

Abstract
Current efforts in developing treatments for diabetes focus on in vitro generation of functional β-cells for cell replacement therapies; however, these attempts have only been partly successful because factors involved in islet formation remain incompletely understood. The embryonic pancreas, which gives rise to β-cells, undergoes early epithelial rearrangements, including transient stratification of an initially monolayered epithelium, followed by microlumen formation and later resolution into branches. Within the epithelium, a multipotent progenitor cell (MPC) population is specified, giving rise to three important lineages: acinar, ductal and endocrine. Pdx1 is a transcription factor required for pancreas development and lineage specification; however, few Pdx1 targets that regulate pancreatogenesis have been identified. We find that pancreatic defects in Pdx1−/− embryos initiate at the time when the progenitor pool is specified and the epithelium should resolve into branches. Pdx1−/− microlumen diameters expand aberrantly, resulting in failure of epithelial tubulogenesis and ductal plexus formation. Pdx1−/− epithelial cell proliferation is decreased and the MPC pool is rapidly lost. We identify two conserved Pdx1 binding sites in the epithelial cadherin (E-cad, Cdh1) promoter, and show that Pdx1 directly binds and activates E-cad transcription. In addition, Pdx1 is required in vivo for maintenance of E-cad expression, actomyosin complex activity and cell shape. These findings demonstrate a novel link between regulators of epithelial architecture, specification of pancreatic cell fate and organogenesis.
KEY WORDS: Pancreatic bud, Epithelium, Stratification, Pdx1, Microlumen, Lumen diameter, E-cadherin, Cdh1, β-catenin, Endocrine, pMLC
Summary: Mouse Pdx1 depletion in the pancreas leads to aberrant cell shape, impaired ductal tubulogenesis and loss of the epithelial multipotent progenitor cell population.
INTRODUCTION
Insulin-producing β-cells are crucial for proper blood glucose homeostasis. These cells are of particular interest since diabetes, which affects millions of people worldwide and causes both significant morbidity and mortality, results from the loss of β-cells (Type 1) or defective insulin signaling (Type 2). Progress has been made in driving embryonic stem cells or other pancreatic cell types towards β-cell fate in culture (D'Amour et al., 2005; Kroon et al., 2008; Pagliuca et al., 2014; Rezania et al., 2014; Zhou et al., 2008); however, this remains an inefficient and poorly understood process. Elucidating normal β-cell ontogeny in the developing pancreas will instruct in vitro differentiation protocols and/or in vivo β-cell regeneration programs, paving the road for development of therapies for diabetic patients.
The study of pancreatic development has been the focus of many research groups in the past few decades and it has elucidated the step-wise process by which β-cells emerge from the pancreatic epithelium, involving both cell-autonomous transcriptional events and cell-cell signaling from surrounding mesoderm (Ahlgren et al., 1996; Arda et al., 2013; Pan and Wright, 2011). Although the roles of many factors involved in these events have been elucidated, there remain significant gaps in our understanding. In particular, little is known regarding the initial events within the progenitor pancreatic epithelium that set in motion the proper allocation and specification of β-cells.
The pool of β-cell progenitors is set aside early during development and their number dictates the ultimate mass of the pancreas (Stanger et al., 2007). Pancreatic lineages emerge from a common endodermal epithelium surrounded by mesodermal mesenchyme, with which it exchanges significant molecular crosstalk. However, the architecture and dynamics of this early niche for progenitors is poorly understood. We and others found that the epithelium undergoes several dramatic changes, including a transient stratification, rosette formation and de novo microlumen formation, followed by epithelial resolution and branch formation (Hick et al., 2009; Kesavan et al., 2009; Villasenor et al., 2010). Hence, for a brief time, the pancreatic bud consists of an outer layer of semi-polarized (‘cap’) cells and internal unpolarized (‘body’) cells. In this stratified epithelium, microlumens fuse, giving rise to a complex ductal plexus that subsequently remodels into a hierarchical tree, with endocrine cells largely delaminating from the central trunk epithelium and acini developing from forming tip domains (Shih et al., 2013). Deleting cell polarity and cytoskeleton regulators causes defects in epithelial remodeling, as well as in the β-cell lineage (Kesavan et al., 2009; Petzold et al., 2013). Questions arise as to how the different lineages become allocated within the epithelium and whether the 3D architecture of the progenitor epithelium impacts β-cell neogenesis.
Identifying ‘stem’ or ‘progenitor’ cells capable of giving rise to endocrine cells, within the early bud or appearing via induced transdifferentiation has been the focus of many efforts (Lysy et al., 2013; Schiesser and Wells, 2014). In 2007, lineage tracing studies identified ‘multipotent progenitor cells' (MPCs) in the early pancreatic epithelium that gave rise to all three lineages – endocrine, acinar and ductal. MPCs were characterized by co-expression of pancreas-specific transcription factor 1a (Ptf1a), carboxypeptidase A1 (CPA1) and c-myc in peripheral epithelial ‘tip’ domains (Pan et al., 2013; Stanger et al., 2007; Zhou et al., 2007) and shown to be multipotent prior to the secondary transition. After embryonic day (E) 12.5, as the epithelium begins to resolve into monolayer branches, MPCs become restricted to the acinar lineage. Therefore, the stratified epithelium of the early pancreatic bud constitutes a potential MPC niche, about which we know very little.
Growth and morphogenesis of the pancreatic bud into a ramifying gland requires the transcription factor pancreatic duodenal homeobox1 (Pdx1). Pdx1, in turn, regulates other transcription factors required for pancreatic cell fates, including Ptf1a and NK6 homeobox1 (Nkx6.1) (Arda et al., 2013; Seymour and Sander, 2011; Shih et al., 2013), and ablation of Pdx1 results in complete pancreas agenesis and lethality at birth (Hale et al., 2005; Jonsson et al., 1994; Offield et al., 1996). Pdx1 is expressed in the foregut endoderm at E8.5 (Villasenor et al., 2008) and in both dorsal and ventral pancreatic buds by E9.5. By late gestation, Pdx1 expression becomes restricted to endocrine cells and later exclusively to β-cells (Guo et al., 2013; Wescott et al., 2009). Although Pdx1 is required for the expression of insulin, developmental targets are only now being identified (Khoo et al., 2012; Oliver-Krasinski et al., 2009; Raum et al., 2015). One report speculated that Pdx1 might regulate cell adhesion (Ahlgren et al., 1996) and another found binding of Pdx1 to the adherens junction E-cadherin (E-cad, Cdh1) promoter in adult islets (Khoo et al., 2012), but no studies have directly addressed its possible roles regulating epithelial morphogenesis or a niche that could support β-cell development.
Here, we characterize the structural abnormalities observed in the Pdx1-null pancreas. We show that Pdx1−/− pancreatic epithelium initially undergoes normal stratification and microlumen formation, but that it displays defects in elaboration of the epithelial tree, including failure of fine lumen diameter maintenance and ductal plexus complexity. In addition, cells within the stratified bud fail to proliferate normally and the MPC pool is rapidly lost. We show that these events are accompanied by a significant decrease in the expression of the cellular adhesion molecules E-cad and β-catenin. Furthermore, we identify E-cad as a novel Pdx1 transcriptional target. We show that Pdx1 directly binds to and activates the E-cad promoter. We also show that pathways downstream of E-cad known to regulate apical constriction, which are necessary for lumen formation and maintenance, are disrupted in the absence of Pdx1. Together, these defects result in the formation of a single, large central lumen encased by a monolayered, duct-like epithelium. Significantly, this study strongly suggests a requirement for three-dimensional architecture in the proper establishment of pancreatic cell fate.
RESULTS
Pancreatic agenesis in Pdx1−/− embryos initiates at the onset of epithelial destratification
To characterize timing of developmental failure of the Pdx1−/− pancreatic bud [Pdx1lacZ/lacZ (Offield et al., 1996)], we analyzed stage-matched littermate buds from E10.0-E14.5 Pdx1+/− and Pdx1−/− embryos. Embryonic midguts (antrum of the stomach, pancreatic bud and duodenum) were dissected and stained for β-galactosidase (Fig. S1). As previously shown (Hale et al., 2005; Jonsson et al., 1994; Offield et al., 1996), we found that Pdx1+/− dorsal pancreatic buds grow and expand rapidly throughout late gestation, whereas in Pdx1−/− they developed normally to about E10.5 (ventral bud never evaginates), but were smaller by E11.0 and failed to grow significantly past E11.5. The Pdx1+/− epithelium was unaffected at all embryonic stages examined, as shown by normal expression of pancreatic markers, such as Nr5a2 and E-cad (Fig. S2).
