |
Binding in vitro |
Reference | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Expt 1 |
Expt 2 |
Expt 3 |
Total |
|||||||||
| Mock | DENV | Mock | DENV | Mock | DENV | Mock | DENV | |||||
| Cold shock domain- containing protein E1 |
CSDE1 | 0 | 0 | 0 | 4 | 0 | 3 | 0 | 7 | INF | X | 12 |
| Nuclease-sensitive element-binding protein 1 |
YBX1 | 0 | 5 | 0 | 4 | 2 | 5 | 2 | 14 | 7.0 | X | 10 |
| Nucleolin | NCL | 2 | 11 | 3 | 6 | 0 | 2 | 5 | 19 | 3.8 | X | 12 |
| Eukaryotic translation initiation factor 4B |
EIF4B | 0 | 4 | 5 | 10 | 3 | 12 | 8 | 26 | 3.3 | ||
| Heterogeneous nuclear ribonucleoproteins C1/C2 |
HNRNPC | 0 | 0 | 2 | 2 | 0 | 3 | 2 | 5 | 2.5 | X | 33 |
| Putative RNA- binding protein 3 |
RBM3 | 3 | 4 | 2 | 8 | 0 | 0 | 5 | 12 | 2.4 | ||
| Polyadenylate- binding protein 1 |
PABPC1 | 0 | 0 | 3 | 4 | 0 | 3 | 3 | 7 | 2.3 | X | 11 |
| Heterogeneous nuclear ribonucleoproteins A2/B1 |
HNRNPA2B1 | 2 | 5 | 4 | 5 | 0 | 4 | 6 | 14 | 2.3 | X | 10 |
| Actin, alpha skeletal muscle |
ACTA1 | 0 | 0 | 0 | 2 | 4 | 7 | 4 | 9 | 2.3 | ||
| Cellular nucleic acid- binding protein |
CNBP | 3 | 8 | 5 | 5 | 2 | 9 | 10 | 22 | 2.2 | X | 12 |
| Single-stranded DNA-binding protein, mitochondrial |
SSBP1 | 0 | 2 | 2 | 2 | 0 | 0 | 2 | 4 | 2.0 | ||
| Plasminogen activator inhibitor 1 RNA-binding protein |
SERBP1 | 0 | 0 | 4 | 6 | 0 | 2 | 4 | 8 | 2.0 | ||
The sum of unique spectra identified in all three experiments was used to calculate a ratio representing the enrichment in infected samples (INF, infinite). Proteins listed are those that were identified by a unique-spectrum count ≥2-fold higher in infected samples than in mock-infected controls in at least two of three independent experiments.
With the exception of alpha-actin (ACTA1), each of the candidate proteins listed in Table 2 has been reported to possess RNA-binding activity (10, 21, 23–32). Further, seven of the proteins have been reported to bind DENV RNA in vitro (10–12, 33). Three of the candidate host proteins have reported roles in modulating DENV replication, although not necessarily through the direct binding of vRNA (10, 33, 34).
Validation of RNA-protein interactions by RNA immunoprecipitation.
We next sought to validate selected candidate interactions using the independent method of RNA immunoprecipitation (RIP). For RIP validation, we first tested a panel of antibodies for their performance in a standard IP assay. Antibodies that were effective for IP were used for RIP in three independent experiments. Huh7 cells were infected with DENV NGC at an MOI of 1 for 30 h. Infected cells were lysed and incubated with a matched isotype control antibody or an antibody specifically directed against the host protein of interest. Specific immunoprecipitation of the protein of interest was confirmed by Western blot analysis (Fig. 2A). Total RNA was purified from the immunoprecipitated material, and the levels of DENV RNA were analyzed using reverse transcription-quantitative PCR (RT-qPCR). The presence of an interaction between vRNA and each protein of interest was evaluated based on an enrichment of vRNA in the experimental IP sample relative to the isotype control (Fig. 2B). As a negative control, RIP was performed with an antibody directed against claudin-2, a cellular protein for which no RNA-binding activity has been reported.
FIG 2 .

Validation of RNA-protein interactions by RNA immunoprecipitation. Huh7 cells were infected with DENV2 NGC at an MOI of 1. Infected cell lysates were incubated with an isotype control antibody or an antibody directed against the indicated cellular proteins. Bound material was captured on Dynabeads protein G for analysis. (A) Western blot analysis of immunoprecipitated material. Input represents 10% of total input sample. IgG lanes represent 20% of immunoprecipitated protein. (B) RT-qPCR analysis of immunoprecipitated RNA. Statistical significance relative to DENV measured by Student’s t test: *, P ≤ 0.05; ◆, P ≤ 0.005; ●, P ≤ 0.0005.
Western blot analysis of protein present in the input (10% of total) relative to the IP (20% of total) samples demonstrated that under each circumstance, the protein of interest was immunoprecipitated by its specific antibody and not by the corresponding isotype control (Fig. 2A). Total RNA was isolated from a fraction of the same IP samples and RT-qPCR was performed to assess the levels of vRNA present. We also assessed the enrichment of two other abundant cellular RNAs with partial cytoplasmic localization, 18S rRNA and U1 spliceosomal RNA. Data are presented as the degree (n-fold) of enrichment of the indicated RNAs present in the specific IP sample relative to the isotype control for each antibody (Fig. 2B). The data indicate that for each candidate RBP tested, there was a significant (≥10-fold) enrichment of DENV RNA in the specific IP sample relative to the isotype control. This is in contrast to the claudin-2-negative control RIP, which was not enriched for any RNA tested. With the exception of SERBP1, each RBP tested immunoprecipitated with significantly more vRNA than either 18S or U1 RNAs. These data show that the majority of the interactions identified using the antisense-mediated affinity purification method can be validated using an independent assay for RNA-protein interactions in the context of infection.
Effect of vRNA-associated RBPs on DENV replication.
