

Abstract
Background
Adult human airway smooth muscle (ASM) produce cytokines involved in recruitment and survival of leukocytes within airway walls. Cytokine generation by adult ASM is glucocorticoid-sensitive. Whether developing lung ASM produces cytokines in a glucocorticoid-sensitive fashion is unknown.
Methods
Cultured fetal human ASM cells stimulated with TNF-α (0–20 ng/ml) were incubated with TNF-α receptor-blocking antibodies, fluticasone (1 and 100 nm), or vehicle. Supernatants and cells were assayed for the production of CCL5, CXCL10, and CXCL8 mRNA and protein and glucocorticoid receptor phosphorylation.
Results
CCL5, CXCL10, and CXCL8 mRNA and protein production by fetal ASM cell was significantly and dose-dependently following TNF-α treatment. Cytokine mRNA and protein production were effectively blocked by TNF-α R1 and R2 receptor neutralizing antibodies but variably inhibited by fluticasone. TNF-α-induced TNF-R1 and R2 receptor mRNA expression was only partially attenuated by fluticasone. Glucocorticoid receptor phosphorylation at serine (Ser) 211 but not at Ser 226 was enhanced by fluticasone.
Conclusion
Production of CCL5, CXCL10, and CXCL8 by fetal ASM appears to involve pathways that are both qualitatively and mechanistically distinct to those described for adult ASM. The findings imply developing ASM has potential to recruit leukocyte into airways and, therefore, of relevance to childhood airway diseases.
Childhood asthma and chronic lung disease of prematurity (CLD) are characterized by airway wall injury, airway inflammation, and airway wall thickening largely due to an increased amount of airway wall smooth muscle (ASM) (1–4). However, mechanisms of airway injury and pattern of inflammation in these disorders are distinct (5,6). Childhood asthma is characterized by increased numbers of airway eosinophils and mast cells and cytokines such as CCL5, CXCL10, and CXCL8, whereas CLD is characterized by increased numbers of airway neutrophils and increased levels of CXCL8 and CXCL10 (5,6).
In adults, ASM cells have been linked with generation of eosinophil chemo-attractants and survival factors including IL-1β, CXCL8, CCL5, and CXCL10 (7–9). Consequently, ASM cell-mediated inflammation is a recognized treatment target in adult asthma (7–9). Whether ASM cells in children with asthma or CLD are involved in pulmonary inflammation is unknown. Previously, we have shown that unlike adult ASM tissue, developing human ASM is myogenic and that in cell culture, fetal ASM cells are smaller than adult counterparts (10–12). In addition, we have found that fetal ASM proliferation in vitro is relatively resistant to glucocorticoid treatment (10). Age-related phenotype differences imply that pharmacological responses observed in adult ASM may not extrapolate to neonatal or pediatric ASM.
Synthetic glucocorticoid (GC) drugs are commonly used to dampen airway inflammation in children with asthma and CLD (13,14). However, protracted therapy with GC drugs in CLD is associated with serious and life-long sequelae, specifically, neurological handicap (14,15). While it may be possible to refine use of GC drugs in childhood respiratory disorders and so reduce the risk of side effects, there is little data about their effects and mechanism of action in developing lung tissue such as ASM. In this study, we show that generation of TNF CCL5, CXCL8, and CXCL10 fetal human ASM is significantly increased by TNF-α stimulation. Moreover, we show that TNF-α-induced cytokine production is only partially inhibited by fluticasone treatment, demonstrating that developing ASM cells have a somewhat reduced sensitivity to GC drugs. Our findings may help explain the clinical observation that synthetic GC therapy in children with asthma or CLD has limited efficacy, and points to a potential mechanism for further exploration to overcome limitations of GC treatment.
Results
Fluticasone Inhibits CXCL8, CCL5, and CXCL10 Production by TNF-α Induced Fetal ASM
Supernatants from unstimulated fetal ASM cells contained CXCL8 and CXCL10 and, in lower concentrations, CCL5 (Figure 1a). Compared to fetal ASM cells treated with vehicle alone, treatment of cells with TNF-α (0, 1, 4, or 20 ng/ml) resulted in a dose-dependent increase in production of all three cytokines. Concentrations of CXCL10, CXCL8, and CCL5 in supernatants bathing cells stimulated with 20 ng/ml TNF-α were (mean ± SEM) 9,273 ± 680, 6,112 ± 537, and 3,809 ± 419 pg/ml, respectively, and significantly greater than found in supernatants from unstimulated cells (P < 0.01 for each cytokine). Concentrations of CXCL8 and CXCL10 appeared to plateau with increasing doses of TNF-α; there was no evidence of a plateau effect with CCL5 (Figure 1). We also assessed the effect of fluticasone on TNF-α-induced chemokine production. Fluticasone at concentrations of 1 and 100 nmol/l reduced TNF-α (20 ng/ml) induced CXCL10, CXCL8, and CCL5 (Figure 1b–d respectively). Fluticasone (100 nmol/l) treatment reduced CXCL10, CXCL8, and CCL5 production by 50, 25, and 85% respectively compared to fetal ASM cells treated with TNF-α alone, P < 0.01 for each cytokine compared to cells not treated with fluticasone.
Figure 1.
