

Abstract
Changes in blood glucose concentration alter autonomic function in a manner consistent with altered neural activity in brain regions controlling digestive processes, including neurons in the brain stem nucleus tractus solitarii (NTS), which process viscerosensory information. With whole cell or on-cell patch-clamp recordings, responses to elevating glucose concentration from 2.5 to 15 mM were assessed in identified GABAergic NTS neurons in slices from transgenic mice that express EGFP in a subset of GABA neurons. Single-cell real-time RT-PCR was also performed to detect glutamic acid decarboxylase (GAD67) in recorded neurons. In most identified GABA neurons (73%), elevating glucose concentration from 2.5 to 15 mM resulted in either increased (40%) or decreased (33%) neuronal excitability, reflected by altered membrane potential and/or action potential firing. Effects on membrane potential were maintained when action potentials or fast synaptic inputs were blocked, suggesting direct glucose sensing by GABA neurons. Glucose-inhibited GABA neurons were found predominantly in the lateral NTS, whereas glucose-excited cells were mainly in the medial NTS, suggesting regional segregation of responses. Responses were prevented in the presence of glucosamine, a glucokinase (GCK) inhibitor. Depolarizing responses were prevented when KATP channel activity was blocked with tolbutamide. Whereas effects on synaptic input to identified GABAergic neurons were variable in GABA neurons, elevating glucose increased glutamate release subsequent to stimulation of tractus solitarius in unlabeled, unidentified neurons. These results indicate that GABAergic NTS neurons act as GCK-dependent glucose sensors in the vagal complex, providing a means of modulating central autonomic signals when glucose is elevated.
Keywords: brain stem, glucokinase, membrane potential, vagus nerve
glucose-sensing neurons are found in several brain regions associated with metabolic regulation, particularly in the hypothalamus and brain stem (Balfour et al. 2006; Briski et al. 2009; Dunn-Meynell et al. 2002; Oomura et al. 1969; Wan and Browning 2008; Yang et al. 1999). Included in this population are neurons of the brain stem dorsal vagal complex (DVC), which regulate parasympathetic processes driven by the vagus nerve, including digestive, cardiorespiratory, and metabolic functions (Laubie et al. 1983; Siemers et al. 1982; Suzuki et al. 1997; Travagli et al. 1991, 2006; Zhu et al. 2010; Zsombok and Smith 2009). Within the DVC, glucose sensing in the nucleus tractus solitarii (NTS) participates in the regulation of feeding and blood glucose (Lam et al. 2010; Ritter et al. 1981; Ritter et al. 2000). Glucose can also potentiate vagally mediated input to the NTS (Wan and Browning 2008), implying that visceral afferent signaling to the brain may be modulated in the context of plasma glucose levels. Furthermore, hepatic gluconeogenesis relies on central regulatory control that is mediated by the vagus nerve, and in vivo studies support a critical role of the DVC in the homeostatic control of blood glucose and autonomic function (Lam et al. 2010; Obici et al. 2002; Pocai et al. 2005a, 2005b; Ritter et al. 1981; Ritter et al. 2000; Sakaguchi and Shimojo 1984). Glucose concentration in the brain changes in a narrow range under normal physiological conditions but can change drastically in metabolic disorders (De Vries et al. 2003; Starr et al. 2003). Areas outside the blood-brain barrier, including the NTS (Merchenthaler 1991), may be particularly sensitive to dynamic changes in plasma glucose concentration. Since changes in blood glucose concentration result in altered gastric and other visceral functions, a sensitive and flexible glucose-sensing mechanism in the DVC may underlie central responses to altered glucose levels.
It has long been known that subsets of NTS neurons, in particular those in caudal parts of the nucleus, respond to varying glucose concentrations by altering their action potential (AP) firing (Mizuno and Oomura 1984). Acute hypoglycemia can increase or decrease membrane potential in small subsets of DVC neurons, effects that probably involve a glucokinase (GCK)-dependent modulation of ATP-sensitive potassium (KATP) channels (Balfour et al. 2006; Wan and Browning 2008). Although only a subset of NTS neurons are glucose sensitive, visceral responsiveness to altered glucose concentration in the NTS can have profound effects on visceral motor activity. Application, for example, of high glucose concentrations (15–30 mM) to the DVC rapidly increases intragastric pressure and inhibits gastric motility in vivo in rats, and motor neurons in the dorsal motor nucleus of the vagus (DMV) undergo a commensurate KATP- and Cl−-dependent hyperpolarization when similarly high glucose concentrations are applied in vitro (Ferreira et al. 2001). Inhibitory, GABAergic NTS neurons make extensive synaptic connections with gastric-related DMV neurons (Davis et al. 2004; Travagli et al. 1991), and GABA release in the DMV potently influences vagal motor activity (Gao and Smith 2010; Travagli et al. 2006). Because the inhibitory effect of glucose on motor neuron activity appears to involve glucose-dependent modulation of upstream, synaptically connected inhibitory neurons, we tested the hypothesis that glucose directly alters the membrane potential of identified GABA neurons in the NTS, correlating expression of glutamic acid decarboxylase (GAD67) with electrical responses to elevated glucose concentrations in mice.
MATERIALS AND METHODS
Animals.
Male and female transgenic mice expressing enhanced green fluorescent protein (EGFP) in the subpopulation of GABAergic neurons that coexpress somatostatin (i.e., GIN mice) (Glatzer et al. 2007; Oliva et al. 2000) were used in this study. Mice were bred in house from homozygous founder mice [FVB-Tg (GadGFP) 4570Swn/J; The Jackson Laboratories, Bar Harbor, ME], and the colony was maintained under a normal 14:10-h light-dark cycle. Water and food were available ad libitum. All procedures conformed to National Institutes of Health guidelines and were approved by the University of Kentucky Animal Care and Use Committee.
Brain slice preparation.
On-cell and whole cell patch-clamp recordings were made with brain stem slices prepared from male and female GIN mice, 3–8 wk of age. Animals were deeply anesthetized by isoflurane inhalation (IsoThesia; Henry Schein, Melville, NY) and decapitated while anesthetized. The brain was rapidly removed and blocked to isolate the brain stem and then glued to a sectioning stage. Transverse (coronal) brain stem slices (300 μm) containing the caudal NTS near the level of the area postrema (i.e., ±600 μm rostral and caudal to area postrema) were made in ice-cold, oxygenated (95% O2-5% CO2) artificial cerebrospinal fluid (ACSF) with a vibrating microtome (Vibratome Series 1000; Technical Products, St. Louis, MO). The ACSF contained (in mM) 124 NaCl, 3 KCl, 2 CaCl2, 1.3 MgCl2, 1.4 NaH2PO4, 26 NaHCO3, and 2.5 glucose (pH 7.15–7.3); osmolality was adjusted to 290–310 mosmol/kgH2O with sucrose. High-glucose ACSF (15 mM) was made by equimolar substitution of NaCl or sucrose. Slices were incubated for an equilibration period for ≥1 h in warmed (30–33°C), oxygenated ACSF prior to recording.
Electrophysiology.
A single brain slice was transferred to a recording chamber mounted on a fixed stage under an upright microscope (BX51WI; Olympus, Melville, NY), where it was continually perfused by warmed (30–33°C), oxygenated ACSF. EGFP-labeled NTS neurons were targeted for recording under a ×40 water-immersion objective with fluorescence and infrared-differential interference contrast (IR-DIC) optics, as previously described (Gao et al. 2009; Glatzer et al. 2007; Glatzer and Smith 2005; Williams and Smith 2006). For recordings from EGFP-labeled NTS neurons, initial visualization was made briefly under epifluorescence with a fluorescein isothiocyanate (FITC) filter set (excitation filter wavelengths: 450–490 nm).
