

Abstract
The enteric nervous system (ENS) develops from neural crest cells that migrate along the intestine, differentiate into neurons and glia, and pattern into two plexuses within the gut wall. Inductive interactions between epithelium and mesenchyme regulate gut development, but the influence of these interactions on ENS development is unknown. Epithelial-mesenchymal recombinations were constructed using avian hindgut mesenchyme and non-intestinal epithelium from the bursa of Fabricius. These recombinations led to abnormally large and ectopically positioned ganglia. We hypothesized that sonic hedgehog (Shh), a secreted intestinal epithelial protein not expressed in the bursa, mediates this effect. Inhibition of Shh signaling, by addition of cyclopamine or a function-blocking antibody, resulted in large, ectopic ganglia adjacent to the epithelium. Shh overexpression, achieved in ovo using Shh-encoding retrovirus and in organ culture using recombinant protein, led to intestinal aganglionosis. Shh strongly induced the expression of versican and collagen type IX, whereas cyclopamine reduced expression of these chondroitin sulfate proteoglycans that are known to be inhibitory to neural crest cell migration. Shh also inhibited enteric neural crest-derived cell (ENCC) proliferation, promoted neuronal differentiation, and reduced expression of Gdnf, a key regulator of ENS formation. Ptc1 and Ptc2 were not expressed by ENCCs, and migration of isolated ENCCs was not inhibited by Shh protein. These results suggest that epithelial-derived Shh acts indirectly on the developing ENS by regulating the composition of the intestinal microenvironment.
KEY WORDS: Enteric nervous system, Sonic hedgehog, Patched, Ptch, Epithelium, Extracellular matrix, Enteric neural crest cells, Hirschsprung disease, Chick, Quail, Mouse
Highlighted article: Gut epithelium-derived sonic hedgehog acts indirectly on the developing chick enteric nervous system by regulating the composition of the intestinal microenvironment.
INTRODUCTION
Gut development relies on interactions among cell types originating from the three germ layers: endoderm, which gives rise to epithelium; mesoderm, which forms the smooth muscle, endothelial cells and connective tissues; and ectoderm, which gives rise to the enteric nervous system (ENS). Enteric neural crest-derived cells (ENCCs) emigrate from the vagal level of the neural tube, turn ventrally to colonize the foregut, and undergo massive proliferation as they continue caudally to populate the embryonic gut (Yntema and Hammond, 1954; Burns and Le Douarin, 2001). In the developing hindgut, vagal crest-derived cells join neural crest cells originating from the sacral level of the neural tube to form the ENS (Burns and Le Douarin, 1998; Nagy et al., 2007, 2012). The ENS consists of two ganglionated plexuses, myenteric and submucosal, which are composed of multiple types of neurons and glia arranged as concentric rings and are responsible for regulating the function of the gut, including peristalsis. Congenital abnormalities of the ENS cause severe intestinal disorders, such as Hirschsprung disease (HSCR) (Goldstein et al., 2013), which is characterized by the absence of enteric ganglia along a variable length of distal intestine. Other ENS abnormalities include hyperganglionosis, ectopic ganglia, and hypoganglionosis, which are often associated with intestinal dysmotility (Kapur, 2000).
ENS development relies on reciprocal interactions between ENCCs and their environment that are crucial for ENCC survival, migration, proliferation, patterning and differentiation. Deficiency in the ENCC receptors Ret or EdnrB, for example, or their respective mesenchymal ligands Gdnf or Edn3 leads to intestinal aganglionosis (Goldstein and Nagy, 2008; Lake and Heuckeroth, 2013). Previous studies have shown that extracellular matrix (ECM) proteins in the gut mesenchyme regulate ENCC migration and differentiation (Wu et al., 1999; Breau et al., 2009; Druckenbrod and Epstein, 2009; Nagy et al., 2009; Akbareian et al., 2013; Raghavan et al., 2013). This mesenchymal environment is highly influenced during gut organogenesis by the developing epithelium (Roberts, 2000). One would therefore expect that epithelial abnormalities would influence ENS development by altering these inductive interactions. Diffusible factors produced by gut epithelium, including netrin and hedgehog (Hh) proteins, are potential candidates. Netrin 1 promotes the radial migration of ENCCs from the myenteric to submucosal region and also prevents premature apoptosis of ENCCs (Jiang et al., 2003). The role of Hh proteins is less clear. A genome-wide association study on HSCR reported genetic variants in Ptc1, supporting a potential role for the Hh pathway in the etiology of aganglionosis (Ngan et al., 2011). Mice deficient in sonic hedgehog (Shh) have increased enteric neurons and abnormally distributed ganglia. Deletion of indian hedgehog (Ihh) leads to segmental aganglionosis in rodents (Ramalho-Santos et al., 2000). Shh;Ihh double mutants display a major reduction in neuronal numbers in the stomach (Mao et al., 2010). In addition, targeted deletion of the G protein-coupled receptor for Shh, smoothened (Smo), decreases enteric neuron number, while overexpression of the downstream transcription factor Gli1 causes aganglionosis (Yang et al., 1997; Huang et al., 2013).
In order to clarify how Shh signaling controls ENS patterning, we used the avian embryo and applied a variety of techniques, including tissue recombination, organ culture, retrovirus-mediated gene overexpression, and cell migration assays, confirming our observations in the mouse embryonic gut. In the absence of Shh-expressing epithelium, large and ectopic enteric ganglia develop. When Shh is overexpressed, aganglionosis ensues. These phenotypes do not result from a direct effect of Shh on ENCCs since the receptors Ptc1 and Ptc2 are not expressed on these cells. Rather, it appears that the effect of Shh on the ENS is mediated through the ECM, whereby Shh induces proteins that inhibit ENCC migration.