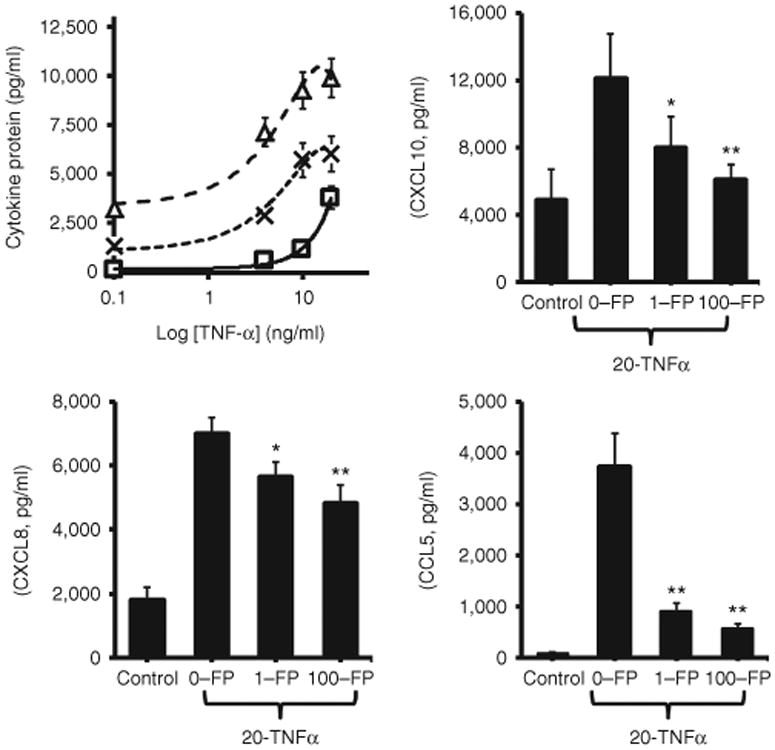
Fluticasone inhibits TNF-α-induced CXCL10, CXCL8, and CCL5 production by fetal human airway smooth muscle (ASM) cells. Quiescent fetal ASM cells generated small amounts of CXCL10 (Δ), CXCL8 (X), and CCL5 (□), protein (panel a). Stimulation with TNF-α resulted in a dose-dependent increase in production of all three cytokines. Fluticasone (FP) attenuated TNF-α (20 ng/ml) stimulated production of CXCL10, CXCL8, and CCL5 by 50, 25, and 85% respectively (panels b–d). Note fetal ASM cells were treated with vehicle or TNF-α following pretreatment (1 h) with vehicle or fluticasone. Supernatant chemokine concentrations were determined by enzyme-linked immunosorbent assay as described in the Methods section and expressed in picograms per milliliter ± SEM of three separate experiments, *P < 0.05, **P < 0.01.
Fluticasone Inhibits CXCL8, CCL5, and CXCL10 mRNA Expression by TNF-α Stimulated Fetal ASM
TNF-α stimulation (0, 1, 4, or 20 ng/ml) induced a dose-dependent increase in expression of CXCL8 and CXCL10 and CCL5 mRNA by fetal ASM cells compared to fetal ASM cells treated with vehicle alone). Expression of CXCL10, CXCL8, and CCL5 mRNA by cells stimulated with 20 ng/ml TNF-α was significantly greater than by vehicle-treated cells (P < 0.01 for each cytokine). Pretreatment of fetal ASM with 1 or 100 nmol/l fluticasone reduced TNF-α-induced expression of cytokine mRNA (Figure 2). For all three cytokines, P < 0.01 comparing fetal ASM cells treated with 100 nmol/l fluticasone and those treated with vehicle. CXCL8 and CXCL10 mRNA expression was reduced by ≥ 50% only in cells treated with 100 nmol/l fluticasone. In contrast, treatment of fetal ASM with 1 nmol/l fluticasone resulted in a 51% relative reduction in expression of CCL5 mRNA (Figure 2), with no further reduction in CCL5 mRNA expression observed in cells treated with 100 nmol/l fluticasone.
Figure 2.
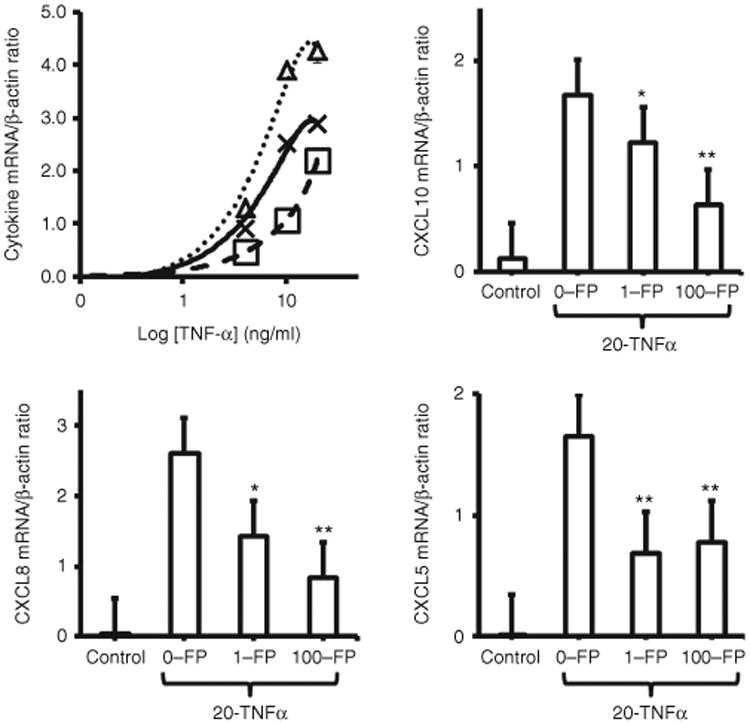
Fluticasone inhibits TNF-α-induced CXCL10, CXCL8, and CCL5 mRNA expression by fetal human airway smooth muscle (ASM) cells. Quiescent fetal ASM cells expressed little CXCL10 (Δ), CXCL8 (X), and CCL5 (□) mRNA under basal conditions (panel a). Stimulation with TNF-α-induced dose-dependent increases in mRNA expression of all three cytokines. Fluticasone (FP, 1 and 100 nmol/l) significantly reduced TNF-α-induced cytokine mRNA expression (panels b–d). Note fetal ASM cells were treated with vehicle or TNF-α (1–20 ng/ml) following pretreatment (1 h) with vehicle or fluticasone. Cytokine mRNA expression was determined 6 h following TNF-α stimulation by RT-PCR as described in the Methods section and expressed as cytokine/β-actin mRNA ratio {plus minus} S.E.M. of three separate experiments, *P < 0.05, **P < 0.01.