On-cell and whole cell patch-clamp recordings were obtained in the NTS with pipettes pulled from borosilicate glass (Garner Glass, Claremont, CA; open tip resistance 4–6 MΩ) with a Sutter P-87 horizontal puller (Sutter Instrument, Novato, CA). The location of each recorded cell was noted and recovered by staining for biocytin (0.2% added to internal recording solution) (Horikawa and Armstrong 1988). Pipettes were filled with a solution containing (in mM) 130 K+-gluconate (or Cs+-gluconate), 1 NaCl, 5 EGTA, 10 HEPES, 1 MgCl2, 1 CaCl2, 3 KOH (or CsOH), and 2–4 ATP (pH 7.15–7.3).
Electrophysiological signals were recorded with a MultiClamp 700B amplifier (Molecular Devices, Sunnyvale, CA), low-pass filtered at 3 kHz, and recorded onto a PC-style computer (Digidata 1440A, Molecular Devices) with pCLAMP 10.2 software (Molecular Devices). Series resistance was monitored throughout the recordings, and data were used for analysis if the series resistance remained <30 MΩ (mean = 17.0 ± 0.7 MΩ) and changed by ≤20% during the recording. Once a recording was obtained, cells were allowed to equilibrate for ∼10 min before the solution with a high glucose concentration (15 mM) was bath applied for 10 min before return to 2.5 mM glucose solution (washout).
Resting membrane potential was measured in current-clamp mode by directly measuring the mean membrane potential over a 30-s period. In neurons with spontaneous APs, membrane potential was measured over 30 s by averaging periods between spikes. Membrane potential was corrected for liquid junction potential post hoc (−8 mV). Resting potential was determined by monitoring the voltage at which no current was injected (I = 0). Electrical stimulation of primary afferent input was made with a concentric bipolar stimulating electrode (125-μm diameter; FHC; Bowdoinham, ME) placed over the tractus solitarius (TS). Evoked excitatory postsynaptic currents (eEPSCs), spontaneous EPSCs (sEPSCs), and tetrodotoxin (TTX)-resistant (i.e., miniature) EPSCs (mEPSCs) were examined at a holding potential of −65 mV, and inhibitory postsynaptic currents [IPSCs; spontaneous (s)IPSCs and miniature (m)IPSCs] were examined at 0 mV with pipettes containing Cs-gluconate to block K+ currents, thereby improving voltage control and reducing noise.
All drugs were bath applied until a steady state was reached (∼10 min). Drugs used included the GCK inhibitor glucosamine (5 μM), the KATP channel blocker tolbutamide (200 μM), the NMDA receptor antagonist AP5 (50 μM), the AMPA/KA receptor antagonist CNQX (10 μM), the GABAA receptor blocker picrotoxin (100 μM), and the Na+ channel blocker TTX (1 μM). AP5, CNQX, and picrotoxin were received from Sigma-Aldrich (St. Louis, MO). Tolbutamide was received from R&D Systems (Minneapolis, MN). TTX was received from Alomone Labs (Jerusalem, Israel). Glucosamine was received from MP Biomedical (Santa Ana, CA).
Histology.
After recording, slices were fixed with 4% paraformaldehyde in 0.15 M sodium phosphate buffer overnight at 4°C (pH 7.4). After three rinses with 0.01 M phosphate-buffered saline (PBS), slices were immersed in avidin conjugated to Texas red (1:400; Vector Laboratories, Burlingame, CA) in PBS containing 0.5% Triton X-100 and incubated for 4 h at room temperature to identify biocytin-filled neurons. Slices were then rinsed three times with PBS, mounted on glass slides, and coverslipped in Vectashield (Vector Laboratories) to reduce photooxidation during visualization. Cells labeled with biocytin during a recording and/or with EGFP were identified with an Olympus BX40 microscope, and images were captured with a Spot RT camera (Diagnostic Instruments, Sterling Heights, MI) using filters for the two fluorescent dyes (Fig. 1).
Fig. 1.
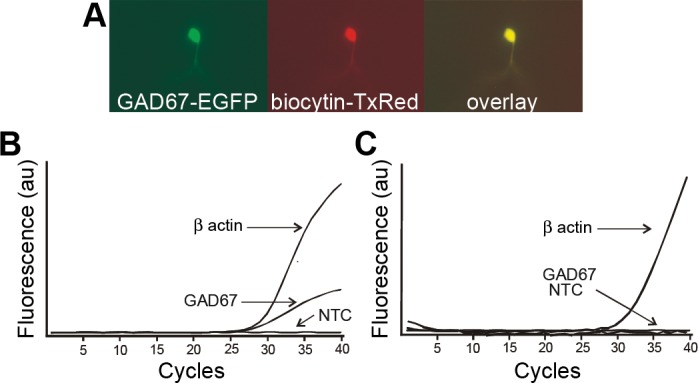
Identification of GABAergic neurons in the nucleus tractus solitarii (NTS). A: visual identification of an enhanced green fluorescent protein (EGFP)-labeled GABA neuron (GAD67-EGFP) in the NTS under a FITC filter set. The same neuron was labeled with avidin-Texas red (TX-Red) subsequent to being filled with biocytin during patch-clamp recording. Overlay of the images confirms that the EGFP-labeled cell was recorded and filled with biocytin. B: representative amplification curves from single-cell, real-time RT-PCR of the contents of an EGFP-expressing neuron, showing exponential increase in β-actin and GAD67 amplicons. C: representative amplification curves from a recorded NTS neuron that did not express EGFP. The neuron expressed β-actin but not GAD67. NTC, no-template control; au, arbitrary units.
Single-cell RT-PCR.
To ensure that the EGFP-expressing neurons were GABAergic, single-cell RT-PCR was performed after electrophysiological recording. The cell contents of the patch-clamp recording pipette were immediately harvested into reverse transcription (RT) reaction mixture after recordings. The RT mixture was comprised of 1× reaction buffer, 200 nM deoxynucleotide triphosphate (dNTP) mix, 40 U of Optizyme Ribonuclease Inhibitor, 200 U of Moloney murine leukemia virus reverse transcriptase (all from Fisher Scientific, Hampton, NH), and 2.5 μM random nonamers (Sigma-Aldrich) in a total volume of 15 μl. The RT mix was prepared without enzymes and stored at 4°C throughout the electrophysiological experiments. Once the cell content was harvested, samples were stored at −80°C until cDNA was created. Only at this point (the creation of cDNA) were the enzymes added to the RT mix, to avoid their degradation. The RT reaction was incubated at 42°C for at least 90 min, followed by inactivation of the enzyme at 95°C for 5 min.
The oligonucleotides used for PCR targeted β-actin (verifying the presence of constituent mRNA) and GAD67 (identifying GABAergic cells) and are shown in Table 1. Cells were required to be positive for β-actin expression in order to be considered for analysis. Twelve microliters of PCR-mix containing 1× reaction buffer, 300 μM dNTP mix, 2.5 mM MgCl2, each primer-probe set at 400 nM, and 2.5 U of Taq DNA polymerase (all from Sigma-Aldrich) was combined with 3 μl of cDNA. Reaction mixtures were placed into an ABI 7500 real-time PCR system (Applied Biosystems, Foster City, CA) at 95°C for 2 min and cycled 40 times through 95°C for 20 s, 60°C for 20 s, and 72°C for 10 s. Fluorescence was monitored during the annealing step of each cycle. β-Actin and GAD67 were tested on five putative motor neurons from the DMV; all were positive for β-actin and none for GAD67. Control samples were run that contained no cDNA (NTC) (Fig. 1).
Table 1.