We previously showed that normal pancreatic epithelium undergoes a rapid, transient stratification early in its development, becoming maximally stratified between E10.0 and E11.0, when microlumens emerge and connect into a lumen plexus (Villasenor et al., 2010). Microlumens are identified by expression of Muc-1 or other apical markers along a visible cavity at the center of rosettes of cells with apically constricted ends. Despite a high degree of variability in size and shape, most microlumens in the pancreas at E10.5 are under 20 µm in diameter. We noted that this stage coincides with the initiation of observed Pdx1−/− defects, so we assessed stratification in Pdx1−/− buds. At E10.0, the epithelium appeared indistinguishable between controls and Pdx1−/−, presenting an average of four epithelial cell layers thick (Fig. S3A,C,E). However, by E10.5, Pdx1+/− buds developed on average six cell layers, whereas Pdx1−/− still only displayed four cell layers (Fig. S3B,B′,D-E). Decrease of relative cell numbers in the mutant was apparent by E11.5 (Fig. S4A-B′), with approximately half the number of cap cells in Pdx1−/− buds (66.0±2.9 compared with 122.9±4.7 in Pdx1+/−) and one quarter the number of internal cells when compared with Pdx1+/− (257.5±13.2 in Pdx1+/− compared with 67.2±4.1 in Pdx1−/−) (Fig. S4C,D).
Microlumens in the Pdx1−/− bud expand to form an epithelial cyst
Since defects were evident early in Pdx1−/− pancreatic epithelium, we characterized microlumen formation and coalescence. Analysis of whole-mount immunofluorescent pancreata stained for E-cad and Mucin1 (Muc1) showed that microlumens in control buds rapidly increased in number, but remained of constant size (Fig. 1A-H). Pdx1+/− epithelium had 270±22 microlumens at E10.5, with a maximum number at E11.0 of 334±49 (Fig. 1I,K,M). As the bud developed, these microlumens connected into a fine plexus of narrow lumens and individual microlumens could not be counted thereafter. By E11.5, 150±30 microlumens and at E12.5, 111±10 microlumens were found in Pdx1+/− buds (Fig. 1A-D,M). By contrast, in Pdx1−/− buds, the number of microlumens decreased over time after E10.5, with an average of 263±34, 115±5 at E11.0, 35±2 at E11.5 and by E12.5, it was further reduced to 9±2 (Fig. 1E-H,M). Imaris surface reconstructions of whole-mount pancreata showed that microlumens initially formed and continued to emerge until E10.5, in both Pdx1+/− and Pdx1−/− buds, with rosette formation and microlumens in equal numbers and similar size (Fig. 1I,K). However, by E11.5, mutant lumens significantly expanded (Fig. 1J,L,N and Movies 1 and 2). We note that Pdx1−/− bud defects were variable, although images shown are representative and quantifications reflect observed phenotypes.
Fig. 1.

In the absence of Pdx1, the pancreatic ductal plexus transforms into a bag-like cyst. (A-H) Z-stack images of whole-mount tissues stained with Muc1 to visualize lumens, reconstructed using 3D projection (ImageJ). (I-L) Imaris surface reconstruction of lumens (Muc1, red; E-cad, green). Note that whereas E10.5 microlumen size is similar in Pdx1+/− and Pdx1−/− buds (I,K), lumen sizes become expanded in Pdx1−/− by E11.5 (L) when compared with Pdx1+/− (J). Representative microlumens included for comparison (insets, panels J,L). (M) Quantification of number of microlumens at different stages. (N) Comparison of microlumen diameter as lumens start to fuse. ns, not statistically significant, *P<0.05, **P<0.01, #P<0.001 (Student's t-test). Scale bars: 50 μm in A-H; 25 μm in I-L.
We also performed analyses on sections to further understand the Pdx1−/− luminal phenotype. Similar to whole mounts, small microlumens of comparable sizes and shapes were found at E10.5 in Pdx1+/− and Pdx1−/− buds (Fig. 2A,E). However, by E11.5-E12.5, instead of a complex luminal network of epithelial tubes as observed in Pdx1+/−, Pdx1−/− microlumens enlarged and formed a progressively simpler plexus consisting of fewer connections, which were later eliminated. By E13.5, Pdx1−/− buds displayed a single large cystic lumen (Fig. 2B-D,F-H). The average lumen diameter in Pdx1+/− at E13.5 was 9.184±1.1 μm at the widest point, while that of Pdx1−/− buds was 48.99±2.2 μm (Fig. 1N and Fig. 2D,H). Maintenance of narrow lumen diameters thus failed in the Pdx1−/− epithelium.
Fig. 2.

Pdx1−/− pancreatic buds display epithelial lumens with progressively expanded diameters after E11.5. Sections through pancreatic buds show epithelial lumen diameters at different stages in (A-D) Pdx1+/− and (E-H) Pdx1−/− embryos. Insets are close-up images of representative lumens, and yellow lines indicate lumens measured at widest diameter (Muc1, red) visible. Note normal microlumens at E10.5 (A,E), and cystic lumen in Pdx1−/− at E13.5 (H), compared with Pdx1+/− (D). Scale bars: 20 μm.
Pdx1−/− pancreatic buds are smaller because of decreased proliferation, not cell death
To assess whether the decrease in size over time in Pdx1−/− buds was due to cell death, we examined cleaved caspase 3 and TUNEL immunostaining on cryosectioned E10.5-E13.5 pancreata (Fig. S5A-F and data not shown). Quantification showed that there are low levels of cell death at these stages in normal pancreas. Similarly, Pdx1−/− pancreata displayed few cells stained for cell death markers. Using cleaved caspase 3 antibody staining, at E10.5 we observed 1.4±0.4% positive cells out of total Pdx1+/− epithelial cells and 1.6±0.2% in Pdx1−/−, at E11.5 we observed 1.6±0.2% apoptotic cells in Pdx1+/− and 1.6±0.3% in Pdx1−/−, and at E12.5 we observed 1.2±0.1% positive cells in Pdx1+/− and 1.5±0.2% in Pdx1−/− (Fig. S5G). Loss of Pdx1 therefore does not result in epithelial cell death.
We then investigated whether proliferative capacity was decreased in Pdx1−/− buds by staining for phospho-histone H3 (pHH3). We found no difference in proliferation between heterozygotes and nulls at E10.5 (Fig. 3A,D,G) (13.6±0.5% pHH3+ cells in Pdx1+/− and 12.8±0.4% pHH3+ cells in Pdx1−/− epithelium). However, fewer pHH3+ cells were observed in Pdx1−/− buds at E11.5 and E12.5 (Fig. 3B,C,E-G). The difference in proliferation at these stages, between Pdx1+/− and Pdx1−/− buds, is largely because fewer internal cells undergo mitosis, whereas Pdx1−/− peripheral cap cells exhibited similar low pHH3+ positivity. At E11.5, Pdx1+/− buds had 8.9±1.2% pHH3+ cap and 12.6±1.0% pHH3+ internal cells, whereas Pdx1−/− had 7.5±1.0% pHH3+ cap and 5.3±0.9% pHH3+ internal cells. By E12.5, proliferation had significantly decreased in both newly forming peripheral cap/tip domains and internal/trunk cells in Pdx1−/− buds, with the difference more striking in trunk cells (2.9±0.3% Pdx1−/− compared with 7.1±0.4% Pdx1+/− in tips and 3.8±0.6% Pdx1−/− compared with 12.0±0.9% Pdx1+/− in trunks).
Fig. 3.

The Pdx1−/− epithelium has decreased proliferative capacity over time. (A-F) Images of sections through pancreatic buds at E10.5 and E11.5 (pHH3, red; E-cad, green; DAPI, blue). Scale bars: 20 μm. (G) Quantification of pHH3+ cells in the E10.5 epithelium shows no difference between Pdx1+/− and Pdx1−/−. Comparison of pHH3+ cells at E11.5 shows decreased proliferation in internal cells, but not cap cells, of the Pdx1−/− bud. By E12.5, the Pdx1−/− epithelium exhibits decreased proliferation in both cap and internal cells. ns, not statistically significant; #P<0.001 (Student's t-test).
These findings are interesting because previous reports (Zhou et al., 2007) show that proliferating cells fueling pancreas growth primarily reside in tip cell domains after E12.5. We subsequently verified that at early stages in the pancreas, more proliferation occurred within internal body cells with pHH3 stains on wild-type (WT) pancreas in 0.5 day increments from E10.5-E13.5 (Fig. S6A-G). At E10.5-E12.5, mitosis was indeed more frequent in the internal/trunk cell domain (6.8-9.4% compared with 2.5-6.1% in cap/tip domain). By E13.5, however, we observed a distinct shift in proliferative regionalization, as tip cells displayed increased pHH3 positivity (5.1% tip compared with 1.8% in trunk cells). Our findings show that early growth primarily occurs in the heart of the stratified epithelium and that the shift in proliferation from internal to external cells was correlated with epithelial resolution and branching initiation.
Multipotent progenitor cells (MPCs) are lost in the Pdx1−/− pancreatic bud
To determine whether progenitors were affected in Pdx1−/− pancreata, we examined MPC marker expression. We performed immunostaining for CPA1, Ptf1a and Nr5a2 on E11.5 and E12.5 bud sections (Fig. 4). Although CPA1 staining was evident in heterozygote littermates at E11.5 and E12.5, it was absent from Pdx1−/− epithelium at both stages (Fig. 4A,D and G,J). Similarly, although the MPC marker Ptf1a was present in E11.5 Pdx1+/− and Pdx1−/− buds (Fig. 4B,E), its expression had ceased in Pdx1−/− buds by E12.5 (Fig. 4H,K). By contrast, Nr5a2 continued to be expressed in Pdx1−/− buds at both stages (Fig. 4C and F,I and L). We confirmed these results through both semi-quantitative PCR and qPCR and observed that in Pdx1−/− buds, Cpa1 transcripts were reduced, whereas Ptf1a transcripts were absent, compared with controls (Fig. 4M,N).