RESULTS
Shh and Ptc1 are expressed in the developing gut during ENS development
At E6 (HH28), the preganglionic hindgut epithelium expresses Shh, as shown by in situ hybridization (Fig. 1A) and immunofluorescence (Fig. 1B). Fixation with Histochoice increased the intensity of staining, showing a gradient deposition of Shh protein in the subepithelial mesenchyme (Fig. 1B, inset). In situ hybridization of adjacent sections revealed transcripts of the Shh receptor Ptc1 in the mesenchyme under the luminal epithelium (Fig. 1C). When migrating ENCCs reach the distal hindgut at E8 (HH34), Ptc1 is expressed underneath the epithelium as well as in a second circumferential ring (Fig. 1D). This outer mesenchymal Ptc1 expression appeared to overlap the presumptive submucosal plexus, but Ptc1 in situ hybridization (Fig. 1D,E) combined with p75 (Ngfr) immunofluorescence to mark ENCCs (Fig. 1F) (Young et al., 1998; Nagy et al., 2012) showed no overlap, and therefore no Ptc1 expression by ENCCs.
Fig. 1.

Expression of Shh and Ptc1 in the developing chick hindgut. Expression of Shh and Ptc1 was determined in E6 (A-C) and E8 (D,E) hindgut. In situ hybridization shows Shh transcript (A) in the epithelium, with a gradient of protein expression seen by immunofluorescence extending into the subepithelial mesenchyme (B, magnified view in inset; dashed line marks the epithelial basement membrane). Ptc1 in situ shows expression in the subepithelial mesenchyme (C). Double staining of E8 hindgut by Ptc1 in situ (D,E) and p75 immunofluorescence (F) shows that Ptc1 does not overlap with p75+ ENCCs (E,F, magnified from boxed area in D). Outlined areas in E mark p75+ submucosal ganglia in F. Transverse sections of E13 bursa and hindgut show that Ptc1 (G) and Shh (H) are expressed only in the gut, whereas E-cadherin is expressed in the epithelia of both organs (I). Bursa lacks Hu+ enteric neurons (J). ep, epithelium; mes, mesenchyme.
Shh and Ptc1 expression were assessed at later stages of hindgut development. At E13 (HH39), Ptc1 continues to be expressed by the subepithelial mesenchyme (Fig. 1G), and not the epithelium itself (Fig. 1G, inset). Shh is expressed specifically by the gut epithelium (Fig. 1H) and not the epithelium of the bursa of Fabricius (Fig. 1H), which is positive for E-cadherin (Fig. 1I). Similarly, hindgut contains Hu+ enteric neurons, whereas the adjacent bursa does not (Fig. 1J). At E16, Ptc1 is restricted to the lamina propria, between the epithelium and muscularis mucosae, and is not expressed in the submucosa (Fig. S1A). After hatching, Ptc1 expression in the colon remains limited to the lamina propria (Fig. S1B,C). At no stages examined was Ptc1 expression seen in the ENS. Expression of Ptc2 was also examined by in situ hybridization and its expression did not overlap with p75+ ENCCs (Fig. S2).
Signaling from the hindgut epithelium is crucial for ENS development
Given the observation that the ENS-containing hindgut epithelium expresses Shh, whereas the bursa has neither ENS nor Shh expression, we hypothesized that the gut epithelium is crucial for normal ENS development, as previously suggested for the stomach (Sukegawa et al., 2000). To study the effect of hindgut and bursa epithelial signaling on ENS development, chick-quail tissue recombination experiments were performed. The epithelium of the bursa of Fabricius from E9 (HH35) chick embryos was separated from the underlying mesenchyme and recombined with E5 (HH27) quail hindgut mesenchyme (Fig. S3A). E9 bursa epithelium was used because the epithelial anlagen of this organ develops at E8, later than the hindgut epithelium. To ensure epithelial-mesenchymal adherence, recombinants were embedded into a three-dimensional collagen gel overnight (Fig. S3B) (Nagy and Goldstein, 2006a). Staining with QCPN confirms the quail origin of the mesenchyme (Fig. S3C). Tissue recombinations were implanted into the coelomic cavity of E3 (HH19) chick embryos (Fig. S3D), a model that allows host neural crest cells to colonize the transplanted intestine (Nagy and Goldstein, 2006b). After 9 days, the graft was removed for immunohistochemistry and the results compared with control recombinations in which E5 quail hindgut mesenchyme was recombined with E5 chicken hindgut epithelium. QCPN immunostaining showed that the mesenchyme is derived from quail (Fig. 2A,E) and 8F3 staining confirmed that both epithelium and ENS are chicken derived (Fig. 2B,F).
Fig. 2.

Hindgut epithelium regulates enteric ganglion size and patterning. Chick-quail chimeras were generated by recombining E5 quail hindgut mesenchyme (HGmes) and E6 chick hindgut epithelium (HGep) (A-D; n=11) or E9 chick bursa epithelium (BFep) (E-H; n=11). Recombinants were implanted into E3 chick coelom for 9 days. QCPN immunostaining confirms that the mesenchyme is quail derived (A,E), while 8F3 (B,F) immunostaining reveals the chick origin of the epithelium, enteric ganglia and blood vessels (B, arrowheads). When gut epithelium is replaced by bursa epithelium, enteric ganglia are larger and closer to the epithelium (F,G) than in control recombinations (B,C). This was confirmed quantitatively (I,J). The muscularis propria also appears thickened in bursa epithelium recombinations (H), confirmed by quantitative analysis showing increased thickness of the gut wall (K) and muscularis propria (L), the latter comprising a greater proportion of the wall than in controls (M). Muscularis mucosae appears closer to the epithelium in bursa recombinants (H) than in the control (D, the dotted line marks epithelial basement membrane). ep, epithelium; mp, myenteric plexus; smp, submucosal plexus; SMA, alpha-smooth muscle actin. *P<0.05, **P<0.01, ***P<0.001.
