

Abstract
Hydrocarbons are worldwide-distributed pollutants that disturb various ecosystems. The aim of this study was to characterize the short-lapse dynamics of soil microbial communities in response to hydrocarbon pollution and different bioremediation treatments. Replicate diesel-spiked soil microcosms were inoculated with either a defined bacterial consortium or a hydrocarbonoclastic bacterial enrichment and incubated for 12 weeks. The microbial community dynamics was followed weekly in microcosms using Illumina 16S rRNA gene sequencing. Both the bacterial consortium and enrichment enhanced hydrocarbon degradation in diesel-polluted soils. A pronounced and rapid bloom of a native gammaproteobacterium was observed in all diesel-polluted soils. A unique operational taxonomic unit (OTU) related to the Alkanindiges genus represented ∼0.1% of the sequences in the original community but surprisingly reached >60% after 6 weeks. Despite this Alkanindiges-related bloom, inoculated strains were maintained in the community and may explain the differences in hydrocarbon degradation. This study shows the detailed dynamics of a soil bacterial bloom in response to hydrocarbon pollution, resembling microbial blooms observed in marine environments. Rare community members presumably act as a reservoir of ecological functions in high-diversity environments, such as soils. This rare-to-dominant bacterial shift illustrates the potential role of a rare biosphere facing drastic environmental disturbances. Additionally, it supports the concept of “conditionally rare taxa,” in which rareness is a temporary state conditioned by environmental constraints.
INTRODUCTION
Soil microbial communities are key players in nutrient cycling and have an active role in the transformation and mineralization of natural compounds and pollutants (1). Microbial communities are largely affected by natural or anthropogenic disturbances, such as pH change (2), metal pollution (3), pesticide application (4), and hydrocarbon pollution (5). Hydrocarbons are organic pollutants of major concern due to their worldwide distribution, persistence, and toxicity. Biological treatments for the cleanup of hydrocarbon-polluted soils are cost-effective and environmentally sustainable (6). Bioremediation strategies include the manipulation of environmental variables in order to enhance hydrocarbon degradation by native microorganisms (biostimulation) or amendment with hydrocarbonoclastic strains (bioaugmentation). Microbial communities play a key role during bioremediation, either by directly degrading pollutants or interacting with inoculated strains.
Hydrocarbonoclastic bacteria belonging to the phyla Actinobacteria, Proteobacteria, and Firmicutes have widely been reported. After hydrocarbon pollution, bacteria from several genera, including Gordonia and Rhodococcus (Actinobacteria), Sphingomonas, Acinetobacter, and Pseudomonas (Proteobacteria), and Bacillus and Exiguobacterium (Firmicutes) are usually enriched (5–8). However, the magnitude and dynamics of microbial community shifts are dependent on the environment (e.g., soil type, freshwater, and seawater), pollution time, hydrocarbon composition (e.g., different crude distillates and specific hydrocarbon), and remediation treatment (9–12).
Obligate hydrocarbonoclastic bacteria (OHCB) constitute a special marine group that is able to use a reduced substrate spectrum as a carbon source, particularly hydrocarbons. OHCB includes the Gammaproteobacteria genera Alcanivorax, Thalassolituus, Marinobacter, Cycloclasticus, Oleiphilus, and Oleispira (13). OHCB illustrate the adaptation of microbial communities to hydrocarbon pollution. OHCB members are present at low levels in pristine marine environments, but their populations increase rapidly after oil spills (9, 13–15). Last decade, high-throughput sequencing revealed that most environments bear a very large proportion of underrepresented taxa, called collectively the rare biosphere or microbial seed bank (16). Rare taxa are important for the maintenance of diversity, and they contribute to pulses of ecosystem activity (17). Additionally, their populations may vary significantly after environmental disturbances and contribute to changes in microbial community diversity, leading to the concept of “conditionally rare taxa” (18). OHCB represent an example of conditionally rare taxa, whose blooms reflect trait selection after drastic environmental changes. In oil spills, hydrocarbons are a huge source of carbon used only by few taxa that may return to low relative abundance in the absence of oil.
In soil, only small bacterial shifts have been reported in response to environmental disturbances. The dynamics of hydrocarbon-responding taxa are generally more diverse, less pronounced, soil dependent, and case specific (5–7, 11, 12). However, detailed temporal studies are missing. In the present study, the short-term soil microbial community dynamics during hydrocarbon biodegradation was examined. We assessed two bioaugmentation strategies using hydrocarbonoclastic strains previously isolated in our laboratory in order to determine the fate of inoculated strains and their relationship with native microbial communities. Sequencing of the 16S rRNA gene provided temporal snapshots of the microbial community structure and dynamics, revealing a bloom of a native rare member and the maintenance of the inoculated strains in low abundance.
MATERIALS AND METHODS
Bacterial strains and enrichments.
Pseudomonas sp. strain DN34, Pseudomonas sp. strain DN36, Acinetobacter sp. strain AF53, and Acinetobacter sp. strain AA64 were isolated from two soil samples collected next to a petroleum refinery located at the Aconcagua River estuary (32°55′S 71°29′W) in December 2008 and February 2010. Bacterial strains were isolated by enrichment in Bushnell-Haas (BH) broth (1 g liter−1 KH2PO4, 1 g liter−1 K2HPO4, 1 g liter−1 NH4NO3, 0.2 g liter−1 MgSO4, 0.02 g liter−1 CaCl2, 0.05 g liter−1 FeCl3 [pH 7.0]) using a polycyclic aromatic hydrocarbon (PAH) as the sole carbon source at 28°C and with 100 mg liter−1 cycloheximide in order to inhibit eukaryotic cell growth. Strains DN34 and DN36 were isolated after enrichment using naphthalene (0.5 g liter−1) as a sole carbon source (19). Strains AF53 and AA64 were isolated after enrichment using fluorene (0.25 g liter−1) and anthracene (0.25 g liter−1), respectively (19), as a sole carbon source. Isolates were obtained by spread plating on BH agar with each hydrocarbon and subsequently purified by streak plating. Bacterial isolates were stored in 15% (vol/vol) glycerol at −80°C.
The use of a hydrocarbon as a sole carbon source was determined in 96-well microplates, as described previously (20), with modifications. Briefly, each isolate was grown in BH broth with one hydrocarbon as the sole carbon source. Growth was determined spectrophotometrically by measuring the reduction of the tetrazolium salt 2,3,5-triphenyl-2H-tetrazolium chloride into a colored formazan by bacterial metabolic activity. For growth curves, isolates were grown in BH broth with succinate (10 mM), pyruvate (10 mM), glucose (5 mM), or sucrose (2.5 mM). Diesel tolerance was tested in 20 g of sterile soil in the absence or presence of diesel (50, 100, and 150 g kg−1 of dry soil), inoculated with each isolate (∼1011 CFU g−1 of dry soil), and incubated at room temperature. CFU were counted in tryptic soy agar (TSA) plates. Strain identification was achieved by 16S rRNA gene PCR amplification with the 27f and 1492r primers and sequencing (3). Phylogenetic trees were based on comparative 16S rRNA gene sequences retrieved from the NCBI database. Sequences were aligned using Clustal W, and phylogenetic trees were constructed with the maximum-likelihood method using the MEGA6 software (www.megasoftware.net). Branch consistency was calculated by bootstrap estimations (1,000 replicates).
Bacterial enrichments (ENRs) were obtained from a soil sample collected in January 2010 by 10 consecutive subcultures (1-week intervals) in BH broth with diesel (10 g liter−1) and cycloheximide (100 mg liter−1) at 30°C. The 10th subculture was stored in 15% (vol/vol) glycerol at −80°C. The identification of bacteria composing the enrichments was done by Illumina sequencing, as described below.