TNF-α Receptor Blockade Abrogates TNF-α Stimulated CXCL8, CCL5, and CXCL10 Protein and mRNA Expression by Fetal ASM
CXCL8, CCL5, and CXCL10 mRNA expression and protein production by TNF-α stimulated fetal ASM cells was effectively abolished by pretreatment of cells with anti-TNFR1 and/or anti-TNFR2 antibody (Figure 3). Protein and mRNA production of all three cytokines was significantly lower in fetal ASM cells incubated with both blocking antibodies prior to TNF-α stimulation compared to cells stimulated with TNF-α and pretreated with vehicle, P < 0.01 for all comparisons. Pretreatment with anti-TNFR1 or anti-TNFR2 antibody both abolished TNF-α stimulated CXCL10 CXCL8 and CCL5 mRNA expression and protein production as effectively as pretreatment with anti-TNFR1 and anti-TNFR2 antibodies, P > 0.05 for all comparisons. In the absence of TNF-α stimulation, protein and mRNA production of all three cytokines was significantly higher in fetal ASM cells incubated with either blocking antibody compared to cells treated with vehicle P < 0.05 for all comparisons.
Figure 3.
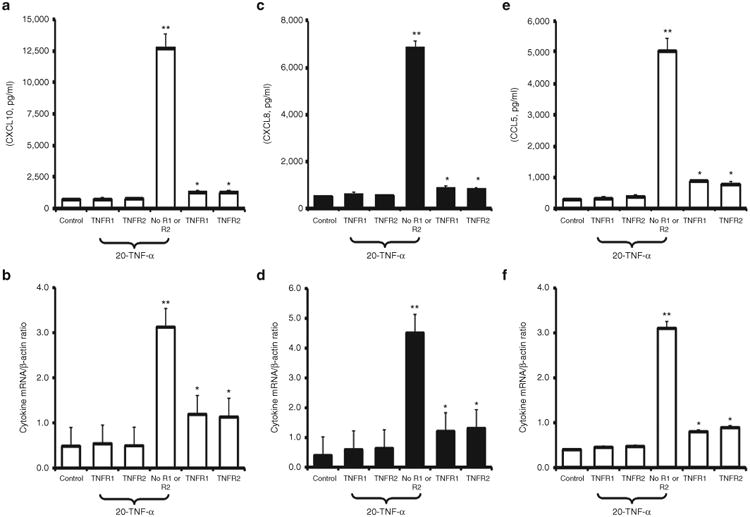
TNFR1 and TNFR2 blocking antibodies inhibit TNF-α-induced production of CXCL10, CXCL8, and CCL5 protein and mRNA by fetal human airway smooth muscle (ASM) equally well. Pretreatment of fetal ASM cells with TNFR1 or TNFR2 blocking antibodies reduced TNF-α stimulated (20 ng/ml) CXCL10 (panels a,b) CXCL8 (panels c,d), and CCL5 (panels e,f) protein production and mRNA expression equally effectively and to levels that were not significantly different to those observed with “untreated” (control) cells. In the absence of blocking antibodies, TNF-α (20 ng/ml) significantly increased cytokine protein production and mRNA expression. In the absence of TNF-α stimulation, TNFR1 and TNFR2 blocking antibodies significantly increased cytokine protein production and mRNA expression. Supernatant bathing cells collected for cytokine estimation 24 h after stimulation with TNF-α and, in separate experiments, total cell RNA was collected 6 h following stimulation with TNF-α. Results are expressed as protein concentration or cytokine/β-actin mRNA ratio ± SEM of experiments involving three different fetal human ASM cell lines, *P < 0.05, **P < 0.01.
TNF-α and Fluticasone Stimulation Upregulate TNF-R1 and TNF-R2 mRNA Expression
Expression of TNF-R1 and TNF-R2 mRNA by fetal ASM cells increased significantly following treatment with TNF-α (20 ng/ml) and TNF-α (20 ng/ml) plus fluticasone (100 nmol/l), P < 0.05 for both comparisons, Figure 4. Stimulation of fetal ASM cells with fluticasone alone was not associated with a statistically significant increase in either TNFR-1 or -2 mRNA expression. Fluticasone treatment significantly inhibited TNF-α-induced TNF-R1 mRNA expression (P < 0.05) but not TNF-α-induced TNF-R2 mRNA expression.
Figure 4.
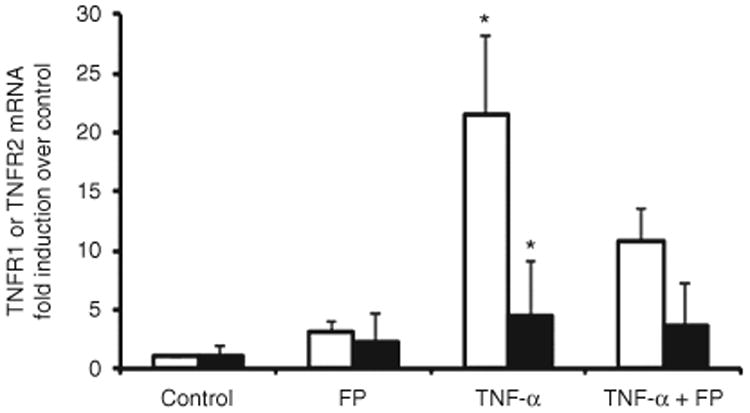
TNF-α induces production of TNFR1 and TNFR2 mRNA by fetal human airway smooth muscle (ASM). TNF-α (20 ng/ml) significantly increased TNFR1 (white bars) and TNFR2 (black bars) mRNA expression. In the absence of TNF-α stimulation (vehicle treatment), cells express small amounts of TNFR and TNFR2 mRNA. Fluticasone partially inhibited TNFR1 and TNFR2 mRNA. Results are expressed as fold induction (cytokine/glyceraldehyde 3-phosphate dehydrogenase mRNA ratio) relative to vehicle-treated (control) cells ± SEM of experiments involving three different fetal human ASM cell lines, *P < 0.05.