Sequences of oligonucleotides
| Oligonucleotides | |
|---|---|
| Mouse β-actin fwd | 5′ GGAGAGCATAGCCCTCGTAG 3′ |
| Mouse β-actin rev | 5′ GCCATGTACGTAGCCATCC 3′ |
| Mouse β-actin probe | 5′ HEX-CTGGTCGTACCACAGGCATTGTG-BHQ1 3′ |
| Mouse GAD67 fwd | 5′ CCGTTCTTAGCTGGAAGCAG 3′ |
| Mouse GAD67 rev | 5′ GTCTTGTGAGCGCCTTCAG 3′ |
| Mouse GAD67 probe | 5′ TX RED-CCGGCGCACAGAGACCGACTTCT-BHQ2 3′ |
NCBI reference sequences used for primer-probe design: NM_007393.2 (β-actin), NM_008077.3 (GAD67), NM_010292.4 (GCK).
Data analysis.
Recordings were analyzed with Clampfit 10.2 (Molecular Devices) and Mini Analysis 6.0.7 (Synaptosoft, Decatur, GA) software. Changes in membrane potential were considered significant if a change of at least ±3 mV was detected. For analysis of postsynaptic currents, 2 min of continuous recording under each condition was used to assess mean EPSC or IPSC frequency, amplitude, 10–90% rise time, and decay time constant for each glucose concentration. The Kolmogorov-Smirnov (K-S) intra-assay test was used to determine statistical significance of changes in EPSC or IPSC frequency within a recording. Only eEPSC responses that were of relatively constant latency (total interstimulus latency range < 0.5 ms) were assessed. Amplitude ratios of eEPSC responses (eEPSC2/eEPSC1) to paired-pulse stimulation of the TS (30-ms pairing) were determined by measuring means of at least 10 responses. A repeated-measures ANOVA with Tukey's post hoc test was used to determine whether neurons recovered from the effects of glucose on membrane potential. A Mann-Whitney U-test was used to determine whether location predicted neuron response. A Wilcoxon matched-pairs signed-rank test was used to examine significance for paired-pulse ratios. For all other data, a paired, two-tailed Student's t-test was used to determine statistical significance (Graphpad Prism, San Diego, CA). Statistical significance for all measures was set at P < 0.05.
RESULTS
Identification of recorded NTS neurons.
Membrane potential and AP firing recordings were made from a total of 45 NTS neurons, identified by their expression of EGFP in slices from 30 mice. When possible, neurons were also recovered with biocytin to ensure colocalizaton of EGFP with the biocytin injected intracellularly during recording (Fig. 1A). In addition to EGFP localization, phenotypic identity of NTS neurons recorded from slices in five animals was assessed by using single-cell quantitative RT-PCR to confirm the expression of GAD67 in EGFP-expressing neurons (n = 12; Fig. 1B). In all but one case, GAD67 expression was confirmed. Non-EGFP-expressing neurons (n = 19) were also tested to ensure that negative expression values could be attained. In these cases, 10 cells were positive for β-actin but negative for GAD67 (Fig. 1C) and 9 unlabeled (i.e., no EGFP expression) neurons contained GAD67 transcript. A GABAergic phenotype was confirmed in EGFP-expressing neurons and also in some neurons that did not express EGFP, consistent with previous findings in this mouse strain (Oliva et al. 2000).
Effects of 15 mM glucose on membrane potential of GAD67-expressing neurons.
In 35 EGFP-expressing neurons, the effect of elevating glucose concentration from 2.5 mM to 15 mM on membrane potential and firing frequency was examined with either cell-attached (i.e., on cell; n = 5) or whole cell (n = 30) recordings. Three types of neuronal response were identified. In some neurons (12/30 cells; 40%), the resting membrane potential was depolarized by glucose (Fig. 2). Elevating glucose concentration depolarized the resting membrane potential in these neurons by 9.0 ± 2.2 mV, from a mean of −54.0 ± 3.4 mV in 2.5 mM glucose to −45.0 ± 3.2 mV in 15 mM glucose (n = 12; P = 0.002; Fig. 2C). These effects were reversed on reinstatement of 2.5 mM glucose (i.e., washout; n = 8). In neurons where APs were present, the elevated glucose resulted in a significant increase in AP firing, including in seven cells recorded in whole cell configuration and three cells recorded in on-cell configuration (0.7 ± 0.3 in 2.5 mM glucose vs. 1.2 ± 0.3 Hz in 15 mM glucose; n = 10; P = 0.002). The membrane potential in a second population of neurons (10/30; 33%) hyperpolarized after increasing glucose concentration (−6.2 ± 0.7 mV; Fig. 3). These neurons were hyperpolarized from a resting membrane potential of −53.1 ± 4.8 mV in 2.5 mM glucose to −59.4 ± 5.1 mV in 15 mM glucose (n = 10; P = 0.000007; Fig. 3C). These effects also reversed on return to 2.5 mM glucose in all neurons examined (n = 8). In neurons where APs were present, including on-cell recordings, elevating glucose concentration resulted in a significant decrease in AP firing (2.6 ± 1.4 vs. 2.0 ± 1.3 Hz; n = 8; P = 0.002). Although baseline AP frequency (in 2.5 mM glucose) was numerically lower in glucose-excited versus glucose-inhibited neurons (0.7 ± 0.3 vs. 2.6 ± 1.4 Hz), there was no statistical difference between these groups (P > 0.05). A third group of neurons (27%) did not significantly respond to the elevation of glucose. Resting membrane potential in these neurons was −45.6 ± 5.6 mV in 2.5 mM glucose and −46.0 ± 5.7 mV in 15 mM glucose (n = 8; P = 0.4). Most identified GABAergic neurons in the NTS were therefore either excited or inhibited by elevated glucose concentration, with a minority of cells being unresponsive.
Fig. 2.

Membrane responses of glucose-excited neurons. A: action potential (AP) activity at resting membrane potential in an EGFP-expressing glucose-excited neuron. Left: 2.5 mM glucose. Center: 15 mM glucose. Right: 15 min after return to 2.5 mM glucose (15-min wash). B: mean and SE of AP firing rate for EGFP-labeled neurons in 2.5 and 15 mM glucose (n = 10). C: resting membrane potential of EGFP-labeled, glucose-excited neurons in 2.5 mM and 15 mM glucose (n = 12) in normal recording conditions (nACSF) and when AP-dependent activity was blocked with tetrodotoxin (TTX). Asterisks in B and C indicate significant difference from controls (paired t-test; P < 0.05).
Fig. 3.

Membrane responses of glucose-inhibited neurons. A: AP activity at resting membrane potential in an EGFP-expressing glucose-inhibited neuron. Left: 2.5 mM glucose. Center: 15 mM glucose. Right: 15 min after return to 2.5 mM glucose (Wash). B: AP firing rate for EGFP-labeled neurons in 2.5 and 15 mM glucose (n = 8). C: resting membrane potential of EGFP-labeled, glucose-inhibited neurons in 2.5 mM and 15 mM glucose (n = 10) under normal recording conditions (nACSF) and in the presence of TTX. Asterisks in B and C indicate significant difference from controls (paired t-test; P < 0.05).
Effects of 15 mM glucose on EGFP-expressing neurons are postsynaptic.
To determine whether the changes in membrane potential in EGFP-labeled neurons in the NTS required synaptic input or AP firing, TTX was added to the ACSF to block APs and AP-dependent synaptic activity. In the presence of TTX (1 μM), 11 of 15 neurons responded to the elevated glucose concentration. These included five neurons (33%) that depolarized (6.6 ± 1.1 mV; P = 0.002; Fig. 2C) and six neurons (40%) that were hyperpolarized (−9.4 ± 2.8 mV; P = 0.002; Fig. 3C). There was no difference in resting membrane potential (P = 0.2) between neurons that were depolarized or hyperpolarized by increased glucose concentration. In all neurons examined during washout, the effects of elevated glucose levels were reversed after 10-min reinstatement of ACSF containing 2.5 mM glucose. The effects on membrane potential of elevated glucose concentration therefore did not require AP-dependent neurotransmission.