Soil microcosm experiments.
Surface soil (0 to 15 cm depth) was collected from a site without hydrocarbon pollution history and with abundant vegetation located in Laguna Verde (33°11′S, 71°71′W), Valparaíso, Chile, in September 2012. Stones, vegetation fragments, insects, and earthworms were removed using gloves. The soil was dried at room temperature for 1 day, homogenized, and sieved through 4.75-mm and 2-mm meshes. Physicochemical soil properties were determined at the soil laboratory at the Agronomy Faculty, Pontificia Universidad Católica de Valparaíso, Quillota, Chile, according to protocols established by the Chilean National Institute of Agriculture and Livestock Research (INIA) (21, 22). Briefly, organic matter was quantified by weight loss at 360°C. Nitrogen content was determined by digestion with sulfuric acid, salicylic acid, potassium sulfate, copper sulfate, and titanium dioxide and titration. Potassium and sodium contents were determined by extraction with ammonium acetate and atomic emission spectroscopy (AES). Zinc, manganese, iron, and copper were quantified by diethylenetriaminepentaacetic acid (DTPA) extraction and atomic absorption spectroscopy (AAS) (21). Soil pH and electrical conductivity were determined in an aqueous soil suspension with a pH meter and conductometer, respectively. Calcium and magnesium contents were determined by extraction with ammonium acetate and AAS. Boron was extracted with calcium chloride and the content determined by the azomethine H colorimetric method. Phosphorus was extracted with calcium bicarbonate and the content determined by the molybdenum blue method (22).
The microcosm setup comprised five experimental sets in triplicate: abiotic control (CA), nonpolluted control (C0), diesel-polluted control (C1), diesel-polluted and bioaugmented with the defined bacterial consortium (E1), and diesel-polluted and bioaugmented with bacterial enrichments (E2) (Table 1). Each microcosm contained 300 g (dry weight) of soil and was sampled weekly for 12 weeks. For the CA group, the soil was autoclaved three times, incubated with sterile Milli-Q water for 48 h at 30°C, and autoclaved twice. Sterility was assessed by the absence of microbial growth on TSA plates. CA, C1, E1, and E2 soils were spiked with diesel (30 g kg−1 dry soil) at time zero and after 8 weeks. The bacterial consortium and enrichments were inoculated in E1 and E2 soils at time zero and after 3 weeks. For E1, all four isolates were grown independently in BH broth with succinate (40 mM) at 30°C. The four cultures were pulled in equal bacterial numbers together and washed 3 times with sterile Milli-Q water. A bacterial suspension (108 to 109 CFU g−1 dry soil) was added to the E1 microcosms. For E2, an ENR glycerol stock was grown in BH broth with diesel (10 g liter−1) at 30°C. The cells were washed, suspended in sterile Milli-Q water, and inoculated (108 to 109 CFU g−1 dry soil) at weeks 0 (ENR.0) and 3 (ENR.3). Moisture was maintained at 19 to 24% by adjustment with sterile Milli-Q water. To mimic natural conditions, temperature was not controlled and ranged between 15 and 24°C.
TABLE 1.
Experimental design of microcosm assays
| Microcosm | Treatment | Diesela | Inoculumb | Objective | GC-FID | Illumina |
|---|---|---|---|---|---|---|
| CA | Abiotic control | + | − | Control diesel loss by abiotic factors | + | − |
| C0 | Without treatment | − | − | Control temporal changes in community | − | + |
| C1 | Polluted | + | − | Control degradation by native community | + | + |
| E1 | Bioaugmented 1 (consortium) | + | + | Additional degradation by four isolates | + | + |
| E2 | Bioaugmented 2 (enrichment) | + | + | Additional degradation by enrichments | + | + |
Diesel amendments at 0 and 8 weeks.
Inoculations at 0 and 3 weeks.
DNA extraction and sequencing of 16S rRNA gene.
Total community DNA was prepared from 0.5 g of dry soil using the PowerSoil DNA isolation kit (Mo Bio, Carlsbad, CA, USA), according to the manufacturer's instructions. Mechanical disruption was performed with the FastPrep-24 bead beater (MP Biomedicals, Santa Ana, CA, USA). The V4 region of the 16S rRNA gene was PCR amplified with the primers 515F (GTGCCAGCMGCCGCGGTAA) and 806R (GGACTACHVGGGTWTCTAAT) and sequenced on the MiSeq (Illumina) platform at the Argonne National Laboratory (Lemont, IL, USA), as described previously (23). In total, 183 samples were multiplexed in one sequencing run, including replicate soil microcosms and the ENR.0 and ENR.3 samples.
Microbial community analysis.
Forward and reverse sequences in raw data were assembled with PANDAseq (24), yielding sequences of 253 ± 5 nucleotides (nt) in length. After that, QIIME was used throughout the analyses (25). Sequences were demultiplexed, quality filtered, and grouped into 97% identity operational taxonomic units (OTUs) using the subsampled open-reference OTU-picking strategy (26). Chimeric sequences were checked with the script identify_chimeric_seqs.py from MacQIIME 1.9.0. OTU clustering and taxonomic assignment were achieved with uclust (27) and RDP Classifier (28), respectively, against the Greengenes 13_8 database (29). A phylogenetic neighbor-joining tree (30) based on PyNAST alignment of OTU sequences (31) was used to calculate phylogenetic α- and β-diversity metrics. Finally, the generated OTU table was the input for subsequent analyses. The same pipeline was applied to soil microcosms and the ENR.0 and ENR.3 community samples.
In order to study the OTUs corresponding to inoculated strains, the 16S rRNA gene sequences of the four isolates and ENR were analyzed by BLAST in the E1 and E2 data sets, respectively. Best matches (>99% identity) were filtered out and their sequences were subjected to a BLAST search against the C0 and C1 data sets. Sequences matching the C0 or C1 data sets (i.e., native community members) were discarded.
For the identification of the dominant OTU, sequences belonging to Moraxellaceae were extracted from the OTU table. Sequences classified as f__Moraxellaceae;g__, f__Moraxellaceae;g__Acinetobacter, and f__Moraxellaceae;g__Alkanindiges were filtered out, keeping only the ones classified as f__Moraxellaceae;Other. Sequences of the dominant OTU in this reduced data set were extracted from the representative set of sequences in the QIIME files (rep_set.fna) and identified by BLAST at the NCBI site.
Quantification of diesel components.
In a 50-ml polypropylene tube, 1 g of soil was mixed with 1 g of anhydrous (NH4)2SO4, 2 g of gravel (2- to 5-mm diameter, previously cleansed and washed with solvent), and 10 ml of n-hexane-dichloromethane (1:3 [vol/vol]). The tubes were shaken vigorously by hand until complete homogenization, incubated for 15 min in orbital agitation (vortexing) at maximum speed, sonicated for 15 min in an ultrasonic bath, and centrifuged at 4,000 × g for 10 min. The supernatant was transferred to a new tube. Extraction was repeated twice, and the three fractions were pulled together, filtered through a filter paper, and stored in hermetically sealed vials at −20°C until analysis. Moisture was measured in a moisture meter (Sartorius MA35) to determine the dry weight. Quantification was performed with a gas chromatograph coupled to a flame ionization detector (GC-FID) (PerkinElmer Clarus 680). Each extract (0.5 μl) was injected into a DB5 column (30 m, 0.25-mm diameter; PerkinElmer) with helium as the carrier gas. The injector and detector temperatures were 320°C and 295°C, respectively. Flow-pulse injection was used to increase sensibility. The flow rate was 10 ml min−1, and the flow was splitless during injection. After 0.5 min, a 1:30 split was opened and the flow decreased to 2 ml min−2 until it reached 1 ml min−1 and was held constant. The thermal profile was 5 min at 50°C, first ramp of 20°C min−1 until 150°C, second ramp of 5°C min−1 until 280°C, third ramp of 50°C min−1 until 310°C, and maintenance at 310°C for 3 min. Standard curves were constructed with the DRO-1 standard (Dr. Ehrenstorfer GmbH, Augsburg, Germany), which includes n-alkanes of C10–C25 chain lengths.