Fluticasone Induces Phosphorylation of Glucocorticoid Receptor at Serine-211 But Not Serine-226 Residues in Fetal ASM
Fluticasone treatment (1 and 100 nmol/l) of fetal ASM cells led to a significant increase in phosphorylated GR-S211 P < 0.05, but not GR-S226, P > 0.05, compared to control cells (vehicle only). TNF-α stimulation in combination with fluticasone had a synergistic effect on GR-(S211) and GR-(S226) phosphorylation (Figure 5), P < 0.05 compared to cells treated with fluticasone alone.
Figure 5.
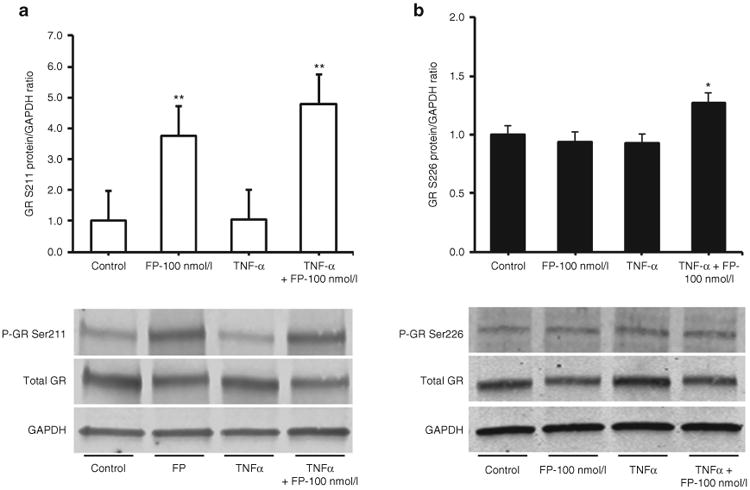
Fluticasone phosphorylates serine 211 (P-GR211) but not serine 226 (P-GR226) of the glucocorticoid receptor in fetal airway smooth muscle (ASM). Fluticasone treatment of fetal ASM significantly increased phosphorylation of glucocorticoid receptors (GR) at Serine 211 (panel a) but had no effect on phosphorylation of the Ser 226 residue (panel b). Cotreatment of fetal ASM cells with Fluticasone and TNF-α resulted in a significant increase in P-GR Ser 211 (panel a) and P-GR Ser 211 (panel b). Quiescent fetal ASM cells were treated with fluticasone (1 and 100 nmol/l, FP) and TNF-α (20 ng/ml) or vehicle (control). Scanning densitometry of three representative immunoblots normalized over the area density of the corresponding glyceraldehyde 3-phosphate dehydrogenase content. The results are expressed as the fold increase over basal values. *P < 0.05, **P < 0.01.
Discussion
In this study, we present novel data that fetal human ASM cells generate CXCL8, CXCL10, and CCL5 cytokines in response to the proinflammatory cytokine TNF-α. Moreover, high-dose fluticasone did not abolish TNF-α stimulated CXCL8, CXCL10, or CCL5 production by fetal ASM cells. These observations extend the potential of developing human ASM to generation of proinflammatory mediators and provide important new information about the differential sensitivity of developing ASM to GC drugs.
Interestingly, fetal ASM cells were found to express detectable amounts of CXCL8, CXCL10, and CCL5 mRNA and protein under basal conditions, a feature that has also been reported in adult ASM cells (16,17). Treatment with TNF-α treatment resulted in a concentration-dependent increase in cytokine mRNA and protein production. The magnitude of chemokine production (pg/ml) by TNF-α was CXCL10 > CXCL8 > CCL5 (see Figure 1). The different dose–response relationships suggest qualitative and/or quantitative differences in CXCL8, CXCL10, and CCL5 cell-signaling pathways in fetal ASM stimulated with TNF-α.
Both TNFR1 and TNFR2 appear to be equally involved in mediating TNF-α-induced chemokine production by fetal human ASM. Treatment with either TNFR1- or TNFR2-blocking antibody abolished cytokine production. Unstimulated fetal ASM cells constitutively expressed small amounts of TNFR1 and TNFR 2 mRNA. Expression of receptor mRNA's was markedly increased by TNF-α and only partially inhibited by fluticasone. These findings contrast with studies performed in adult human ASM in which TNF-α signaling is predominantly mediated by activation of TNFR1 (18,19). Thus, TNFR1/2 signaling is a striking characteristic trait that distinguishes developing human ASM from mature human ASM. Regulation of TNFR1 and TNFR2 expression are poorly defined (20); regulation of TNFR1 and TNFR2 expression is poorly defined (20,21); TNFR1 appears to be constitutively expressed by most cells whereas constitutive expression of TNFR2 is restricted to certain cell lineages, e.g., T-cells, oligodendrocytes, and endothelial cells (21–25). Clearly, further studies are needed to understand whether these receptors play a role in airway development and to define signaling pathways linked to TNFR mRNA and protein expression under inflammatory conditions.
