

Supplemental Digital Content is available in the text.
Keywords: apolipoprotein A-I, carboxyl-terminal cleavage, chymase, endothelial cells, inflammatory, mast cell, proteases
Abstract
Objective—
Apolipoprotein A-I (apoA-I) has been shown to possess several atheroprotective functions, including inhibition of inflammation. Protease-secreting activated mast cells reside in human atherosclerotic lesions. Here we investigated the effects of the neutral proteases released by activated mast cells on the anti-inflammatory properties of apoA-I.
Approach and Results—
Activation of human mast cells triggered the release of granule-associated proteases chymase, tryptase, cathepsin G, carboxypeptidase A, and granzyme B. Among them, chymase cleaved apoA-I with the greatest efficiency and generated C-terminally truncated apoA-I, which failed to bind with high affinity to human coronary artery endothelial cells. In tumor necrosis factor-α–activated human coronary artery endothelial cells, the chymase-cleaved apoA-I was unable to suppress nuclear factor-κB–dependent upregulation of vascular cell adhesion molecule-1 (VCAM-1) and to block THP-1 cells from adhering to and transmigrating across the human coronary artery endothelial cells. Chymase-cleaved apoA-I also had an impaired ability to downregulate the expression of tumor necrosis factor-α, interleukin-1β, interleukin-6, and interleukin-8 in lipopolysaccharide-activated GM-CSF (granulocyte-macrophage colony-stimulating factor)– and M-CSF (macrophage colony-stimulating factor)–differentiated human macrophage foam cells and to inhibit reactive oxygen species formation in PMA (phorbol 12-myristate 13-acetate)–activated human neutrophils. Importantly, chymase-cleaved apoA-I showed reduced ability to inhibit lipopolysaccharide-induced inflammation in vivo in mice. Treatment with chymase blocked the ability of the apoA-I mimetic peptide L-4F, but not of the protease-resistant D-4F, to inhibit proinflammatory gene expression in activated human coronary artery endothelial cells and macrophage foam cells and to prevent reactive oxygen species formation in activated neutrophils.
Conclusions—
The findings identify C-terminal cleavage of apoA-I by human mast cell chymase as a novel mechanism leading to loss of its anti-inflammatory functions. When targeting inflamed protease-rich atherosclerotic lesions with apoA-I, infusions of protease-resistant apoA-I might be the appropriate approach.
Circulating high-density lipoprotein (HDL) comprises a spectrum of lipoproteins ranging from nascent discoidal to mature spherical particles, the former having preβ- and the latter α-electrophoretic mobility.1 Irrespective of their shape, size, or composition, all HDL particles contain either a single copy or multiple copies of apolipoprotein A-I (apoA-I), a polypeptide with an apparent molecular weight of 28 000 kDa. Both lipid-free apoA-I and the nascent lipid-poor preβ-HDL are the primary acceptors of cholesterol effluxed via the ATP-binding cassette transporter A1 (ABCA1) from macrophage foam cells,2 and so play critical roles in promoting reverse cholesterol transport in vivo. Although the circulating blood contains only minute amounts of preβ-HDL, these particles are enriched in human interstitial fluids.3 This appears also to apply to the arterial intimal fluid, with a concentration of HDL almost 40% of that in plasma, and in which most of the HDL particles have a density comparable to the very high–density lipoprotein subclass and contain only apoA-I.4
Current data suggest that by regulating cellular cholesterol homeostasis, HDL can also regulate inflammatory responses in various types of cells that have been activated by proinflammatory stimuli in the arterial wall.5 Importantly, proinflammatory activation of the endothelium is regarded critical for the initiation and progression of atherosclerosis. Mechanistically, dysfunctional endothelium may arise when activated endothelial cells (ECs) express the vascular cell adhesion molecule-1 (VCAM-1) or the intercellular adhesion molecule-1 that trigger leukocyte adhesion to the activated ECs.6 Both lipid-free apoA-I and HDL particles have been shown to exert potent anti-inflammatory effects on activated cultured ECs of human, bovine, or murine origin7–9 and also on other cell types involved in atherogenesis, such as human monocytes10 and monocyte-derived macrophages.11,12 The anti-inflammatory actions of apoA-I and HDL have been shown to involve attenuation of nuclear factor-κB (NF-κB) activation in various types of human ECs when they are exposed to proinflammatory stimuli, such as tumor necrosis factor (TNF-α), lipopolysaccharide (LPS), or palmitic acid.8,13–15 ApoA-I exhibits anti-inflammatory functions also in vivo, as demonstrated by injecting into rabbits apoA-I in the lipid-free form, or as a component of discoidal reconstituted HDL (rHDL) or of mature spherical HDL.16,17
In atherosclerotic lesions, the infiltrating inflammatory cells include mast cells, which upon activation and ensuing degranulation release neutral serine proteases, among them chymase and tryptase, both capable of cleaving the various apolipoproteins present in HDL particles.18 Importantly, mast cell chymase efficiently cleaves lipid-free apoA-I and depletes preβ-HDL particles, and so blocks their ability to promote ABCA1-dependent cholesterol efflux from macrophage foam cells in vitro and in vivo.19–22 Here we hypothesized that proteolytic cleavage of apoA-I by chymase could also influence its anti-inflammatory activities. Our data demonstrate that C-terminal cleavage of apoA-I by mast cell chymase impairs its ability to suppress proinflammatory responses in vitro and in vivo. Similarly, plasmin-treated apoA-I also failed to exert anti-inflammatory action on activated human coronary artery endothelial cells (HCAECs) in culture. The present study discloses a pathophysiological link between proteolytic cleavage of apoA-I and vascular inflammation.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
Proteolysis Impairs the Ability of ApoA-I to Attenuate TNF-α-Induced VCAM-1 Expression in HCAECs
HCAECs were preincubated with lipid-free apoA-I, then exposed to TNF-α, and the expression of VCAM-1 protein, as a cell surface marker of proinflammatory activation,23 was analyzed by flow cytometry. Stimulation with TNF-α shifted the fluorescence peak from left to right, indicating upregulation of VCAM-1, whereas pretreatment with apoA-I partially prevented it, as indicated by the lesser right-shift of the fluorescence peak (Figure 1A). To evaluate the effect of proteolysis on this anti-inflammatory function of apoA-I, we incubated apoA-I with recombinant human chymase and compared the abilities of the untreated and chymase-treated apoA-I to inhibit TNF-α-dependent VCAM-1 expression. As shown in Figure 1B, addition of increasing concentrations of untreated apoA-I to the cultured HCAECs suppressed VCAM-1 upregulation in a dose-dependent fashion, with maximal suppression observed at 50 μg/mL. In contrast, chymase-treated apoA-I totally failed to prevent the TNF-α-dependent induction of VCAM-1 expression.
Figure 1.
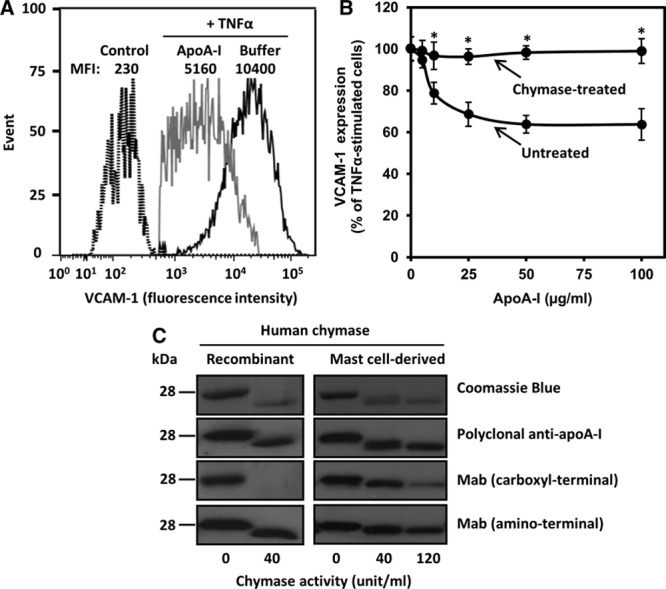
Chymase treatment abolishes the anti-inflammatory activity of apolipoprotein A-I (apoA-I) on activated human coronary artery endothelial cells (HCAECs). A, HCAECs were preincubated for 16 h in the absence (buffer) or presence of apoA-I (50 μg/mL) and then activated with tumor necrosis factor-α (TNF-α; 10 ng/mL) for 5 h. Nonactivated cells were incubated in medium alone for 21 h (control). Cell surface vascular cell adhesion molecule-1 (VCAM-1) protein was determined by flow cytometry. Data are representative of 6 independent experiments. B, ApoA-I (1 mg/mL) was treated for 6 h in the absence (untreated) or the presence (chymase-treated) of chymase (0.5 μg=40 BTEE units/mL). HCAECs were preincubated with increasing concentrations of the untreated or chymase-treated apoA-I and then activated with tumor necrosis factor-α (TNF-α), as described in A. TNF-α-induced VCAM-1 surface protein expression was analyzed by flow cytometry and expressed as percentage of its expression levels in TNF-α-activated cells preincubated in the absence of apoA-I, which was set as 100%. Data represent the means±SD from 3 to 4 independent experiments performed in duplicate. *P<0.01 denotes statistical significance between cells preincubated with untreated or chymase-treated apoA-I. C, ApoA-I was incubated with the indicated activities (BTEE units) of recombinant human chymase or chymase-containing human mast cell–conditioned medium for 6 h, after which the incubation was stopped by adding soybean trypsin inhibitor. Proteins in the incubation mixtures were resolved in 12.5% SDS-PAGE and detected by Coomassie Blue or immunoblotted with anti-human apoA-I polyclonal antibody or with anti-human apoA-I monoclonal antibodies recognizing either a C-terminal (amino acids 211–220) or an N-terminal (amino acids 2–8) region of apoA-I. MFI indicates median fluorescence intensity.