Nucleotide sequence accession numbers.
Sequences were submitted to the NCBI nucleotide database under the accession numbers KT368948 (Pseudomonas sp. DN34), KT368949 (Pseudomonas sp. DN36), KT368950 (Acinetobacter sp. AF53), and KT368951 (Acinetobacter sp. AA64). Soil sample sequences were submitted to the NCBI BioProject database with the project identification no. PRJNA291526 and the sample accession numbers SAMN03960136 to SAMN03960301 (microcosm soil samples), SAMN03963309 (ENR.0), and SAMN03963310 (ENR.3).
RESULTS AND DISCUSSION
Bacterial isolates and enrichments.
In this study, two bioaugmentation strategies were applied in highly diesel-polluted soil: (i) inoculation with a defined bacterial consortium and (ii) inoculation with a bacterial enrichment. The bacterial consortium was composed of Pseudomonas sp. strains DN34 and DN36 and Acinetobacter strains AF53 and AA64. Pseudomonas sp. strains DN34 and DN36 are phylogenetically related to Pseudomonas stutzeri species (see Fig. S1 in the supplemental material). Acinetobacter sp. AF53 is phylogenetically related to Acinetobacter venetianus RAG-1, whereas Acinetobacter sp. AA64 is closely related to Acinetobacter tandoii DSM 14970 (see Fig. S1). These four hydrocarbon-degrading strains used short-chain (n-octane), long-chain (n-hexadecane), and cyclic (cyclohexane) alkanes for growth as sole carbon sources (see Table S1 in the supplemental material). In addition, both Pseudomonas strains degrade naphthalene, fluorene, and phenanthrene, whereas Acinetobacter sp. strain AA64 degrades anthracene (see Table S1). These four bacterial strains tolerated up to 15 g kg−1 of diesel in soil (see Fig. S2 in the supplemental material). Diverse Pseudomonas strains capable of degrading alkanes and polycyclic aromatic hydrocarbons (PAHs) have been characterized (32). Acinetobacter strains involved in the degradation of alkanes and aromatic compounds, such as salicylate and anilines, have been reported (32, 33). However, only a few Acinetobacter strains capable of degrading PAHs (naphthalene, fluorene, and biphenyl) have been described (33, 34).
The compositions of the first (ENR.0) and second (ENR.3) bacterial enrichments used for inoculation were determined by V4 region sequencing of the 16S rRNA gene. Two dominant Pseudomonadaceae populations comprised >98% of ENR.0, whereas ENR.3 was composed mainly of Acinetobacter (83.9%), Rhizobiaceae (11.1%), and Moraxellaceae (1.9%) (see Table S2 in the supplemental material). Although several hydrocarbonoclastic taxa may be present in a polluted environment, bacteria with higher growth rates may become dominant during enrichment, and the slow-growing bacteria may be eventually washed out. The absence of Actinobacteria in these enrichments is probably related to their low growth rates under these conditions (35, 36). Illumina sequencing of the PCR 16S rRNA gene amplicons was useful for the identification of dominant ENR.0 and ENR.3 taxa and the analyses of their dynamics during soil bioaugmentation.
Microbial community dynamics during diesel pollution and bioaugmentation.
For microbial community analyses, the V4 region of 16S rRNA genes was PCR amplified and sequenced. Although quantification by a PCR amplicon sequencing approach is widely used in microbial ecology (16, 18, 23), it may bias community composition based on the amplification efficiency of the primers. After assembly of forward and reverse sequences, demultiplexing, and OTU picking (97% similarity), the data set consisted of 3,700,542 sequences distributed in 183 samples across the four experimental sets (C0, C1, E1, and E2, each in triplicate). Samples were rarefied to 9,600 sequences, and the 17 samples with <9,600 sequences were discarded, yielding 3,698,288 sequences distributed in 67,155 OTUs present in 166 samples. Rarefaction curves did not survey the full extent of the diversity, as measured by either richness or phylogenetic diversity. However, both metrics show higher α-diversity in nonpolluted (C0) than in replicate diesel-polluted (C1, E1, and E2) microcosms (see Fig. S3 in the supplemental material). Pronounced decreases in richness and phylogenetic diversity during the first 6 weeks in diesel-polluted sets were observed (Fig. 1). The fast decrease in α-diversity may indicate the selection of the few community members able to degrade or tolerate hydrocarbons due to the toxicity of diesel components or their degradation products (5, 7).
FIG 1.
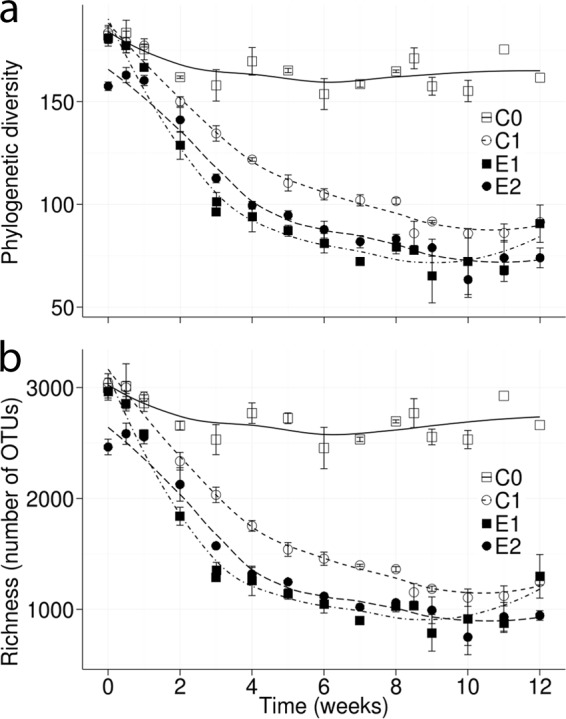
Effect of diesel pollution and bioremediation treatments on microbial community α-diversity dynamics in replicate soil microcosms. Diesel amendment produced a drastic drop in α-diversity in polluted microcosms C1, E1, and E2, measured as phylogenetic diversity (a) and richness (b). Diversity in the unpolluted microcosm C0 remained close to the initial values. Additional microbial community structure changes were not observed after the second diesel amendment at week 8. Each point is an average of the results from three independent experiments. The vertical bars indicate standard deviations.
The β-diversity dynamics was assessed by principal coordinate analysis (PCoA) based on weighted UniFrac distance (Fig. 2), unweighted UniFrac distance (see Fig. S4a in the supplemental material), and Bray-Curtis dissimilarity measurements (see Fig. S4b in the supplemental material). Similar patterns were observed in all cases: communities from diesel-amended C1, E1, and E2 soils diverged from the nonpolluted C0 soil community in a time-dependent manner. The three metrics used are based on different data aspects and can be more or less effective for revealing gradients or clustering patterns (37). UniFrac calculates the fraction of the branch length unique to a sample in a phylogenetic tree constructed from each pair of samples, providing a phylogenetic metric of community distance (2). The unweighted UniFrac measurement is a qualitative distance that accounts for only the presence or absence of taxa. The higher similarity of communities observed at time zero is expected (see Fig. S4a), given that inoculated strains did not add novel divergent OTUs to the E1 and E2 data sets. On the other hand, weighted UniFrac distance and Bray-Curtis dissimilarity measurements are quantitative, i.e., they account for the relative abundance of each OTU. In both cases, the proportion of inoculated strains affected the measurement, resulting in more distant initial communities. Taking into account both UniFrac and Bray-Curtis measurements, PCoA plots suggest that differences in microbial dynamics are based exclusively on the presence or absence of diesel.