In recombinations using bursa epithelium, large and ectopic Hu+ ganglia were located abnormally close to the epithelium, in notable contrast to the control (Fig. 2C,G). As shown in Fig. 2I, the distance from the epithelial basement membrane to the submucosal ganglia was significantly shorter in the presence of bursa epithelium (56±25 µm) than in controls using gut epithelium (83±20 µm). Submucosal ganglia were also markedly larger (92±41 µm versus 35±17 µm; Fig. 2J). Staining for alpha-smooth muscle actin (SMA) showed a significant difference in muscle thickness (Fig. 2D,I). The total thickness of the gut wall and of the muscularis propria were both markedly increased in bursa recombinants (Fig. 2K,L), with the muscularis propria comprising a greater proportion of the total gut wall (Fig. 2M). In bursa epithelial recombinants, the muscularis mucosae was also located immediately under the epithelium (Fig. 2H), much closer than normal (Fig. 2D), suggesting loss of the normal lamina propria.
Inhibition of Shh signaling leads to intestinal hyperganglionosis
Given the absence of Shh expression in bursa epithelium and the abnormal ENS development in bursa epithelial recombinants, we hypothesized that Shh is a crucial epithelium-derived factor during ENS formation. To test this, we modulated Shh activity in explanted embryonic intestine. E5 chick gut was cultured for 3 days in the presence of Shh protein (2 µg/ml) or cyclopamine (2 µM). Wholemount and longitudinal sections were stained with Hu antibody (Fig. 3). Normally, by E5 enteric neurons have only migrated to the level of the distal midgut (Fig. 3A, arrow), but reach the distal hindgut by E8 (Fig. 3B, arrow). Similarly, E5 intestine cultured for 3 days demonstrates full ENCC colonization of the hindgut (Fig. 3C). By contrast, when cultured in the presence of excess Shh protein, the hindgut remained aganglionic (Fig. 3D,I), whereas addition of cyclopamine to inhibit Shh signaling led to complete colonization and the formation of large, ectopic ganglia (Fig. 3E,J).
Fig. 3.
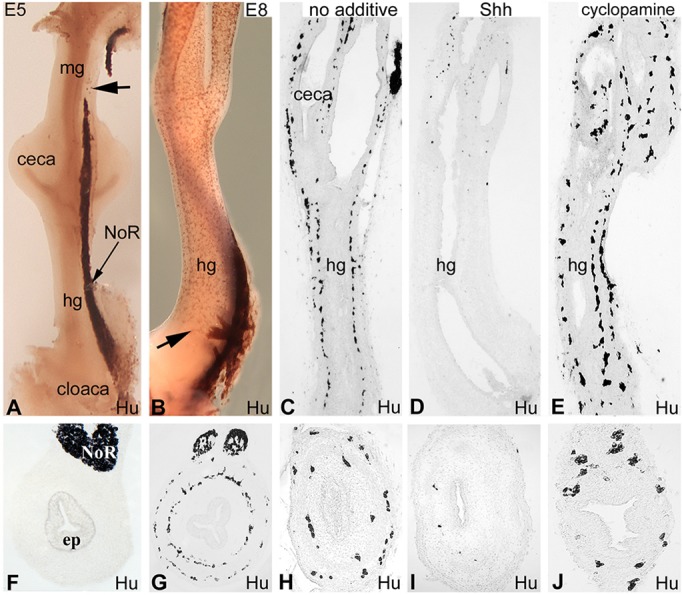
Shh overexpression leads to hindgut aganglionosis. At E5, the ENCC wavefront is in the distal midgut (A, arrow), whereas at E8 ENCCs have reached the distal hindgut (B, arrow). Chick E5 gut was cultured in collagen gel for 3 days in the absence of additives (C), with Shh protein (D), or with cyclopamine (E). Longitudinal sections are shown (A-E), with corresponding transverse sections through the mid-hindgut shown beneath (F-J). Addition of Shh inhibits ENCC colonization of the hindgut (D,I), whereas inhibition of Shh signaling leads to ectopic and large ganglia (E,J). n=27. ep, epithelium; hg, hindgut; mg, midgut; NoR, nerve of Remak.
The effect of Shh inhibition on ENCC migration could result from changes in ENCC proliferation, survival or differentiation. We examined cell proliferation by measuring BrdU incorporation into p75+ ENCCs at the migratory wavefront in the presence of Shh protein or cyclopamine. In control, non-treated hindguts, BrdU+ cells were primarily seen in the submucosal mesenchyme and in 29.7±8.7% of p75+ cells (Fig. 4A). Shh treatment increased the proliferation of mesenchymal cells (Fig. 4B), but had no effect on the rate of ENCC proliferation (22.9±11.4%). By contrast, cyclopamine dramatically increased the proliferation of p75+ ENCCs (Fig. 1C; 52.8±14.8%) compared with both of the other groups (Fig. 4D), suggesting that inhibition of Shh signaling promotes ENCC proliferation in the hindgut. To determine whether Shh induces ENCC apoptosis, we examined the expression of activated caspase 3, a marker of programmed cell death. There was no caspase 3 expression in any of the treatment groups (Fig. 4E-G). Apoptosis was occasionally identified in the gut mesenchyme, and was readily seen in the E8 dorsal sensory ganglia (Fig. 4D), which is known to contain apoptotic neural crest cells and therefore served as a positive control.