Pretreatment of fetal ASM with fluticasone partially and differentially inhibited TNF-α-induced CCL5, CXCL8, and CXCL10 mRNA and protein generation (see Figure 2). These observations contrast with the effects of TNFR1/2 blockade in fetal ASM and studies showing > 95% inhibition of TNF-α stimulated CCL5 and CXCl10 production by adult human ASM pretreated with fluticasone (10 nmol/l) (26). We therefore investigated whether TNF-α alters fluticasone signaling in fetal ASM. Indeed, western blotting data demonstrated that fluticasone phosphorylates GR at Ser211 but not Ser226 in agreement with our previous data and that of other groups using adult ASM cells (20,27,28). More importantly, although TNF-α treatment alone did not alter GR-phosphorylation at Ser211 or Ser226, treatment with TNF-α and fluticasone led to increased phosphorylation at GR-Ser226 but not at GR-Ser211. The implications of these observations are unclear. Phosphorylation of Ser211 is essential for gene-specific GR transactivation (20,27,28). In contrast, GR-Ser226 phosphorylation is associated with reduced GR function (20,27,28). Thus, in fetal ASM, it is possible that phosphorylation at GR-Ser226 induced by TNF-α/fluticasone cotreatment antagonizes the anti-inflammatory action of fluticasone. Although this mechanism could account for the failure of fluticasone to abrogate TNF-α-induced cytokine production by fetal ASM, it does not explain why production of CXCL8, CXCL10, and CCL5 by fetal ASM is differentially sensitive to fluticasone. The latter is most likely explained by studies showing that TNF-α activates a number of parallel intracellular signaling pathways involving multiple transcription factors (NF-κ-B, activating protein-1, nuclear factor of activated T cells) and MAP kinases (29). Reciprocal inhibitory mechanisms, e.g., competition for transcriptional coactivators, interference with transrepression activities, and post-translational modifications (30) may also contribute to differential effect of GCs on TNF-α induced chemokine production.
The experimental set-up described in this work was designed to study the inflammatory and pharmacological responses of ASM in neonatal and pediatric airways. However, the cells were cultured from prenatal lungs. The data also therefore imply that ASM has the potential to participate in airway inflammation in utero (31). Consequently, prenatal insults such as chorioamnionitis and maternal smoking may contribute to the development of neonatal chronic lung disease by stimulating ASM proinflammatory responses. It is possible that fetal ASM are involved in the alteration of airway responses associated with environmental exposures during fetal life (32,33) by secreting cytokines under these conditions.
Our study opens the possibility of using this model to better investigate the inflammatory mechanisms linked to childhood asthma and CLD. How closely a fetal ASM cell culture model simulates responses of ASM in children with asthma or CLD is an important and presently unanswerable question. The model cannot replicate the numerous cell-cell and cell-mediator interactions that ultimately define ASM responses in vivo. Nonetheless, the data shows the capacity of developing ASM cells to recruit eosinophils and T-cells (CCL5), neutrophils (CXCL8) and mast cells (CXCL10) into airways and that potent corticosteroids such as fluticasone, at high dose, partially inhibit mediators of leucocyte recruitment such as CCL5, CXCl8, and CXCL10. However, these data also suggest that corticosteroid action in developing ASM may differ from that in adults. Further investigations will be needed to further elucidate the effects of corticosteroids on developing airway smooth muscle in order to determine how steroids should be used to blunt inflammatory effects in neonatal and pediatric asthma.
Methods
Fetal human cell cultures (N = 6 different fetal lungs) were placed in primary culture using fetal trachealis muscle/lung tissue (8–14 wk gestation, see Figure 6) provided by The MRC-Wellcome Trust Human Developmental Biology Resource Group, Newcastle University & University College London. The use of human fetal tissue in our institution was approved by the Leicestershire, Northamptonshire and Rutland Research Ethics Committee-1 (Ref No. 08/H0406/68). Primary cultures were established and cells maintained in culture and passaged as required using previously described techniques (25,34). Cell lines were between passage 4 and 8 and displayed “hill and valley” morphology typical of smooth muscle with greater than 90% cells positive for SM-α-actin (Figure 6). Fetal human ASM were propagated using Dulbecco's modified Eagle's medium containing 10% fetal calf serum supplemented with 1% pen/strep, 1% glutamine, 1% sodium pyruvate, and 1% nonessential amino acids solution. For experiments, cells were growth starved for 24 h in Dulbecco's modified Eagle's medium containing 0.5% fetal calf serum and supplements. All cell reagents were purchased from Sigma-Aldrich, Poole, UK.
Figure 6.
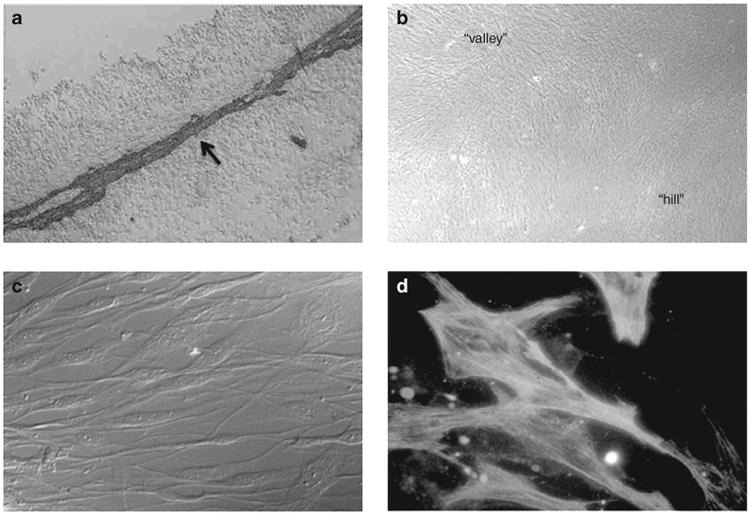
Culture of fetal human airway smooth muscle (ASM) cells. Fetal human tracheal ASM cells were cultured from fetal trachealis muscle tissue, which strongly expresses SM-α-actin (a). The trachealis muscle (indicated by arrow) is visualized using SM-α-actin antibody and the chromopgen 3,3′-diaminobenzidine tetrahydrochloride (a = tracheal lumen). In monlolayer cell culture, the cells display “hill and valley” morphology, characteristic of cultured smooth muscle cells (b). Cultured cells are elongate and bipolar appearance figure (c) and express Sm-α-actin filaments (visualized using Sm-α-actin antibody/fluorescein isothiocyanate), which are best seen as crossing cel nuclei (d).