Analysis of the apoA-I degradation products by Western blotting indicated that chymase completely depleted the full-length apoA-I and generated a large-sized polypeptide band which had lost immunoreactivity against a monoclonal antibody recognizing a C-terminal epitope encompassing the amino acid residues 211–220 (Figure 1C, left). In agreement with a previous report, chymase did not modify apoA-I reactivity with a monoclonal antibody specific for the N-terminal region (amino acids 2–8).20 A similar degradation pattern was observed when apoA-I was treated with conditioned culture medium in which human mast cells had been activated to degranulate and release a complex mixture of granule neutral proteases, including chymase.24 Accordingly, incubation of apoA-I with such preconditioned medium resulted in loss of C-terminal reactivity in a concentration-dependent manner (Figure 1C, right). We further determined the individual contribution of each of the 5 released proteases (chymase, tryptase, cathepsin G, carboxypeptidase A3, and granzyme B) to apoA-I degradation. For this purpose, specific protease inhibitors were used. Regarding inhibition of the 2 chymotryptic enzymes, that is, chymase and cathepsin G, the chymase inhibitor specifically inhibited chymase, whereas the cathepsin G inhibitor was found to inhibit both chymase and cathepsin G. We observed that selective inhibition of chymase almost completely blocked apoA-I degradation, whereas the combined inhibition chymase and cathepsin G completely blocked it. In contrast, inhibition of tryptase, carboxypeptidase A3, or granzyme B only slightly inhibited proteolysis or had no effect (Figure I in the online-only Data Supplement). These results indicated that among the neutral proteases released by activated human mast cells, chymase was the main protease responsible for the mast cell–dependent loss of the anti-inflammatory function of apoA-I in HCAECs. This finding prompted us to perform subsequent experiments with chymase alone.
Lipidation of ApoA-I Renders It Partly Resistant to Chymase-Dependent Loss of Its Anti-Inflammatory Effects on HCAECs
To examine the effect of apoA-I lipidation on the ability of chymase to attenuate its anti-inflammatory properties, we studied apoA-I-containing reconstituted HDL ((A-I)rHDL) particles that have been widely used to mimic the naturally occurring preβ-HDL.25 For this purpose, we prepared 3 species of discoidal (A-I)rHDL with increasing amounts of phospholipids and constant amounts of cholesterol and apoA-I: rHDL-1, rHDL-2, and rHDL-3 (Table I in the online-only Data Supplement). Similar to proteolysis of lipid-free apoA-I with chymase, proteolysis of lipidated apoA-I generated a polypeptide band with an apparent molecular weight slightly lower than that of intact apoA-I (Figure 2A, top). Yet, the degree of lipidation was inversely related to the degree of proteolysis (Figure 2A, bottom), suggesting that lipidation progressively masked the chymase cleavage sites of apoA-I. Next, we examined the effect of chymase on the anti-inflammatory properties of the various (A-I)rHDL preparations. When activated HCAECs were preincubated with any of the untreated (A-I)rHDL species, the levels of VCAM-1 mRNA and protein were reduced to levels comparable to those observed when lipid-free apoA-I was used (Figure 2B, top and middle). Moreover, any of the untreated (A-I)rHDL reduced adhesion of THP-1 monocytes to the activated HCAECs to the levels observed in the nonactivated control cells (Figure 2B, bottom). Cleavage of apoA-I in (A-I)rHDLs affected the ability of these particles to inhibit surface VCAM-1 protein expression, and it appeared that the stronger apoA-I degradation (rHDL-1>rHDL-2>rHDL-3), the weaker its inhibitory effect on VCAM-1 protein expression (P<0.05 for trend). However, all of the proteolyzed rHDLs had lost to a similar extent their capacity to reduce VCAM-1 mRNA and monocyte adhesion to TNF-α-activated HCAECs (Figure 2B, top and bottom), suggesting that even a minor degree of proteolysis may have significantly affected particle integrity and the specific conformational characteristics of apoA-I in (A-I)rHDL particles that were essential for these particular apoA-I-dependent effects. Importantly, analysis with nondenaturing electrophoresis revealed that incubation of (A-I)rHDLs with chymase resulted in significant changes in protein staining pattern and particle size distribution (Figure IIA and IIB in the online-only Data Supplement). Consistent with previous reports,20,26 and in contrast to lipid-free apoA-I and rHDLs, apoA-I in mature HDL particles isolated from human plasma (HDL2 and HDL3) was poorly degraded by chymase, and the particles maintained their ability to inhibit TNF-α-induced VCAM-1 expression, despite treatment with chymase (Figure IIIA and IIIB in the online-only Data Supplement). It is plausible that the insensitivity of apoA-I in mature HDL particles is because of inaccessibility of proteases to the apolipoprotein, which is partially embedded in the surface phospholipid layer of the particles. Interestingly, neither chymase nor cathepsin G, but rather α-1 antitrypsin, has been found to be a component of total (unfractionated) HDL and the HDL3 fraction derived from circulating plasma or from atherosclerotic carotid tissue,27 and so providing an additional explanation for the protease insensitivity of apoA-I in these particles.
Figure 2.
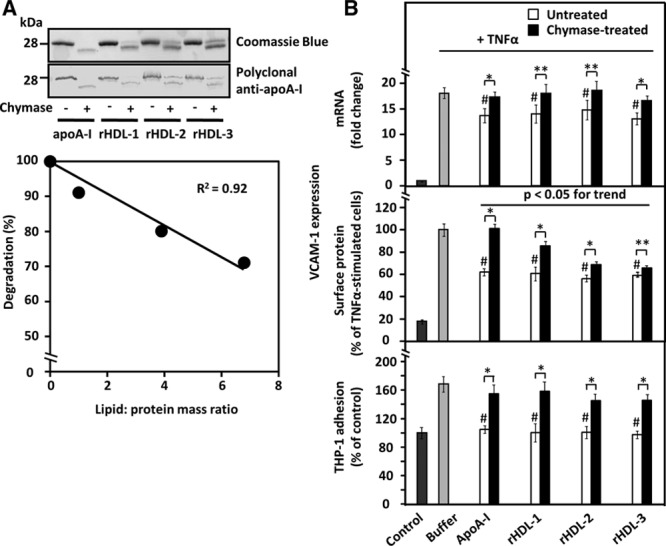
Effect of apolipoprotein A-I (apoA-I) lipidation on its sensitivity to proteolysis and on the ability of untreated and chymase-treated apoA-I to influence vascular cell adhesion molecule-1 (VCAM-1) expression in and THP-1 adhesion to tumor necrosis factor-α (TNF-α)-activated human coronary artery endothelial cells (HCAECs). A, Top, ApoA-I and discoidal apoA-I-containing reconstituted high-density lipoprotein ((A-I)rHDLs) with 3 different degrees of lipidation (see Table I in the online-only Data Supplement) were incubated in the absence or presence of recombinant chymase, and the proteins in the incubation mixtures were analyzed, as described in Figure 1. Bottom, ApoA-I degradation was expressed as a percentage of the intensity of the intact band in the proteolyzed samples relative to the corresponding untreated sample and plotted against the lipid/protein mass ratio of the various (A-I)rHDL preparations. B, HCAECs were preincubated for 16 h with the various untreated or chymase-treated apoA-I or (A-I)rHDL species (50 μg/mL, each) and then activated with tumor necrosis factor-α (TNF-α), as described in Figure 1. Nonactivated cells (control) and TNF-α-activated cells preincubated in only medium (buffer) acted as references. Vascular cell adhesion molecule-1 (VCAM-1) mRNA levels (top, fold change relative to control) and surface protein expression (middle, % of buffer) were evaluated in the TNF-α-activated cells. In a separate experiment, HCAECs that had been preincubated with the various apoA-I-containing preparations were further incubated for 1 h with fluorescently labeled THP-1cells, and the fluorescence of HCAECs-bound THP-1 cells was then measured (bottom). The fluorescence signal of the TNF-α-activated HCAECs was expressed as percentage of the basal signal from the nonactivated cells (control) which was set as 100%. Data shown in each panel represent the means±SD from 3 to 4 independent experiments performed in triplicate wells. **P<0.05; *P<0.01; #P<0.01 (untreated apoA-I-containing preparations vs buffer).