To test whether the host proteins found to bind vRNA play a role in mediating efficient DENV replication, we performed siRNA-mediated gene silencing followed by infection. Huh7 cells were transfected with a pool of four unique gene-specific siRNAs per target or with a nonspecific control siRNA (NS control). Silencing was confirmed 48 h posttransfection by Western blot analysis of total protein (see Fig. S1A in the supplemental material). The relative level of silencing was determined by quantification of the signal for each protein compared to beta-actin in the same sample using LI-COR Image Studio software (see Fig. S1B). Three of the gene targets, ACTA1, CNBP, and SSBP1, were not appreciably silenced by these siRNA pools and thus were excluded from subsequent infection assays.
For the analysis of virus replication, siRNA-transfected cells were infected with DENV NGC at an MOI of 1 for 24 h. The levels of virus replication were then measured by expression of viral envelope protein, vRNA accumulation, and production of infectious virus particles (Fig. 3 to 5).
FIG 3 .

High-content imaging of DENV infected cells. Huh7 cells were transfected with the indicated siRNAs and infected with DENV2 NGC at an MOI of 1 for 24 h. (A) Representative images used for high-content imaging. Nuclei are stained with Hoechst. Infected cells are indicated by the Alexa Fluor 488 signal from indirect immunofluorescent staining using a primary antibody recognizing the viral envelope protein. (B) Quantification of nuclei per condition by high-content imaging analysis. Data are representative of 4 independent experiments. Error bars represent standard errors of the mean (SEM) for three individual wells. (C) Percentage of cells infected measured by high-content imaging of cells expressing viral envelope protein. Statistical significance measured by Student’s t test: *, P ≤ 0.05; **, P ≤ 0.005; ***, P ≤ 0.0005.
FIG 4 .
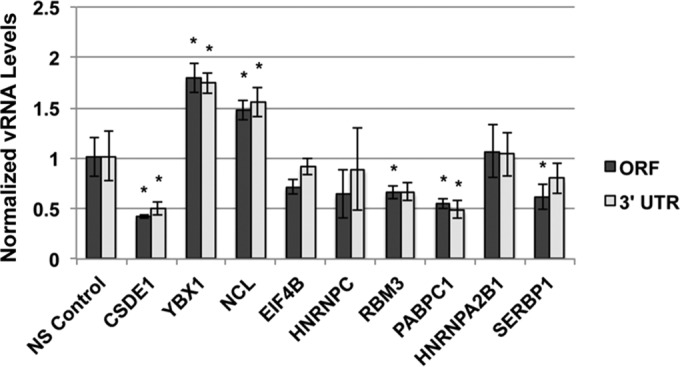
Assessment of the role of host RBPs on vRNA levels. Huh7 cells were transfected with nonsilencing (NS) control siRNA or siRNA pools containing 4 unique siRNAs per target. Cells were infected with DENV 48 h posttransfection at an MOI of 1. Total cell-associated RNA was harvested 24 h postinfection, and levels of v RNA accumulation were measured by RT-qPCR using primers that amplify a region in the middle of the viral coding sequence (ORF) or in the 3′ UTR. Statistical significance measured by Student’s t test: *, P ≤ 0.05.
FIG 5 .

Measurement of viral titer after host RBP depletion. Huh7 cells were transfected with nonsilencing (NS) control siRNA or siRNA pools containing 4 unique siRNAs per target. Cells were infected with DENV 48 h posttransfection at an MOI of 1. Supernatants were collected 24 h postinfection, and viral titer was determined by focus formation assay. Statistical significance measured by Student’s t test: *, P ≤ 0.05; **, P ≤ 0.005.
First, high-content imaging was used to quantify the number of infected cells based on the expression of viral envelope protein. Cells were fixed, permeabilized, and stained for envelope protein using indirect immunofluorescence. Representative images of infected cells after silencing of each candidate RBP are shown in Fig. 3A. Each image represents a single field per well; for the quantification of the percentage of cells infected, 16 fields per well were analyzed. The presence of envelope protein is indicated by Alexa Fluor 488 signal (green), and nuclei are stained with Hoechst (blue). As an indirect measure of potential effects of each siRNA pool on cell viability or proliferation, the number of nuclei per well was evaluated (Fig. 3B). We found that the only treatment condition that resulted in a statistically significant change (P = 0.04) in the number of cells present was treatment with siRNAs targeting RBM3, which resulted in a modest but reproducible increase in the number of cells per well. None of the siRNA pools resulted in a significant decrease in the number of cells, suggesting that there was no significant toxicity or inhibition of proliferation induced as a result of siRNA treatment.
Quantification of the high-content imaging data is shown in Fig. 3C. At an MOI of 1, approximately 50% of the cells became infected in cultures transfected with the NS control. In contrast, we found that siRNA-mediated gene silencing resulted in a significant decrease in the percentage of infected cells for six of the nine candidate RBPs tested. While this reduction was modest in some cases, it was found to be reproducible across several independent experiments (see Table S2 in the supplemental material).
We next assessed the accumulation of vRNA by RT-qPCR using primers that amplify a region within the open reading frame (ORF) of the viral genome. Relative levels of vRNA were determined by normalization to GAPDH and are expressed relative to the value for the NS control condition (Fig. 4). We found that siRNA-mediated gene silencing resulted in a significant change in the level of vRNA accumulation in infected cells for six of the nine RBPs tested. Four of the RBPs that were required for establishing infection as measured by the expression of envelope protein were also required for the efficient accumulation of vRNA (CSDE1, RBM3, PABPC1, and SERBP1). Although not statistically significant, the levels of vRNA were also reduced in cells depleted of EIF4B and HNRNPC. Silencing of YBX1 and NCL both resulted in an increase in the accumulation of vRNA relative to the NS control. Use of a different set of qPCR primers designed to amplify a region in the 3′ UTR of the vRNA yielded similar results, suggesting that the differences observed were not simply due to the region of the vRNA amplified.