Fig. 4.
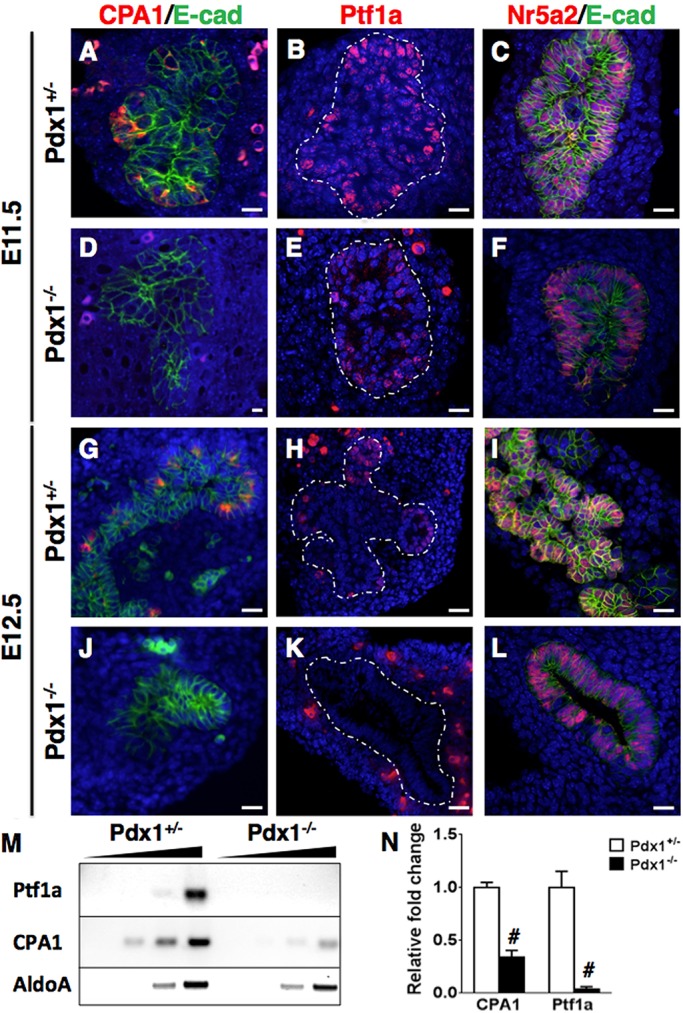
Progenitor pool is impaired early in the Pdx1−/− pancreas. (A-L) MPC marker expression analyzed in the absence of Pdx1. CPA1 (red) is absent at both E11.5 and E12.5 (compare D,J with A,G), whereas Ptf1a (red) is reduced by E12.5 (K compared with H) in the Pdx1−/− epithelium. Nr5a2 is present at both stages (F,L) in Pdx1-null epithelium. Scale bars: 20 μm. (M) Semiquantitative PCR was performed on cDNA isolated from single E12.5 pancreatic buds for Cpa1 and Ptf1a. Cpa1 transcript levels are decreased in the Pdx1−/− bud, and Ptf1a is completely absent by this stage. First lane for each individual set of samples per genotype is a water blank stopped after 36 cycles, and second to fourth lanes correspond to cDNA containing reactions stopped at 30, 33 and 36 cycles, respectively. (N) qPCR analysis performed on cDNA isolated from single E11.5 buds (n=5 for each genotype, in triplicate) show that Cpa1 and Ptf1a transcripts are significantly lower in the Pdx1−/− pancreas, compared with Pdx1+/−. #P<0.001 (Student's t-test).
Similarly, other markers of pancreatic progenitors were reduced in the absence of Pdx1. In WT pancreatic buds, SRY-box 9 (Sox9) is expressed broadly in the epithelium, albeit at different intensities in different cells, with both Sox9hi and Sox9lo cells observed. Sox9hi cells are believed to be bipotential cells that give rise to endocrine and ductal cells (Kopp et al., 2011; Lynn et al., 2007). We found that whereas Sox9 staining is present throughout E11.5 Pdx1−/− pancreatic buds, Sox9hi cells were reduced by 30% (Fig. S7A-B′). Analysis of overall intensity of Sox9 staining at E12.5 (Fig. S7C,D) confirmed decreased levels of Sox9 in Pdx1−/− buds, although Sox9 transcript levels were unchanged. This finding suggests that the bipotent Sox9hi progenitor population is reduced in the absence of Pdx1.
E-cadherin and β-catenin are downregulated in Pdx1−/− pancreatic epithelium
Given that epithelial morphology was significantly disrupted in Pdx1−/− buds, we visualized the epithelium using the classical adhesion components E-cad and β-catenin (β-cat). While the Pdx1−/− epithelium generally remained cohesive, we nonetheless observed a marked reduction of E-cad (Fig. 5A,C), β-cat (Fig. 5B,D) and α-catenin (α-cat, data not shown) expression by immunofluorescence at E11.5. However, staining in the esophagus (where Pdx1 is not normally expressed) was unaffected (Fig. 5E-H). In E10.5 Pdx1−/− buds, levels appeared relatively normal, suggesting that E-cad expression was lost at the onset of destratification. Quantification of pixel intensity of E-cad and β-cat determined that levels of adhesion molecules in the absence of Pdx1 were approximately half that obtained in Pdx1+/− (Fig. 5I).
Fig. 5.
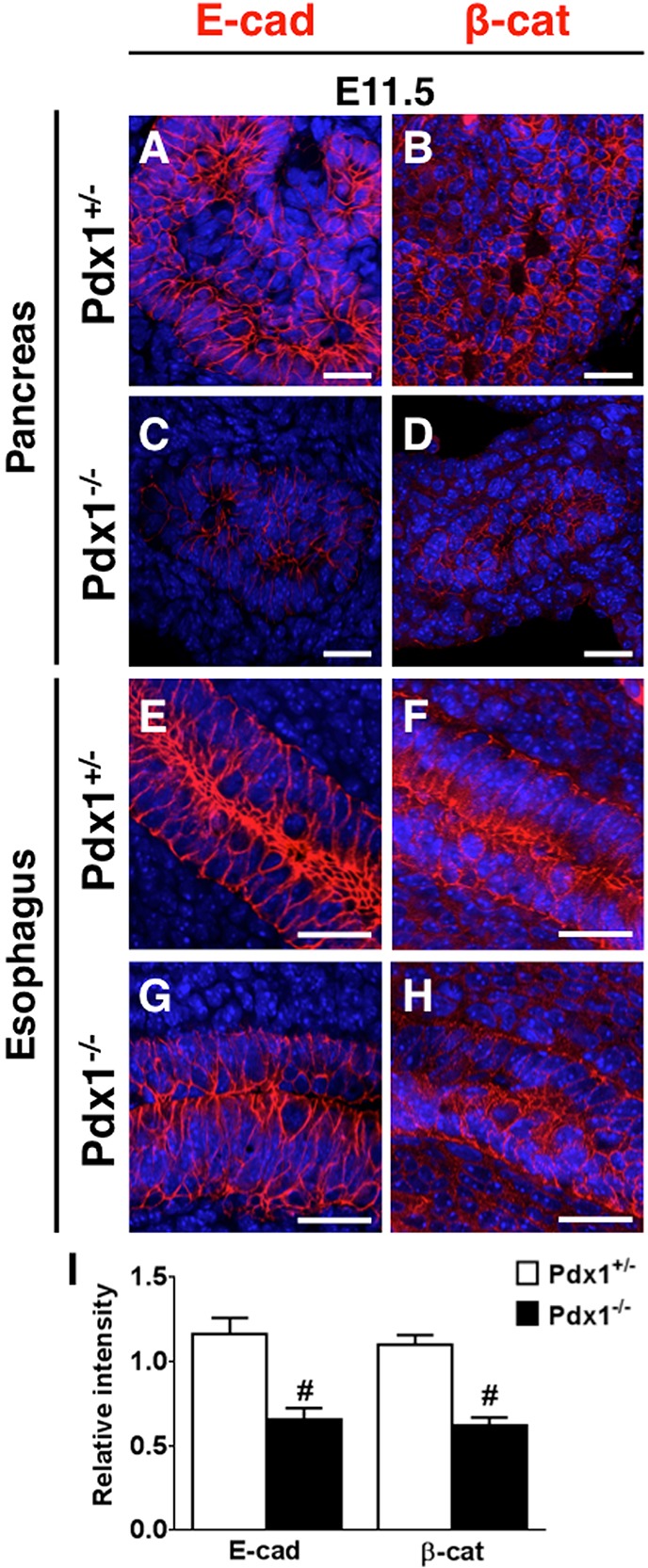
Expression of E-cadherin and β-catenin is decreased in the Pdx1−/− pancreatic epithelium. (A-D) Pancreas sections stained for E-cad and β-cat (in red) and DAPI (in blue). (E-H) Esophagus sections from the same slides show that E-cad and β-cat staining is not affected in tissues not expressing Pdx1. Scale bars: 25 μm. (I) Pixel intensity quantifications of E-cad and β-cat in pancreas of Pdx1+/− and Pdx1−/−. #P<0.001 (Student's t-test).