Fig. 4.
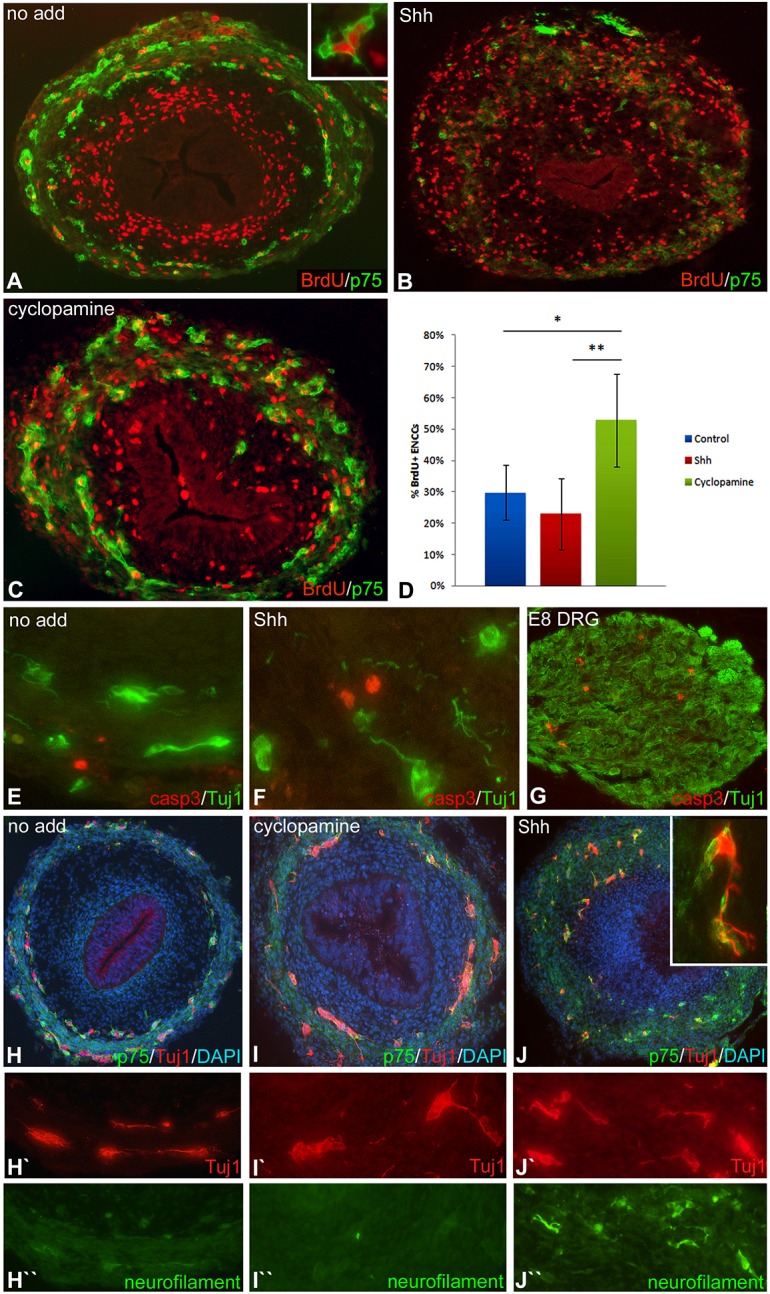
Shh overexpression inhibits ENCC proliferation and promotes neuronal differentiation. Chick E5 hindgut was cultured for 3 days with or without Shh protein or cyclopamine. The rate of ENCC proliferation without additive (A, example of BrdU+ p75+ ENCC shown in inset) was 29%, and was markedly reduced by Shh protein (B) and increased by cyclopamine (C), as shown quantitatively (D). Cultured guts were stained for activated caspase 3 and Tuj1, and no apoptotic enteric neurons were observed in control (E) or Shh-treated (F) intestine. Apoptosis was seen in an E8 dorsal root ganglion (DRG) used as a positive control (G). Tuj1 and neurofilament, which are markers of early and late neuronal differentiation, respectively, show that Tuj1+ neurons are present in all treatment groups (H-J′), while neurofilament is increased following Shh treatment (H″-J″). *P<0.002, **P<0.0004.
Shh promotes neuronal differentiation in the hindgut
To determine whether Shh induces premature neuronal differentiation, thus accounting for the aganglionic phenotype, Tuj1 and neurofilament double immunofluorescence was performed. In the chick, the distal gut is colonized by p75+ ENCCs at E8, and immediately followed by neuronal and glial differentiation. Tuj1 antibody marks early differentiating neurons, whereas neurofilament expression identifies later neurons (Nagy et al., 2012). After culturing E5 intestine for 3 days with no additive or with cyclopamine, Tuj1+ cells are found in both enteric plexuses along the length of the gut, but neurofilament is not yet expressed (Fig. 4H-I″). In Shh-treated cultures, Tuj1+ cells were present only at the level of the cecum, consistent with hindgut aganglionosis. Interestingly, the majority of these Tuj1+ cells coexpressed neurofilament (Fig. 4H-J), suggesting that Shh might induce premature differentiation of wavefront cells, leading to aganglionosis.