FIG 2.
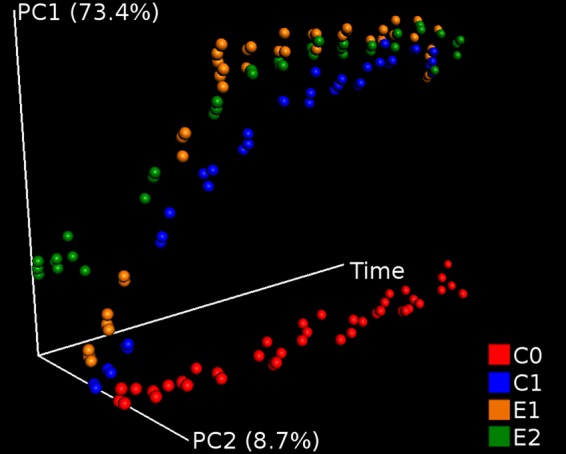
Effect of diesel pollution and bioremediation treatments on microbial community β-diversity dynamics in replicate soil microcosms. A PCoA was based on weighted UniFrac distances among the community composition of the samples. Communities from diesel-polluted microcosms C1, E1, and E2 displayed a different dynamic than that of the community from the unpolluted microcosm C0.
Proteobacteria and Actinobacteria were the dominant phyla in the soil bacterial communities at time zero, both representing >50% of the sequences in all experimental sets (see Fig. S5a in the supplemental material). Each phylum represented approximately 25% of the sequences in nonpolluted C0 soil. In replicate diesel-polluted C1 microcosms, Proteobacteria and Actinobacteria represented 24% and 38% of the sequences, respectively. In replicate bioaugmented microcosms, the phyla Proteobacteria and Actinobacteria represented 29% and 34% of the in E1 soils, respectively, whereas these phyla represented 48% and 26% of the E2 soils, respectively. Because all replicate microcosms were prepared with the same soil, the differences observed could be explained by the specific treatment for each microcosm. Higher proteobacterial proportions in E1 and E2 replicate microcosms are probably associated with inoculated strains, especially in E2, in which the Pseudomonadaceae of ENR.0 represented ∼30% of the sequences. The presence of diesel differentiates C1 and C0 soil microcosms and also may bias DNA extraction and PCR amplification (38). C0 soil microcosms were useful for assessing the temporal microbial community changes not attributable to diesel or bacterial inoculation. In all replicate microcosms at time zero, Acidobacteria, Planctomycetes, and Verrucomicrobia sequences represented 5 to 11% of the sequences, whereas Bacteroidetes, Chloroflexi, Firmicutes, and Gemmatimonadetes represented <5% of the sequences. Archaeal sequences were <1% (<100 of 9,600 sequences) in all samples, and they could not be analyzed consistently. The primers amplified bacterial and archaeal 16S rRNA gene V4 regions (39), although a bias toward a more efficient amplification of bacterial sequences cannot be discarded (40). Archaea are in general less represented in soils, ranging from 0 to 10% of the bacterial/archaeal communities, as determined by either PCR-based techniques or microarrays (39, 41). Some archaea are able to use hydrocarbons as a carbon source (42), but their population usually decreases after pollution, and their ecological role regarding hydrocarbon degradation has scarcely been studied (41, 43).
From rare to dominant: a pronounced bloom of a native soil member.
A marked increase in the proportion of Proteobacteria occurred in diesel-polluted soils after the second week, whereas other phyla decreased, especially Actinobacteria (see Fig. S5a in the supplemental material). The relative abundance of Proteobacteria increased during incubation, but the phylogenetic diversity of this phylum decreased 2-fold in diesel-polluted soils (Fig. 3), indicating that only a few proteobacterial members were enriched. The members that increased belonged to the class Gammaproteobacteria (see Fig. S5b in the supplemental material), the order Pseudomonadales (see Fig. S5c in the supplemental material), and the family Moraxellaceae (see Fig. S5d in the supplemental material). The RDP Classifier performed the taxonomic assignment based on a confidence threshold of 0.8, which should retrieve an accurate assignment at the genus level for the V4 region (28). However, when RDP Classifier is not able to classify a sequence with 80% confidence, it assigns the upper taxonomic level to that sequence. Changing the confidence threshold did not improve classification accuracy (data not shown). In this case, the dominant OTU could not be classified with 80% confidence to any Moraxellaceae genus and was classified as f__Moraxellaceae;Other in QIIME's output (Fig. 4). Moraxellaceae was composed of 2,785 OTUs in the data set. Surprisingly, the entire bloom was due to a unique OTU, whereas the other 2,784 OTUs experienced only negligible variations over time. This OTU was represented by approximately 5 sequences per sample (<0.1%) in the original community in CA, C1, E1, and E2 soil microcosms at time zero and reached >5,000 sequences (>60%) at 6 weeks after diesel amendment (see Fig. S6 in the supplemental material). In order to further identify this dominant OTU, a representative sequence was extracted from the data set and identified by BLAST against the NCBI 16S rRNA sequence database. The best match was reached with Alkanindiges illinoisensis GTI MVAB Hex1T (ATCC PTA-4839T = DSM 15370T), with 97% identity. A. illinoisensis GTI MVAB Hex1T is an obligate hydrocarbonoclastic strain able to use several hydrocarbons and Tween surfactants as sole carbon sources, and it was isolated from an oilfield in Illinois (44). Alkanindiges species play an active role in biological foaming and the floating hydrophobic biomass in activated sludge from wastewater treatment plants (45). An increase in an Alkanindiges-related population after nutrient addition in landfarming treatment of subarctic soils polluted with aged hydrocarbons has been reported (46). The results of our study suggest that an Alkanindiges-related population may be enriched in soil after amendment with abundant hydrophobic hydrocarbons. Alkanindiges strains have also been isolated from lettuce plants (47) and clinical samples (48). In this study, the Alkanindiges strain observed after diesel amendment was originally present in unpolluted soil with abundant vegetation located in Laguna Verde at the south of Valparaíso, Chile. This study and previous reports suggest that Alkanindiges is a genus as ubiquitous as Acinetobacter. The isolation of this interesting blooming bacterium will be useful for characterizing its metabolic capabilities and studying its application in bioremediation.
FIG 3.
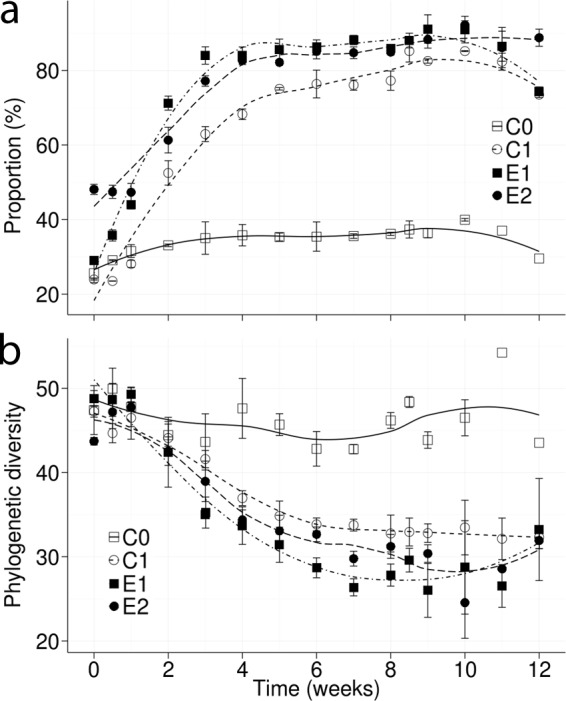
Effect of diesel amendment and bioremediation treatments on proteobacterial dynamics in replicate soil microcosms. The increase in relative community proportion (a) contrasted with the drop in phylogenetic diversity (b) in diesel-polluted microcosms C1, E1, and E2. Additional microbial community structure changes were not observed after the second diesel amendment at week 8. Each point is an average of the results from three independent experiments. The vertical bars indicate standard deviations.