Effects of elevating glucose were determined in another group of neurons in the presence of the ionotropic glutamate and GABA receptor antagonists AP5 (50 μM), CNQX (10 μM), and picrotoxin (100 μM), which block all fast neurotransmission in the NTS (Smith et al. 1998). In the presence of these antagonists, elevating glucose depolarized the membrane potential of 5 of 14 neurons (36%; 11.1 ± 5.4 mV; Fig. 4). In 4 of 14 neurons (28%), elevating glucose concentration hyperpolarized the membrane potential by 5.4 ± 0.9 mV. There was no effect of glucose elevation in the remaining five neurons. The effect of elevating glucose concentration therefore did not require glucose modulation of synaptic transmission and appeared to involve direct effects on the recorded GABA neurons.
Fig. 4.

Graphical summary of membrane responses to elevated glucose: % of neurons that responded to elevated glucose concentration in normal recording conditions (nACSF; n = 22), when APs were blocked with TTX (n = 15), when fast synaptic receptor activity was blocked with NMDA, AMPA/KA, and GABAA receptor antagonists [AP5, CNQX, and picrotoxin (PTX), respectively; n = 14], in the presence of the glucokinase blocker glucosamine (GA; n = 8), or in the presence of the KATP channel blocker tolbutamide (TOL; n = 7).
Spatial distribution of neurons responding to increased glucose concentration.
Figure 5 shows an example of the typical distribution of EGFP-expressing neurons within the DVC and responses of individual neurons to increased glucose concentration. There was a statistically significant topography to the distribution of cellular responses in NTS GABA neurons (Fig. 5B; Mann-Whitney, P = 0.04). The medial NTS (mNTS) contained more neurons that responded to elevated glucose with a depolarization/increase in AP firing (60%) compared with the lateral NTS (lNTS; 20%). Conversely, the lNTS contained more neurons that responded with a hyperpolarization/decrease in AP firing (60%) compared with the mNTS (23%). With some exceptions, mNTS neurons tended to be depolarized, whereas lNTS neurons tended to be hyperpolarized, with increased glucose concentration.
Fig. 5.
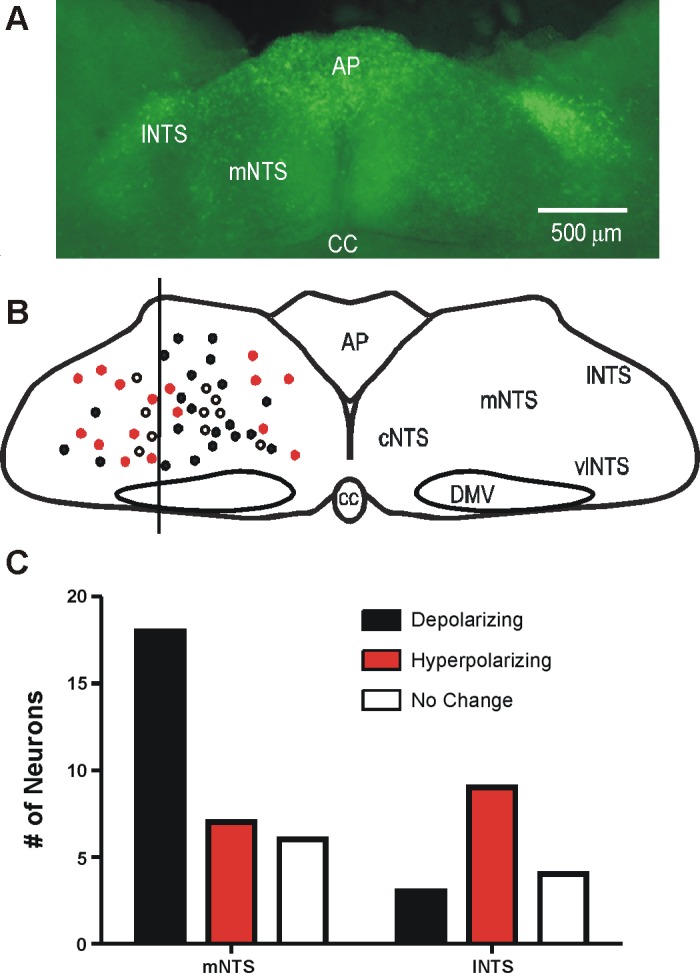
Distribution of EGFP-expressing GABAergic neurons in the dorsal vagal complex (DVC). A: image of the DVC (300-μm slice) at the level of the area postrema illustrates the distribution of EGFP-labeled neurons in the DVC. B: distribution of glucose-sensitive neurons recorded for this study (n = 45). Filled circles mark the position of glucose-excited GABA neurons; open circles indicate glucose-inhibited GABA neurons; red circles mark neurons with no response to glucose. C: plot of the number of neurons responding as a function of their relative location in the NTS. AP, area postrema; CC, central canal; cNTS, mNTS, lNTS, and vNTS: central, medial, lateral, and ventral NTS; DMV, dorsal motor nucleus of the vagus.
Effect of glucokinase inhibition.
To confirm GCK's role in the ability of EGFP-expressing NTS neurons to respond to glucose concentration change, slices were pretreated with glucosamine (5 mM), a GCK inhibitor, prior to elevation of glucose concentration to 15 mM. In the presence of glucosamine, the membrane potential was unchanged by increasing glucose concentration in seven of eight cells (−0.7 ± 0.5 mV; n = 8; P = 0.2; Fig. 4). One neuron was mildly hyperpolarized (−3 mV) in the presence of elevated glucose. Thus blocking GCK activity prevented the effects of increased glucose concentration on membrane potential, consistent with previous reports demonstrating GCK expression in glucose-responsive neurons of the vagal complex (Balfour et al. 2006).
Effects of KATP channel blockade.
To determine the involvement of KATP channels on the effects of elevated glucose, tolbutamide (200 μM) was preapplied before glucose was elevated to 15 mM (n = 7). In most of these neurons (n = 5; 71%), membrane potential was unchanged when glucose concentration was increased (−0.6 ± 0.6 mV change; P > 0.05). The two (29%) remaining neurons responded to the increase in glucose concentration with a hyperpolarization (−5.35 ± 1.56 mV); no neurons were depolarized. Thus tolbutamide prevented the depolarizing effects of elevated glucose concentration on membrane potential (Fig. 4), suggesting that blocking KATP channels prevented the depolarization, consistent with previous findings in unidentified neurons (Balfour et al. 2006).
EPSCs evoked from tractus solitarius.