Misexpression of Shh changes ECM patterning in the hindgut
Although Shh has a major influence on ENS development, the absence of Shh receptor expression on ENCCs indicates that the effect is mediated indirectly via alterations in the microenvironment. We determined how modulating Shh signaling affects the expression of ECM proteins known to be permissive or inhibitory to neural crest cell migration: collagen I, III, VI, IX, XVIII, laminin, fibronectin, versican, tenascin, agrin, heparan sulfate and chondroitin sulfate proteoglycan (CSPG). Fibronectin is uniformly expressed throughout the gut mesenchyme, with intense fibrillar staining in the inner mesenchymal layer. Presence of Shh or cyclopamine did not significantly alter this expression pattern (Fig. 5A-D). Similarly, the expression of collagen III, VI and XVIII, laminin, agrin and heparan sulfate was not significantly altered (data not shown). In E8 hindgut, collagen I is normally expressed in the outer mesenchyme (Fig. 5E,F). By contrast, E5 hindgut cultured for 3 days in the presence of Shh or cyclopamine showed an altered distribution pattern, with collagen I fibers uniformly distributed throughout the gut wall (Fig. 5G).
Fig. 5.

Altering Shh expression in the gut modifies ECM patterning. Expression of several ECM proteins was examined in chick E8 hindgut (A,E,I,M) and compared with E5 guts cultured for 3 days in the presence of no additive (B,F,J,N), Shh protein (C,G,K,O) or cyclopamine (D,H,L,P). The expression of fibronectin (A-D), collagen I (E-H), collagen IX (I-L) and versican V0/V2 (M-P) in transverse sections through the mid-hindgut is shown. ep, epithelium; NoR, nerve of Remak.
The most dramatic ECM effects, however, were observed with members of the CSPG family, which are known inhibitors of neural crest cell migration (Ring et al., 1996; Dutt et al., 2006). We observed significant Shh-induced changes in collagen IX, CS-56 (which marks the glycosaminoglycan portion of CSPG core protein) and versican isoforms [including immunostaining with anti-GAGβ or anti-GAGα protein domain-specific antibodies and an antibody that recognizes common epitopes on the core protein in the V0 and V2 isoforms (Landolt et al., 1995; Zanin et al., 1999; Dutt et al., 2006, 2011)]. As shown in Fig. 5, collagen IX and versican V0/V2 are normally expressed strongly in the inner mesenchymal layer, just beneath the epithelium. CS-56 has a similar expression pattern (Fig. S4B). After Shh treatment, both collagen IX and versican V0/V2 were dramatically upregulated and distributed throughout the mesenchyme (Fig. 5K,O). The same pattern was observed with isoform-specific antibodies to versican (data not shown). By contrast, cyclopamine treatment markedly reduced collagen IX and versican, limiting them to a small ring of subepithelial mesenchyme (Fig. 5L,P).
To confirm the effect of Shh inhibition on ECM expression, Shh activity was also inhibited with a function-blocking antibody (5E1; Ericson et al., 1996) and results compared with control guts treated with a non-specific monoclonal antibody. p75 expression revealed large, ectopically positioned ganglia following 5E1 treatment (Fig. S4E), as observed with cyclopamine (Fig. 3J). Treatment with 5E1 reduced the expression of CS-56, collagen IX and versican V2 proteins, restricting them to the inner mesenchyme (Fig. S4F-H). The radial extent of versican expression was quantitatively analyzed (Fig. S4I) in guts treated with no additive, Shh protein, cyclopamine and 5E1 and confirmed the immunohistochemical results shown in Fig. 5O,P and Fig. S4H. The extent of versican inhibition following 5E1 (Fig. S4H) is less pronounced than following cyclopamine (Fig. 5P), which is likely to be due to the more potent Shh inhibition achieved with cyclopamine, as noted previously (Liu et al., 2004).
Given the important role of mesenchymal Gdnf and Edn3 during ENS development, we used quantitative RT-PCR to determine their transcript levels in E5 guts cultured in the presence or absence of Shh. Shh significantly reduced Gdnf expression compared with control, whereas Edn3 expression was unchanged (Fig. 6).
Fig. 6.
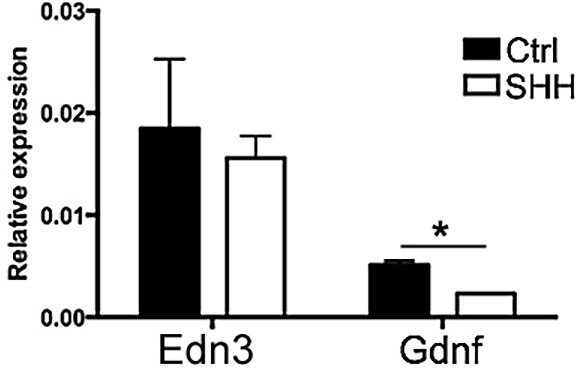
Shh overexpression reduces levels of Gdnf transcript. The relative expression of Edn3 and Gdnf genes in E5 guts cultured for 3 days in the presence of no additive or 2 µg/ml Shh recombinant protein was determined by qRT-PCR. Whereas Edn3 expression was not affected, the expression of Gdnf was markedly reduced in the presence of Shh. *P<0.01.