Pdx1 regulates expression of E-cadherin, but not β-catenin
We therefore asked whether Pdx1 might play a direct role in the regulation of E-cad and β-cat. Using the Evolutionary Conservation of Genomes (ECR) browser (Ovcharenko et al., 2004) and performing sequence alignments with consensus Pdx1 binding sequence CATYAS, we identified two conserved predicted binding sites for Pdx1 in the E-cad promoter and five conserved predicted binding sites in the β-cat (Ctnnb1) promoter (Fig. 6B,D,F). Pdx1 was previously observed to bind the promoters of E-cad and β-cat in adult β-cells, via a chromatin immunoprecipitation using anti-Pdx1 in both mouse and human adult pancreas (Khoo et al., 2012).
Fig. 6.

Pdx1 binds to and activates the E-cad promoter. (A) Luciferase assays show that Pdx1 transcriptionally activates both a 2.8 kb and a 300 bp fragment of the WT E-cad promoter (Prom) fused to a luciferase reporter gene. (B) Schematic showing two predicted Pdx1 binding sites (consensus sequence CATYAS) in the E-cad WT 2.8 kb and 300 bp promoter fragments. (C) When predicted binding sites in the 300 bp E-cad promoter fragment are mutated, reporter activity decreases. (D) Schematic showing promoter fragments analyzed in C, with the WT E-cad 300 bp promoter fragment with and without Pdx1 binding site point mutations. (E) Addition of Pdx1 protein to the 2.3 kb β-cat promoter did not increase luciferase reporter activity. (F) Schematic comparing fragments used for luciferase assays in E; 2.8 kb E-cad promoter fragment has two predicted Pdx1 binding sites and the 2.3 kb β-cat promoter has five predicted Pdx1 binding sites. (G) E-cad and β-cat transcript levels from individual E12.5 pancreatic buds were reduced using semi-quantitative PCR in the Pdx1−/− pancreas (three different primer pairs, n=3/genotype, in triplicate); β-cat levels remained unchanged. Pdx1 and aldolaseA (AldoA) primer sets were used as controls. (H) qPCR analysis shows that E-cad transcripts are reduced in Pdx1−/− buds at E11.5, whereas β-cat transcripts were unaffected (n=3/genotype, in triplicate, 6-12 repeats). ns, not statistically significant; **P<0.01 (Student's t-test).
To determine whether Pdx1 was capable of activating transcription of E-cad and β-cat, we performed luciferase assays in HEK293T cells (which normally do not express Pdx1). We found that Pdx1 bound to the E-cad promoter and activated expression of E-cad, using both 2.8 kb and 300 bp promoter fragments (both containing predicted Pdx1 binding sites). Increasing concentrations of Pdx1 protein resulted in a corresponding increase in luciferase reporter activity (Fig. 6A,B). However, mutation of binding sites in the 300 bp E-cad promoter fragment, either individually or combined, hampered luciferase reporter activity, with the highest luminescence signal corresponding to less than half that seen with the WT promoter fragment (Fig. 6C,D). We also assayed a 2.3 kb fragment of the β-cat promoter that encompassed five predicted Pdx1 binding sites, but found that increasing Pdx1 concentration did not activate luciferase (Fig. 6E,F). These results suggested that Pdx1 directly regulates E-cad in the developing pancreas.
To further examine this possibility, we carried out semi-quantitative RT-PCRs and qPCRs on Pdx1+/+, Pdx1+/− and Pdx1−/− pancreatic epithelium to assess E-cad transcripts. In the absence of Pdx1, we found a significant decrease in E-cad mRNA, but not β-cat mRNA (Fig. 6G,H, Fig. S8). Conversely, we used HEK293T cells to test whether increased levels of Pdx1 might promote an increase in the E-cad protein level. In this context, we found a modest but measurable increase in E-cad protein in transfected cells (Fig. S9), further suggesting a positive, regulatory relationship between Pdx1 and E-cad.
Epithelial reacquisition of apical polarity during microlumen formation is not affected
Given the dramatic differences in epithelial organization in Pdx1+/− versus Pdx1−/− buds, and our finding that Pdx1 binds and activates the E-cad promoter, we examined the cellular events that might result in loss of lumen diameter maintenance. We previously showed that epithelial cells acquire apicobasal polarity following stratification, undergoing apical constriction and rosette formation and resulting in microlumen formation (Villasenor et al., 2010). Given the defects in maintenance of microlumen diameter in Pdx1−/− buds, we assessed cell polarity determinants at stages when the epithelium is stratified and forming microlumens. Immunostaining for F-actin (phalloidin), ZO-1, GM-130, atypical protein kinase C (aPKC), Par3 and ezrin showed no evident defects in apical polarity when comparing Pdx1+/− and Pdx1−/− buds, either early at E10.5 or during resolution at E12.5 (Figs S10, S11 and data not shown).
Pdx1−/− epithelial cells fail to maintain apical constriction
Although apical polarization of Pdx1−/− epithelial cells was grossly normal, we assessed cellular morphology. We speculated that inability to maintain cell shape might impact maintenance of luminal diameter. By E10.0, stratified pancreatic epithelial cells undergo something akin to apical constriction, changing their morphology from cuboidal to ‘bottle’ shaped (Villasenor et al., 2010). Groups of cells coordinately adopt this shape to generate rosettes that open microlumens at their centers, as described in forming kidney tubules (Lienkamp et al., 2012) and zebrafish gut epithelium (Horne-Badovinac et al., 2001).
We found that Pdx1+/− pancreatic epithelial cells acquired this ‘bottle shape’ and formed normal microlumens by E10.5 (Fig. 7A-A″). However, although Pdx1−/− cells developed apically constricted ends around E10.5, they did not maintain this bottle morphology over time (Fig. 7B-B″). Pdx1−/− cells suffered rapid loss of apical constriction and abnormal cell morphology. We used ImageJ to quantify cell shape parameters, including area, circularity and the apico-basal surface ratio. We found that Pdx1−/− cells were generally larger (67.51±2.38 µm2 in Pdx1+/− compared with 95.30±3.35 µm2 in Pdx1−/−) (Fig. 7C) and adopted a columnar, rather than bottle-shaped, morphology (as per ImageJ circularity assessment, where values equal to 1.0 indicate an ideal circular shape). Pdx1+/− epithelial cells exhibited an average circularity value of 0.69±0.01, whereas Pdx1−/− measurements indicated linear morphology with a value of 0.56±0.01 (Fig. 7D). Measurements of the apico-basal surface ratio showed that Pdx1+/− cells were significantly more constricted at their apical surface (area less than half that of basal surface, ratio=0.48±0.03) (Fig. 7E). However, Pdx1−/− epithelial cells showed altered cell shape at E11.5 and E12.5, with cells almost equal in their apical and basal surface lengths (ratio=0.88±0.05).
Fig. 7.

Cell shape is altered in the Pdx1−/− pancreas by E12.5. (A-B″) E12.5 pancreas sections stained for epithelial marker β-cat (in red), apical polarity determinant aPKC (in green) and DAPI (in blue). Squares in A and B indicate regions shown at high magnification in A′,B′, and those in A′ and B′ indicate regions shown in A″,B″. White outlines indicate cells used for morphometric analysis. (Contrast is enhanced in the red channel to allow for visualization of cell shape in Pdx1−/−.) Only cells with defined apical and basolateral domains were used for analysis. Scale bars: 20 μm. (C-E) Cell morphology parameters assessed indicate that Pdx1−/− cells have a larger area, are less circular and have a higher apical:basal surface ratio, demonstrating that these cells do not apically constrict. #P<0.001 (Student’s t-test).
Regulators of apical constriction downstream of E-cadherin are absent in Pdx1−/− pancreata
In order to determine a cause for expansion of apical membranes and increased lumen diameters in Pdx1−/− pancreata, we examined components downstream of E-cad and β-cat in the classical cadherin signaling pathway. Specifically, we assessed the actomyosin machinery, as it is known to regulate apical constriction. Phospho-myosin light chain (pMLC) activates myosin II complexes, acting as a molecular ratchet, preventing relaxation of constriction (Ebrahim et al., 2013; Martin and Goldstein, 2014; Mason and Martin, 2011). Disruption of actin-myosin has been shown to disrupt apical constriction of epithelial cells (Solon et al., 2009). We found that, overall, pMLC was present in both Pdx1+/− and Pdx1−/− pancreatic epithelium at E10.5 (Fig. 8A,D). However, by E11.5-E12.5, it became reduced in Pdx1−/− pancreatic epithelium, but not nearby blood vessels (Fig. 8B,E,C,F). High-magnification imaging shows equivalent pMLC expression at the apical surface of rosettes in both Pdx1+/− and Pdx1−/− epithelium at E10.5 (Fig. 8G,J). By E11.5 and E12.5, pMLC staining was markedly reduced in remaining abnormal rosettes visible in Pdx1−/− cells, indicating loss of this molecular clutch (Fig. 8H,I,K,L) and was absent from other epithelial regions.