Most GABAergic NTS neurons receive input from primary viscerosensory afferents (Bailey et al. 2008; Glatzer et al. 2007), and glucose alters glutamate release from viscerosensory afferents in a subset of unidentified NTS neurons (Wan and Browning 2008). The effect of elevating glucose concentration on primary viscerosensory afferent input to GABAergic neurons was determined. Paired electrical stimulation of the TS resulted in eEPSCs in EGFP-labeled and unlabeled NTS neurons. In all neurons, poststimulus latency of the eEPSC was relatively constant (total interresponse latency variability < 500 μs); the amplitude of the second eEPSC was smaller than the first, typical of putative second-order viscerosensory neurons (Andresen and Mendelowitz 1996; Doyle and Andresen 2001; Glatzer et al. 2007; Smith et al. 1998). The amplitude of the first TS-eEPSC (eEPSC1) was unchanged by elevated glucose concentrations in identified GABAergic NTS neurons (−104 ± 14 pA in 2.5 mM glucose vs. −92 ± 9 pA in 15 mM glucose; P > 0.05; n = 5; Fig. 6). Paired stimulation of the TS at a pairing interval of 30 ms resulted in a paired-pulse amplitude response ratio (PPr) of 0.63 ± 0.09 in the presence of 2.5 mM glucose and 0.79 ± 0.11 in the presence of 15 mM glucose (P > 0.05; n = 5). Although the overall ratio was unchanged, the PPr was increased by >15% (n = 4; P < 0.05) or decreased (n = 1) in individual EGFP-labeled neurons. In those neurons with increased PPr, the ratio change was due to a significant increase in the amplitude of the second eEPSC (eEPSC2; P < 0.05).
Fig. 6.

Glucose modulation of evoked excitatory postsynaptic currents (eEPSCs) in EGFP-expressing and non-EGFP-expressing NTS neurons. A: representative traces showing responses to paired tractus solitarius (TS) stimulation in an EGFP-labeled mNTS neuron. B: responses to paired TS stimulation in a non-EGFP-labeled neuron. TS stimulation resulted in EPSCs with relatively constant latency to the stimulus. C: increasing glucose concentration from 2.5 to 15 mM did not significantly alter the amplitude of the first eEPSC (eEPSC1) in EGFP-labeled neurons (n = 5; P > 0.05), whereas the eEPSC1 amplitude was significantly increased in nonlabeled neurons (n = 6; *P < 0.05). D: no significant changes were seen in the paired-pulse ratio (PPr) across the population of EGFP-labeled neurons, although changes in individual recordings could be measured.
In unidentified, non-EGFP-labeled NTS neurons, elevating glucose concentration from 2.5 to 15 mM resulted in an increase in the amplitude of eEPSC1 in all neurons from this group, from −196 ± 36 to −248 ± 42 pA (P < 0.05; n = 6; Fig. 6). Paired-pulse stimulation of TS resulted in a PPr of 0.34 ± 0.04 in 2.5 mM glucose and 0.38 ± 0.03 in 15 mM glucose (P > 0.05; n = 6; Fig. 6), with the PPr being increased by ≥15% (n = 3), decreased (n = 1), or unchanged (n = 2) in unidentified NTS neurons. Whereas glutamate release subsequent to TS stimulation was increased in unidentified NTS neurons, consistent with a previous report (Wan and Browning 2008), TS-evoked EPSCs were not consistently increased in identified GABAergic neurons, although an effect on PPr due to increased amplitude of eEPSC2 was often observed.
Effects of elevated glucose concentration on spontaneous neurotransmitter release.
Figure 7 illustrates the effects of elevating glucose concentration on sEPSCs, mEPSCs, sIPSCs, and mIPSCs. Elevating glucose concentration was without significant effect on sEPSC frequency (P = 0.3) or amplitude (P = 0.09) in eight EGFP-expressing NTS neurons. Significant within-cell effects on sEPSC frequency were observed, however, in the majority of neurons (P < 0.02; K-S test; Fig. 7); the direction of change (i.e., increase or decrease) did not correlate with effects on whole cell current or membrane potential. In the presence of TTX (1 μM, n = 7), mEPSC frequency and amplitude were also unaffected overall (P > 0.05) but could be decreased, increased, or unchanged in individual EGFP-labeled neurons, independent of any effect on membrane potential or whole cell current change. Results from analysis of sIPSCs and mIPSCs similarly indicated that increasing glucose concentration resulted in no overall effect on frequency or amplitude of sIPSCs (n = 7; P > 0.05 for both measures) or mIPSCs (n = 6; P > 0.05 for both measures), but increasing glucose concentration altered either or both measurements within individual neurons (P < 0.02; K-S test; Fig. 7). Thus effects on EPSCs and IPSCs were observed in most neurons, but generalizable effects on synaptic event frequency or amplitude were not detected in identified GABAergic NTS neurons.
Fig. 7.

Modulation of synaptic input to EGFP-expressing NTS neurons. A: representative traces of miniature (m)EPSCs recorded in a GABAergic NTS neuron in the presence of 2.5 and 15 mM glucose. B: no differences were observed in mean spontaneous (s)EPSC (n = 8) or mEPSC (TTX; n = 7) frequency. C: glucose-induced changes in mean spontaneous (sIPSC) or miniature (mIPSC) inhibitory postsynaptic current amplitude were also not detected. D: graphical representation of significant changes in mEPSC frequency in individual neurons, determined by the Kolmogorov-Smirnov test (K-S test; P < 0.02). E: representative trace of mIPSCs recorded in a GABAergic NTS neuron in the presence of 2.5 and 15 mM glucose. F and G: no differences in mean frequency (F) or amplitude (G) were observed for either sIPSCs (n = 7) or mIPSCs (n = 6). H: graphical representation of significant changes in mEPSC frequency in individual neurons (K-S test; P < 0.02). No significant mean changes were detected, but synaptic input to most individual neurons was significantly enhanced or inhibited by increased glucose concentration.
DISCUSSION
Here, single-cell real-time TaqMan PCR, a highly sensitive and specific means of detecting mRNA content (Bustin and Mueller 2005; Mackay et al. 2002), was used in concert with EGFP expression in transgenic mice to verify the presence of GAD67 in recorded NTS neurons and identify their GABAergic phenotype. Elevating glucose concentration from 2.5 to 15 mM resulted in altered membrane responses in 73% of identified GABAergic neurons in the NTS under normal recording conditions. The response could be either excitatory or inhibitory (i.e., depolarizing or hyperpolarizing) and was accompanied by either increased or decreased AP frequency. The change in membrane potential was retained in the presence of TTX, which blocked AP-dependent neurotransmitter release, or a mixture of ionotropic glutamate and GABA receptor antagonists, indicating that the effect occurred in the absence of any change in fast, ionotropic receptor-mediated synaptic neurotransmission. Previous reports have described glucose-induced changes in membrane potential or AP firing in only a minority (20–40%) of unidentified NTS neurons in vitro (Balfour et al. 2006; Mizuno and Oomura 1984; Wan and Browning 2008). Although responses in other neuronal phenotypes remain possible, the large percentage of glucose-responsive GABAergic neurons implies that changes in glucose concentration in the NTS potently affect inhibitory, GABAergic neurons.
In pancreatic beta cells, the GCK enzyme (hexokinase IV) facilitates the phosphorylation of intracellular glucose and other hexoses (Kang et al. 2004). During the process, ATP is released, which can influence membrane potential via ubiquitous KATP channels. Expression of GCK can thus mediate neuronal responses to glucose that affect membrane electrical activity (Briski et al. 2009; Dunn-Meynell et al. 2002), including in the DVC (Balfour et al. 2006; Ferreira et al. 2001), where closure of the KATP channels and consequent membrane depolarization is the assumed mechanism underlying the response of glucose-excited neurons. Excitatory effects of elevated glucose in the present study were prevented by the KATP channel blocker tolbutamide, consistent with a role for KATP channel-mediated depolarization in glucose-excited GABAergic NTS neurons. Although tolbutamide depolarized NTS neurons when intracellular ATP concentration was 2–4 mM (Williams and Smith 2006), it is possible that the intracellular ATP concentration used here (2 mM) diminished KATP channel-dependent effects of glucose in some neurons, potentially leading to an underestimation of glucose effects. Although both depolarizing and hyperpolarizing responses were sensitive to GCK inhibition, the hyperpolarization was not prevented by the KATP channel blocker, suggesting that a different mechanism may contribute to the response. Uncovering the mechanism of glucose-induced hyperpolarization will require further analysis, but modulation of BK channels has been hypothesized to be involved in other brain areas (Shanley et al. 2002).