Finally, virus present in culture supernatants, which is the most relevant measure of biological importance, was titrated to measure the production of infectious virus particles under conditions of siRNA-mediated depletion of each RBP (Fig. 5). Silencing of CSDE1, YBX1, HNRNPC, and PABPC1 resulted in a significant reduction in virus titer compared to the NS control. In contrast, depletion of NCL resulted in an increase in virus titer, suggesting an overall antiviral role for nucleolin during DENV replication in Huh7 cells.
Taken together, these results demonstrate that in each assay, more than half of the host RBPs found to be specifically associated with DENV RNA in infected cells play a biologically relevant role in mediating efficient virus replication. The results of the virus replication assays across several independent experiments are summarized in Table S2 in the supplemental material.
DISCUSSION
In summary, we used an approach that relies on UV cross-linking followed by antisense-mediated affinity purification and mass spectrometry to identify proteins that are associated with DENV RNA in the context of infected cells. A set of stringent selection criteria was applied to the mass spectrometry data in an effort to reduce the occurrence of false positives and identify only those proteins that are likely to have been identified as a result of a direct association with viral RNA. We then used three independent assays for measuring virus replication to functionally validate effects of individual RBPs on the DENV life cycle. Using this approach, we showed that at least half of the proteins found to associate with DENV RNA using the antisense-mediated affinity purification method likely play some role in DENV replication.
The affinity purification technique employed here is associated with significant levels of background, likely due to the fact that the antisense oligonucleotides used all exhibit partial complementarity to cellular nucleic acids and thus are expected to purify cellular RNP complexes. In an effort to eliminate this background from our analysis and identify proteins that are likely to specifically associate with vRNA, we used mock-infected cells as a negative control. We then applied a set of stringent selection criteria to the mass spectrometry data to assemble a candidate list of proteins bound to vRNA. Because of the background present and the stringent criteria used, only 12 of the 233 proteins identified by mass spectrometry were considered for further validation (Table 2; also, see Table S1 in the supplemental material). It is likely, however, that other proteins in the more extensive list (see Table S1) also specifically associate with vRNA and modulate virus infection.
Of the 12 proteins that met our stringent selection criteria, 6 were previously reported to bind DENV RNA in vitro (Table 2) (10–12). Based on the available data, we are unable to definitively conclude whether the approach described here is an improvement over simpler in vitro strategies for identifying biologically relevant interactions between vRNA and host proteins. Such a determination would require large-scale validation of interactions identified in vitro, coupled with functional assays to establish whether these interactions are important for infection. Nonetheless, our data suggest that the in vivo method will reveal interacting proteins not observed using conventional in vitro purification techniques.
We used siRNA-mediated gene silencing to investigate a potential role for vRNA-associated RBPs in mediating efficient DENV replication. Using three independent assays to measure virus replication, we showed that more than half of the RBPs tested appeared to be involved in some aspect of the viral life cycle. This degree of functional validation is consistent with the results of a similar study identifying RBPs that are associated with poliovirus RNA (35). In some cases, the effect of RBP silencing on the efficiency of virus replication, although statistically significant, is subtle. This could be explained in part by incomplete knockdown of protein levels under the conditions of siRNA transfection used. It is also possible that there is functional redundancy among host RBPs, such that inhibition of one RBP that plays a role in virus replication can be compensated for by another in its absence. The siRNA-mediated gene silencing experiments described here suggest a role for individual RBPs, either direct or indirect, in mediating DENV replication. However, these results do not demonstrate whether a physical interaction between a given RBP and vRNA is required for the effect observed after RBP depletion.
Of the host factors identified, YBX1, NCL, and HNRNPC have been reported to be involved in DENV replication in the context of infection (10, 33, 34). The results we obtained upon silencing of HNRNPC are consistent with those previously observed in Huh7 cells, where HNRNPC was required for efficient virus replication (33). In contrast, YBX1 was previously reported to play an antiviral role in MEF cells derived from YBX1-knockout mice; however, we found that YBX1 appears to have a proviral function in Huh7 cells (10). This difference could be due to distinct roles for YBX1 in mouse fibroblasts and human hepatoma cells. Alternatively, it is possible that different phenotypes are observed under conditions of partial silencing versus genetic knockout of YBX1, a possibility that we are currently investigating. Our results also differ from those previously observed upon depletion of NCL in HEK 293 cells, where NCL was found to be required for the production of infectious virus, perhaps through alterations in capsid structure (34). It is not surprising to observe cell type-specific differences in the requirements for host proteins during virus replication; therefore, it is difficult to assess the significance, if any, of these discrepancies.
We used three independent measures of virus replication to assess the potential role of individual RBPs in DENV infection. Taken together, these data can provide insight into which phase of the viral life cycle may be affected by depletion of individual host proteins, especially in cases where silencing had opposing effects on virus replication in different assays. For example, depletion of YBX1 had no apparent effect on the percentage of cells found to express viral envelope protein (Fig. 3C), yet it resulted in an increase in the accumulation of vRNA (Fig. 4). This increase in the accumulation of cell-associated vRNA corresponded with an overall decrease in the titer of extracellular virus (Fig. 5). This phenotype suggests a defect that occurs late in the viral life cycle and could be explained by an inhibition of genome packaging or virion egress, phases of DENV replication about which little is known. A more detailed analysis of these late events, in the absence of YBX-1, would be required to formally address these possibilities.
Based on the known functions of individual RBPs found to be required for efficient DENV replication, specific hypotheses can be formed regarding the potential mechanisms by which these host proteins act to exert their effect on the viral life cycle. CSDE1 (unr) is a well-characterized RNA-binding protein that binds to DENV UTR RNA in vitro and was identified in a genome-wide RNA interference (RNAi) screen as a factor necessary for efficient replication of DENV in Drosophila cells (12, 36). CSDE1 is involved in cap-independent translation initiation of both cellular and viral RNAs, including rhinovirus and poliovirus RNA (37–40). Translation of DENV RNA is initiated predominantly by a cap-dependent mechanism, although cap-independent translation initiation has been demonstrated under conditions in which cap-dependent initiation is inhibited (41). Even under conditions that favor cap-dependent translation initiation of DENV RNA, it is possible that CSDE1 is involved in the translation, synthesis, or stability of vRNA through its RNA chaperone activity. For example, binding of CSDE1 to either DENV UTR could modify the structure of the vRNA, leading to the recruitment of other factors that ultimately enhance translation.