Proteolysis of ApoA-I by Plasmin or Cathepsin S Impairs Its Anti-Inflammatory Effects on HCAECs
We evaluated the effect of other proteases known to be present in atherosclerotic lesions, namely plasmin and cathepsin S,28,29 on the anti-inflammatory activity of apoA-I in cultured HCAECs. Similar to chymase, plasmin cleaved the C-terminal region, resulting in the formation of a fragment containing the N-terminal region, whereas cathepsin S completely degraded apoA-I (Figure IVA in the online-only Data Supplement). Importantly, similar to chymase, treatment of apoA-I with either of the 2 proteases resulted in complete loss of its ability to inhibit VCAM-1 expression in TNF-α-activated HCAECs and to prevent the adhesion of THP-1 monocytes to these cells (Figure IVB in the online-only Data Supplement).
Chymase Treatment of ApoA-I Impairs Its Ability to Attenuate NF-κB Activation, to Bind to HCAECs, and to Prevent Transendothelial Migration of THP-1 Monocytes
Next we examined the effect of chymase treatment of lipid-free apoA-I on NF-κB activation, a dominant signal transduction pathway upregulating a variety of chemokines and adhesion molecules in human ECs.30 Pretreatment of HCAECs with untreated apoA-I significantly attenuated TNF-α-mediated NF-κB activation, as indicated by suppression of nuclear translocation of the p65 subunit of NF-κB (Figure 3A), a finding in agreement with previous reports.13,14 In contrast, treatment with chymase abolished the inhibitory effect of apoA-I on NF-κB activation, indicating that the intactness of apoA-I is a prerequisite for the inhibition of this signaling pathway. To further investigate the mechanisms involved in the chymase-dependent loss of the anti-inflammatory effects of apoA-I, we compared binding of untreated and chymase-treated 125I-labeled apoA-I to HCAECs. We found that chymase treatment severely blunted (≥80%) the high-affinity binding of apoA-I to HCAECs (Figure 3B), strongly suggesting that reduced interaction of apoA-I with the cell membrane was necessary for loss of the anti-inflammatory effect. The above finding also agrees with the observed strong reduction of specific binding to bovine aortic endothelial cells of C-terminal deletion mutants of apoA-I.31
Figure 3.
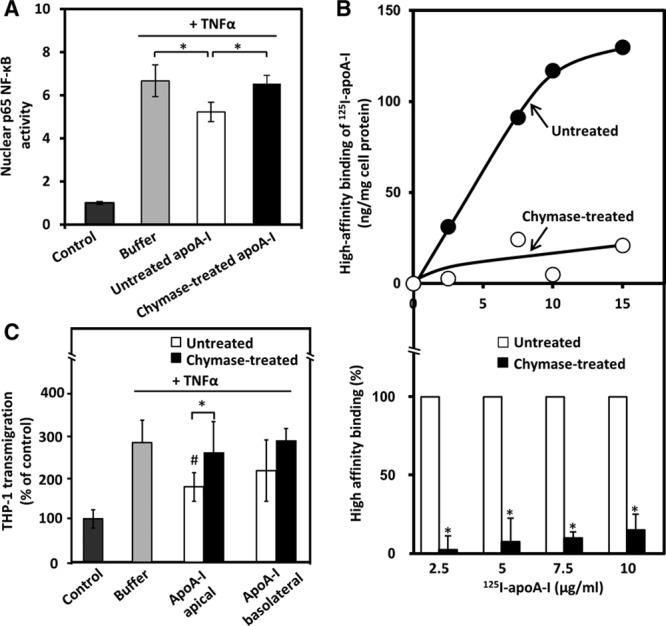
Proteolysis of apolipoprotein A-I (apoA-I) impairs its ability to attenuate nuclear factor-κB (NF-κB) activation to bind to human coronary artery endothelial cells (HCAECs) and to prevent transmigration of THP-1 monocytes across HCAEC monolayer. A, HCAECs were incubated for 16 h with untreated or chymase-treated apoA-I and then activated with tumor necrosis factor-α (TNF-α) for 15 min. Nonactivated cells (control) and TNF-α-activated cells preincubated in only medium (buffer) acted as a reference. Nuclear NF-κB p65 activity was determined by measuring nuclear translocation of the NF-κB p65 subunit. The basal activity of the nonactivated HCAECs (control) was set as 1. Data represent the means±SD from triplicate wells and are representative of 2 independent experiments. *P<0.05. B, ApoA-I was labeled with 125I and then treated with chymase as described in Figure 1B. HCAECs were incubated with the indicated concentrations of 125I-labeled untreated or chymase-treated apoA-I in the absence or presence of a 40-fold excess of untreated apoA-I for 2 h at 4°C. High-affinity binding of 125I-apoA-I to HCAECs was calculated by subtracting the values of the nonspecific binding from the total binding. A representative pair of binding curves is shown in the top panel. Data (mean±SD) from 4 independent experiments from duplicate wells are expressed as percentages of untreated apoA-I (bottom). *P<0.01. C, Confluent HCAEC monolayers grown on a transwell insert were first incubated with untreated or chymase-treated apoA-I added either to the apical or basolateral compartment, then activated with TNF-α for 5 h, and finally incubated with fluorescently labeled THP-1 cells added to the apical compartment. Migration across the endothelial monolayer of the cells was quantified by measuring the fluorescence signal of the transmigrated cells. Migration in the absence of any additions was designated as 100% (control). Data shown represent the means±SD from 3 independent experiments performed in triplicate wells. *P<0.05; #P<0.05 (untreated apoA-I vs buffer).
We further examined the effect of untreated and chymase-treated apoA-I on transmigration of THP-1 monocytes across monolayers of TNF-α-activated HCAECs using the transwell system, which mimics the human endothelium in vivo. We found that, regardless of whether adding to the apical or basolateral compartment, chymase-treated apoA-I failed to attenuate THP-1 transmigation across the activated HCAECs (Figure 3C). Interestingly, untreated apoA-I significantly prevented the endothelial transmigration of THP-1 when placed in the apical compartment, whereas a slight, but nonsignificant decrease was observed when apoA-I was added to the basolateral compartment. The smaller effect of untreated apoA-I could be possibly because of much lower basolateral than apical binding of apoA-I, which may be partly caused by sterical hindrance binding to ECs because of the presence of a porous membrane and coated matrix in the transwell insert.32
Chymase Treatment of ApoA-I Reduces Its Ability to Inhibit Expression of Proinflammatory Genes in HCAECs and in Macrophages and to Induce Cholesterol Efflux From Macrophage Foam Cells
We next examined the effect of chymase-treated lipid-free apoA-I on selected proinflammatory genes in TNF-α-activated HCAECs. TNF-α strongly increased mRNA levels of intercellular adhesion molecule-1, cyclooxygenase-2 (COX-2), and interleukin (IL)-8, although only slightly affected that of IL-6 (Figure 4A–4D). Treatment of HCAECs before activation with untreated, but not with chymase-treated, apoA-I significantly decreased the TNF-α-dependent induction of COX-2, IL-6, and IL-8 mRNA expression. In contrast, neither untreated nor chymase-treated apoA-I had any effect on intercellular adhesion molecule-1 expression. We also determined the effect of the chymase-treated apoA-I on the expression of cytokines in LPS-activated macrophage foam cells. For this purpose, human monocytes were allowed to differentiate into macrophages either in the presence of GM-CSF (granulocyte-macrophage colony-stimulating factor) or M-CSF (macrophage colony-stimulating factor) to obtain the GM-Mac and M-Mac subpopulations, which have their counterparts in human atherosclerotic lesions,33 and finally incubated with acetyl-LDL. To avoid any LPS binding to apoA-I,34 the foam cells were preincubated with untreated or chymase-treated apoA-I, then extensively washed, and finally challenged with LPS. Similar to the effects observed in HCAECs, preincubation with the untreated apoA-I, but not with the chymase-treated apoA-I, significantly suppressed LPS-induced upregulation of mRNA levels of proinflammatory cytokines, namely of TNF-α, IL-1β, IL-6, and IL-8 in both macrophage foam cell subtypes (Figure 4E–4L). To investigate how the loss of the anti-inflammatory effect of apoA-I by chymase modification was related to the known chymase-dependent loss of the cholesterol efflux ability of apoA-I, we also determined cholesterol efflux capacity of chymase-treated apoA-I in the GM-Mac- and M-Mac-derived foam cells. Consistent with previous studies,19,20 treatment of apoA-I with chymase decreased its ability to induce cholesterol efflux from macrophage foam cells (Figure 4M). Taken together, cleavage of apoA-I with chymase impaired both its anti-inflammatory and cholesterol efflux-inducing activities.
Figure 4.