In humans, fasting plasma glucose ≥ 7.0 mM (126 mg/dl) or 2-h plasma glucose ≥ 11.1 mM (200 mg/dl) is considered mild hyperglycemia but can be unnoticed, whereas when blood glucose levels rise to 15 mM (270 mg/dl) symptoms of hyperglycemia become apparent (Rehman et al. 2011). The glucose concentration used in the present study may reflect frank hyperglycemia, whereas concentrations typically used to sustain healthy slices in electrophysiological experiments (i.e., 10–11 mM) likely represent elevated, but not “diabetic,” conditions. Of note, the nonfasted blood glucose concentration in mice is relatively higher than in rats or humans (Bach et al. 2015; Yang et al. 1997; Zsombok et al. 2011). Our results suggest an important role for GCK in GABAergic NTS neurons when glucose levels are raised to hyperglycemic levels, and GCK is postulated to function optimally when the extracellular glucose level is in a range of 5–20 mM (Iynedjian 2009). Like area postrema, much of the NTS has abundant fenestrated capillaries and is considered to be largely outside the blood-brain barrier (Gross et al. 1990; Merchenthaler 1991). As such, it is possible that when plasma glucose increases, glucose levels are less well buffered in this region than in brain areas with an intact blood-brain barrier, where interstitial glucose concentration is relatively low (De Vries et al. 2003). Additionally, disease status with elevated glucose level can change the permeability of the blood-brain barrier, increasing direct access of NTS neurons to blood glucose (Starr et al. 2003). The physiological level of glucose in the cerebrospinal fluid is typically below 5 mM but increases quickly with elevated blood glucose levels, and it can be permanently high in metabolic disorders (Yang et al. 1999, 2004). Glucose-sensing GABAergic neurons in the NTS may act as direct glucose sensors in the brain, relaying information related to large elevations in glucose concentration to other metabolic control areas (Zsombok and Smith 2009). The present findings are consistent with longstanding evidence that glucose-sensing neurons in the NTS participate in the glucose-induced regulation of feeding and blood glucose concentration (Ritter et al. 1981; Ritter et al. 2000), and they support a prominent role of GABAergic NTS neurons in this response.
Both glucose-excited and glucose-inhibited neurons have been identified in the vagal complex (Balfour et al. 2006; Mizuno and Oomura 1984; Yettefti et al. 1995), but the subregional location of these responses has not been described previously, and a subset of GABA neurons was excited by hypoglycemia (Lamy et al. 2014). Here, identified GABA neurons could be either excited or inhibited by increased glucose concentration, and the direction of the response (i.e., excitatory or inhibitory) was loosely associated with the location of the cell within the nucleus, suggesting the possibility that different responses might be associated with viscerotopic organization. Acute hyperglycemia in the vagal complex affects the central components of gastric vagal reflexes by enhancing a Cl− current in gastric-projecting motor neurons in the DMV (Ferreira et al. 2001), consistent with the hypothesis that glucose excites GABAergic mNTS neurons with intact projections to the DMV. The present data support and extend these conclusions to identify mNTS GABA neurons as being directly depolarized by elevated glucose. In addition to projecting to gastric-related DMV cells (Ferreira et al. 2001; Glatzer et al. 2003), neurons in the mNTS are thought to receive primarily gastrointestinal and baroreceptor inputs, serving as central components of the vago-vagal reflex. Reflex responses seem likely to be modulated, therefore, in the context of glucose concentration effects on NTS neuron activity, independent of any effect of glucose on primary afferent terminals.
Neurons in the lNTS modulate cardiopulmonary functions, which can also be affected by changes in glucose concentration (Yeh et al. 2008), via connections with the caudal ventrolateral medulla (Kubo et al. 1985; Laubie et al. 1983; Siemers et al. 1982; Suzuki et al. 1997; Travers 1988). Neurons in the lNTS region also receive viscerosensory inputs from the esophagus, which participate in esophageal-gastric reflexes (Rogers et al. 2003). The predominantly hyperpolarizing response of lNTS neurons to glucose elevation might be expected to reduce inhibition of parasympathetic preganglionic vagal motor neurons controlling esophageal or cardiorespiratory functions, in concert with the increased inhibition of gastric-related DMV cells. Alternatively, selective activation of a subset of glucose-inhibited, GABAergic NTS neurons increased vagal firing rate (Lamy et al. 2014), possibly because of activation of local-circuit synapses within the NTS that regulate DMV neuron activity (Babic et al. 2011; Glatzer and Smith 2005). Identification of glucose effects in the context of regulation of specific viscera deserves further study. Activity of viscera-specific vagal circuitry seems likely to occur, however, in the context of glucose concentration.
The effect of elevating glucose on overall synaptic input to NTS neurons was inconsistent, being increased, decreased, or unchanged, and could reflect effects on intact local circuit synaptic connections within the NTS (Glatzer et al. 2007; Herman et al. 2009). A previous study found that acute glucose elevation increased glutamate release onto unidentified NTS neurons by acting on primary viscerosensory afferent terminals (Wan and Browning 2008), which we also observed. In identified GABA neurons, however, a similar glucose-induced increase in glutamate release was not observed after TS activation. An increase in PPr was detected in most GABA neurons, however, and this was due to increased amplitude of the second eEPSC, suggesting a form of short-term plasticity in the response. Glutamate released from primary viscerosensory afferents onto NTS neurons is typically reduced as the frequency of afferent firing increases (Andresen and Mendelowitz 1996; Doyle and Andresen 2001; Glatzer et al. 2007; Smith et al. 1998). Modulation of the amplitude of this frequency-dependent adaptation may be related to control of vagal reflexes, as opposed to ongoing primary afferent activity (Andresen et al. 2004). Primary viscerosensory input to GABAergic NTS neurons would thus be expected to be mainly affected by hyperglycemia under conditions of sustained afferent activation, but not necessarily with phasic viscerosensory afferent activation that occurs during brief activation of vagal afferents. Conversely, glucose may modulate vagal input to unidentified NTS neurons in a phasic, event-specific manner. The manner in which these apparently differential responses to glucose affect vagal reflex control of specific viscera remains to be determined. It is evident, however, that glucose regulation of GABAergic neurons can modulate both tonic and reflexive vagal activity through direct actions on the neurons as well as at primary afferent synaptic input.
Conclusions.
The present results suggest that most GABAergic neurons in the NTS are either excited or inhibited by elevating glucose concentration. Both responses were independent of synaptic input and required GCK activity. The depolarization occurs via a KATP channel-mediated mechanism, whereas the hyperpolarization did not appear to require KATP channels. mNTS GABA neurons, which presumably mediate glucose-responsive inhibitory connections between the NTS and gastric-projecting DMV neurons, were mainly depolarized, which could account for the inhibition of gastric motor neurons and altered gastric motility when glucose is elevated acutely in the DVC (Ferreira et al. 2001). Conversely, inhibition by glucose of GABAergic lNTS neurons likely to participate in cardiorespiratory regulation or esophageal-gastrointestinal coordination would tend to disinhibit downstream targets. Emerging data suggest the importance of a gastric-brain stem-hepatic circuit in regulating systemic glucose metabolism (Cheung et al. 2009; Lam et al. 2010; Rasmussen et al. 2012), which seems likely to involve glucose-sensing, GABAergic NTS neurons identified here. Glucose-responsive GABAergic neurons in the NTS are therefore situated to control multiple autonomic systems responsible for feeding, digestion, and metabolism in the context of plasma glucose levels.