TNF-α Stimulation and Treatment of Fetal ASM With Fluticasone
Fetal ASM cell were seeded at a density of 1 × 106/well into a 24-well plate, left to adhere overnight, and then growth arrested for 24 h. The next day the cells were treated with vehicle ± fluticasone (1–100 nm) for 1 h and then stimulated with TNF-α (10 ng/ml).
Enzyme-Linked Immunosorbent Assay for CXCL8, CCL5, and CXCL10
Cytokine levels were measured in media collected from fetal human ASM cells first treated with vehicle ± fluticasone (1–100 nm) for 1 h and then stimulation of TNF-α. Measurement of cytokine concentration in ASM supernatants was performed by enzyme-linked immunosorbent assay kits and according to the manufacturer's instructions (CCL5 or CXCL-10 DuoSet enzyme-linked immunosorbent assay from R&D Systems, Abingdon, UK; CXCL-8 OptEIA enzyme-linked immunosorbent assay from BD Biosciences, Oxford UK).
TNF-α-Receptor-1, -2, CXCL-8, CCL5, and CXCL-10 mRNA
Total ASM cell mRNA was isolated at 24 h (TNFR1 and TNFR 2) after stimulation either with vehicle, TNF-α, luticasone or using TNF-α plus fluticasone “TRI-reagent” (Sigma-Aldrich). Real-time PCR was performed using the LightCycler 480 system (Roche Diagnostics, Mannheim, Germany) and gene-specific primers purchased from Eurofins MWG Operon, Huntsville, AL:
p75TNFR1: Fwd 5′ GGGCAAGATAACGCAC 3′. Rev 5′ AGAGATGGCTACGAGG 3′.
p55TNFR2: Fwd 5′ C AGTACCGGCATTATTGG3′. Rev 5′ CCGTTGGTAGCGATACA 3′.
hsGAPDH: Fwd 5′ AAGGTGAAGGTCGGAGTCAACGGATT3′. Rev 5′ hsGAPDH-R1 CCATGGAATTTGCCATGGGAGGAAT3′.
CXCL10: Fwd 5′ GGATGGACCACACAGAGGCTGC 3′. Rev 5′ GCCCCTTGGGAGGATGGCAGT 3′.
CCL5: Fwd 5′GGCAGCCCTCGCTGTCATCC 3′. Rev 5 ′GCCC TTCAAGGAGCGGGTGG 3′.
CXCL8: Fwd 5′ AGACAGCAGAGCACACAAGC 3′. Rev 5′ AGGAAGGCTGCCAAGAGAG 3′.
β-actin: Fwd 5′ GCTCGTCGTCGACAACGGCTC-3′. Rev 5′ CAAACATGATCTGGGTCATCTTCTC 3′
The PCR amplification reactions were prepared in “SYBR-Green PCR Mastermix” (Sigma Aldrich) according to manufacturer's instructions. The amplification step was run for 20 cycles, after optimization of cycling conditions, and the amount of specific PCR product was estimated by the second derivative maximum method. The Lightcycler Data Analysis (MRCMEG, Roche Diagnostics) package was used to analyze the data and to quantify cytokine mRNA quantification. Gene expression was normalized to either β-actin (CXCL10, CCL5, and CXCL8) or glyceraldehyde 3-phosphate dehydrogenase (TNFR1 and TNFR2) mRNA expression.
Glucocorticoid Receptor Phosphorylation
Growth-arrested fetal ASM cell were treated with vehicle ± fluticasone (1–100 nm) for 1 h and then stimulated with TNF-α (10 ng/ml). At 6 h post-TNF-α stimulation, the culture media was aspirated and the cells washed in ice-cold PBS containing protease inhibitors (200 μmol/l Na3VO4, 2 mmol/l phenylmethylsulfonyl fluoride). The cells were then lysed by scraping in ice-cold extraction buffer containing 20 mmol/l Tris, pH7.5, 150 mmol/l NaCl, 1 mmol/l ethylenediamine tetraacetic acid, 1 mmol/l ethyleneglycol tetraacetic acid, 1% Triton X-100, 2.5 mmol/l sodium pyrophosphate, 1 mmol/l-glycerolphosphate, 2 mmol/l Na3VO4, 1 μg/ml leupeptin, and 2 mmol/l phenylmethylsulfonyl fluoride. The cell protein homogenate was loaded (10 μg/lane) on 10% acrylamide gels, separated using SDS-PAGE and then transferred to a nitrocellulose membrane. Each was then immersed in PBS containing Tween 20 (0.1%) and dried nonfat milk (3%) for 1 h to block nonspecific binding sites. Thereafter, each blot was incubated with a rabbit anti-human polyclonal GR, GR-ser211, or GR-226 antibody (1:1,000, 1:500, and 1:500 respectively; Cell Signalling Technology, Danvers, MA) overnight at 4 °C. Presence of GR protein was detected using a goat anti-rabbit IgG horseradish peroxidase-conjugated secondary antibody (1:10,000 or 1:5,000; Santa Cruz Biotechnology Dallas, TX) and visualized by enhanced chemilluminescence (Amersham-Pharmacia, Amersham, UK). To control for differences in loading, blots were stripped and reprobed with a monoclonal antibody directed against mouse glyceraldehyde 3-phosphate dehydrogenase protein to (1:2,500, Sigma Chemical, Poole, UK) and detected with the goat anti-mouse IgG horseradish peroxidase-conjugated secondary antibody (1:2,500; Santa Cruz Biotechnology). All other reagents were purchased from Sigma-Aldrich.