FIG 4.

Effect of diesel amendment and bioremediation treatments on relative abundance of genus-level taxon dynamics in replicate soil microcosms. Diesel amendment at week 0 strongly altered the relative proportions of genera. Members belonging to the family Moraxellaceae rapidly increased in number in response to diesel. The second diesel amendment at week 8 did not introduce additional alterations to the microbial community structure.
Most environments are dominated by low-abundance taxa (i.e., the rare biosphere), in which rare is usually defined as members present in ≤0.1% of a microbial community (16, 49). It is well known that the microbial community structure can rapidly change across space and time, reflecting a fast growth or dispersal of rare or dormant taxa. This concept involves the idea that “everything is everywhere, but the environment selects” (16). A drastic environmental disturbance, such as pollution or fire, generates a selective sweep causing huge changes in community structure and diversity (50), which is in agreement with the phylogenetic diversity drop observed after diesel amendment (Fig. 3). Microbial community assembly after a disturbance can be driven by niche (i.e., selection of traits by environmental constraints) or neutral (i.e., dispersal of microorganisms from one environment to another) processes. Both are important, depending on the type and intensity of the disturbance and the ecosystem examined (50). However, when dispersal is limited (i.e., the microcosm is a closed environment), the niche process becomes obviously dominant. Therefore, the bacterial bloom observed in this study can be explained exclusively by trait selection, probably by the ability to use hydrocarbon as a carbon source at a high growth rate. Rare-to-abundant microbial shifts have been reported in aquatic environments in response to seasonal transition (51) or changes in salinity and dissolved organic carbon (52). Marine OHCB bloom after oil spills is a well-known phenomenon, in which specific taxa may bloom several days after hydrocarbon pollution (13). The model OHCB Alcanivorax borkumensis is present in low abundance in marine environments but may reach 80 to 90% of the hydrocarbonoclastic community after hydrocarbon pollution (14). This bacterial bloom phenomenon was described using culture-dependent techniques during the emblematic Exxon Valdez oil spill (15). However, the taxonomic and metabolic features of blooming bacteria were elucidated by only genome sequencing (14). The genera Alcanivorax and Thalassolituus are main alkane degraders, whereas Cycloclasticus has mainly been associated with PAH degradation. Cycloclasticus strains have been reported in seawater as part of the rare members that bloomed after phenanthrene addition and have become key hydrocarbon degraders (49). Therefore, OHCB are examples of conditionally rare taxa, a concept proposed based on the dynamics of rare taxa in other environments but not in soil (18). Soil microbial community dynamics in response to hydrocarbon pollution is more diverse and niche specific than microbial dynamics in seawater, probably due to the limited dispersal compared with that in marine environments. Different soil types may respond differentially following pollution with the same PAH (i.e., phenanthrene), whereas the same soil may develop different community structures in response to related PAHs (e.g., naphthalene, phenanthrene, and pyrene) (53, 54). The organic matter content is probably a major physicochemical soil feature inducing differential responses (55).
Most studies indicate that proteobacteria and actinobacteria are the major hydrocarbon degraders in soils. Sphingomonadaceae, Caulobacteraceae, and Rhodospirillaceae (Alphaproteobacteria), Alcaligenaceae, Comamonadaceae, and Burkholderiaceae (Betaproteobacteria), Pseudomonadaceae and Xanthomonadaceae (Gammaproteobacteria), and Nocardiaceae (Actinobacteria), among others, have been reported to be enriched during hydrocarbon degradation (5, 55, 56). Bacteroidetes may become enriched in creosote-contaminated soils, in which PAHs are main pollutants (7). However, all microbial shifts have been described in long-term periods, and the short-lapse dynamics have been overlooked. Ding et al. (54) analyzed the community 21 and 63 days after spiking with naphthalene (20 g kg−1 dry soil), whereas Bell et al. (55) analyzed the community composition 4 weeks after amendment with diesel (5.5 g kg−1 dry soil). Yang et al. (56) reported shifts in several genera in permafrost soils 8 weeks after amendment with diesel (300 g kg−1 dry soil). Nonetheless, the shifts were less pronounced, and none of the soils included Moraxellaceae members. A detailed report on short-lapse soil community dynamics affecting the selection of antibiotic-resistant bacteria showed that significant shifts occurred 2 to 5 weeks after manure amendments (57). Nonetheless, the shifts represented <10% of the total community in all cases. To our knowledge, this study is the first report of such a pronounced and rapid bloom of rare bacteria in soil. These results depict the role of rare members in the adaptation of microbial communities facing severe environmental disturbances.
Survival of inoculated strains and effectiveness of bioaugmentation strategies.
Diesel amendments were performed in CA, C1, E1, and E2 replicate soil microcosms at time zero and after 8 weeks. Bacterial consortium and enrichments (108 to 109 CFU g−1 dry soil) were inoculated in replicate soil microcosms at time zero and after 3 weeks in E1 and E2 soils, respectively (Table 1). Inoculation of the strains in the E1 and E2 microcosms was followed by filtering of the corresponding sequences from the data sets. The inoculated strains in E1 microcosms were monitored by pairs: the 16S rRNA V4 sequence from inoculated Acinetobacter isolates (strains AF53 and AA64 have identical V4 sequences) and the 16S rRNA V4 sequence from Pseudomonas isolates (strains DN34 and DN36 possess identical V4 sequences). Sequences corresponding to inoculated Acinetobacter and Pseudomonas spp. were close to ∼5% and 2% of the sequences, respectively (Fig. 5a). As expected, the second inoculation at the third week increased the proportions of Acinetobacter and Pseudomonas in the community. During the incubation, Acinetobacter isolates maintained close to initial values (5%) until week 12, whereas Pseudomonas isolates decreased close to 1% toward week 12.
FIG 5.
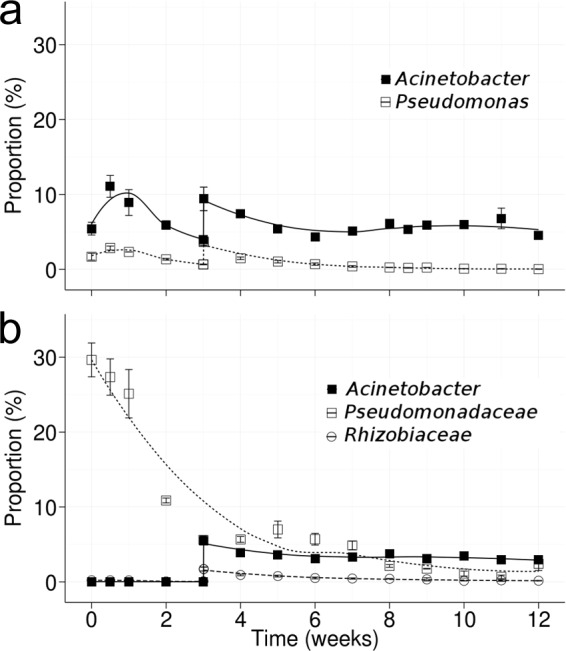
Dynamic of the inoculated bacterial strains in bioaugmented E1 and E2 microcosms. Strains were inoculated at weeks 0 and 3 and remained until week 12 in both E1 (a) and E2 (b) microcosms. Each point is an average of the results from three independent experiments. The vertical bars indicate standard deviations.