Retrovirus-mediated overexpression of Shh induces aganglionosis in vivo
To test whether the effect of Shh on ENS development could be recapitulated in vivo, Shh was misexpressed in avian embryos using a replication-competent retrovirus (RCAS) expressing chick Shh. RCAS virus was injected into the presumptive hindgut mesoderm at E2 (HH10-12), as previously described (Goldstein et al., 2005). The gross morphology of RCAS-Shh-infected embryos differed markedly from normal by E5, when extra digits could be seen in the hindlimb (Fig. 7A; normal embryos have four digits at this stage), as previously reported (Riddle et al., 1993). By E8, 3C2 antibody, which recognizes the gag protein P19 of RCAS (Potts et al., 1987), demonstrated extensive viral expression throughout the gut wall (Fig. 7B). Importantly, Hnk1-positive ENCCs were absent from the hindgut mesenchyme where Shh is overexpressed (Fig. 7C), consistent with Shh-induced aganglionosis, as seen in organ culture (Fig. 3D,I).
Fig. 7.

Virus-mediated in ovo misexpression of Shh disrupts ENS and ECM development. Injection of RCAS-Shh into the chick E2 presumptive hindgut mesoderm leads to hindlimb polydactyly (A, digits numbered) and tailbud malformation by E5 (A, arrow). By E8, RCAS is expressed throughout the hindgut, as detected by 3C2 antibody (B), and associated with severe ENS disruption (C). Since in ovo injection was lethal by E7-E9 (n=75), explanted E5 guts were infected with RCAS-Shh ex vivo (D, arrows denote Fast Green-labeled RCAS injection) and cultured for 9 days on an E9 CAM (E). RCAS (F) and Shh (G) were strongly expressed throughout the gut, and associated with hypoganglionosis, abnormal radial patterning of ENCCs (H) and altered smooth muscle development (I). RCAS-Shh infection significantly expanded collagen IX (J). Immunostaining of control E14 colon for Hu (K), SMA (L) and collagen IX (M) is shown for comparison. ep, epithelium; mp, myenteric plexus; smp, submucosal plexus.
Since 80% of embryos die by E8 after RCAS-Shh infection, E5 midgut/hindgut explants were injected with RCAS-Shh ex vivo, and transplanted onto a chick chorioallantoic membrane (CAM) for 9 days (Fig. 7D,E). Successful viral replication was confirmed with 3C2 immunostaining (Fig. 7F), and robust Shh overexpression was confirmed using Shh-specific antibody (Fig. 7G). ENCC colonization of the Shh-overexpressing hindgut was markedly diminished, with severe hypoganglionosis in the proximal colon (Fig. 7H) and aganglionosis distally. Furthermore, enteric ganglia proximal to the aganglionic segment were small, sparse and ectopic, with no organized patterning into discrete submucosal and myenteric plexuses (Fig. 7H), as compared with the wild-type control (Fig. 7K). Also, Hu+ enteric neurons were seen immediately adjacent to the epithelium (Fig. 7H, arrow), where they normally do not occur. Smooth muscle development was severely disorganized (Fig. 7I), as compared with the normal patterning of the three muscle layers (muscularis mucosae, circular and longitudinal) at this stage (Fig. 7L). Interestingly, collagen IX expression was dramatically increased upon Shh overexpression (Fig. 7J), as compared with the wild-type control (Fig. 7M). Versican showed a similar change (data not shown).
Shh induces collagen IX expression
Collagen IX is normally expressed by the gut, and not bursa, mesenchyme (Fig. 8A). Shh-expressing epithelium was isolated from E6 chick hindgut and recombined with E9 chick bursa mesenchyme (Fig. 8B). Recombinants were cultured in the E3 chick coelom for 7 days and collagen IX immunostaining performed. The presence of hindgut epithelium induced the expression of collagen IX in the bursa mesenchyme (Fig. 8C). To test whether Shh is responsible for inducing this expression, Affi-Gel beads soaked in Shh protein were implanted into E9 bursa primordia and cultured on a CAM for 9 days (Fig. 8D). Shh was detected in the surrounding bursa mesenchyme (Fig. 8E), where a ring of collagen IX immunoreactivity was seen (Fig. 8F), suggesting that Shh induces the expression of this ECM protein.
Fig. 8.

Shh signaling induces collagen IX expression. Collagen IX is expressed by the gut mesenchyme, but not the bursa (A). Intestinal epithelium from chick E6 hindgut was transplanted into E9 bursa mesenchyme from which the epithelium had been removed (B), and cultured for 10 days in an E3 chick coelom. Hindgut epithelium induces ectopic collagen IX expression in the surrounding bursa mesenchyme (C). Shh-soaked beads were implanted into E9 chick bursa and cultured on the CAM for 7 days (D). Shh protein expression in and around the bead is observed (E), with induction of collagen IX in the mesenchyme (F, arrows). BF, bursa of Fabricius; CAM, chorioallantoic membrane; ep, epithelium; hg, hindgut.
Shh does not act directly on ENCCs to inhibit their migration
Given the significant changes in ECM expression induced by Shh, and the lack of Ptc1 expression by ENCCs, our results suggest that Shh does not directly affect ENCC development, but rather disrupts the microenvironment and indirectly leads to aganglionosis. To determine whether Shh has a direct effect on ENCC migration, E8 midgut was cultured with Gdnf (10 ng/ml) or with both Gdnf (10 ng/ml) and Shh (2 µg/ml) for 48 h (Fig. 9). As expected, Gdnf led to the extensive migration of ENCCs into the surrounding collagen gel (Fig. 9A), and Shh inhibited this Gdnf-mediated migratory effect (Fig. 9B). Transverse sections of the gut confirmed the presence of Tuj1+ enteric neurons in the surrounding gel following Gdnf treatment (Fig. 9C), but not when Shh was added (Fig. 9D). Importantly, versican expression was significantly upregulated by Shh (Fig. 9E,F).