Statistical Analyses
Results are presented as means ± SEM or ± SD values, where indicated. Comparisons between groups were performed using the unpaired Student's t-test or one-way ANOVA. A probability of <0.05 was considered statistically significant. Statistical analyses were conducted using the Prism computer program by GraphPad Software, La Jolla, CA.
Acknowledgments
Statement of Financial Support: This study was supported by National Institutes of Health, Bethesda, USA T32 HL 105355 and F32 HL123075 (A.D.B.); R01 HL 056470 and R01 HL 088029 (Y.S.P.).
Disclosure: The human embryonic and fetal material was provided by the Joint MRC (grant # G0700089)/Wellcome Trust (grant # GR082557) Human Developmental Biology Resource (http://hdbr.org), Newcastle and London, United Kingdom.
Footnotes
Author Contributions: All authors contributed to the conception and design of the study, analysis, and interpretation of data. All authors have contributed to writing the article and have given their final approval of the version to be published.
References
- 1.Hislop AA, Haworth SG. Airway size and structure in the normal fetal and infant lung and the effect of premature delivery and artificial ventilation. Am Rev Respir Dis. 1989;140:1717–26. doi: 10.1164/ajrccm/140.6.1717. [DOI] [PubMed] [Google Scholar]
- 2.O'Reilly R, Ullmann N, Irving S, et al. Increased airway smooth muscle in preschool wheezers who have asthma at school age. J Allergy Clin Immunol. 2013;131:1024–32. 1032.e1–16. doi: 10.1016/j.jaci.2012.08.044. [DOI] [PubMed] [Google Scholar]
- 3.Regamey N, Ochs M, Hilliard TN, et al. Increased airway smooth muscle mass in children with asthma, cystic fibrosis, and non-cystic fibrosis bronchiectasis. Am J Respir Crit Care Med. 2008;177:837–43. doi: 10.1164/rccm.200707-977OC. [DOI] [PubMed] [Google Scholar]
- 4.Sward-Comunelli SL, Mabry SM, Truog WE, Thibeault DW. Airway muscle in preterm infants: changes during development. J Pediatr. 1997;130:570–6. doi: 10.1016/s0022-3476(97)70241-5. [DOI] [PubMed] [Google Scholar]
- 5.Fitzpatrick AM, Higgins M, Holguin F, Brown LA, Teague WG National Institutes of Health/National Heart, Lung, and Blood Institute's Severe Asthma Research Program. The molecular phenotype of severe asthma in children. J Allergy Clin Immunol. 2010;125:851–857.e18. doi: 10.1016/j.jaci.2010.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ambalavanan N, Carlo WA, D'Angio CT, et al. Eunice Kennedy Shriver National Institute of Child Health and Human Development Neonatal Research Network. Cytokines associated with bronchopulmonary dysplasia or death in extremely low birth weight infants. Pediatrics. 2009;123:1132–41. doi: 10.1542/peds.2008-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ammit AJ, Lazaar AL, Irani C, et al. Tumor necrosis factor-alpha-induced secretion of RANTES and interleukin-6 from human airway smooth muscle cells: modulation by glucocorticoids and beta-agonists. Am J Respir Cell Mol Biol. 2002;26:465–74. doi: 10.1165/ajrcmb.26.4.4681. [DOI] [PubMed] [Google Scholar]
- 8.Hirst SJ, Walker TR, Chilvers ER. Phenotypic diversity and molecular mechanisms of airway smooth muscle proliferation in asthma. Eur Respir J. 2000;16:159–77. doi: 10.1034/j.1399-3003.2000.16a28.x. [DOI] [PubMed] [Google Scholar]
- 9.Pang L, Knox AJ. Synergistic inhibition by beta(2)-agonists and corticosteroids on tumor necrosis factor-alpha-induced interleukin-8 release from cultured human airway smooth-muscle cells. Am J Respir Cell Mol Biol. 2000;23:79–85. doi: 10.1165/ajrcmb.23.1.3985. [DOI] [PubMed] [Google Scholar]
- 10.Pandya HC. MD thesis. University of London Press; Chapter 6: Evaluation of the effect of hydrogen peroxide, antioxidants and dexamethasone on fetal bovine serum-stimulated fetal human ASM cell proliferation in Investigations of fetal human airway smooth muscle: potential mechanisms of abnormal airway wall modelling in chronic lung disease of preterm infants, 2001. [Google Scholar]
- 11.Pandya HC, Innes J, Hodge R, Bustani P, Silverman M, Kotecha S. Spontaneous contraction of pseudoglandular-stage human airspaces is associated with the presence of smooth muscle-alpha-actin and smooth muscle-specific myosin heavy chain in recently differentiated fetal human airway smooth muscle. Biol Neonate. 2006;89:211–9. doi: 10.1159/000089797. [DOI] [PubMed] [Google Scholar]
- 12.Snetkov VA, Pandya H, Hirst SJ, Ward JP. Potassium channels in human fetal airway smooth muscle cells. Pediatr Res. 1998;43(4 Pt 1):548–54. doi: 10.1203/00006450-199804000-00019. [DOI] [PubMed] [Google Scholar]
- 13.Doyle LW, Ehrenkranz RA, Halliday HL. Dexamethasone treatment in the first week of life for preventing bronchopulmonary dysplasia in preterm infants: a systematic review. Neonatology. 2010;98:217–24. doi: 10.1159/000286210. [DOI] [PubMed] [Google Scholar]
- 14.Pruteanu AI, Chauhan BF, Zhang L, Prietsch SO, Ducharme FM. Inhaled corticosteroids in children with persistent asthma: dose-response effects on growth. Cochrane Database Syst Rev. 2014;7:CD009878. doi: 10.1002/14651858.CD009878.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barrington KJ. The adverse neuro-developmental effects of postnatal steroids in the preterm infant: a systematic review of RCTs. BMC Pediatr. 2001;1:1. doi: 10.1186/1471-2431-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Che W, Parmentier J, Seidel P, et al. Corticosteroids inhibit sphingosine 1-phosphate-induced interleukin-6 secretion from human airway smooth muscle via mitogen-activated protein kinase phosphatase 1-mediated repression of mitogen and stress-activated protein kinase 1. Am J Respir Cell Mol Biol. 2014;50:358–68. doi: 10.1165/rcmb.2013-0208OC. [DOI] [PubMed] [Google Scholar]