Inoculated strains in E2 soils corresponded to sequences matching enrichment cultures ENR.0 and ENR.3 but absent in C0 and C1 (i.e., native community members). The Pseudomonadaceae of ENR.0 represented approximately 30% of the sequences in E2 at time zero, but their proportion markedly decreased to 5% after 3 weeks and to 1 to 2% after 8 weeks (Fig. 5b). ENR.0 members were probably less tolerant to the diesel concentration used in the soil microcosms. An ENR.0 inoculum was prepared in minimal broth with 10 g liter−1 diesel, but when exposed to 30 g kg−1 diesel in soil, its members probably did not tolerate the higher diesel concentration, and their population rapidly decreased after diesel amendment. ENR.3 used in the second inoculation at week 3 was composed mainly of members of Acinetobacter and Rhizobiaceae. Acinetobacter sequences from ENR.3 represented close to 3% of the sequences until week 12, similar to the stable maintenance observed for Acinetobacter sp. strains AF53 and AA64 in the E1 microcosm (Fig. 5). Rhizobiaceae sequences were close to 2% at week 3 and decreased to 0.2% toward the 12th week.
Diesel (30 g kg−1 dry soil) was added to the CA, C1, E1, and E2 replicate microcosms at time zero and at week 8. Degradation of C11–C15 n-alkanes was more efficient in the E1 and E2 microcosms than in C1 soil after the first pollution (weeks 0 to 8). The remaining C11–C20 n-alkanes at the eighth week were significantly (P < 0.05) lower in bioaugmented E1 and E2 (63%) microcosms than in diesel-polluted control C1 (81%) soils (Fig. 6). Quantification of C21–C25 and C26–C30 n-alkanes showed high dispersion, and no significant (P < 0.05) differences were observed among the C1, E1, and E2 microcosms (not shown). Hydrocarbon hydrophobicity is proportional to molecule size, and larger hydrocarbons tend to be more tightly adsorbed to soil particles (58). Nonetheless, the high dispersion of >C20 n-alkanes could not be explained by tighter adsorption to soil due to high extraction efficiencies (100%) for all n-alkanes (data not shown). However, the total diesel extraction efficiency was >87%, suggesting that adsorption may have affected the extraction of other diesel components (e.g., naphthenes, branched alkanes, and PAHs). Adsorption may also explain the oscillating values in the abiotic control, where the successive autoclave treatments may change soil physicochemical properties (59). However, values close to the initial 100% indicate that loss by abiotic factors was minimal. No significant differences were observed in hydrocarbon degradation after a second diesel pollution during weeks 8 to 12 (see Fig. S7 in the supplemental material), probably due to nutrient limitation. The recommended molar C:N:P ratio is 100:10:1 (60). However, in this study, the molar C:N:P ratio was 100:4:0.8 in the original soil, 100:2.5:0.4 after the first diesel amendment, and ∼100:1.8:0.3 after the second amendment (see Table S3 in the supplemental material). Despite the high diesel concentration that emulated highly polluted soil (61, 62), the small number of bacterial inoculations (2 times), nutrient limitation, and nonregulated temperature, degradation was significantly higher in both bioaugmented microcosms than in the noninoculated C1 soil after 8 weeks. Future studies should focus on additional field conditions.
FIG 6.
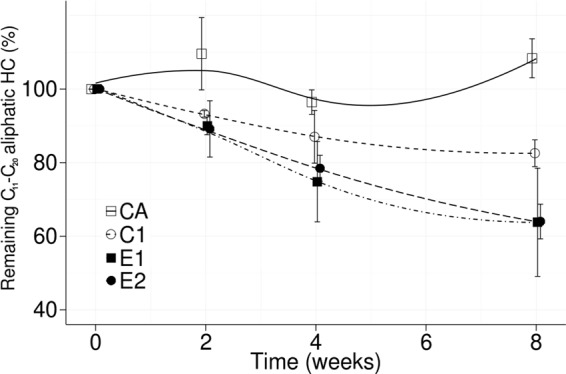
Effect of bioaugmentation on degradation of diesel C11–C20 n-alkanes in replicate soil microcosms. Higher degradation of C11–C20 n-alkanes was observed in bioaugmented microcosms E1 and E2 than in the control microcosms C1 and CA after 8 weeks. Each point is an average of the results from three independent experiments. The vertical bars indicate standard deviations.
Concluding remarks and perspectives.
In the present study, a single rare member became dominant in the soil after a strong disturbance imposed by diesel pollution. To our knowledge, there are no previous reports describing such a pronounced bloom of a rare community member in soil. This study shows that after a drastic environmental change, microbial community dynamics in soil is similar to the reported microbial dynamics in aquatic ecosystems. In addition, the long-term fate of inoculated strains and the microbial community response to subsequent pollution are topics of special relevance. Might bioaugmentation introduce novel conditionally rare taxa? If inoculated strains become part of the rare biosphere, might they bloom after forthcoming pollution events? The bloom described in this study is in agreement with the conditionally rare taxon concept, in which rare taxa remain at low levels until favorable environmental conditions allow their pronounced bloom in the ecosystem.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge Beatriz Cámara for her valuable comments on the manuscript.
S.F. and M.S. conceived and designed the experiments. S.F. and B.B. performed the experiments. S.F., B.B., J.G.C., and M.S. analyzed the data. S.F. and M.S. contributed reagents/materials. S.F., J.G.C., and M.S. wrote the paper.
Funding Statement
This work was supported by CONICYT Ph.D. fellowship (to S.F. and B.B.), Fulbright and PUCV scholarships for a research internship (to S.F.), and CONICYT-AT24100177 (to S.F.) and PIIC-USM-2012 (to S.F.) grants.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02625-15.