Fig. 8.
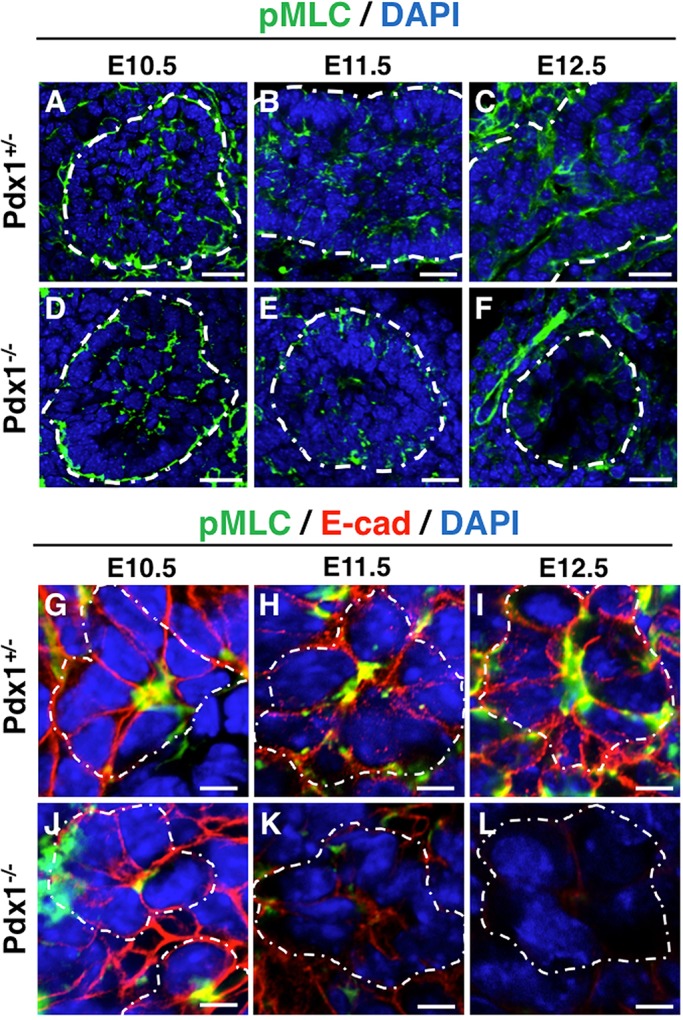
Apical pMLC is present in early Pdx1−/− epithelial cells, but decreases significantly by E12.5. (A-F) Sections stained for pMLC (in green) and DAPI (in blue). Note decreased level of pMLC in the Pdx1−/− epithelium at E11.5 (E) and marked decrease by E12.5 (F), whereas levels in the mesenchyme (especially blood vessels) are unchanged. White dotted outlines in A-F indicate basal surface of epithelium. (G-L) Close-up images of rosettes show that the pMLC decreases apically in the Pdx1−/− bud after E11.5 (K,L). White dotted outlines in G-L indicate rosettes. Scale bars: 20 μm in A-F; 5 μm in G-L.
Indeed, loss of Pdx1, E-cad and pMLC was accompanied by marked loss of rosette structures between E10.5 and E12.5, as well as altered rosette morphology. By E12.5, we found far fewer rosettes per section (50% fewer at E11.5 and 75% fewer at E12.5), as well as rosettes containing fewer and larger epithelial cells (Fig. S12). Together, these findings suggest that cells lacking Pdx1 are unable to maintain the constriction of their apical surface and undergo deleterious changes in cell shape, leading to an overall failure in morphogenesis of the pancreas epithelial tree.
DISCUSSION
Here, we report the architectural defects of the pancreatic epithelium that arise in the classical model of pancreatic failure – the Pdx1−/− mutant – and we identify E-cadherin as a direct target of Pdx1. Specifically, we report that defects in the Pdx1−/− pancreatic epithelium culminate in defective epithelial tubulogenesis, failed bud expansion and blocked lineage differentiation. We propose that a deeper understanding of the early pancreatic epithelium is needed, because dynamic morphological and architectural changes occur at a time when multipotent progenitors (MPCs) are set aside (Stanger et al., 2007; Villasenor et al., 2010; Zhou et al., 2007). How they arise and whether their microenvironment, or niche, is crucial to their fates remain unknown. In this study, we show that the early Pdx1−/− pancreatic epithelium initially develops normally, stratifying and opening microlumens in a manner similar to WT littermates. However, defects appear when the epithelium would normally begin to destratify into a monolayered tree, to generate fine tubular branches. Failures in expansion and fusion of lumens ultimately yield a cystic, monolayered epithelial ductule. Defects are correlated with reduction in cellular adhesion (decreased adherens junction components E-cad and β-cat), decreased proliferation and failure of maintenance of epithelial lumen diameter (Fig. 9). In addition, we implicate the actomyosin machinery, downstream of E-cad, in modulating cell shape during microlumen formation. In the absence of Pdx1 and E-cad, MLC activity is lost along with apical constriction of rosette cells and morphogenesis is disrupted. Together, these findings suggest that establishment of the pancreatic MPC population is likely to depend on proper 3D epithelial architecture.
Fig. 9.

Pdx1 regulates adhesion, cell shape and fate, driving epithelial morphogenesis in the developing pancreatic bud. (A) Our data support a molecular model that places Pdx1 as an activator of E-cad transcription in the developing pancreas. Signaling downstream of E-cad, α-cat and β-cat further regulates cytoskeletal architecture (such as the actomyosin complex) and transcription factors that drive proliferation and differentiation. (B) In the early pancreatic bud, microlumens form within the stratified epithelium and are characterized by the apical accumulation of pMLC (among other known polarity determinants and apical markers, such as Muc1, aPKC, ZO-1). E-cad regulates cellular adhesion, as well as lumen size within rosettes through activity of its downstream effector pMLC, which maintains apical constriction. Regulation of microlumen size is crucial for establishment of a fine ductal network as the pancreatic epithelium resolves. In Pdx1−/− buds, regulation of lumen size is lost because of decreases in E-cad and pMLC, and loss of apical constriction in epithelial cells. This ultimately leads to formation of a single, large lumen in Pdx1−/− pancreata.
Pdx1 is essential for pancreatic development and its crucial role has long been recognized, because deletion of Pdx1 results in pancreatic agenesis in mice and in humans (Ahlgren et al., 1996; Stoffers et al., 1997b). However, further understanding of its impact is needed because it remains unclear why the pancreatic epithelium fails to develop in its absence. Furthermore, point mutations in PDX1 in patients are associated with maturity-onset diabetes of the young (MODY4) or increased susceptibility to gestational diabetes (Gragnoli et al., 2005; Stoffers et al., 1997a). Although the Pdx1−/− pancreas was originally reported as failing to develop, yielding only a residual ‘ductule’, little was understood about the dynamics or underpinnings of this failure (Ahlgren et al., 1997; Offield et al., 1996). Why does the early pancreatic bud fail to develop? Conditional ablation of Pdx1 using a Tet-off system showed that epithelial branching and specification of pancreatic lineages depended on the precise timing of Pdx1 expression (Hale et al., 2005; Holland et al., 2002). Inactivation of Pdx1 at late stages of development lead to mild pancreatic defects, whereas early deletion prior to destratification (at E11.5) results in major architectural abnormalities and absence of functional pancreatic lineages (Hale et al., 2005). While these findings suggested a role for Pdx1 in architecture of the pancreatic epithelium, the dynamics or causes for this failure were not examined.
What downstream molecular pathways are disrupted in the absence of Pdx1? Surprisingly, despite more than two decades of intense interest by the field, only a handful of developmental targets of Pdx1 have been identified. Pdx1 was initially identified as Xlhbox8 and its expression was shown to be restricted to the pancreatic anlagen and duodenum (Wright et al., 1989). Pdx1 was later characterized under the name IPF1 (insulin promoting factor) because it transactivates the insulin promoter (Ohlsson et al., 1993) and was shown to maintain the expression of Ptf1a in acinar cells, ensuring exocrine differentiation (Pan and Wright, 2011). In addition, Pdx1 interacts with Meis1 to downregulate keratin 19, and forms complexes with Pbx1b and Meis2 to control elastase transcription (Deramaudt et al., 2006; Swift et al., 1998). More recent ChIP studies carried out in the embryonic pancreas also identified Ngn3, Pax6 and Tshz1 as direct targets (Oliver-Krasinski et al., 2009; Raum et al., 2015). However, beyond these targets, it has been unclear what pathways Pdx1 controls during pancreas morphogenesis and how this impacts pancreatic cell lineages.