- 17.Sutcliffe A, Kaur D, Page S, et al. Mast cell migration to Th2 stimulated airway smooth muscle from asthmatics. Torax. 2006;61:657–62. doi: 10.1136/thx.2005.056770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amrani Y, Lazaar AL, Hoffman R, Amin K, Ousmer S, Panettieri RA., Jr Activation of p55 tumor necrosis factor-alpha receptor-1 coupled to tumor necrosis factor receptor-associated factor 2 stimulates intercellular adhesion molecule-1 expression by modulating a thapsigargin-sensitive pathway in human tracheal smooth muscle cells. Mol Pharmacol. 2000;58:237–45. doi: 10.1124/mol.58.1.237. [DOI] [PubMed] [Google Scholar]
- 19.Amrani Y, Ammit AJ, Panettieri RA., Jr Tumor necrosis factor receptor (TNFR) 1, but not TNFR2, mediates tumor necrosis factor-alpha-induced interleukin-6 and RANTES in human airway smooth muscle cells: role of p38 and p42/44 mitogen-activated protein kinases. Mol Pharmacol. 2001;60:646–55. [PubMed] [Google Scholar]
- 20.Itoh M, Adachi M, Yasui H, Takekawa M, Tanaka H, Imai K. Nuclear export of glucocorticoid receptor is enhanced by c-Jun N-terminal kinase-mediated phosphorylation. Mol Endocrinol. 2002;16:2382–92. doi: 10.1210/me.2002-0144. [DOI] [PubMed] [Google Scholar]
- 21.Puimège L, Libert C, Van Hauwermeiren F. Regulation and dysregulation of tumor necrosis factor receptor-1. Cytokine Growth Factor Rev. 2014;25:285–300. doi: 10.1016/j.cytogfr.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 22.Baxter FO, Came PJ, Abell K, et al. IKKbeta/2 induces TWEAK and apoptosis in mammary epithelial cells. Development. 2006;133:3485–94. doi: 10.1242/dev.02502. [DOI] [PubMed] [Google Scholar]
- 23.Dopp JM, Mackenzie-Graham A, Otero GC, Merrill JE. Differential expression, cytokine modulation, and specific functions of type-1 and type-2 tumor necrosis factor receptors in rat glia. J Neuroimmunol. 1997;75:104–12. doi: 10.1016/s0165-5728(97)00009-x. [DOI] [PubMed] [Google Scholar]
- 24.Choi SJ, Lee KH, Park HS, Kim SK, Koh CM, Park JY. Differential expression, shedding, cytokine regulation and function of TNFR1 and TNFR2 in human fetal astrocytes. Yonsei Med J. 2005;46:818–26. doi: 10.3349/ymj.2005.46.6.818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Middlebrook AJ, Lebsack T, DeLuca D. TNF-alpha mediated modulation of T cell development and exacerbation of in vitro T1DM in fetal thymus organ culture. J Autoimmun. 2007;29:134–45. doi: 10.1016/j.jaut.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 26.Banerjee A, Damera G, Bhandare R, et al. Vitamin D and glucocorticoids differentially modulate chemokine expression in human airway smooth muscle cells. Br J Pharmacol. 2008;155:84–92. doi: 10.1038/bjp.2008.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blind RD, Garabedian MJ. Differential recruitment of glucocorticoid receptor phospho-isoforms to glucocorticoid-induced genes. J Steroid Biochem Mol Biol. 2008;109:150–7. doi: 10.1016/j.jsbmb.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bouazza B, Krytska K, Debba-Pavard M, et al. Cytokines alter glucocorticoid receptor phosphorylation in airway cells: role of phosphatases. Am J Respir Cell Mol Biol. 2012;47:464–73. doi: 10.1165/rcmb.2011-0364OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sabio G, Davis RJ. TNF and MAP kinase signalling pathways. Semin Immunol. 2014;26:237–45. doi: 10.1016/j.smim.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Bogaert T, De Bosscher K, Libert C. Crosstalk between TNF and glucocorticoid receptor signaling pathways. Cytokine Growth Factor Rev. 2010;21:275–86. doi: 10.1016/j.cytogfr.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 31.Gervasi MT, Romero R, Bracalente G, et al. Midtrimester amniotic fluid concentrations of interleukin-6 and interferon-gamma-inducible protein-10: evidence for heterogeneity of intra-amniotic inflammation and associations with spontaneous early (<32 weeks) and late (>32 weeks) preterm delivery. J Perinat Med. 2012;40:329–43. doi: 10.1515/jpm-2012-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elliot J, Vullermin P, Robinson P. Maternal cigarette smoking is associated with increased inner airway wall thickness in children who die from sudden infant death syndrome. Am J Respir Crit Care Med. 1998;158:802–6. doi: 10.1164/ajrccm.158.3.9709055. [DOI] [PubMed] [Google Scholar]
- 33.Stick SM, Burton PR, Gurrin L, Sly PD, LeSouëf PN. Effects of maternal smoking during pregnancy and a family history of asthma on respiratory function in newborn infants. Lancet. 1996;348:1060–4. doi: 10.1016/s0140-6736(96)04446-7. [DOI] [PubMed] [Google Scholar]
- 34.Pandya HC, Snetkov VA, Twort CH, Ward JP, Hirst SJ. Oxygen regulates mitogen-stimulated proliferation of fetal human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1220–30. doi: 10.1152/ajplung.00268.2001. [DOI] [PubMed] [Google Scholar]