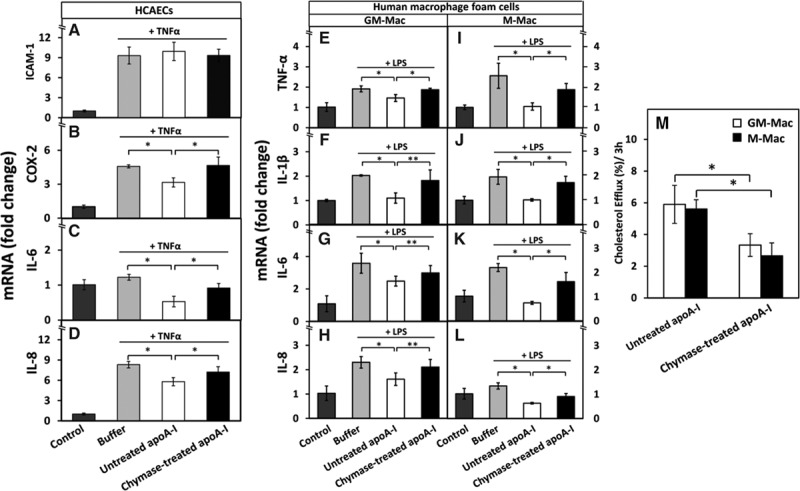
Proteolysis of apolipoprotein A-I (apoA-I) reduces its ability to downregulate the expression of proinflammatory genes in human coronary artery endothelial cells (HCAECs) and in human GM-Mac and M-Mac macrophage subpopulations and to promote cholesterol efflux from the macrophages. A-D, HCAECs were preincubated with untreated or chymase-treated apoA-I and then activated with tumor necrosis factor-α (TNF-α), as described in Figure 1. Nonactivated cells (control) and TNF-α-activated cells preincubated in only medium (buffer) acted as a reference. The mRNA levels of intercellular adhesion molecule-1 (ICAM-1), cyclooxygenase-2 (COX-2), interleukin (IL)-6, and IL-8 in HCAECs were measured by quantitative real-time polymerase chain reaction (qRT-PCR) and expressed as fold changes relative to the control cells. Data represent the means±SD from 3 independent experiments performed in duplicate. *P<0.01. E-L, Human monocyte-derived macrophages were differentiated in the presence of GM-CSF (granulocyte-macrophage colony-stimulating factor) or M-CSF (macrophage colony-stimulating factor) into GM-Mac and M-Mac subtypes, respectively, and then converted into foam cells by incubation with acetyl low density lipoprotein (LDL). The cells were then incubated for 3 h with untreated or chymase-treated apoA-I (50 μg/mL, each), washed, and activated for 3 h with lipopolysaccharide (LPS). Nonactivated cells (control) and LPS-activated cells preincubated in only medium (buffer) acted as references. LPS-induced mRNA levels of TNF-α, IL-1β, IL-6, and IL-8 in GM-Mac and M-Mac foam cells were evaluated by qRT-PCR and expressed as fold changes relative to the control cells. *P<0.01; **P<0.05. M, GM-Mac- and M-Mac-derived foam cells were incubated with untreated or chymase-treated apoA-I (50 μg/mL, each) for 3 h, the media were collected, centrifuged to remove cellular debris, and the radioactivity of each supernatant was determined by liquid scintillation counting. Cells were solubilized, and radioactivity was determined in the cell lysates. Cholesterol efflux was calculated as dpmmedium/(dpmcells+dpmmedium)×100. Data represent the means±SD from triplicate wells and are representative of 2 independent experiments. *P<0.01.
Chymase Treatment of ApoA-I Reduces Its Anti-Inflammatory Properties In Vivo
To learn whether chymase would also affect the anti-inflammatory properties of lipid-free apoA-I in vivo, we used the well-established mouse model of LPS-induced inflammation, in which apoA-I has been shown to exert its anti-inflammatory properties.7,35,36 We found that LPS administration remarkably increased the levels of 2 circulating proinflammatory mediators (TNF-α and IL-1β). Concurrent treatment of mice with both LPS and lipid-free apoA-I significantly reduced the level of TNF-α and IL-1β (Figure 5A and 5B), consistent with the well-known anti-inflammatory effect of apoA-I on LPS-induced inflammation in vivo.35 Importantly, concurrent treatment of mice with LPS and chymase-treated apoA-I failed to significantly diminish the levels of either TNF-α or IL-1β, indicating that the proteolytic degradation of apoA-I by chymase impaired its anti-inflammatory properties (Figure 5A and 5B). To obtain further insight into the mechanisms by which chymase impaired anti-inflammatory properties of apoA-I in vivo, we determined endotoxin activity using the limulus amebocyte lysate assay. In line with previous studies demonstrating that both HDL and apoA-I exert their anti-inflammatory properties in vivo via their ability to bind to LPS and neutralize endotoxin,35–37 we found that apoA-I strongly neutralized LPS endotoxin activity in vitro (Figure 5C). In sharp contrast, chymase-treated apoA-I had totally lost the ability to neutralize endotoxin activity.
Figure 5.
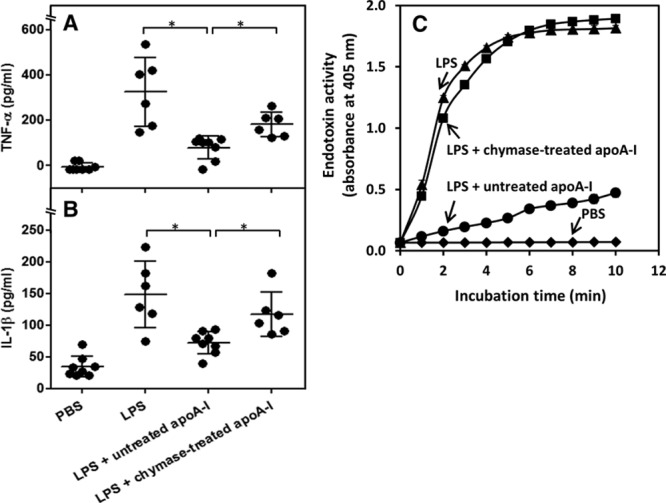
Proteolysis of apolipoprotein A-I (apoA-I) impairs its ability to inhibit lipopolysaccharide (LPS)-induced inflammation in vivo and to neutralize endotoxin activity of LPS in vitro. Mice (6–8 per group) were randomized to receive vehicle (PBS), LPS (1 mg/kg), LPS (1 mg/kg) plus apoA-I (10 mg/kg), or LPS (1 mg/kg) plus chymase-treated apoA-I (10 mg/kg). After 3 h, serum was collected, and TNF-α (A) and IL-1β (B) concentrations were measured. Data are means±SD, *P<0.05. C, LPS (1 μg/mL) was mixed with untreated apoA-I and chymase-treated apoA-I (10 μg/mL) and endotoxin activity was measured with kinetic colorimetric limulus amebocyte lysate (LAL) assay. Data represent the means±SD from triplicate wells and are representative of 2 independent experiments.
Chymase Treatment Does Not Blunt the Anti-Inflammatory Effects of D-4F on HCAECs, Neutrophils, and Macrophages
The apoA-I mimetic peptide L-4F exerts potent anti-inflammatory effects by reducing the chemotactic activity of human aortic ECs for monocytes.38 Given the cleavage specificity of chymase, a chymotryptic protease, and the arrangement of aromatic amino acid residues in the 4F structure that determines its functional properties,39 we considered that chymase could cleave L-4F, so resulting in its functional inactivation. As a control, we used D-4F, which is resistant to cleavage by mammalian proteases.40 Both untreated L-4F and D-4F decreased VCAM-1 protein expression in TNF-α-activated HCAECs to a similar extent (Figure 6A). However, when chymase-treated L-4F and D-4F were compared, we found that the former had lost, but the latter had maintained its anti-inflammatory effect. Because the remarkable anti-inflammatory properties of both 4F species appear to be accounted for their ability to bind proinflammatory oxidized lipids,38 we also evaluated the effect of chymase on their antioxidant effects in PMA (phorbol 12-myristate 13-acetate)–activated neutrophils. We found that, although apoA-I and both mimetic peptides significantly diminished superoxide production by the activated neutrophils in a dose-dependent manner, chymase treatment fully abolished the antioxidant effect of apoA-I and L-4F, but not that of D-4F (Figure 6B). Similar to findings observed with HCAECs and neutrophils, both untreated and chymase-treated D-4F significantly decreased to a similar extent the mRNA levels of the proinflammatory cytokines (IL-1β, IL-6, and IL-8) in LPS-activated macrophage foam cells of the 2 phenotypes (GM-Mac and M-Mac) (Figure 6C–CJ). Interestingly, unlike apoA-I, D-4F failed to decrease mRNA levels of TNF-α in either macrophage phenotype.
Figure 6.