We found that epithelial tubulogenesis was severely disrupted in the absence of Pdx1. We also serendipitously noted a sharp reduction in E-cad, as it is a common marker used to visualize epithelial cells. We speculated that architectural molecules such as E-cad might represent potential direct targets of Pdx1. Analysis of published ChIP-on-chip and ChIP-Seq data obtained by other groups revealed that E-cad and β-cat promoters are bound by Pdx1 in isolated adult β-cells (Khoo et al., 2012; Teo et al., 2015), making these promising candidates. Our data provides further evidence that E-cad is a direct target of Pdx1.
How might loss of E-cad influence morphogenesis of the pancreatic epithelium? We found that cell polarity was unaffected in Pdx1+/− pancreatic buds, as mutants showed normal expression of all apical markers examined between E10.5 and E12.5 (aPKC, ezrin, ZO-1). However, we found significant differences in maintenance of epithelial cell shape during morphogenesis, as Pdx1−/− epithelial cells lost their bottle cell morphology after E10.5, taking on a more cuboidal appearance. Measurements of the apical membrane in Pdx1−/− buds at time points after lumen formation (E11.5) revealed significant expansion. We propose that inappropriate expansion of the apical membrane alters overall morphogenesis of the pancreatic tubular epithelium.
The question arises as to whether the normally small apical membrane observed lining WT microlumens is the result of constriction of that surface (previously larger and juxtaposed to other neighboring epithelial cells), building of that surface de novo or simply a change in cell shape driven by aggregation of tight junctions within rosettes. Although our data do not resolve these possibilities, we did observe a significant loss of apical pMLC in Pdx1−/− epithelial cells. The actin-myosin cytoskeleton at the apical membrane has also been dubbed a ‘subcellular ratchet to reduce apical area incrementally’ (Martin et al., 2009). Mutations affecting the formation of the actomyosin complex result in ineffective periodic constriction and relaxation, without a concomitant reduction in the size of the apical domain (Solon et al., 2009) and loss of normal cell shape during embryogenesis (Blanchard et al., 2010). Importantly, E-cad loss of function also causes failure in apical constriction, and a decreased tension between cells that disrupts signaling to the apical constriction machinery, including pMLC (Ebrahim et al., 2013). Disruption of adherens junctions by laser ablation in individual cells also leads to loss of apical constriction in neighboring affected cells (Jayasinghe et al., 2013). It is also possible that Pdx1 directly regulates the actomyosin machinery in addition to E-cad, because we observe Pdx1 consensus binding sites within the MLC promoter; however, future studies will be needed to examine this possibility. Our present study suggests that microlumen and plexus formation occur, at least in part, through Pdx1-coordinated apical constriction and that Pdx1 thereby plays a role in regulating epithelial architecture.
The coincidence of major epithelial defects with absence of all three lineages in Pdx1−/− pancreata is striking and raises the question as to cause and effect. Does the architecture of the epithelium impact the fate of cells therein? We hypothesized it does, because structural defects observed in the Pdx1−/− pancreas coincide with the timing of specification of the MPCs in the pancreas. In addition, a growing number of studies link development of epithelial architecture to cell fate (Cortijo et al., 2012; Kesavan et al., 2009; Petzold et al., 2013). Indeed, there are fewer progenitors in Pdx1−/− pancreata expressing high levels of Sox9 (Kopp et al., 2011) and none expressing Cpa1. Loss of Ptf1a expression was also observed at the time when expansion and differentiation of the MPCs should occur (staining is present at E11.5 in Pdx1−/−, but absent by E12.5). It is of interest that these markers are rapidly down-regulated only after architectural defects initiate in the Pdx1−/− epithelium. Similarly, proliferative defects also occur at this later time. One can imagine that crucial processes such as the cell cycle or cell fate depend directly on Pdx1, or alternatively, that they are the result of defects in the morphology of a putative epithelial signaling niche. Structural defects in the Pdx1−/− epithelium are therefore correlated with, or possibly contribute to, failure of specification of MPC fate.
Together, this work demonstrates that Pdx1 is important for pancreatic architecture and cell fate through its regulation of molecular machinery that is essential for cell shape and adhesion. Our investigations identified the novel Pdx1 developmental target E-cad. This finding suggests that structural and cell fate defects may arise in the Pdx1−/− epithelium because of defective regulation of apical constriction maintenance downstream of E-cad, including α-cat and pMLC. In addition, these findings raise the possibility that transcriptional control of cell fate is influenced by biomechanical forces resulting from cell-cell adhesion and tension. An important next frontier will be to elucidate how pancreatic fate is regulated at the transcriptional level by the intracellular and extracellular microenvironment. No cell ‘lives on an island’, and we propose that understanding the extrinsic context of neighboring cells and the signals they convey to MPCs in the early bud will be key to understanding how to harness and manipulate endocrine cell fate.
MATERIALS AND METHODS
Mice and embryo handling
Experiments were performed in accordance with protocols approved by the UTSW IACUC as previously described (Villasenor et al., 2010). E10.0-E14.5 embryos were dissected and fixed in 4% PFA in PBS for 3 h at 4°C except for β-galactosidase reaction (see next section).
β-galactosidase reaction
Pancreata were fixed using glutaraldehyde for 15 min, rinsed in PBS and stained for β-galactosidase overnight as previously described (Villasenor et al., 2008). Images were taken with a NeoLumar stereomicroscope (Zeiss) using a DP-70 camera (Olympus).
Immunofluorescence on sections
Fixed pancreata were washed in PBS and cryoprotected in 30% sucrose overnight. Tissues were embedded in Tissue-Tek OCT compound and sectioned at 10 µm on a cryostat. Sections were washed in 1× PBS and blocked for 1 h at room temperature (RT) in 5% serum. Primary antibody incubations were done at 4°C overnight (for antibodies and dilutions, see Table S1). Slides were washed in PBS, incubated in secondary antibody (Jackson ImmunoResearch or Invitrogen) for 1 h at RT and subsequently incubated in DAPI. Slides were rinsed in PBS and mounted using Prolong Gold Mounting Medium (Invitrogen). Images were obtained using either an LSM510 or LSM710 Meta Zeiss confocal. In cases where signal amplification was required, TSA kit 12 (Invitrogen) was used according to the manufacturer's directions.
Whole-mount immunofluorescence
Fixed embryonic guts were dehydrated in methanol and incubated in Dent's bleach (methanol:DMSO:30% H2O2, 4:1:1). Tissues were then rehydrated to PBS and blocked using TNB reagent (PerkinElmer). Embryonic guts were incubated in primary antibodies overnight at 4°C. Tissues were washed in PBS and incubated in secondary antibodies overnight at 4°C and then dehydrated in methanol. Tissues were visualized after clearing in BABB (benzyl alcohol, benzyl benzoate) using an LSM710 Meta Zeiss confocal to take optical sections every 2 µm in tissues from E10.5-E11.5 and every 5 µm in tissues older than E12.5. Quantification of microlumens was performed in at least three pancreata per genotype.
Quantification of cell shape parameters
ImageJ was used to quantify cell morphology in the Pdx1+/− and Pdx1−/− strains at E12.5. The parameters assessed were the circularity, area and the ratio of apical to basal surface. At least three pancreata were analyzed per genotype, with 50 cells analyzed per pancreas. Only cells with defined apical (aPKC+) and basolateral (bud periphery) domains were used for analysis. Average values were calculated and mean differences were tested for statistical significance using the Student's t-test.
E-cadherin and β-catenin promoter cloning and mutagenesis
E-cad (2.8 kb and 300 bp) and β-cat (2.3 kb) promoter fragments encompassing predicted Pdx1 binding sites were amplified from mouse genomic DNA (Clontech) with Accuprime Pfx Supermix polymerase (Life Technologies), using primers (IDT) indicated in Table S2. Promoter fragments were inserted into the SmaI site of the pGL3 Luciferase vector (Promega). Full-length Pdx1 was amplified from plasmid DNA (ThermoScientific, 40045664) using primers indicated in Table S2 and inserted into the XhoI-XbaI sites of the CS2-eGFP vector. Mutations in the 300 bp E-cad promoter fragment were generated by stitching PCR using transition substitution for nucleotides.
Luciferase assays
HEK293T cells were seeded (4×104 cells/well) in DMEM/10% fetal bovine serum (FBS) in 24-well tissue culture plates and transfected with 400 ng plasmid DNA using Fugene6 (Promega) after 24 h. Cells were lysed in passive lysis buffer (Promega) 24 h after transfection and subjected to freeze-thaw at −80°C (Anderson et al., 2009). Luciferase activity was measured by reacting 20 µl lysate with 50 µl luciferase assay buffer (Promega) using a Fluostar Optima microplate reader (BMG Labtech), with normalization to β-galactosidase activity using the FluoReporter lacZ/galactosidase quantitation kit (Life Technologies). Experiments were performed in triplicate and repeated five times. Further details can be found in supplementary Materials and Methods.