Fig. 9.
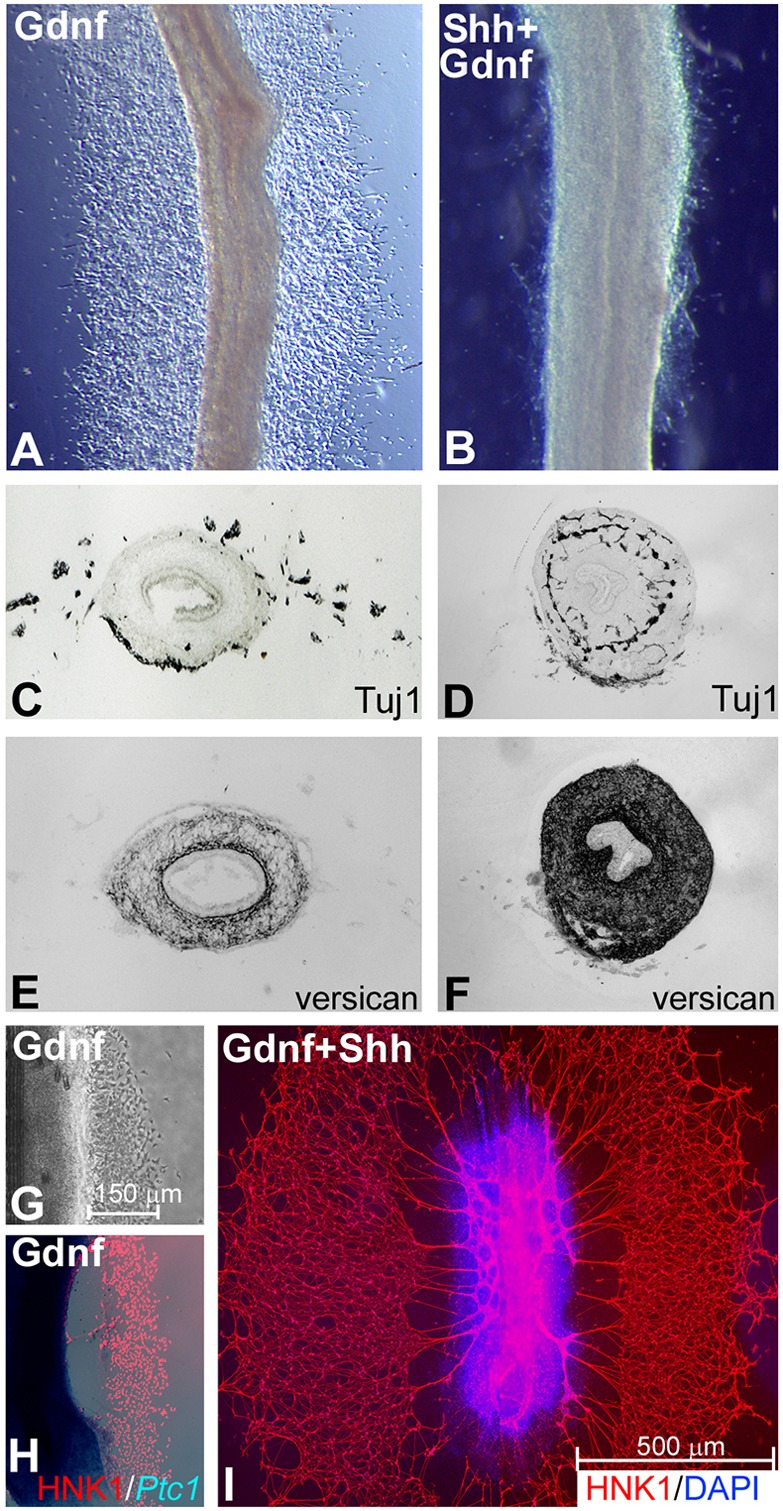
Shh does not act directly on ENCCs. Explanted chick E8 midgut was cultured with Gdnf (A,C,E) or Gdnf and Shh (B,D,F) for 48 h. Gdnf induces ENCCs to migrate out of the gut (A,C), whereas addition of Shh inhibits this effect (B,D). The presence of Shh induces a significant increase in versican expression (F), as compared with Gdnf alone (E). Gdnf-mediated ENCC migration from E6 midgut is robust at 24 h, with the distance of cell migration shown (G). In situ hybridization for Ptc1 expression shows no staining of migrating Hnk1+ neural crest cells (H). Addition of Shh at 24 h, for an additional 48 h, does not inhibit the continued migration of these cells (I).
To distinguish between a direct versus indirect effect of Shh on ENCCs, E6 midgut was cultured on a fibronectin-coated surface for 24 h in the presence of Gdnf, which again led to robust ENCC migration onto the surrounding surface, extending ∼150 µm (Fig. 9G). In situ hybridization with a Ptc1 riboprobe showed that Ptc1 is expressed in the gut, but not in the migrating Hnk1+ neural crest cells (Fig. 9H). After 24 h in the presence of Gdnf, soluble Shh protein was added to the medium and cultures incubated for an additional 48 h. As shown in Fig. 9I, robust ENCC migration continued in the presence of Shh, with cells migrating up to 500 µm (n=10). We conclude that, in the absence of gut mesenchyme, Shh has no direct inhibitory effect on ENCC migration.