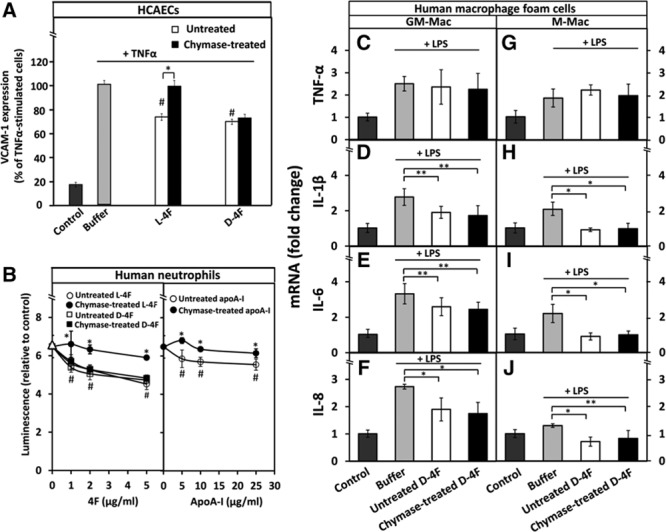
Chymase treatment does not affect the anti-inflammatory effects of D-4F on human coronary artery endothelial cells (HCAECs), neutrophils, and macrophages. A, HCAECs were preincubated with untreated or chymase-treated L-4F and D-4F (50 μg/mL, each) after which the cells were then activated with tumor necrosis factor-α (TNF-α), as described in Figure 1. Nonactivated cells (control) and TNF-α-activated cells preincubated in only medium (buffer) were used as references. Cell-surface expression of vascular cell adhesion molecule-1 (VCAM-1) protein was determined by flow cytometry. Data represent the means±SD from 3 independent experiments performed in duplicate. *P<0.01; #P<0.01 (untreated L-4F or untreated D-4F vs buffer). B, Superoxide radical production by PMA (phorbol 12-myristate 13-acetate)–activated neutrophils was determined after the cells had been preincubated for 5 min with the indicated concentrations of untreated or chymase-treated L-4F, D-4F, or apolipoprotein A-I (apoA-I). PMA-activated neutrophils preincubated in only medium (buffer) were used as a reference (open triangles). Luminescence emitted by nonactivated neutrophils (control) was set as 1. Data are derived from 2 donors and represent the means±SD from 2 independent experiments, each performed in 3 to 6 wells. *P<0.01 (untreated vs chymase-treated L-4F, and untreated vs chymase-treated apoA-I); #P<0.01 (untreated L-4F, D-4F, or apoA-I vs buffer). C-J, Human monocyte–derived macrophages were differentiated into GM-Mac and M-Mac subtypes and converted into foam cells by incubation with acetyl low-density lipoprotein (LDL). The foam cells were then incubated for 3 h with untreated or chymase-treated D-4F (50 μg/mL, each), washed, and activated for 3 h with lipopolysaccharide (LPS). Nonactivated foam cells (control) and LPS-activated foam cells preincubated in only medium (buffer) served as references. LPS-induced mRNA levels of TNF-α, interleukin (IL)-1β, IL-6, and IL-8 in GM-Mac and M-Mac foam cells were evaluated by quantitative real-time polymerase chain reaction (qRT-PCR) and expressed as fold changes relative to the control cells. Data represent the means±SD from triplicate wells and are representative of 2 independent experiments. *P<0.01; **P<0.05.
Discussion
The present study demonstrates that protease-dependent loss of the C-terminal region of apoA-I is sufficient to fully inhibit its anti-inflammatory properties both in vitro and in vivo. The enzymes capable of such cleavage comprised chymase and plasmin, the former known to be secreted by vascular mast cells41 and the latter by vascular smooth muscle cells and ECs.18 The repertoire of cell-derived extracellular proteases present in the human atherosclerotic arterial intima and capable of cleaving the C-terminal domain of apoA-I extends to the members of the matrix metalloproteinase family-3, -7, and -12.18 Altogether, these findings suggest that an inflamed atherosclerotic lesion is a compartment in which protease-dependent attenuation of the anti-inflammatory functions of HDL could take place.
The structure–function relationships involved in the mechanisms determining the anti-inflammatory activities of apoA-I and HDL are manifold. According to current notions, apoA-I suppresses the expression of surface adhesion molecules in ECs in vitro via interaction with ABCA1, which initiates a cascade of intracellular signaling events that are thought to involve lipid efflux–dependent disruption of lipid rafts.5,42 Moreover, apoA-I has been shown to induce COX-2 expression and PGI-2 (prostacyclin) release in unstimulated HUVECs via interaction with ABCA1,43 revealing functional interactions between apoA-I and ABCA1. Furthermore, it was recently shown that the anti-inflammatory and antioxidant effects of the apoA-I mimetic peptide 5A are mediated via the ABCA1 transporter and NF-κB signaling pathways in HCAECs.14 Thus, it is likely that, besides being involved in the transcytotic pathway of lipid-free apoA-I through ECs,32,44 binding of apoA-I to ABCA1 also essentially contributes to the anti-inflammatory actions of lipid-free apoA-I in HCAECs. Interestingly, the C-terminal domain of apoA-I has been shown to be required for endothelial binding and transendothelial transport of lipid-free apoA-I.31 Here, we have demonstrated that cleavage of the C-terminal domain of apoA-I by chymase prevented binding of apoA-I to HCAECs with high affinity and caused its failure to suppress the initiation of the NF-κB-mediated downstream inflammatory signaling in TNF-α-activated HCAECs. Similarly, glycation of apoA-I which modifies the conformation of epitopes present in C-terminal and central regions of apoA-I has been shown to impair its anti-inflammatory effects on HCAECs.13,45
Regarding the effect of chymase treatment of apoA-I on its ability to interact with macrophages, we found that chymase-treated apoA-I had lost its anti-inflammatory effects and its ability to promote macrophage cholesterol efflux from human macrophages. It has been found earlier that loss of apoA-I/ABCA1 interaction not only prevents initiation of cholesterol efflux, but also initiation of the anti-inflammatory effects of apoA-I in these cells,46,47 and moreover that silencing of ABCA1 abolishes the inhibitory effect of apoA-I on the production of TNF-α and other inflammatory cytokines in LPS-activated human macrophages.48 Moreover, apoA-I has been recently found to exert anti-inflammatory effects on peptidoglycan polysaccharide–activated human macrophages by decreasing NF-κB activation in an ABCA1-dependent manner.49 Thus, we infer that the present observation of the loss of the ability of chymase-treated apoA-I to abolish a proinflammatory response of macrophages to LPS treatment actually reflected lost interaction with macrophage ABCA1, similar to the loss of the apoA-I-dependent induction of cholesterol efflux from macrophage foam cells after chymase treatment of apoA-I.19 Thus, a failure to bind to macrophage ABCA1 appears to be the molecular link between the losses of the anti-inflammatory and the cholesterol efflux-inducing capacities of the C-terminally truncated apoA-I because of chymase treatment. Similarly, myeloperoxidase-derived hypochlorous acid, which oxidizes apoA-I, impairs the ability of HDL particles to promote cholesterol efflux and also compromises their anti-inflammatory properties.50 More recently, non-enzymatic glycation, which induces changes in structure and in lipid-binding activity of apoA-I,51 was also observed to impair both the cholesterol efflux capacity and anti-inflammatory properties of apoA-I.13,45 Thus, several apoA-I-modifying factors alter the structure of apoA-I in such a way that, besides affecting adversely its anti-inflammatory properties, they also impair its cholesterol efflux–inducing capacity.
In vivo studies have demonstrated that administration of apoA-I inhibits inflammatory responses in experimental animals, like in rabbits,16,17 mice,35 and rats49 subjected to acute or chronic inflammation. Actually, overexpression of human apoA-I in mice attenuates LPS- or high fat–induced inflammation,7,8 whereas apoA-I deletion results in loss of its anti-inflammatory actions.52 Our in vivo data in mice showed that proteolytic modification of apoA-I by chymase failed to neutralize endotoxin activity, thereby impairing its anti-inflammatory properties in acute inflammation induced by LPS. Similarly, in rabbits with acute vascular inflammation, the anti-inflammatory properties of apoA-I in vivo have been found to be markedly reduced when the animals were infused with glycated apoA-I derived from in vitro modification or from patients with persistent hyperglycemia,13 so providing a relevant link between experimental and clinical studies. Of particular relevance to the present work are some recent findings from other laboratories, which attribute the origins of the C-terminally truncated apoA-I in human plasma to mast cell–secreted chymase or to plasmin generated during thrombolytic treatment of coronary patients with tissue plasminogen activator,53–55 that is, the 2 proteases also studied here. Obviously, the chymase-generated C-terminally truncated apoA-I must result from extracellular activity of chymase, that is, it requires mast cell degranulation with ensuing release of granule-bound chymase. Regarding plasmin-generated C-terminally truncated apoA-I, also endogenously generated plasmin in atherosclerotic lesions may be involved. Thus, given that apoA-I fragments have been detected in human plasma, albeit at low concentrations, as a result of rapid catabolism of C-terminally truncated apoA-I in vivo,56 a small fraction of the lipid-free or lipid-poor apoA-I appears to be specifically proteolyzed in the extracellular compartment of human body. Regarding the generation of chymase-specific apoA-I degradation products in humans, the cardiovascular system could be one source. Thus, mast cells are particularly abundant in human coronary intima, and the number of activated mast cells in atherosclerotic plaques increase with disease progression.57 Activated mast cells have been also demonstrated to accumulate in canine myocardium after ischemia and reperfusion, and moreover, myocardial chymase activity is significantly increased in the acute phase of myocardial infarction in hamsters.58–60 Importantly, the substrates for chymase, that is, the chymase-sensitive small lipid-poor preβ-HDL particles, are enriched in the arterial intimal fluid and also in the peripheral lymphatic fluid, which among other tissues and organs, also drains the coronary arterial wall and the myocardium.3,4 The challenge is to find a suitable animal model in which chymase-specific apoA-I degradation products would reside in the lymphatic fluid draining an ischemic myocardium.