Real-time and semiquantitative PCR
Briefly, total RNA (250 ng) from mouse pancreata was isolated (RNeasy Micro Kit, Qiagen) and treated with DNAse. For RT-PCR, cDNA was generated (iScript cDNA synthesis kit, Bio-Rad) and 1 µl (1/100th bud) of the cDNA and 1 µl MMTaq was used with 0.5 µl primer (20 nM) for analysis. Individual reactions were stopped at 30, 33 and 36 cycles, to assess levels within the linear range (water blanks stopped at 36th cycle). For qPCR, 1 μl cDNA was generated (SuperScript II, Invitrogen) and used for analysis (Power SybrGreen Master Mix, Applied Biosystems on CFX96, Bio-Rad, using 95°C, 30 s; 60°C, 30 s; 72°C, 30 s; 35 cycles). Fluorescence was measured at 72°C. Transcript levels were assessed by threshold cycle (Ct) calibrated to a standard curve generated for each assay using a five-step 1:5 dilution curve of WT mouse E11.5 pancreas and lung cDNA, and were normalized to cyclophilin (Das et al., 2013). All primers listed in Table S3. Data for RT-PCR were collected from three individual pancreatic buds per genotype, in triplicate, and data for qPCR from five individual isolated buds per genotype, in triplicate (6-12 repeats for each gene analyzed). For further details of methods used, see supplementary Materials and Methods.
Transfection for immunofluorescent staining
HEK293T cells were seeded at 1.6×105 cells/well in DMEM/10% FBS onto coverslips coated with fibronectin in six-well tissue-culture plates, then transfected with 1600 ng plasmid DNA (CS2GFP or CS2GFP-Pdx1). After 24 h, medium was removed, cells were fixed in 4% PFA/PBS, washed in 0.1% NP40/PBS (PBSN), blocked in 5% serum for 30 min, incubated for 1 h with primary antibodies (mouse anti-E-cadherin and chicken anti-GFP, Table S1). Cells were rinsed in PBSN, incubated with secondary antibodies (Invitrogen Alexa Fluor 555 donkey anti-mouse and Alexa Fluor 488 goat anti-chicken) for 1 h, washed in PBSN, processed and visualized as above. E-cadherin levels were quantified using the mean gray value function in ImageJ, on 150 individual transfected cells, obtained from three separate transfection and staining assays in which three replicate wells were used for each transfection type. Further details can be found in supplementary Materials and Methods.
Statistics
Data are presented as means+s.e.m. Quantifications were performed on multiple sections from at least three pancreatic buds for each genotype, at each stage. We show representative images chosen from the 5-10 sections analyzed from each bud (each section spans the entire bud diameter). All statistical analysis was performed using two-tailed, unpaired, Student's t-test in GraphPad Prism software. P<0.05 was considered statistically significant.
Acknowledgements
Thanks to the Johnson and MacDonald labs at UT Southwestern Medical Center for aliquots of Ptf1a antibody, as well as the Olson lab for CS2-GFP and pGL3 vectors and HEK293T cells. We are grateful to the Johnson, MacDonald, Olson, Cleaver and Carroll labs for invaluable discussions and assistance.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
All experiments were performed by L.M.-S. O.C. supervised the overall project and contributed to the analysis. L.M.-S. and O.C. wrote the manuscript.
Funding
This work was supported by the National Institutes of Health (NIH) [3R01DK079862 and 5F31DK092098 to L.M.-S. and NIH R01 grant DK079862-01 to O.C.]. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.126755/-/DC1
References
- Ahlgren U., Jonsson J. and Edlund H. (1996). The morphogenesis of the pancreatic mesenchyme is uncoupled from that of the pancreatic epithelium in IPF1/PDX1-deficient mice. Development 122, 1409-1416. [DOI] [PubMed] [Google Scholar]
- Ahlgren U., Pfaff S. L., Jessell T. M., Edlund T. and Edlund H. (1997). Independent requirement for ISL1 in formation of pancreatic mesenchyme and islet cells. Nature 385, 257-260. 10.1038/385257a0 [DOI] [PubMed] [Google Scholar]
- Anderson D. M., Beres B. J., Wilson-Rawls J. and Rawls A. (2009). The homeobox gene Mohawk represses transcription by recruiting the sin3A/HDAC co-repressor complex. Dev. Dyn. 238, 572-580. 10.1002/dvdy.21873 [DOI] [PubMed] [Google Scholar]
- Arda H. E., Benitez C. M. and Kim S. K. (2013). Gene regulatory networks governing pancreas development. Dev. Cell 25, 5-13. 10.1016/j.devcel.2013.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard G. B., Murugesu S., Adams R. J., Martinez-Arias A. and Gorfinkiel N. (2010). Cytoskeletal dynamics and supracellular organisation of cell shape fluctuations during dorsal closure. Development 137, 2743-2752. 10.1242/dev.045872 [DOI] [PubMed] [Google Scholar]
- Cortijo C., Gouzi M., Tissir F. and Grapin-Botton A. (2012). Planar cell polarity controls pancreatic beta cell differentiation and glucose homeostasis. Cell Rep. 2, 1593-1606. 10.1016/j.celrep.2012.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amour K. A., Agulnick A. D., Eliazer S., Kelly O. G., Kroon E. and Baetge E. E. (2005). Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat. Biotechnol. 23, 1534-1541. 10.1038/nbt1163 [DOI] [PubMed] [Google Scholar]
- Das A., Tanigawa S., Karner C. M., Xin M., Lum L., Chen C., Olson E. N., Perantoni A. O. and Carroll T. J. (2013). Stromal-epithelial crosstalk regulates kidney progenitor cell differentiation. Nat. Cell Biol. 15, 1035-1044. 10.1038/ncb2828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deramaudt T. B., Sachdeva M. M., Wescott M. P., Chen Y., Stoffers D. A. and Rustgi A. K. (2006). The PDX1 homeodomain transcription factor negatively regulates the pancreatic ductal cell-specific keratin 19 promoter. J. Biol. Chem. 281, 38385-38395. 10.1074/jbc.M605891200 [DOI] [PubMed] [Google Scholar]
- Ebrahim S., Fujita T., Millis B. A., Kozin E., Ma X., Kawamoto S., Baird M. A., Davidson M., Yonemura S., Hisa Y. et al. (2013). NMII forms a contractile transcellular sarcomeric network to regulate apical cell junctions and tissue geometry. Curr. Biol. 23, 731-736. 10.1016/j.cub.2013.03.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gragnoli C., Stanojevic V., Gorini A., Von Preussenthal G. M., Thomas M. K. and Habener J. F. (2005). IPF-1/MODY4 gene missense mutation in an Italian family with type 2 and gestational diabetes. Metabolism 54, 983-988. 10.1016/j.metabol.2005.01.037 [DOI] [PubMed] [Google Scholar]
- Guo L., Inada A., Aguayo-Mazzucato C., Hollister-Lock J., Fujitani Y., Weir G. C., Wright C. V. E., Sharma A. and Bonner-Weir S. (2013). PDX1 in ducts is not required for postnatal formation of beta-cells but is necessary for their subsequent maturation. Diabetes 62, 3459-3468. 10.2337/db12-1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale M. A., Kagami H., Shi L., Holland A. M., Elsässer H.-P., Hammer R. E. and MacDonald R. J. (2005). The homeodomain protein PDX1 is required at mid-pancreatic development for the formation of the exocrine pancreas. Dev. Biol. 286, 225-237. 10.1016/j.ydbio.2005.07.026 [DOI] [PubMed] [Google Scholar]
- Hick A.-C., van Eyll J. M., Cordi S., Forez C., Passante L., Kohara H., Nagasawa T., Vanderhaeghen P., Courtoy P. J., Rousseau G. G. et al. (2009). Mechanism of primitive duct formation in the pancreas and submandibular glands: a role for SDF-1. BMC Dev. Biol. 9, 66 10.1186/1471-213X-9-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland A. M., Hale M. A., Kagami H., Hammer R. E. and MacDonald R. J. (2002). Experimental control of pancreatic development and maintenance. Proc. Natl. Acad. Sci. USA 99, 12236-12241. 10.1073/pnas.192255099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horne-Badovinac S., Lin D., Waldron S., Schwarz M., Mbamalu G., Pawson T., Jan Y.-N., Stainier D. Y. R. and Abdelilah-Seyfried S. (2001). Positional cloning of heart and soul reveals multiple roles for PKC lambda in zebrafish organogenesis. Curr. Biol. 11, 1492-1502. 10.1016/S0960-9822(01)00458-4 [DOI] [PubMed] [Google Scholar]
- Jayasinghe A. K., Crews S. M., Mashburn D. N. and Hutson M. S. (2013). Apical oscillations in amnioserosa cells: basolateral coupling and mechanical autonomy. Biophys. J. 105, 255-265. 10.1016/j.bpj.2013.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson J., Carlsson L., Edlund T. and Edlund H. (1994). Insulin-promoter-factor 1 is required for pancreas development in mice. Nature 371, 606-609. 10.1038/371606a0 [DOI] [PubMed] [Google Scholar]
- Kesavan G., Sand F. W., Greiner T. U., Johansson J. K., Kobberup S., Wu X., Brakebusch C. and Semb H. (2009). Cdc42-mediated tubulogenesis controls cell specification. Cell 139, 791-801. 10.1016/j.cell.2009.08.049 [DOI] [PubMed] [Google Scholar]