PABPC1 (PABP) also binds DENV RNA in vitro, and its binding site has been mapped to the 3′ UTR (11). There is evidence that binding of PABPC1 to the DENV 3′ UTR stimulates translation of a DENV reporter RNA in cell-free extracts; however, a biological effect of this interaction has not been tested in the context of infection. Interestingly, CSDE1 and PABPC1 cooperate to regulate the translation or stability of several cellular mRNAs (38, 42–44). Therefore, it is possible that these proteins function together to regulate virus replication, for example at the level of translation, which would be consistent with the observation that the levels of virus replication were reduced in all three assays when the expression of either protein was silenced (Fig. 3 to 5).
In conclusion, this study represents the first report of host proteins that physically associate with DENV RNA in vivo. Further, we demonstrated that several of these proteins play a role in mediating virus replication. Because of the highly stringent nature of the criteria used to select proteins for functional validation, it is likely that some of the proteins that did not meet these rigorous criteria (see Table S1 in the supplemental material) are nonetheless genuine interacting partners that may play some role in the viral life cycle. Future studies involving more detailed analysis of individual phases of the viral life cycle in the absence of these proteins or their interactions with vRNA will lead to the elucidation of the molecular mechanisms by which these proteins promote or inhibit virus replication.
MATERIALS AND METHODS
Cells, viruses, and infections.
Huh7 human hepatoma cells and Vero African green monkey kidney cells were maintained in Dulbecco’s modified eagle medium (DMEM) containing 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 µg/ml streptomycin, and 1× modified Eagle medium (MEM) nonessential amino acids in a 37°C humidified incubator containing 5% CO2. DENV2 NGC was a kind gift from Aravinda de Silva (University of North Carolina, Chapel Hill, NC). Virus stocks were propagated in Aedes albopictus C6/36 cells grown at 28°C in RPMI supplemented with 2% FBS, 100 U/ml penicillin, and 100 µg/ml streptomycin. Cells were infected in DMEM containing 2% FBS, 100 U/ml penicillin, 100 µg/ml streptomycin, 1× MEM nonessential amino acids, and 10 mM HEPES for 1 h, washed once with phosphate-buffered saline (PBS), and incubated in fresh medium as indicated in the figure legends.
Oligonucleotides.
Antisense oligonucleotides used for the affinity purification of viral RNP complexes were biotinylated at the 5′ end, followed by addition of a tetraethylene glycol (TEG) spacer, and correspond to sequences present in the DENV2 NGC genome (GenBank accession no. M29095.1). Antisense oligonucleotide sequences and their complementary nucleotide positions in the viral genome are shown in Table 1. The oligonucleotides used for RT-qPCR amplification of DENV RNA were DENV ORF For (5′ GAA ATG GGT GCC AAC TTC AAG GCT 3′) and DENV ORF Rev (5′ TCT TTG TGC TGC ACT AGA GTG GGT 3′) for amplification of nucleotides 5755 to 5892 in the ORF of the viral genome and DENV 3′ UTR For (5′ CGT AGT GGA CTA GCG GTT AGA G 3′) and DENV 3′ UTR Rev (5′ TCT GGT CTT TCC CAG CGT 3′) for amplification of nucleotides 10487 to 10645 in the 3′ UTR of the viral genome. Relative gene expression analysis was performed by normalization to GAPDH amplified with primers GAPDH For (5′ AGC CAC ATC GCT CAG ACA C 3′) and GAPDH Rev (5′ GCC CAA TAC GAC CAA ATC C 3′). Absolute gene expression analysis for RIP was performed using primers 18S For (5′ GTA ACC CGT TGA ACC CCA TT 3′), 18S Rev (5′ CCA TCC AAT CGG TAG TAG CG 3′), U1 For (5′ GGG AGA TAC CAT GAT CAC GAA GGT 3′), and U1 Rev (5′ ATG CAG TCG AGT TTC CCA CA 3′).
Antibodies.
The following antibodies were used for RIP and Western blot analysis: mouse envelope 4G2 (45), mouse beta-actin (SC-47778; Santa Cruz Biotechnology), goat ACTA1 (SC-1616; Santa Cruz Biotechnology), rabbit claudin-2 (51-6100; Life Technologies), rabbit CNBP (AB156420; Abcam), rabbit CSDE1 (AB176584; Abcam), rabbit EIF4B (A301-767A; Bethyl Laboratories), rabbit HNRNPA2B1 (C192416; LSBio), goat HNRNPC1/C2 (SC-10037; Santa Cruz Biotechnology), rabbit NCL (A300-709A; Bethyl Laboratories), rabbit PABP (AB21060; Abcam), rabbit RBM3 (C154720; LSBio), goat SERBP1 (SC-131741; Santa Cruz Biotechnology), rabbit SSBP1 (AB199687; Abcam), rabbit YBX1 (AB12148; Abcam). Secondary antibodies used for imaging on an Odyssey CLx infrared imaging system (LI-COR) were donkey anti-mouse IRDye 680, donkey anti-goat IRDye 800, and goat anti-rabbit IRDye 800.
Affinity purification of DENV RNP complexes.
Affinity purification of DENV RNP complexes was performed as described elsewhere (20). Briefly, Huh7 cells were mock-infected or infected with DENV2 NGC at an MOI of 1 for 30 h. Cells were irradiated at 254 nm with 0.15 J/cm2. Irradiated cells were collected, lysed in lysis buffer (200 mM KCl, 20 mM HEPES [pH 7.2], 2% N-dodecyl-β-d-maltoside, 1% Igepal CA-360, 40 U/ml murine RNase inhibitor [NEB] and 1× protease inhibitor cocktail [Roche]), and processed for affinity purification in batches of 8e7 cells or approximately two 500-cm2 dishes per batch. Protein was stored at −80°C until pooling of like samples and submission for liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis.