GRANTS
This work was supported by National Institutes of Health Grants R01 DK-056132 and R21 HD-079256.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: C.R.B., P.G., and H.X. performed experiments; C.R.B., P.G., H.X., and B.N.S. analyzed data; C.R.B., P.G., and B.N.S. interpreted results of experiments; C.R.B., P.G., H.X., and B.N.S. prepared figures; C.R.B., P.G., and B.N.S. drafted manuscript; C.R.B. and B.N.S. edited and revised manuscript; C.R.B., P.G., H.X., and B.N.S. approved final version of manuscript; P.G. and B.N.S. conception and design of research.
REFERENCES
- Andresen MC, Doyle MW, Bailey TW, Jin YH. Differentiation of autonomic reflex control begins with cellular mechanisms at the first synapse within the nucleus tractus solitarius. Braz J Med Biol Res 37: 549–558, 2004. [DOI] [PubMed] [Google Scholar]
- Andresen MC, Mendelowitz D. Sensory afferent neurotransmission in caudal nucleus tractus solitarius—common denominators. Chem Senses 21: 387–395, 1996. [DOI] [PubMed] [Google Scholar]
- Babic T, Browning KN, Travagli RA. Differential organization of excitatory and inhibitory synapses within the rat dorsal vagal complex. Am J Physiol Gastrointest Liver Physiol 300: G21–G32, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach EC, Halmos KC, Smith BN. Enhanced NMDA receptor-mediated modulation of excitatory neurotransmission in the dorsal vagal complex of streptozotocin-treated, chronically hyperglycemic mice. PLoS One 10: e0121022, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TW, Appleyard SM, Jin YH, Andresen MC. Organization and properties of GABAergic neurons in solitary tract nucleus (NTS). J Neurophysiol 99: 1712–1722, 2008. [DOI] [PubMed] [Google Scholar]
- Balfour RH, Hansen AM, Trapp S. Neuronal responses to transient hypoglycaemia in the dorsal vagal complex of the rat brainstem. J Physiol 570: 469–484, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briski KP, Cherian AK, Genabai NK, Vavaiya KV. In situ coexpression of glucose and monocarboxylate transporter mRNAs in metabolic-sensitive caudal dorsal vagal complex catecholaminergic neurons: transcriptional reactivity to insulin-induced hypoglycemia and caudal hindbrain glucose or lactate repletion during insulin-induced hypoglycemia. Neuroscience 164: 1152–1160, 2009. [DOI] [PubMed] [Google Scholar]
- Bustin SA, Mueller R. Real-time reverse transcription PCR (qRT-PCR) and its potential use in clinical diagnosis. Clin Sci (Lond) 109: 365–379, 2005. [DOI] [PubMed] [Google Scholar]
- Cheung GW, Kokorovic A, Lam CK, Chari M, Lam TK. Intestinal cholecystokinin controls glucose production through a neuronal network. Cell Metab 10: 99–109, 2009. [DOI] [PubMed] [Google Scholar]
- Davis SF, Derbenev AV, Williams KW, Glatzer NR, Smith BN. Excitatory and inhibitory local circuit input to the rat dorsal motor nucleus of the vagus originating from the nucleus tractus solitarius. Brain Res 1017: 208–217, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vries MG, Arseneau LM, Lawson ME, Beverly JL. Extracellular glucose in rat ventromedial hypothalamus during acute and recurrent hypoglycemia. Diabetes 52: 2767–2773, 2003. [DOI] [PubMed] [Google Scholar]
- Doyle MW, Andresen MC. Reliability of monosynaptic sensory transmission in brain stem neurons in vitro. J Neurophysiol 85: 2213–2223, 2001. [DOI] [PubMed] [Google Scholar]
- Dunn-Meynell AA, Routh VH, Kang L, Gaspers L, Levin BE. Glucokinase is the likely mediator of glucosensing in both glucose-excited and glucose-inhibited central neurons. Diabetes 51: 2056–2065, 2002. [DOI] [PubMed] [Google Scholar]
- Ferreira M Jr, Browning KN, Sahibzada N, Verbalis JG, Gillis RA, Travagli RA. Glucose effects on gastric motility and tone evoked from the rat dorsal vagal complex. J Physiol 536: 141–152, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Glatzer NR, Williams KW, Derbenev AV, Liu D, Smith BN. Morphological and electrophysiological features of motor neurons and putative interneurons in the dorsal vagal complex of rats and mice. Brain Res 1291: 40–52, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Smith BN. Tonic GABAA receptor-mediated inhibition in the rat dorsal motor nucleus of the vagus. J Neurophysiol 103: 904–914, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatzer NR, Derbenev AV, Banfield BW, Smith BN. Endomorphin-1 modulates intrinsic inhibition in the dorsal vagal complex. J Neurophysiol 98: 1591–1599, 2007. [DOI] [PubMed] [Google Scholar]
- Glatzer NR, Hasney CP, Bhaskaran MD, Smith BN. Synaptic and morphologic properties in vitro of premotor rat nucleus tractus solitarius neurons labeled transneuronally from the stomach. J Comp Neurol 464: 525–539, 2003. [DOI] [PubMed] [Google Scholar]
- Glatzer NR, Smith BN. Modulation of synaptic transmission in the rat nucleus of the solitary tract by endomorphin-1. J Neurophysiol 93: 2530–2540, 2005. [DOI] [PubMed] [Google Scholar]
- Gross PM, Wall KM, Pang JJ, Shaver SW, Wainman DS. Microvascular specializations promoting rapid interstitial solute dispersion in nucleus tractus solitarius. Am J Physiol Regul Integr Comp Physiol 259: R1131–R1138, 1990. [DOI] [PubMed] [Google Scholar]
- Herman MA, Cruz MT, Sahibzada N, Verbalis J, Gillis RA. GABA signaling in the nucleus tractus solitarius sets the level of activity in dorsal motor nucleus of the vagus cholinergic neurons in the vagovagal circuit. Am J Physiol Gastrointest Liver Physiol 296: G101–G111, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikawa K, Armstrong WE. A versatile means of intracellular labeling: injection of biocytin and its detection with avidin conjugates. J Neurosci Methods 25: 1–11, 1988. [DOI] [PubMed] [Google Scholar]
- Iynedjian PB. Molecular physiology of mammalian glucokinase. Cell Mol Life Sci 66: 27–42, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang L, Routh VH, Kuzhikandathil EV, Gaspers LD, Levin BE. Physiological and molecular characteristics of rat hypothalamic ventromedial nucleus glucosensing neurons. Diabetes 53: 549–559, 2004. [DOI] [PubMed] [Google Scholar]
- Kubo T, Amano H, Misu Y. Caudal ventrolateral medulla. A region responsible for the mediation of vasopressin-induced pressor responses. Naunyn Schmiedebergs Arch Pharmacol 328: 365–372, 1985. [DOI] [PubMed] [Google Scholar]
- Lam CK, Chari M, Su BB, Cheung GW, Kokorovic A, Yang CS, Wang PY, Lai TY, Lam TK. Activation of N-methyl-d-aspartate (NMDA) receptors in the dorsal vagal complex lowers glucose production. J Biol Chem 285: 21913–21921, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamy CM, Sanno H, Labouebe G, Picard A, Magnan C, Chatton JY, Thorens B. Hypoglycemia-activated GLUT2 neurons of the nucleus tractus solitarius stimulate vagal activity and glucagon secretion. Cell Metab 19: 527–538, 2014. [DOI] [PubMed] [Google Scholar]
- Laubie M, Drouillat M, Schmitt H. Nucleus tractus solitarii respiratory neurons in the chemoreceptor pathway activated by almitrine. Eur J Pharmacol 93: 87–93, 1983. [DOI] [PubMed] [Google Scholar]