- Khoo C., Yang J., Weinrott S. A., Kaestner K. H., Naji A., Schug J. and Stoffers D. A. (2012). Research resource: the pdx1 cistrome of pancreatic islets. Mol. Endocrinol. 26, 521-533. 10.1210/me.2011-1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp J. L., Dubois C. L., Schaffer A. E., Hao E., Shih H. P., Seymour P. A., Ma J. and Sander M. (2011). Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development 138, 653-665. 10.1242/dev.056499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroon E., Martinson L. A., Kadoya K., Bang A. G., Kelly O. G., Eliazer S., Young H., Richardson M., Smart N. G., Cunningham J. et al. (2008). Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol. 26, 443-452. 10.1038/nbt1393 [DOI] [PubMed] [Google Scholar]
- Lienkamp S. S., Liu K., Karner C. M., Carroll T. J., Ronneberger O., Wallingford J. B. and Walz G. (2012). Vertebrate kidney tubules elongate using a planar cell polarity-dependent, rosette-based mechanism of convergent extension. Nat. Genet. 44, 1382-1387. 10.1038/ng.2452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn F. C., Smith S. B., Wilson M. E., Yang K. Y., Nekrep N. and German M. S. (2007). Sox9 coordinates a transcriptional network in pancreatic progenitor cells. Proc. Natl. Acad. Sci. USA 104, 10500-10505. 10.1073/pnas.0704054104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lysy P. A., Weir G. C. and Bonner-Weir S. (2013). Making beta cells from adult cells within the pancreas. Curr. Diab. Rep. 13, 695-703. 10.1007/s11892-013-0400-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A. C. and Goldstein B. (2014). Apical constriction: themes and variations on a cellular mechanism driving morphogenesis. Development 141, 1987-1998. 10.1242/dev.102228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A. C., Kaschube M. and Wieschaus E. F. (2009). Pulsed contractions of an actin-myosin network drive apical constriction. Nature 457, 495-499. 10.1038/nature07522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason F. M. and Martin A. C. (2011). Tuning cell shape change with contractile ratchets. Curr. Opin. Genet. Dev. 21, 671-679. 10.1016/j.gde.2011.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offield M. F., Jetton T. L., Labosky P. A., Ray M., Stein R. W., Magnuson M. A., Hogan B. L. and Wright C. V. (1996). PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development 122, 983-995. [DOI] [PubMed] [Google Scholar]
- Ohlsson H., Karlsson K. and Edlund T. (1993). IPF1, a homeodomain-containing transactivator of the insulin gene. EMBO J. 12, 4251-4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver-Krasinski J. M., Kasner M. T., Yang J., Crutchlow M. F., Rustgi A. K., Kaestner K. H. and Stoffers D. A. (2009). The diabetes gene Pdx1 regulates the transcriptional network of pancreatic endocrine progenitor cells in mice. J. Clin. Invest. 119, 1888-1898. 10.1172/JCI37028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovcharenko I., Nobrega M. A., Loots G. G. and Stubbs L. (2004). ECR Browser: a tool for visualizing and accessing data from comparisons of multiple vertebrate genomes. Nucleic Acids Res. 32, W280-W286. 10.1093/nar/gkh355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliuca F. W., Millman J. R., Gürtler M., Segel M., Van Dervort A., Ryu J. H., Peterson Q. P., Greiner D. and Melton D. A. (2014). Generation of functional human pancreatic beta cells in vitro. Cell 159, 428-439. 10.1016/j.cell.2014.09.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan F. C. and Wright C. (2011). Pancreas organogenesis: from bud to plexus to gland. Dev. Dyn. 240, 530-565. 10.1002/dvdy.22584 [DOI] [PubMed] [Google Scholar]
- Pan F. C., Bankaitis E. D., Boyer D., Xu X., Van de Casteele M., Magnuson M. A., Heimberg H. and Wright C. V. E. (2013). Spatiotemporal patterns of multipotentiality in Ptf1a-expressing cells during pancreas organogenesis and injury-induced facultative restoration. Development 140, 751-764. 10.1242/dev.090159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petzold K. M., Naumann H. and Spagnoli F. M. (2013). Rho signalling restriction by the RhoGAP Stard13 integrates growth and morphogenesis in the pancreas. Development 140, 126-135. 10.1242/dev.082701 [DOI] [PubMed] [Google Scholar]
- Raum J. C., Soleimanpour S. A., Groff D. N., Coré N., Fasano L., Garratt A. N., Dai C., Powers A. C. and Stoffers D. A. (2015). Tshz1 regulates pancreatic beta-cell maturation. Diabetes 64, 2905-2914. 10.2337/db14-1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezania A., Bruin J. E., Arora P., Rubin A., Batushansky I., Asadi A., O'Dwyer S., Quiskamp N., Mojibian M., Albrecht T. et al. (2014). Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 32, 1121-1133. 10.1038/nbt.3033 [DOI] [PubMed] [Google Scholar]
- Schiesser J. V. and Wells J. M. (2014). Generation of beta cells from human pluripotent stem cells: are we there yet? Ann. N. Y. Acad. Sci. 1311, 124-137. 10.1111/nyas.12369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour P. A. and Sander M. (2011). Historical perspective: beginnings of the beta-cell: current perspectives in beta-cell development. Diabetes 60, 364-376. 10.2337/db10-1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih H. P., Wang A. and Sander M. (2013). Pancreas organogenesis: from lineage determination to morphogenesis. Annu. Rev. Cell Dev. Biol. 29, 81-105. 10.1146/annurev-cellbio-101512-122405 [DOI] [PubMed] [Google Scholar]
- Solon J., Kaya-Copur A., Colombelli J. and Brunner D. (2009). Pulsed forces timed by a ratchet-like mechanism drive directed tissue movement during dorsal closure. Cell 137, 1331-1342. 10.1016/j.cell.2009.03.050 [DOI] [PubMed] [Google Scholar]
- Stanger B. Z., Tanaka A. J. and Melton D. A. (2007). Organ size is limited by the number of embryonic progenitor cells in the pancreas but not the liver. Nature 445, 886-891. 10.1038/nature05537 [DOI] [PubMed] [Google Scholar]
- Stoffers D. A., Ferrer J., Clarke W. L. and Habener J. F. (1997a). Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat. Genet. 17, 138-139. 10.1038/ng1097-138 [DOI] [PubMed] [Google Scholar]
- Stoffers D. A., Zinkin N. T., Stanojevic V., Clarke W. L. and Habener J. F. (1997b). Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat. Genet. 15, 106-110. 10.1038/ng0197-106 [DOI] [PubMed] [Google Scholar]
- Swift G. H., Liu Y., Rose S. D., Bischof L. J., Steelman S., Buchberg A. M., Wright C. V. E. and MacDonald R. J. (1998). An endocrine-exocrine switch in the activity of the pancreatic homeodomain protein PDX1 through formation of a trimeric complex with PBX1b and MRG1 (MEIS2). Mol. Cell. Biol. 18, 5109-5120. 10.1128/MCB.18.9.5109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo A. K. K., Tsuneyoshi N., Hoon S., Tan E. K., Stanton L. W., Wright C. V. E. and Dunn N. R. (2015). PDX1 binds and represses hepatic genes to ensure robust pancreatic commitment in differentiating human embryonic stem cells. Stem Cell Rep. 4, 578-590. 10.1016/j.stemcr.2015.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villasenor A., Chong D. C. and Cleaver O. (2008). Biphasic Ngn3 expression in the developing pancreas. Dev. Dyn. 237, 3270-3279. 10.1002/dvdy.21740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villasenor A., Chong D. C., Henkemeyer M. and Cleaver O. (2010). Epithelial dynamics of pancreatic branching morphogenesis. Development 137, 4295-4305. 10.1242/dev.052993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wescott M. P., Rovira M., Reichert M., von Burstin J., Means A., Leach S. D. and Rustgi A. K. (2009). Pancreatic ductal morphogenesis and the Pdx1 homeodomain transcription factor. Mol. Biol. Cell 20, 4838-4844. 10.1091/mbc.E09-03-0203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright C. V., Schnegelsberg P. and De Robertis E. M. (1989). XlHbox 8: a novel Xenopus homeo protein restricted to a narrow band of endoderm. Development 105, 787-794. [DOI] [PubMed] [Google Scholar]
- Zhou Q., Law A. C., Rajagopal J., Anderson W. J., Gray P. A. and Melton D. A. (2007). A multipotent progenitor domain guides pancreatic organogenesis. Dev. Cell 13, 103-114. 10.1016/j.devcel.2007.06.001 [DOI] [PubMed] [Google Scholar]
- Zhou Q., Brown J., Kanarek A., Rajagopal J. and Melton D. A. (2008). In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 455, 627-632. 10.1038/nature07314 [DOI] [PMC free article] [PubMed] [Google Scholar]