Finally, considering the demonstrated therapeutic potential of parenterally infused apoA-I in atherosclerotic coronary artery disease,61 the therapeutic potential of an intravenously infused apoA-I-containing rHDL preparation (including the protease-sensitive apoA-I Milano)62,63 or of an apoA-I-mimetic peptide could be locally compromised by extracellular proteolytic activities in the target tissue, notably in an inflamed advanced coronary lesion. This challenge could be overcome by parenteral administration of an apoA-I mimetic peptide, such as the D-4F peptide, in which high anti-atherosclerotic activity is combined with an ability to resist proteolytic attack.
Acknowledgments
We thank Maija Atuegwu, Mari Jokinen, María Arraño de Kivikko, Reija Silvennoinen, and Ilona Kareinen for excellent technical assistance.
Sources of Funding
Wihuri Research Institute is maintained by the Jenny and Antti Wihuri Foundation. This study was also supported by the Academy of Finland (265940 to S.D. Nguyen and K. Öörni and 257545 to M. Jauhiainen), the Finnish Foundation for Cardiovascular Research (K. Maaninka and K. Nurmi), and the Ida Montin Foundation (K. Maaninka).
Disclosures
None.
Supplementary Material
Nonstandard Abbreviations and Acronyms
- (A-I)rHDL
- apoA-I-containing reconstituted HDL
- ABCA1
- ATP-binding cassette transporter A1
- apoA-I
- apolipoprotein A-I
- EC
- endothelial cells
- HCAECs
- human coronary artery endothelial cells
- HDL
- high-density lipoprotein
- IL
- interleukin
- NF-κB
- nuclear factor-κB
- TNFα
- tumor necrosis factor-α
- VCAM-1
- vascular cell adhesion molecule-1
The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.115.306827/-/DC1.
Significance
Activated mast cells are present in human atherosclerotic lesions where they release chymase capable of avidly degrading lipid-free apolipoprotein A-I (apoA-I) and lipid-poor species of high-density lipoprotein. Here, we report that C-terminal cleavage of apoA-I by chymase abolished its anti-inflammatory properties. This is the first demonstration of involvement of a pathophysiologically relevant protease in the regulation of a multitude of anti-inflammatory functions of apoA-I. Because atherosclerotic lesions contain a variety of extracellular proteases capable of C-terminally cleaving apoA-I, local proteolytic inactivation of apoA-I and lipid-poor preβ-high-density lipoprotein particles is likely. Therefore, the use of protease-resistant apoA-I-mimetic peptides may be of special value in the treatment of atherosclerosis.
References
- 1.Asztalos BF, Tani M, Schaefer EJ. Metabolic and functional relevance of HDL subspecies. Curr Opin Lipidol. 2011;22:176–185. doi: 10.1097/MOL.0b013e3283468061. doi: 10.1097/MOL.0b013e3283468061. [DOI] [PubMed] [Google Scholar]
- 2.Kane JP, Malloy MJ. Prebeta-1 HDL and coronary heart disease. Curr Opin Lipidol. 2012;23:367–371. doi: 10.1097/MOL.0b013e328353eef1. doi: 10.1097/MOL.0b013e328353eef1. [DOI] [PubMed] [Google Scholar]
- 3.Miller NE, Olszewski WL, Hattori H, Miller IP, Kujiraoka T, Oka T, Iwasaki T, Nanjee MN. Lipoprotein remodeling generates lipid-poor apolipoprotein A-I particles in human interstitial fluid. Am J Physiol Endocrinol Metab. 2013;304:E321–E328. doi: 10.1152/ajpendo.00324.2012. doi: 10.1152/ajpendo.00324.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith EB. Transport, interactions and retention of plasma proteins in the intima: the barrier function of the internal elastic lamina. Eur Heart J. 1990;11(suppl E):72–81. doi: 10.1093/eurheartj/11.suppl_e.72. [DOI] [PubMed] [Google Scholar]
- 5.Mineo C, Shaul PW. Regulation of signal transduction by HDL. J Lipid Res. 2013;54:2315–2324. doi: 10.1194/jlr.R039479. doi: 10.1194/jlr.R039479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rao RM, Yang L, Garcia-Cardena G, Luscinskas FW. Endothelial-dependent mechanisms of leukocyte recruitment to the vascular wall. Circ Res. 2007;101:234–247. doi: 10.1161/CIRCRESAHA.107.151860b. doi: 10.1161/CIRCRESAHA.107.151860b. [DOI] [PubMed] [Google Scholar]
- 7.Van Linthout S, Spillmann F, Graiani G, Miteva K, Peng J, Van Craeyveld E, Meloni M, Tölle M, Escher F, Subasigüller A, Doehner W, Quaini F, De Geest B, Schultheiss HP, Tschöpe C. Down-regulation of endothelial TLR4 signalling after apo A-I gene transfer contributes to improved survival in an experimental model of lipopolysaccharide-induced inflammation. J Mol Med (Berl) 2011;89:151–160. doi: 10.1007/s00109-010-0690-6. doi: 10.1007/s00109-010-0690-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng AM, Handa P, Tateya S, Schwartz J, Tang C, Mitra P, Oram JF, Chait A, Kim F. Apolipoprotein A-I attenuates palmitate-mediated NF-κB activation by reducing Toll-like receptor-4 recruitment into lipid rafts. PLoS One. 2012;7:e33917. doi: 10.1371/journal.pone.0033917. doi: 10.1371/journal.pone.0033917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D’Souza W, Stonik JA, Murphy A, Demosky SJ, Sethi AA, Moore XL, Chin-Dusting J, Remaley AT, Sviridov D. Structure/function relationships of apolipoprotein a-I mimetic peptides: implications for antiatherogenic activities of high-density lipoprotein. Circ Res. 2010;107:217–227. doi: 10.1161/CIRCRESAHA.110.216507. doi: 10.1161/CIRCRESAHA.110.216507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murphy AJ, Woollard KJ, Hoang A, Mukhamedova N, Stirzaker RA, McCormick SP, Remaley AT, Sviridov D, Chin-Dusting J. High-density lipoprotein reduces the human monocyte inflammatory response. Arterioscler Thromb Vasc Biol. 2008;28:2071–2077. doi: 10.1161/ATVBAHA.108.168690. doi: 10.1161/ATVBAHA.108.168690. [DOI] [PubMed] [Google Scholar]
- 11.Smythies LE, White CR, Maheshwari A, Palgunachari MN, Anantharamaiah GM, Chaddha M, Kurundkar AR, Datta G. Apolipoprotein A-I mimetic 4F alters the function of human monocyte-derived macrophages. Am J Physiol Cell Physiol. 2010;298:C1538–C1548. doi: 10.1152/ajpcell.00467.2009. doi: 10.1152/ajpcell.00467.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yvan-Charvet L, Kling J, Pagler T, Li H, Hubbard B, Fisher T, Sparrow CP, Taggart AK, Tall AR. Cholesterol efflux potential and antiinflammatory properties of high-density lipoprotein after treatment with niacin or anacetrapib. Arterioscler Thromb Vasc Biol. 2010;30:1430–1438. doi: 10.1161/ATVBAHA.110.207142. doi: 10.1161/ATVBAHA.110.207142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nobécourt E, Tabet F, Lambert G, Puranik R, Bao S, Yan L, Davies MJ, Brown BE, Jenkins AJ, Dusting GJ, Bonnet DJ, Curtiss LK, Barter PJ, Rye KA. Nonenzymatic glycation impairs the antiinflammatory properties of apolipoprotein A-I. Arterioscler Thromb Vasc Biol. 2010;30:766–772. doi: 10.1161/ATVBAHA.109.201715. doi: 10.1161/ATVBAHA.109.201715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tabet F, Remaley AT, Segaliny AI, Millet J, Yan L, Nakhla S, Barter PJ, Rye KA, Lambert G. The 5A apolipoprotein A-I mimetic peptide displays antiinflammatory and antioxidant properties in vivo and in vitro. Arterioscler Thromb Vasc Biol. 2010;30:246–252. doi: 10.1161/ATVBAHA.109.200196. doi: 10.1161/ATVBAHA.109.200196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park SH, Park JH, Kang JS, Kang YH. Involvement of transcription factors in plasma HDL protection against TNF-alpha-induced vascular cell adhesion molecule-1 expression. Int J Biochem Cell Biol. 2003;35:168–182. doi: 10.1016/s1357-2725(02)00173-5. [DOI] [PubMed] [Google Scholar]