- Mackay IM, Arden KE, Nitsche A. Real-time PCR in virology. Nucleic Acids Res 30: 1292–1305, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchenthaler I. Neurons with access to the general circulation in the central nervous system of the rat: a retrograde tracing study with fluoro-gold. Neuroscience 44: 655–662, 1991. [DOI] [PubMed] [Google Scholar]
- Mizuno Y, Oomura Y. Glucose responding neurons in the nucleus tractus solitarius of the rat: in vitro study. Brain Res 307: 109–116, 1984. [DOI] [PubMed] [Google Scholar]
- Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med 8: 1376–1382, 2002. [DOI] [PubMed] [Google Scholar]
- Oliva AA Jr, Jiang M, Lam T, Smith KL, Swann JW. Novel hippocampal interneuronal subtypes identified using transgenic mice that express green fluorescent protein in GABAergic interneurons. J Neurosci 20: 3354–3368, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oomura Y, Ono T, Ooyama H, Wayner MJ. Glucose and osmosensitive neurones of the rat hypothalamus. Nature 222: 282–284, 1969. [DOI] [PubMed] [Google Scholar]
- Pocai A, Lam TK, Gutierrez-Juarez R, Obici S, Schwartz GJ, Bryan J, Aguilar-Bryan L, Rossetti L. Hypothalamic KATP channels control hepatic glucose production. Nature 434: 1026–1031, 2005a. [DOI] [PubMed] [Google Scholar]
- Pocai A, Obici S, Schwartz GJ, Rossetti L. A brain-liver circuit regulates glucose homeostasis. Cell Metab 1: 53–61, 2005b. [DOI] [PubMed] [Google Scholar]
- Rasmussen BA, Breen DM, Luo P, Cheung GW, Yang CS, Sun B, Kokorovic A, Rong W, Lam TK. Duodenal activation of cAMP-dependent protein kinase induces vagal afferent firing and lowers glucose production in rats. Gastroenterology 142: 834–843.e3, 2012. [DOI] [PubMed] [Google Scholar]
- Rehman A, Setter SM, Vue MH. Drug-induced glucose alterations. 2. Drug-induced hyperglycemia. Diabetes Spectrum 24: 234–238, 2011. [Google Scholar]
- Ritter R, Slusser PG, Stone S. Glucoreceptors controlling feeding and blood glucose: location in the hindbrain. Science 213: 451–452, 1981. [DOI] [PubMed] [Google Scholar]
- Ritter S, Dinh TT, Zhang Y. Localization of hindbrain glucoreceptive sites controlling food intake and blood glucose. Brain Res 856: 37–47, 2000. [DOI] [PubMed] [Google Scholar]
- Rogers RC, Travagli RA, Hermann GE. Noradrenergic neurons in the rat solitary nucleus participate in the esophageal-gastric relaxation reflex. Am J Physiol Regul Integr Comp Physiol 285: R479–R489, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi T, Shimojo E. Inhibition of gastric motility induced by hepatic portal injections of d-glucose and its anomers. J Physiol 351: 573–581, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanley LJ, Irving AJ, Rae MG, Ashford ML, Harvey J. Leptin inhibits rat hippocampal neurons via activation of large conductance calcium-activated K+ channels. Nat Neurosci 5: 299–300, 2002. [DOI] [PubMed] [Google Scholar]
- Siemers ER, Rea MA, Felten DL, Aprison MH. Distribution and uptake of glycine, glutamate and gamma-aminobutyric acid in the vagal nuclei and eight other regions of the rat medulla oblongata. Neurochem Res 7: 455–468, 1982. [DOI] [PubMed] [Google Scholar]
- Smith BN, Dou P, Barber WD, Dudek FE. Vagally evoked synaptic currents in the immature rat nucleus tractus solitarii in an intact in vitro preparation. J Physiol 512: 149–162, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr JM, Wardlaw J, Ferguson K, MacLullich A, Deary IJ, Marshall I. Increased blood-brain barrier permeability in type II diabetes demonstrated by gadolinium magnetic resonance imaging. J Neurol Neurosurg Psychiatry 74: 70–76, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Takayama K, Miura M. Distribution and projection of the medullary cardiovascular control neurons containing glutamate, glutamic acid decarboxylase, tyrosine hydroxylase and phenylethanolamine N-methyltransferase in rats. Neurosci Res 27: 9–19, 1997. [DOI] [PubMed] [Google Scholar]
- Travagli RA, Gillis RA, Rossiter CD, Vicini S. Glutamate and GABA-mediated synaptic currents in neurons of the rat dorsal motor nucleus of the vagus. Am J Physiol Gastrointest Liver Physiol 260: G531–G536, 1991. [DOI] [PubMed] [Google Scholar]
- Travagli RA, Hermann GE, Browning KN, Rogers RC. Brainstem circuits regulating gastric function. Annu Rev Physiol 68: 279–305, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers JB. Efferent projections from the anterior nucleus of the solitary tract of the hamster. Brain Res 457: 1–11, 1988. [DOI] [PubMed] [Google Scholar]
- Wan S, Browning KN. d-Glucose modulates synaptic transmission from the central terminals of vagal afferent fibers. Am J Physiol Gastrointest Liver Physiol 294: G757–G763, 2008. [DOI] [PubMed] [Google Scholar]
- Williams KW, Smith BN. Rapid inhibition of neural excitability in the nucleus tractus solitarii by leptin: implications for ingestive behaviour. J Physiol 573: 395–412, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Dickson BC, O'Hali W, Kearns H, Wright JR. Functional comparison of mouse, rat, and fish islet grafts transplanted into diabetic nude mice. Gen Comp Endocrinol 106: 384–388, 1997. [DOI] [PubMed] [Google Scholar]
- Yang XJ, Kow LM, Funabashi T, Mobbs CV. Hypothalamic glucose sensor: similarities to and differences from pancreatic beta-cell mechanisms. Diabetes 48: 1763–1772, 1999. [DOI] [PubMed] [Google Scholar]
- Yang XJ, Kow LM, Pfaff DW, Mobbs CV. Metabolic pathways that mediate inhibition of hypothalamic neurons by glucose. Diabetes 53: 67–73, 2004. [DOI] [PubMed] [Google Scholar]
- Yeh HC, Punjabi NM, Wang NY, Pankow JS, Duncan BB, Cox CE, Selvin E, Brancati FL. Cross-sectional and prospective study of lung function in adults with type 2 diabetes: the Atherosclerosis Risk in Communities (ARIC) study. Diabetes Care 31: 741–746, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yettefti K, Orsini JC, el Ouazzani T, Himmi T, Boyer A, Perrin J. Sensitivity of nucleus tractus solitarius neurons to induced moderate hyperglycemia, with special reference to catecholaminergic regions. J Auton Nerv Syst 51: 191–197, 1995. [DOI] [PubMed] [Google Scholar]
- Zhu W, Czyzyk D, Paranjape SA, Zhou L, Horblitt A, Szabo G, Seashore MR, Sherwin RS, Chan O. Glucose prevents the fall in ventromedial hypothalamic GABA that is required for full activation of glucose counterregulatory responses during hypoglycemia. Am J Physiol Endocrinol Metab 298: E971–E977, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zsombok A, Bhaskaran MD, Gao H, Derbenev AV, Smith BN. Functional plasticity of central TRPV1 receptors in brainstem dorsal vagal complex circuits of streptozotocin-treated hyperglycemic mice. J Neurosci 31: 14024–14031, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zsombok A, Smith BN. Plasticity of central autonomic neural circuits in diabetes. Biochim Biophys Acta 1792: 423–431, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]