LC-MS/MS analysis.
Samples in 20 mM Tris (pH 7.5), 1 mM EDTA, 150 mM NaCl, and 0.5 mM dithiothreitol (DTT) were supplemented with 0.1% Rapigest SF acid-labile surfactant (Waters Corp) prior to being reduced with 10 mM DTT for 30 min at 80°C and alkylated with 22 mM iodoacetamide for 45 min at room temperature. Samples were proteolytically digested with 500 ng sequencing-grade modified trypsin (Promega) for 18 h at 37°C. Rapigest surfactant was hydrolyzed with acidification to pH 2.5 with trifluoroacetic acid (TFA) for 2 h at 60°C. Samples were subjected to solid-phase extraction (SPE) on a 1-ml Oasis MCX cartridge (Waters) according to the manufacturer-recommended protocol. Eluted peptides were brought to dryness by using vacuum centrifugation prior to LC-MS/MS.
Samples were resuspended in 10 µl of 1% TFA–2% acetonitrile, and LC-MS/MS was performed on 5 µl of each sample using a nanoAcquity UPLC system (Waters Corp.) coupled to a Thermo QExactive Plus high-resolution accurate-mass tandem mass spectrometer (Thermo) via a nanoelectrospray ionization source. Briefly, the peptides were trapped on a Symmetry C18 300-mm by 180-mm trapping column (5 µl/min at 99.9/0.1 [vol/vol] water-acetonitrile), after which analytical separation was performed using a 1.7-µm Acquity BEH130 C18 column (75 mm by 250 mm; Waters Corp.) using a 90-min gradient of 5 to 40% acetonitrile with 0.1% formic acid at a flow rate of 400 nl/min with a column temperature of 55°C. Data collection on the QExactive Plus mass spectrometer was performed in a data-dependent acquisition (DDA) mode of acquisition with an r value of 70,000 (at m/z 200) and a full MS scan from m/z 375 to 1,600 with a target AGC value of 1 × 106 ions, followed by 10 MS/MS scans at an r value of 17,500 (at m/z 200) at a target AGC value of 5 × 104 ions. A 20-s dynamic exclusion was employed to increase depth of coverage.
Raw LC-MS/MS data files were processed in Proteome Discoverer and then subjected to independent Mascot searches (Matrix Science) against a SwissProt database (all taxonomies) containing both forward and reverse entries for each protein. Search tolerances were 5 ppm for precursor ions and 0.02 Da for product ions using trypsin specificity with up to two missed cleavages. Carbamidomethylation (+57.0214 Da on C) was set as a fixed modification, whereas oxidation (+15.9949 Da on M) and deamidation (+0.98 Da on NQ) were dynamic modifications. All searched spectra were imported into Scaffold (v4.4; Proteome Software), and scoring thresholds were set to achieve a protein false discovery rate of 1.0% using the PeptideProphet algorithm.
RNA immunoprecipitation.
RNA immunoprecipitations were performed as described elsewhere (46), with slight modifications. Huh7 cells were infected with DENV2 NGC at an MOI of 1 for 30 h. Cells were harvested, pelleted, and lysed in approximately one cell pellet volume of lysis buffer (200 mM KCl, 20 mM HEPES [pH 7.2], 2% N-dodecyl-β-d-maltoside, 1% Igepal CA-360, 100 U/ml murine RNase inhibitor [NEB], and 1× protease inhibitor cocktail [Roche]). Fresh lysates were cleared by centrifugation at 12,000 × g at 4°C for 10 min, snap frozen in liquid nitrogen, and stored at −80°C until use. For immunoprecipitation, 1 mg of protein was brought to a volume of 800 µl with immunoprecipitation buffer (50 mM Tris HCl [pH 7.4], 150 mM NaCl, 1 mM MgCl2, and 0.05% Igepal CA-360) and incubated with 5 µg of isotype control or specific antibody for 2 h at 4°C. Bound complexes were captured on 50 µl Dynabeads-protein G (Life Technologies) by rotation at 4°C overnight. Complexes were washed three times in immunoprecipitation buffer, and immunoprecipitated protein and RNA were analyzed by Western blotting and RT-qPCR, respectively.
siRNA transfections.
For siRNA transfections, 1 × 104 Huh7 cells were plated per well in 96-well plates 24 h prior to transfection. Cells were transfected at ~40% confluence with siRNA GeneSolution duplexes (Qiagen) at a final concentration of 10 nM using Lipofectamine RNAiMax (Life Technologies) according to the manufacturer’s protocol.
Immunofluorescence and high-content imaging.
Huh7 cells were infected with DENV2 NGC at an MOI of 1 for 24 h. Cells were washed with PBS, fixed in 4% paraformaldehyde, permeabilized in 0.1% Tween 20, and blocked with 1% normal goat serum. Indirect immunofluorescence staining was performed using mouse monoclonal antienvelope antibody (4G2) followed by secondary goat anti-mouse IgG labeled with Alexa Fluor 488 (Life Technologies). Nuclei were stained with Hoechst. High-content imaging analysis was performed using an Opera Phenix (PerkinElmer). Quantification was performed using data collected from 16 fields per well in 96-well format. Nuclei were counted in the Hoechst channel, and infected cells were identified using cytoplasmic signal in the Alexa 488 channel.
RT-qPCR.
Total RNA was extracted using Trizol (Life Technologies) or the ReliaPrep RNA cell miniprep system (Promega) according to the manufacturer’s protocols. cDNA synthesis was performed using a high-capacity cDNA reverse transcription kit (ABI) according to the manufacturer’s protocol. qPCR was performed on a LightCycler 480II (Roche) using LightCycler 480 SYBR green I master mix (Roche) according to the manufacturer’s protocol.
Focus formation assay.