- 16.Patel S, Di Bartolo BA, Nakhla S, Heather AK, Mitchell TW, Jessup W, Celermajer DS, Barter PJ, Rye KA. Anti-inflammatory effects of apolipoprotein A-I in the rabbit. Atherosclerosis. 2010;212:392–397. doi: 10.1016/j.atherosclerosis.2010.05.035. doi: 10.1016/j.atherosclerosis.2010.05.035. [DOI] [PubMed] [Google Scholar]
- 17.Nicholls SJ, Dusting GJ, Cutri B, Bao S, Drummond GR, Rye KA, Barter PJ. Reconstituted high-density lipoproteins inhibit the acute pro-oxidant and proinflammatory vascular changes induced by a periarterial collar in normocholesterolemic rabbits. Circulation. 2005;111:1543–1550. doi: 10.1161/01.CIR.0000159351.95399.50. doi: 10.1161/01.CIR.0000159351.95399.50. [DOI] [PubMed] [Google Scholar]
- 18.Lee-Rueckert M, Kovanen PT. Extracellular modifications of HDL in vivo and the emerging concept of proteolytic inactivation of preβ-HDL. Curr Opin Lipidol. 2011;22:394–402. doi: 10.1097/MOL.0b013e32834a3d24. doi: 10.1097/MOL.0b013e32834a3d24. [DOI] [PubMed] [Google Scholar]
- 19.Favari E, Lee M, Calabresi L, Franceschini G, Zimetti F, Bernini F, Kovanen PT. Depletion of pre-beta-high density lipoprotein by human chymase impairs ATP-binding cassette transporter A1- but not scavenger receptor class B type I-mediated lipid efflux to high density lipoprotein. J Biol Chem. 2004;279:9930–9936. doi: 10.1074/jbc.M312476200. doi: 10.1074/jbc.M312476200. [DOI] [PubMed] [Google Scholar]
- 20.Lee-Rueckert M, Vikstedt R, Metso J, Jauhiainen M, Kovanen PT. Association of cholesteryl ester transfer protein with HDL particles reduces its proteolytic inactivation by mast cell chymase. J Lipid Res. 2008;49:358–368. doi: 10.1194/jlr.M700392-JLR200. doi: 10.1194/jlr.M700392-JLR200. [DOI] [PubMed] [Google Scholar]
- 21.Lee-Rueckert M, Silvennoinen R, Rotllan N, Judström I, Blanco-Vaca F, Metso J, Jauhiainen M, Kovanen PT, Escola-Gil JC. Mast cell activation in vivo impairs the macrophage reverse cholesterol transport pathway in the mouse. Arterioscler Thromb Vasc Biol. 2011;31:520–527. doi: 10.1161/ATVBAHA.110.221069. doi: 10.1161/ATVBAHA.110.221069. [DOI] [PubMed] [Google Scholar]
- 22.Lee-Rueckert M, Kovanen PT. The mast cell as a pluripotent HDL-modifying effector in atherogenesis: from in vitro to in vivo significance. Curr Opin Lipidol. 2015;26:362–368. doi: 10.1097/MOL.0000000000000224. doi: 10.1097/MOL.0000000000000224. [DOI] [PubMed] [Google Scholar]
- 23.Cook-Mills JM, Marchese ME, Abdala-Valencia H. Vascular cell adhesion molecule-1 expression and signaling during disease: regulation by reactive oxygen species and antioxidants. Antioxid Redox Signal. 2011;15:1607–1638. doi: 10.1089/ars.2010.3522. doi: 10.1089/ars.2010.3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maaninka K, Lappalainen J, Kovanen PT. Human mast cells arise from a common circulating progenitor. J Allergy Clin Immunol. 2013;132:463–469.e3. doi: 10.1016/j.jaci.2013.02.011. doi: 10.1016/j.jaci.2013.02.011. [DOI] [PubMed] [Google Scholar]
- 25.Vucic E, Rosenson RS. Recombinant high-density lipoprotein formulations. Curr Atheroscler Rep. 2011;13:81–87. doi: 10.1007/s11883-010-0141-4. doi: 10.1007/s11883-010-0141-4. [DOI] [PubMed] [Google Scholar]
- 26.Usami Y, Kobayashi Y, Kameda T, Miyazaki A, Matsuda K, Sugano M, Kawasaki K, Kurihara Y, Kasama T, Tozuka M. Identification of sites in apolipoprotein A-I susceptible to chymase and carboxypeptidase A digestion. Biosci Rep. 2013;33:49–56. doi: 10.1042/BSR20120094. doi: 10.1042/BSR20120094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vaisar T, Pennathur S, Green PS, et al. Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J Clin Invest. 2007;117:746–756. doi: 10.1172/JCI26206. doi: 10.1172/JCI26206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lindstedt L, Kovanen PT. Plasmin and kallikrein reduce HDL-induced cholesterol efflux from foam cells. Biochem Biophys Res Commun. 2000;277:552–557. doi: 10.1006/bbrc.2000.3704. doi: 10.1006/bbrc.2000.3704. [DOI] [PubMed] [Google Scholar]
- 29.Lindstedt L, Lee M, Oörni K, Brömme D, Kovanen PT. Cathepsins F and S block HDL3-induced cholesterol efflux from macrophage foam cells. Biochem Biophys Res Commun. 2003;312:1019–1024. doi: 10.1016/j.bbrc.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 30.Denk A, Goebeler M, Schmid S, Berberich I, Ritz O, Lindemann D, Ludwig S, Wirth T. Activation of NF-kappa B via the Ikappa B kinase complex is both essential and sufficient for proinflammatory gene expression in primary endothelial cells. J Biol Chem. 2001;276:28451–28458. doi: 10.1074/jbc.M102698200. doi: 10.1074/jbc.M102698200. [DOI] [PubMed] [Google Scholar]
- 31.Ohnsorg PM, Rohrer L, Perisa D, Kateifides A, Chroni A, Kardassis D, Zannis VI, von Eckardstein A. Carboxyl terminus of apolipoprotein A-I (ApoA-I) is necessary for the transport of lipid-free ApoA-I but not prelipidated ApoA-I particles through aortic endothelial cells. J Biol Chem. 2011;286:7744–7754. doi: 10.1074/jbc.M110.193524. doi: 10.1074/jbc.M110.193524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rohrer L, Cavelier C, Fuchs S, Schlüter MA, Völker W, von Eckardstein A. Binding, internalization and transport of apolipoprotein A-I by vascular endothelial cells. Biochim Biophys Acta. 2006;1761:186–194. doi: 10.1016/j.bbalip.2006.01.009. doi: 10.1016/j.bbalip.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 33.Waldo SW, Li Y, Buono C, Zhao B, Billings EM, Chang J, Kruth HS. Heterogeneity of human macrophages in culture and in atherosclerotic plaques. Am J Pathol. 2008;172:1112–1126. doi: 10.2353/ajpath.2008.070513. doi: 10.2353/ajpath.2008.070513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Henning MF, Herlax V, Bakás L. Contribution of the C-terminal end of apolipoprotein AI to neutralization of lipopolysaccharide endotoxic effect. Innate Immun. 2011;17:327–337. doi: 10.1177/1753425910370709. doi: 10.1177/1753425910370709. [DOI] [PubMed] [Google Scholar]
- 35.Yan YJ, Li Y, Lou B, Wu MP. Beneficial effects of ApoA-I on LPS-induced acute lung injury and endotoxemia in mice. Life Sci. 2006;79:210–215. doi: 10.1016/j.lfs.2006.02.011. doi: 10.1016/j.lfs.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 36.Li Y, Dong JB, Wu MP. Human ApoA-I overexpression diminishes LPS-induced systemic inflammation and multiple organ damage in mice. Eur J Pharmacol. 2008;590:417–422. doi: 10.1016/j.ejphar.2008.06.047. doi: 10.1016/j.ejphar.2008.06.047. [DOI] [PubMed] [Google Scholar]
- 37.Levine DM, Parker TS, Donnelly TM, Walsh A, Rubin AL. In vivo protection against endotoxin by plasma high density lipoprotein. Proc Natl Acad Sci U S A. 1993;90:12040–12044. doi: 10.1073/pnas.90.24.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Lenten BJ, Wagner AC, Jung CL, Ruchala P, Waring AJ, Lehrer RI, Watson AD, Hama S, Navab M, Anantharamaiah GM, Fogelman AM. Anti-inflammatory apoA-I-mimetic peptides bind oxidized lipids with much higher affinity than human apoA-I. J Lipid Res. 2008;49:2302–2311. doi: 10.1194/jlr.M800075-JLR200. doi: 10.1194/jlr.M800075-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.White CR, Datta G, Buck AK, Chaddha M, Reddy G, Wilson L, Palgunachari MN, Abbasi M, Anantharamaiah GM. Preservation of biological function despite oxidative modification of the apolipoprotein A-I mimetic peptide 4F. J Lipid Res. 2012;53:1576–1587. doi: 10.1194/jlr.M026278. doi: 10.1194/jlr.M026278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Navab M, Anantharamaiah GM, Hama S, Garber DW, Chaddha M, Hough G, Lallone R, Fogelman AM. Oral administration of an Apo A-I mimetic peptide synthesized from D-amino acids dramatically reduces atherosclerosis in mice independent of plasma cholesterol. Circulation. 2002;105:290–292. doi: 10.1161/hc0302.103711. [DOI] [PubMed] [Google Scholar]