REFERENCES
- 1.Schloter M, Dilly O, Munch JC. 2003. Indicators for evaluating soil quality. Agric Ecosyst Environ 98:255–262. doi: 10.1016/S0167-8809(03)00085-9. [DOI] [Google Scholar]
- 2.Lauber CL, Hamady M, Knight R, Fierer N. 2009. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl Environ Microbiol 75:5111–5120. doi: 10.1128/AEM.00335-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Altimira F, Yáñez C, Bravo G, González M, Rojas LA, Seeger M. 2012. Characterization of copper-resistant bacteria and bacterial communities from copper-polluted agricultural soils of central Chile. BMC Microbiol 12:193. doi: 10.1186/1471-2180-12-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morgante V, López-López A, Flores C, González M, González B, Vásquez M, Rosselló-Mora R, Seeger M. 2010. Bioaugmentation with Pseudomonas sp. strain MHP41 promotes simazine attenuation and bacterial community changes in agricultural soils. FEMS Microbiol Ecol 71:114–126. doi: 10.1111/j.1574-6941.2009.00790.x. [DOI] [PubMed] [Google Scholar]
- 5.Yergeau E, Sanschagrin S, Beaumier D, Greer CW. 2012. Metagenomic analysis of the bioremediation of diesel-contaminated Canadian high arctic soils. PLoS One 7:e30058. doi: 10.1371/journal.pone.0030058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fuentes S, Méndez V, Aguila P, Seeger M. 2014. Bioremediation of petroleum hydrocarbons: catabolic genes, microbial communities, and applications. Appl Microbiol Biotechnol 81:4781–4794. [DOI] [PubMed] [Google Scholar]
- 7.Viñas M, Sabaté J, Espuny MJ, Solanas AM. 2005. Bacterial community dynamics and polycyclic aromatic hydrocarbon degradation during bioremediation of heavily creosote-contaminated soil. Appl Environ Microbiol 71:7008–7018. doi: 10.1128/AEM.71.11.7008-7018.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang S, Wen X, Jin H, Wu Q. 2012. Pyrosequencing investigation into the bacterial community in permafrost soils along the China-Russia Crude Oil Pipeline (CRCOP). PLoS One 7:e52730. doi: 10.1371/journal.pone.0052730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yakimov MM, Gentile G, Bruni V, Cappello S, D'Auria G, Golyshin PN, Giuliano L. 2004. Crude oil-induced structural shift of coastal bacterial communities of rod bay (Terra Nova Bay, Ross Sea, Antarctica) and characterization of cultured cold-adapted hydrocarbonoclastic bacteria. FEMS Microbiol Ecol 49:419–432. doi: 10.1016/j.femsec.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 10.McKew BA, Coulon F, Osborn AM, Timmis KN, McGenity TJ. 2007. Determining the identity and roles of oil-metabolizing marine bacteria from the Thames estuary, UK. Environ Microbiol 9:165–176. doi: 10.1111/j.1462-2920.2006.01125.x. [DOI] [PubMed] [Google Scholar]
- 11.Hamamura N, Ward DM, Inskeep WP. 2013. Effects of petroleum mixture types on soil bacterial population dynamics associated with the biodegradation of hydrocarbons in soil environments. FEMS Microbiol Ecol 85:168–178. doi: 10.1111/1574-6941.12108. [DOI] [PubMed] [Google Scholar]
- 12.Kaplan CW, Kitts CL. 2004. Bacterial succession in a petroleum land treatment unit. Appl Environ Microbiol 70:1777–1786. doi: 10.1128/AEM.70.3.1777-1786.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yakimov MM, Timmis KN, Golyshin PN. 2007. Obligate oil-degrading marine bacteria. Curr Opin Biotechnol 18:257–266. doi: 10.1016/j.copbio.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 14.Schneiker S, Martins dos Santos VA, Bartels D, Bekel T, Brecht M, Buhrmester J, Chernikova TN, Denaro R, Ferrer M, Gertler C, Goesmann A, Golyshina OV, Kaminski F, Khachane AN, Lang S, Linke B, McHardy AC, Meyer F, Nechitaylo T, Pühler A, Regenhardt D, Rupp O, Sabirova JS, Selbitschka W, Yakimov MM, Timmis KN, Vorhölter F-J, Weidner S, Kaiser O, Golyshin PN. 2006. Genome sequence of the ubiquitous hydrocarbon-degrading marine bacterium Alcanivorax borkumensis. Nat Biotechnol 24:997–1004. doi: 10.1038/nbt1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Atlas RM, Hazen TC. 2011. Oil biodegradation and bioremediation: a tale of the two worst spills in U.S. history. Environ Sci Technol 45:6709–6715. doi: 10.1021/es2013227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lynch MD, Neufeld JD. 2015. Ecology and exploration of the rare biosphere. Nat Rev Microbiol 13:217–229. doi: 10.1038/nrmicro3400. [DOI] [PubMed] [Google Scholar]
- 17.Aanderud Z, Jones S, Fierer N, Lennon JT. 2015. Resuscitation of the rare biosphere contributes to pulses of ecosystem activity. Front Microbiol 6:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shade A, Jones SE, Caporaso JG, Handelsman J, Knight R, Fierer N, Gilbert JA. 2014. Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. mBio 5(4):e01371-14. doi: 10.1128/mBio.01371-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuentes S. 2014. Biaugmentation of hydrocarbon-polluted soils and its effect on microbial communities. Ph.D. thesis Universidad Técnica Federico Santa María, Valparaíso, Chile. [Google Scholar]
- 20.Dinamarca A, Cereceda-Balic F, Fadic X, Seeger M. 2007. Microbial simazine-degrading cells in soil: most-probable-number enumeration and tetrazolium salt detection in microtiter plates. Int Microbiol 10:209–215. [PubMed] [Google Scholar]
- 21.Sadzawka A. 1990. Métodos de análisis de suelos. Instituto de Investigaciones Agropecuarias, Serie Actas INIA, Santiago, Chile. [Google Scholar]
- 22.Sadzawka A, Carrasco MA, Grez R, Mora MDLL, Flores H, Neaman A. 2006. Métodos de análisis recomendados para los suelos de Chile. Instituto de Investigaciones Agropecuarias, Serie Actas INIA, Santiago, Chile. [Google Scholar]
- 23.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R. 2012. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD. 2012. PANDAseq: paired-end assembler for Illumina sequences. BMC Bioinformatics 13:31. doi: 10.1186/1471-2105-13-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rideout JR, He Y, Navas-Molina JA, Walters WA, Ursell LK, Gibbons SM, Chase J, McDonald D, Gonzalez A, Robbins-Pianka A, Clemente JC, Gilbert JA, Huse SM, Zhou H-W, Knight R, Caporaso JG. 2014. Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. PeerJ 2:e545. doi: 10.7717/peerj.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 28.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. 2012. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6:610–618. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Price MN, Dehal PS, Arkin AP. 2010. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. 2010. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26:266–267. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Hamme J, Singh A, Ward OP. 2003. Recent advances in petroleum microbiology. Microbiol Mol Biol Rev 67:503–549. doi: 10.1128/MMBR.67.4.503-549.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thangaraj K, Kapley A, Purohit HJ. 2008. Characterization of diverse Acinetobacter isolates for utilization of multiple aromatic compounds. Bioresour Technol 99:2488–2494. doi: 10.1016/j.biortech.2007.04.053. [DOI] [PubMed] [Google Scholar]
- 34.Oh YK, Ryu BH, Bae KC, Bin JH. 1989. Biodegradation of naphthalene by Acinetobacter calcoaceticus R-88. J Korean Agric Chem Soc 32:315–320. [Google Scholar]
- 35.Molina MC, González N, Bautista LF, Sanz R, Simarro R, Sánchez I, Sanz JL. 2009. Isolation and genetic identification of PAH degrading bacteria from a microbial consortium. Biodegradation 20:789–800. doi: 10.1007/s10532-009-9267-x. [DOI] [PubMed] [Google Scholar]
- 36.Ciric L, Philp JC, Whiteley AS. 2010. Hydrocarbon utilization within a diesel-degrading bacterial consortium. FEMS Microbiol Lett 303:116–122. doi: 10.1111/j.1574-6968.2009.01871.x. [DOI] [PubMed] [Google Scholar]