Supernatant medium from infected cells was serially diluted and used to infect confluent monolayers of Vero cells. After an infection period of 1 h, cells were washed, and infection medium was replaced with overlay containing 0.5% tragacanth (Sigma), 1× Eagle’s modified essential medium (EMEM) (Lonza), 5% FBS, and 10 mM HEPES. After 72 h, cells were washed with PBS, fixed in 4% paraformaldehyde, permeabilized in 0.1% Tween 20, and blocked with 1% normal goat serum. Indirect immunofluorescence staining was performed using mouse monoclonal antienvelope antibody (4G2) followed by secondary goat anti-mouse IgG labeled with Alexa Fluor 488 (Life Technologies). Foci were counted by visualization with an epifluorescence microscope.
SUPPLEMENTAL MATERIAL
Validation of siRNA knockdown. Huh7 cells were transfected with nonsilencing control siRNA (NS control) or siRNA pools containing 4 unique siRNAs per target (knockdown). Total protein was harvested 48 h posttransfection, and the level of silencing was assessed by Western blot analysis. (A) Western blot images using the indicated antibodies. (B) Quantitation of protein levels relative to the nonsilencing control. Error bars represent SEM of three individual wells. Results are representative of two independent experiments. Download
Proteins identified by mass spectrometry. Ribonucleoprotein complexes were purified from infected cells using biotinylated antisense oligonucleotides complementary to the dengue viral genome. Mock-infected cell lysates were processed in parallel as negative controls. Proteins in affinity-purified samples were identified by LC-MS/MS. The numbers of unique peptides and spectra identified in three independent experiments are shown. The sum of the numbers of spectra for all three experiments and the ratio of numbers of spectra in infected versus mock-infected samples are indicated. “INF” indicates the ratio for proteins that were present only in infected samples.
Summary of virus replication after siRNA-mediated silencing of RNA-binding proteins. Levels of virus replication for each of the three assays used are expressed relative to the nonsilencing control along with corresponding standard deviation across the indicated number of samples. P values were determined by Student’s t test comparing siRNA-treated samples to nonsilencing (NS) control samples. Blue, P ≤ 0.05; pink, P ≤ 0.005; green, P ≤ 0.0005.
ACKNOWLEDGMENTS
We thank Arthur Moseley (Duke University) for helpful discussion regarding mass spectrometry. We thank Rafael Campos and Holly Heimsath for assistance in harvesting cells. We also thank Daniel Engel (University of Virginia) for helpful suggestions regarding this work.
This work was supported by funding from NIH grant RO1AI101431 to Mariano Garcia-Blanco and NIH grant F32AI109834 to Stacia L. Phillips. The funding agency had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Citation Phillips SL, Soderblom EJ, Bradrick SS, Garcia-Blanco MA. 2016. Identification of proteins bound to dengue viral RNA in vivo reveals new host proteins important for virus replication. mBio 7(1):e01865-15. doi: 10.1128/mBio.01865-15.
REFERENCES
- 1.Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF, George DB, Jaenisch T, Wint GRW, Simmons CP, Scott TW, Farrar JJ, Hay SI. 2013. The global distribution and burden of dengue. Nature 496:504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Daffis S, Szretter KJ, Schriewer J, Li J, Youn S, Errett J, Lin T, Schneller S, Zust R, Dong H, Thiel V, Sen GC, Fensterl V, Klimstra WB, Pierson TC, Buller RM, Gale M, Shi P, Diamond MS. 2010. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 468:452–456. doi: 10.1038/nature09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bidet K, Garcia-Blanco M. 2014. Flaviviral RNAs: weapons and targets in the war between virus and host. Biochem J 462:215–230. doi: 10.1042/BJ20140456. [DOI] [PubMed] [Google Scholar]
- 4.Moon SL, Anderson JR, Kumagai Y, Wilusz CJ, Akira S, Khromykh AA, Wilusz J. 2012. A noncoding RNA produced by arthropod-borne flaviviruses inhibits the cellular exoribonuclease XRN1 and alters host mRNA stability. RNA 18:2029–2040. doi: 10.1261/rna.034330.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clarke BD, Roby JA, Slonchak A, Khromykh AA. 2015. Functional non-coding RNAs derived from the flavivirus 3′ untranslated region. Virus Res 206:53–61. doi: 10.1016/j.virusres.2015.01.026. [DOI] [PubMed] [Google Scholar]
- 6.Pijlman GP, Funk A, Kondratieva N, Leung J, Torres S, van der Aa L, Liu WJ, Palmenberg AC, Shi P, Hall RA, Khromykh AA. 2008. A highly structured, nuclease-resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell Host Microbe 4:579–591. doi: 10.1016/j.chom.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 7.Bidet K, Dadlani D, Garcia-Blanco MA. 2014. G3BP1, G3BP2 and CAPRIN1 are required for translation of interferon stimulated mRNAs and are targeted by a dengue virus non-coding RNA. PLoS Pathog 10:e1004242. doi: 10.1371/journal.ppat.1004242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manokaran G, Finol E, Wang C, Gunaratne J, Bahl J, Ong EZ, Tan HC, Sessions OM, Ward AM, Gubler DJ, Harris E, Garcia-Blanco MA, Ooi EE. 2015. Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 350:217–221 doi: 10.1126/science.aab3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang R, Hsu T, Chen Y, Liu S, Tsai Y, Lin Y, Chen Y, Fan Y. 2013. Japanese encephalitis virus non-coding RNA inhibits activation of interferon by blocking nuclear translocation of interferon regulatory factor 3. Vet Microbiol 166:11–21. doi: 10.1016/j.vetmic.2013.04.026. [DOI] [PubMed] [Google Scholar]