- 41.Kovanen PT. Chymase-containing mast cells in human arterial intima: implications for atherosclerotic disease. Heart Vessels. 1997;(suppl 12):125–127. [PubMed] [Google Scholar]
- 42.Prosser HC, Ng MK, Bursill CA. The role of cholesterol efflux in mechanisms of endothelial protection by HDL. Curr Opin Lipidol. 2012;23:182–189. doi: 10.1097/MOL.0b013e328352c4dd. doi: 10.1097/MOL.0b013e328352c4dd. [DOI] [PubMed] [Google Scholar]
- 43.Liu D, Ji L, Tong X, Pan B, Han JY, Huang Y, Chen YE, Pennathur S, Zhang Y, Zheng L. Human apolipoprotein A-I induces cyclooxygenase-2 expression and prostaglandin I-2 release in endothelial cells through ATP-binding cassette transporter A1. Am J Physiol Cell Physiol. 2011;301:C739–C748. doi: 10.1152/ajpcell.00055.2011. doi: 10.1152/ajpcell.00055.2011. [DOI] [PubMed] [Google Scholar]
- 44.Cavelier C, Rohrer L, von Eckardstein A. ATP-binding cassette transporter A1 modulates apolipoprotein A-I transcytosis through aortic endothelial cells. Circ Res. 2006;99:1060–1066. doi: 10.1161/01.RES.0000250567.17569.b3. doi: 10.1161/01.RES.0000250567.17569.b3. [DOI] [PubMed] [Google Scholar]
- 45.Hoang A, Murphy AJ, Coughlan MT, Thomas MC, Forbes JM, O’Brien R, Cooper ME, Chin-Dusting JP, Sviridov D. Advanced glycation of apolipoprotein A-I impairs its anti-atherogenic properties. Diabetologia. 2007;50:1770–1779. doi: 10.1007/s00125-007-0718-9. doi: 10.1007/s00125-007-0718-9. [DOI] [PubMed] [Google Scholar]
- 46.Tang C, Liu Y, Kessler PS, Vaughan AM, Oram JF. The macrophage cholesterol exporter ABCA1 functions as an anti-inflammatory receptor. J Biol Chem. 2009;284:32336–32343. doi: 10.1074/jbc.M109.047472. doi: 10.1074/jbc.M109.047472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yvan-Charvet L, Welch C, Pagler TA, Ranalletta M, Lamkanfi M, Han S, Ishibashi M, Li R, Wang N, Tall AR. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 2008;118:1837–1847. doi: 10.1161/CIRCULATIONAHA.108.793869. doi: 10.1161/CIRCULATIONAHA.108.793869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yin K, Deng X, Mo ZC, Zhao GJ, Jiang J, Cui LB, Tan CZ, Wen GB, Fu Y, Tang CK. Tristetraprolin-dependent post-transcriptional regulation of inflammatory cytokine mRNA expression by apolipoprotein A-I: role of ATP-binding membrane cassette transporter A1 and signal transducer and activator of transcription 3. J Biol Chem. 2011;286:13834–13845. doi: 10.1074/jbc.M110.202275. doi: 10.1074/jbc.M110.202275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu BJ, Ong KL, Shrestha S, Chen K, Tabet F, Barter PJ, Rye KA. Inhibition of arthritis in the Lewis rat by apolipoprotein A-I and reconstituted high-density lipoproteins. Arterioscler Thromb Vasc Biol. 2014;34:543–551. doi: 10.1161/ATVBAHA.113.302832. doi: 10.1161/ATVBAHA.113.302832. [DOI] [PubMed] [Google Scholar]
- 50.Pirillo A, Uboldi P, Catapano AL. Dual effect of hypochlorite in the modification of high density lipoproteins. Biochem Biophys Res Commun. 2010;403:447–451. doi: 10.1016/j.bbrc.2010.11.053. doi: 10.1016/j.bbrc.2010.11.053. [DOI] [PubMed] [Google Scholar]
- 51.Calvo C, Talussot C, Ponsin G, Berthézène F. Non enzymatic glycation of apolipoprotein A-I. Effects on its self-association and lipid binding properties. Biochem Biophys Res Commun. 1988;153:1060–1067. doi: 10.1016/s0006-291x(88)81336-6. [DOI] [PubMed] [Google Scholar]
- 52.Wang W, Xu H, Shi Y, Nandedkar S, Zhang H, Gao H, Feroah T, Weihrauch D, Schulte ML, Jones DW, Jarzembowski J, Sorci-Thomas M, Pritchard KA., Jr Genetic deletion of apolipoprotein A-I increases airway hyperresponsiveness, inflammation, and collagen deposition in the lung. J Lipid Res. 2010;51:2560–2570. doi: 10.1194/jlr.M004549. doi: 10.1194/jlr.M004549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Usami Y, Matsuda K, Sugano M, Ishimine N, Kurihara Y, Sumida T, Yamauchi K, Tozuka M. Detection of chymase-digested C-terminally truncated apolipoprotein A-I in normal human serum. J Immunol Methods. 2011;369:51–58. doi: 10.1016/j.jim.2011.04.002. doi: 10.1016/j.jim.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 54.Eberini I, Gianazza E, Breghi L, Klugmann S, Calabresi L, Gomaraschi M, Mombelli G, Brusoni B, Wait R, Sirtori CR. Apolipoprotein A-I breakdown is induced by thrombolysis in coronary patients. Ann Med. 2007;39:306–311. doi: 10.1080/07853890701288760. doi: 10.1080/07853890701288760. [DOI] [PubMed] [Google Scholar]
- 55.Gomaraschi M, Ossoli A, Vitali C, Pozzi S, Vitali Serdoz L, Pitzorno C, Sinagra G, Franceschini G, Calabresi L. Off-target effects of thrombolytic drugs: apolipoprotein A-I proteolysis by alteplase and tenecteplase. Biochem Pharmacol. 2013;85:525–530. doi: 10.1016/j.bcp.2012.11.023. doi: 10.1016/j.bcp.2012.11.023. [DOI] [PubMed] [Google Scholar]
- 56.Schmidt HH, Remaley AT, Stonik JA, Ronan R, Wellmann A, Thomas F, Zech LA, Brewer HB, Jr, Hoeg JM. Carboxyl-terminal domain truncation alters apolipoprotein A-I in vivo catabolism. J Biol Chem. 1995;270:5469–5475. doi: 10.1074/jbc.270.10.5469. [DOI] [PubMed] [Google Scholar]
- 57.Kaartinen M, Penttilä A, Kovanen PT. Accumulation of activated mast cells in the shoulder region of human coronary atheroma, the predilection site of atheromatous rupture. Circulation. 1994;90:1669–1678. doi: 10.1161/01.cir.90.4.1669. [DOI] [PubMed] [Google Scholar]
- 58.Frangogiannis NG, Perrard JL, Mendoza LH, Burns AR, Lindsey ML, Ballantyne CM, Michael LH, Smith CW, Entman ML. Stem cell factor induction is associated with mast cell accumulation after canine myocardial ischemia and reperfusion. Circulation. 1998;98:687–698. doi: 10.1161/01.cir.98.7.687. [DOI] [PubMed] [Google Scholar]
- 59.Frangogiannis NG, Lindsey ML, Michael LH, Youker KA, Bressler RB, Mendoza LH, Spengler RN, Smith CW, Entman ML. Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation. 1998;98:699–710. doi: 10.1161/01.cir.98.7.699. [DOI] [PubMed] [Google Scholar]
- 60.Jin D, Takai S, Yamada M, Sakaguchi M, Kamoshita K, Ishida K, Sukenaga Y, Miyazaki M. Impact of chymase inhibitor on cardiac function and survival after myocardial infarction. Cardiovasc Res. 2003;60:413–420. doi: 10.1016/s0008-6363(03)00535-2. [DOI] [PubMed] [Google Scholar]
- 61.Nissen SE, Tsunoda T, Tuzcu EM, Schoenhagen P, Cooper CJ, Yasin M, Eaton GM, Lauer MA, Sheldon WS, Grines CL, Halpern S, Crowe T, Blankenship JC, Kerensky R. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA. 2003;290:2292–2300. doi: 10.1001/jama.290.17.2292. doi: 10.1001/jama.290.17.2292. [DOI] [PubMed] [Google Scholar]
- 62.Calabresi L, Tedeschi G, Treu C, Ronchi S, Galbiati D, Airoldi S, Sirtori CR, Marcel Y, Franceschini G. Limited proteolysis of a disulfide-linked apoA-I dimer in reconstituted HDL. J Lipid Res. 2001;42:935–942. [PubMed] [Google Scholar]
- 63.Favari E, Gomaraschi M, Zanotti I, Bernini F, Lee-Rueckert M, Kovanen PT, Sirtori CR, Franceschini G, Calabresi L. A unique protease-sensitive high density lipoprotein particle containing the apolipoprotein A-I(Milano) dimer effectively promotes ATP-binding cassette A1-mediated cell cholesterol efflux. J Biol Chem. 2007;282:5125–5132. doi: 10.1074/jbc.M609336200. doi: 10.1074/jbc.M609336200. [DOI] [PubMed] [Google Scholar]