- 37.Kuczynski J, Liu Z, Lozupone C, Mcdonald D, Fierer N, Knight R. 2010. Microbial community resemblance methods differ in their ability to detect biologically relevant patterns. Nat Methods 7:813–819. doi: 10.1038/nmeth.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petric I, Philippot L, Abbate C, Bispo A, Chesnot T, Hallin S, Laval K, Lebeau T, Lemanceau P, Leyval C, Lindström K, Pandard P, Romero E, Sarr A, Schloter M, Simonet P, Smalla K, Wilke B-M, Martin-Laurent F. 2011. Inter-laboratory evaluation of the ISO standard 11063 “Soil quality–method to directly extract DNA from soil samples.” J Microbiol Methods 84:454–460. [DOI] [PubMed] [Google Scholar]
- 39.Bates ST, Berg-Lyons D, Caporaso JG, Walters WA, Knight R, Fierer N. 2011. Examining the global distribution of dominant archaeal populations in soil. ISME J 5:908–917. doi: 10.1038/ismej.2010.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pinto AJ, Raskin L. 2012. PCR biases distort bacterial and archaeal community structure in pyrosequencing datasets. PLoS One 7:e43093. doi: 10.1371/journal.pone.0043093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liang Y, Li G, Van Nostrand JD, He Z, Wu L, Deng Y, Zhang X, Zhou J. 2009. Microarray-based analysis of microbial functional diversity along an oil contamination gradient in oil field. FEMS Microbiol Ecol 70:168–177. [DOI] [PubMed] [Google Scholar]
- 42.Le Borgne S, Paniagua D, Vazquez-Duhalt R. 2008. Biodegradation of organic pollutants by halophilic bacteria and archaea. J Mol Microbiol Biotechnol 15:74–92. doi: 10.1159/000121323. [DOI] [PubMed] [Google Scholar]
- 43.Röling WFM, de Brito Couto IR, Swannell RPJ, Head IM. 2004. Response of archaeal communities in beach sediments to spilled oil and bioremediation. Appl Environ Microbiol 70:2614–2620. doi: 10.1128/AEM.70.5.2614-2620.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bogan BW, Sullivan WR, Kayser KJ, Derr KD, Aldrich HC, Paterek JR. 2003. Alkanindiges illinoisensis gen. nov., sp. nov., an obligately hydrocarbonoclastic, aerobic squalane-degrading bacterium isolated from oilfield soils. Int J Syst Evol Microbiol 53:1389–1395. doi: 10.1099/ijs.0.02568-0. [DOI] [PubMed] [Google Scholar]
- 45.Klein AN, Frigon D, Raskin L. 2007. Populations related to Alkanindiges, a novel genus containing obligate alkane degraders, are implicated in biological foaming in activated sludge systems. Environ Microbiol 9:1898–1912. doi: 10.1111/j.1462-2920.2007.01307.x. [DOI] [PubMed] [Google Scholar]
- 46.Chang W, Whyte L, Ghoshal S. 2011. Comparison of the effects of variable site temperatures and constant incubation temperatures on the biodegradation of petroleum hydrocarbons in pilot-scale experiments with field-aged contaminated soils from a cold regions site. Chemosphere 82:872–878. doi: 10.1016/j.chemosphere.2010.10.072. [DOI] [PubMed] [Google Scholar]
- 47.Williams TR, Marco ML. 2014. Phyllosphere microbiota composition and microbial community transplantation on lettuce plants grown indoors. mBio 5(4):e01564-14. doi: 10.1128/mBio.01564-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Woo PCY, Tse H, Lau SKP, Leung K-W, Woo GKS, Wong MKM, Ho C-M, Yuen K-Y. 2005. Alkanindiges hongkongensis sp. nov. A novel Alkanindiges species isolated from a patient with parotid abscess. Syst Appl Microbiol 28:316–322. [DOI] [PubMed] [Google Scholar]
- 49.Sauret C, Séverin T, Vétion G, Guigue C, Goutx M, Pujo-Pay M, Conan P, Fagervold SK, Ghiglione J-F. 2014. “Rare biosphere” bacteria as key phenanthrene degraders in coastal seawaters. Environ Pollut 194:246–253. doi: 10.1016/j.envpol.2014.07.024. [DOI] [PubMed] [Google Scholar]
- 50.Ferrenberg S, O'Neill SP, Knelman JE, Todd B, Duggan S, Bradley D, Robinson T, Schmidt SK, Townsend AR, Williams MW, Cleveland CC, Melbourne BA, Jiang L, Nemergut DR. 2013. Changes in assembly processes in soil bacterial communities following a wildfire disturbance. ISME J 7:1102–1111. doi: 10.1038/ismej.2013.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alonso-Sáez L, Zeder M, Harding T, Pernthaler J, Lovejoy C, Bertilsson S, Pedrós-Alió C. 2014. Winter bloom of a rare betaproteobacterium in the Arctic Ocean. Front Microbiol 5:425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sjöstedt J, Koch-Schmidt P, Pontarp M, Canbäck B, Tunlid A, Lundberg P, Hagström A, Riemann L. 2012. Recruitment of members from the rare biosphere of marine bacterioplankton communities after an environmental disturbance. Appl Environ Microbiol 78:1361–1369. doi: 10.1128/AEM.05542-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ní Chadhain SM, Norman RS, Pesce KV, Kukor JJ, Zylstra GJ. 2006. Microbial dioxygenase gene population shifts during polycyclic aromatic hydrocarbon biodegradation. Appl Environ Microbiol 72:4078–4087. doi: 10.1128/AEM.02969-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ding G-C, Heuer H, Zühlke S, Spiteller M, Pronk GJ, Heister K, Kögel-Knabner I, Smalla K. 2010. Soil type-dependent responses to phenanthrene as revealed by determining the diversity and abundance of polycyclic aromatic hydrocarbon ring-hydroxylating dioxygenase genes by using a novel PCR detection system. Appl Environ Microbiol 76:4765–4771. doi: 10.1128/AEM.00047-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bell TH, Yergeau E, Maynard C, Juck D, Whyte LG, Greer CW. 2013. Predictable bacterial composition and hydrocarbon degradation in Arctic soils following diesel and nutrient disturbance. ISME J 7:1200–1210. doi: 10.1038/ismej.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang S, Wen X, Zhao L, Shi Y, Jin H. 2014. Crude oil treatment leads to shift of bacterial communities in soils from the deep active layer and upper permafrost along the China-Russia crude oil pipeline route. PLoS One 9:e96552. doi: 10.1371/journal.pone.0096552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Udikovic-Kolic N, Wichmann F, Broderick NA, Handelsman J. 2014. Bloom of resident antibiotic-resistant bacteria in soil following manure fertilization. Proc Natl Acad Sci U S A 111:15202–15207. doi: 10.1073/pnas.1409836111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baboshin MA, Golovleva LA. 2012. Aerobic bacterial degradation of polycyclic aromatic hydrocarbons (PAHs) and its kinetic aspects. Microbiology 81:639–650. doi: 10.1134/S0026261712060021. [DOI] [PubMed] [Google Scholar]
- 59.Lotrario JB, Stuart BJ, Lam T, Arands RR, O'Connor OA, Kosson DS. 1995. Effects of sterilization methods on the physical characteristics of soil: implications for sorption isotherm analyses. Bull Environ Contam Toxicol 54:668–675. [DOI] [PubMed] [Google Scholar]
- 60.Leys NM, Bastiaens L, Verstraete W, Springael D. 2005. Influence of the carbon/nitrogen/phosphorus ratio on polycyclic aromatic hydrocarbon degradation by Mycobacterium and Sphingomonas in soil. Appl Microbiol Biotechnol 66:726–736. doi: 10.1007/s00253-004-1766-4. [DOI] [PubMed] [Google Scholar]
- 61.Roldán-Martín A, Calva-Calva G, Rojas-Avelizapa N, Díaz-Cervantes MD, Rodríguez-Vázquez R. 2007. Solid culture amended with small amounts of raw coffee beans for the removal of petroleum hydrocarbon from weathered contaminated soil. Int Biodeter Biodegr 60:35–39. doi: 10.1016/j.ibiod.2006.10.008. [DOI] [Google Scholar]
- 62.Brennerova MV, Josefiova J, Brenner V, Pieper DH, Junca H. 2009. Metagenomics reveals diversity and abundance of meta-cleavage pathways in microbial communities from soil highly contaminated with jet fuel under air-sparging bioremediation. Environ Microbiol 11:2216–2227. doi: 10.1111/j.1462-2920.2009.01943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.