- 10.Paranjape SM, Harris E. 2007. Y box-binding protein-1 binds to the dengue virus 3′-untranslated region and mediates antiviral effects. J Biol Chem 282:30497–30508. doi: 10.1074/jbc.M705755200. [DOI] [PubMed] [Google Scholar]
- 11.Polacek C, Friebe P, Harris E. 2009. Poly(A)-binding protein binds to the non-polyadenylated 3′ untranslated region of dengue virus and modulates translation efficiency. J Gen Virol 90:687–692. doi: 10.1099/vir.0.007021-0. [DOI] [PubMed] [Google Scholar]
- 12.Ward AM, Bidet K, Yinglin A, Ler SG, Hogue K, Blackstock W, Gunaratne J, Garcia-Blanco MA. 2011. Quantitative mass spectrometry of DENV-2 RNA-interacting proteins reveals that the DEAD-box RNA helicase DDX6 binds the DB1 and DB2 3′ UTR structures. RNA Biol 8:1173–1186. doi: 10.4161/rna.8.6.17836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Nova-Ocampo M, Villegas-Sepúlveda N, del Angel RM. 2002. Translation elongation factor-1alpha, La, and PTB interact with the 3′ untranslated region of dengue 4 virus RNA. Virology 295:337–347. doi: 10.1006/viro.2002.1407. [DOI] [PubMed] [Google Scholar]
- 14.García-Montalvo BM, Medina F, del Angel RM. 2004. La protein binds to NS5 and NS3 and to the 5′ and 3′ ends of dengue 4 virus RNA. Virus Res 102:141–150. doi: 10.1016/j.virusres.2004.01.024. [DOI] [PubMed] [Google Scholar]
- 15.Gomila RC, Martin GW, Gehrke L. 2011. NF90 binds the dengue virus RNA 3′ terminus and is a positive regulator of dengue virus replication. PLoS One 6:e16687. doi: 10.1371/journal.pone.0016687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lei Y, Huang Y, Zhang H, Yu L, Zhang M, Dayton A. 2011. Functional interaction between cellular p100 and the dengue virus 3′ UTR. J Gen Virol 92:796–806. doi: 10.1099/vir.0.028597-0. [DOI] [PubMed] [Google Scholar]
- 17.Yocupicio-Monroy RME, Medina F, Reyes-del Valle J, del Angel RM. 2003. Cellular proteins from human monocytes bind to dengue 4 virus minus-strand 3′ untranslated region RNA. J Virol 77:3067–3076. doi: 10.1128/JVI.77.5.3067-3076.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yocupicio-Monroy M, Padmanabhan R, Medina F, del Angel RM. 2007. Mosquito La protein binds to the 3′ untranslated region of the positive and negative polarity dengue virus RNAs and relocates to the cytoplasm of infected cells. Virology 357:29–40. doi: 10.1016/j.virol.2006.07.042. [DOI] [PubMed] [Google Scholar]
- 19.Ong CC, Lam SK, AbuBakar S. 1998. Cellular proteins bind to the 3′ and 5′ untranslated regions of dengue 2 virus genome. Malays J Pathol 20:11–17. [PubMed] [Google Scholar]
- 20.Phillips SL, Garcia-Blanco MA, Bradrick SS. 2015. Antisense-mediated affinity purification of dengue virus ribonucleoprotein complexes from infected cells. Methods doi: 10.1016/j.ymeth.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann B, Strein C, Davey N, Humphreys D, Preiss T, Steinmetz L, Krijgsveld J, Hentze M. 2012. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 149:1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 22.Wagenmakers AJM, Reinders RJ, van Venrooij WJ. 1980. Cross-linking of mRNA to proteins by irradiation of intact cells with ultraviolet light. Eur J Biochem 112:323–330. doi: 10.1111/j.1432-1033.1980.tb07207.x. [DOI] [PubMed] [Google Scholar]
- 23.Baltz A, Munschauer M, Schwanhäusser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M, Wyler E, Bonneau R, Selbach M, Dieterich C, Landthaler M. 2012. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell 46:674–690. doi: 10.1016/j.molcel.2012.05.021. [DOI] [PubMed] [Google Scholar]
- 24.Moran-Jones K, Wayman L, Kennedy DD, Reddel RR, Sara S, Snee MJ, Smith R. 2005. hnRNP A2, a potential ssDNA/RNA molecular adapter at the telomere. Nucleic Acids Res 33:486–496. doi: 10.1093/nar/gki203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grange T, de Sa CM, Oddos J, Pictet R. 1987. Human mRNA polyadenylate binding protein: evolutionary conservation of a nucleic acid binding motif. Nucleic Acids Res 15:4771–4787. doi: 10.1093/nar/15.12.4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heaton JH, Dlakic WM, Dlakic M, Gelehrter TD. 2001. Identification and cDNA cloning of a novel RNA-binding protein that interacts with the cyclic nucleotide-responsive sequence in the type-1 plasminogen activator inhibitor mRNA. J Biol Chem 276:3341–3347. doi: 10.1074/jbc.M006538200. [DOI] [PubMed] [Google Scholar]
- 27.Derry JMJ, Kerns JA, Francke U. 1995. RBM3, a novel human gene in Xp11.23 with a putative RNA-binding domain. Hum Mol Genet 4:2307–2311. doi: 10.1093/hmg/4.12.2307. [DOI] [PubMed] [Google Scholar]
- 28.Neubauer G, King A, Rappsilber J, Calvio C, Watson M, Ajuh P, Sleeman J, Lamond A, Mann M. 1998. Mass spectrometry and EST-database searching allows characterization of the multi-protein spliceosome complex. Nat Genet 20:46–50. doi: 10.1038/1700. [DOI] [PubMed] [Google Scholar]
- 29.Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. 2015. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518:560–564. doi: 10.1038/nature14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Milburn SC, Hershey JW, Davies MV, Kelleher K, Kaufman RJ. 1990. Cloning and expression of eukaryotic initiation factor 4B cDNA: sequence determination identifies a common RNA recognition motif. EMBO J 9:2783–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ishikawa F, Matunis MJ, Dreyfuss G, Cech TR. 1993. Nuclear proteins that bind the pre-mRNA 3′ splice site sequence r(UUAG/G) and the human telomeric DNA sequence:d(TTAGGG)n. Mol Cell Biol 13:4301–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jacquemin-Sablon H, Triqueneaux G, Deschamps S, le Maire M, Doniger J, Dautry F. 1994. Nucleic acid binding and intracellular localization of unr, a protein with five cold shock domains. Nucleic Acids Res 22:2643–2650. doi: 10.1093/nar/22.13.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dechtawewat T, Songprakhon P, Limjindaporn T, Puttikhunt C, Kasinrerk W, Saitornuang S, Yenchitsomanus P, Noisakran S. 2015. Role of human heterogeneous nuclear ribonucleoprotein C1/C2 in dengue virus replication. Virol J 12:14. doi: 10.1186/s12985-014-0219-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Balinsky CA, Schmeisser H, Ganesan S, Singh K, Pierson TC, Zoon KC. 2013. Nucleolin interacts with the dengue virus capsid protein and plays a role in formation of infectious virus particles. J Virol 87:13094–13106. doi: 10.1128/JVI.00704-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lenarcic EM, Landry DM, Greco TM, Cristea IM, Thompson SR. 2013. Thiouracil cross-linking mass spectrometry: a cell-based method to identify host factors involved in viral amplification. J Virol 87:8697–8712. doi: 10.1128/JVI.00950-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sessions OM, Barrows NJ, Souza-Neto JA, Robinson TJ, Hershey CL, Rodgers MA, Ramirez JL, Dimopoulos G, Yang PL, Pearson JL, Garcia-Blanco MA. 2009. Discovery of insect and human dengue virus host factors. Nature 458:1047–1050. doi: 10.1038/nature07967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hunt SL, Hsuan JJ, Totty N, Jackson RJ. 1999. unr, a cellular cytoplasmic RNA-binding protein with five cold-shock domains, is required for internal initiation of translation of human rhinovirus RNA. Genes Dev 13:437–448. doi: 10.1101/gad.13.4.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mitchell SA, Brown EC, Coldwell MJ, Jackson RJ, Willis AE. 2001. Protein factor requirements of the Apaf-1 internal ribosome entry segment: roles of polypyrimidine tract binding protein and upstream of N-ras. Mol Cell Biol 21:3364–3374. doi: 10.1128/MCB.21.10.3364-3374.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Evans JR, Mitchell SA, Spriggs KA, Ostrowski J, Bomsztyk K, Ostarek D, Willis AE. 2003. Members of the poly (rC) binding protein family stimulate the activity of the c-myc internal ribosome entry segment in vitro and in vivo. Oncogene 22:8012–8020. doi: 10.1038/sj.onc.1206645. [DOI] [PubMed] [Google Scholar]
- 40.Tinton S, Schepens B, Bruynooghe Y, Beyaert R, Cornelis S. 2005. Regulation of the cell-cycle-dependent internal ribosome entry site of the PITSLRE protein kinase: roles of Unr (upstream of N-ras) protein and phosphorylated translation initiation factor eIF-2alpha. Biochem J 385:155–163. doi: 10.1042/BJ20040963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edgil D, Polacek C, Harris E. 2006. Dengue virus utilizes a novel strategy for translation initiation when cap-dependent translation is inhibited. J Virol 80:2976–2986. doi: 10.1128/JVI.80.6.2976-2986.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duncan KE, Strein C, Hentze MW. 2009. The SXL-UNR corepressor complex uses a PABP-mediated mechanism to inhibit ribosome recruitment to msl-2 mRNA. Mol Cell 36:571–582. doi: 10.1016/j.molcel.2009.09.042. [DOI] [PubMed] [Google Scholar]
- 43.Patel GP, Ma S, Bag J. 2005. The autoregulatory translational control element of poly(A)-binding protein mRNA forms a heteromeric ribonucleoprotein complex. Nucleic Acids Res 33:7074–7089. doi: 10.1093/nar/gki1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chang T-, Yamashita A, Chen CY, Yamashita Y, Zhu W, Durdan S, Kahvejian A, Sonenberg N, Shyu AB. 2004. UNR, a new partner of poly(A)-binding protein, plays a key role in translationally coupled mRNA turnover mediated by the c-fos major coding-region determinant. Genes Dev 18:2010–2023. doi: 10.1101/gad.1219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Henchal EA, Gentry MK, McCown JM, Brandt WE. 1982. Dengue virus-specific and flavivirus group determinants identified with monoclonal antibodies by indirect immunofluorescence. Am J Trop Med Hyg 31:830–836. [DOI] [PubMed] [Google Scholar]
- 46.Keene JD, Komisarow JM, Friedersdorf MB. 2006. RIP-Chip: the isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat Protoc 1:302–307. doi: 10.1038/nprot.2006.47. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Validation of siRNA knockdown. Huh7 cells were transfected with nonsilencing control siRNA (NS control) or siRNA pools containing 4 unique siRNAs per target (knockdown). Total protein was harvested 48 h posttransfection, and the level of silencing was assessed by Western blot analysis. (A) Western blot images using the indicated antibodies. (B) Quantitation of protein levels relative to the nonsilencing control. Error bars represent SEM of three individual wells. Results are representative of two independent experiments. Download
Proteins identified by mass spectrometry. Ribonucleoprotein complexes were purified from infected cells using biotinylated antisense oligonucleotides complementary to the dengue viral genome. Mock-infected cell lysates were processed in parallel as negative controls. Proteins in affinity-purified samples were identified by LC-MS/MS. The numbers of unique peptides and spectra identified in three independent experiments are shown. The sum of the numbers of spectra for all three experiments and the ratio of numbers of spectra in infected versus mock-infected samples are indicated. “INF” indicates the ratio for proteins that were present only in infected samples.
Summary of virus replication after siRNA-mediated silencing of RNA-binding proteins. Levels of virus replication for each of the three assays used are expressed relative to the nonsilencing control along with corresponding standard deviation across the indicated number of samples. P values were determined by Student’s t test comparing siRNA-treated samples to nonsilencing (NS) control samples. Blue, P ≤ 0.05; pink, P ≤ 0.005; green, P ≤ 0.0005.