Shh signaling has a similar effect on mouse ENS development
To determine whether the role of Shh in avian ENS development is conserved in mammals, we first characterized the temporal pattern of colorectal ENS development in the mouse using the p75 antibody. At E12.5 the migratory wavefront is in the mid-hindgut (Fig. 10A). Similar to the chick embryo, Ptc1 and p75 double-immunofluorescence staining at E12.5 showed that Ptc1 is expressed in the inner mesenchyme and not in p75+ ENCCs (Fig. 10B). When E12.5 hindguts were cultured for 48 h with no additive (Fig. 10C) or with cyclopamine (Fig. 10D), ENCCs completed their migration into the distal hindgut. Expression of the CSPG marker CS-56 was reduced in the outer mesenchyme of cyclopamine-treated guts compared with the control (Fig. 10C,D). With addition of Shh protein, the distal hindgut remained aganglionic and exhibited expansion of the CS-56 domain (Fig. 10E), consistent with our avian results.
Fig. 10.

Shh overexpression leads to aganglionosis and modifies CS-56 immunostaining in the mouse hindgut. At E12.5, p75+ ENCCs have migrated into the mid-hindgut (A, section 2), and the distal hindgut is still aganglionic (A, section 3). ENCCs do not co-immunostain with Ptc1 (B). After 48 h in culture (n=15), ENCCs complete their migration into the distal hindgut and CS-56 immunostaining is observed (C). Addition of cyclopamine to the cultured gut does not inhibit ENCC migration, but reduces CS-56 immunostaining in the outer mesenchyme (D). Addition of Shh inhibits ENCC colonization, leading to distal aganglionosis, and increases CS-56 immunostaining throughout the mesenchyme (E).
DISCUSSION
Development of a normal ENS is important for gut function. In HSCR, ENCCs fail to complete rostrocaudal migration, leaving the distal intestine aganglionic. ENS development relies on the migration, proliferation, differentiation and patterning of ENCCs into two ganglionated plexuses within the submucosal and intermyenteric regions of the gut. These events require tightly regulated interactions between ENCCs and their microenvironment. Although many of the factors involved have been identified (Goldstein et al., 2013), their precise mechanism of action remains unknown. In the stomach, tissue recombinations showed that removal of the gastric epithelium results in increased enteric neuronal density (Sukegawa et al., 2000), suggesting that the epithelium inhibits ENCC proliferation. We tested this using epithelial-mesenchymal recombinations by replacing the normal hindgut epithelium with a non-intestinal epithelium from the bursa of Fabricius, a lymphoid organ derived from the cloaca. In these recombinations, ENCCs migrate close to the epithelium, forming ectopic and abnormally large ganglia adjacent to the epithelial basement membrane, demonstrating the importance of the hindgut epithelium during ENS patterning and suggesting that it might contain an inhibitory factor that is both chemorepulsive to prevent inward migration of ENCCs and antimitogenic to limit ganglion size.
We previously showed that Shh is expressed by the gut epithelium but not the bursa (Nagy and Oláh, 2010). Sukegawa et al. (2000) showed not only that removing the gastric epithelium results in hyperganglionosis, but also that inhibiting Shh leads to increased neuronal numbers and ectopic ganglia. We hypothesized that Shh is a candidate epithelium-derived factor responsible for patterning the hindgut ENS. In Shh−/− mice, the intestine contains increased numbers of neurons that differentiate ectopically under the epithelium and project abnormal extensions into the villi (Ramalho-Santos et al., 2000; Jin et al., 2015). Deletion of Gas1, a Hh cell-surface receptor, or Gnaz, an intracellular effector for Gas1, leads to similar phenotypes, with mislocalized enteric ganglia and increased enteric neuronal numbers (Biau et al., 2013; Jin et al., 2015). Conversely, overexpression of Gli1 leads to a patchy absence of enteric ganglia in the gut (Yang et al., 1997). When Shh is added to intestinal explants, ganglion formation is impaired and enteric neurons are scattered sparsely throughout the gut wall (Fu et al., 2004). These data support an inhibitory role for Shh on ENCC proliferation, cell migration, ganglion formation and axon extension. However, addition of Shh to mouse enteric neurospheres increased ENCC proliferation (Fu et al., 2004), and addition of cyclopamine to zebrafish embryos inhibited ENCC proliferation and led to intestinal aganglionosis (Reichenbach et al., 2008). These inconsistencies suggest a complex role for Hh activity during ENS development.
We took advantage of the versatility of the avian embryo to test the role of Shh in ENS development using a variety of experimental approaches, including organ culture, tissue recombination, chick-quail chimera, CAM grafting and retrovirus-mediated gene overexpression in ovo. We find that inhibiting Hh signaling, either with cyclopamine or with a function-blocking antibody, results in hyperganglionosis, whereas Shh overexpression causes aganglionosis. The failure of ENCCs to complete their colonization can result from abnormalities in cell migration, proliferation or differentiation. We find that Shh overexpression significantly decreases ENCC proliferation and promotes their premature differentiation into neurons, accounting for the aganglionic phenotype. Retrovirus-mediated overexpression of Shh in vivo confirms these results, leading to distal aganglionosis with severe hypoganglionosis in the proximal colon. Our results corroborate previous observations and reveal an essential role for Shh signaling in regulating ENCC proliferation, differentiation, migration and patterning (Fu et al., 2004; Biau et al., 2013).
The effects of a secreted ligand, such as Shh, would presumably be mediated through a receptor expressed by the target cell, but whether ENCCs express Shh receptors has been unclear. Fu et al. (2004) and Ngan et al. (2011) both reported expression of Ptc1 by ENCCs, with the latter showing that ENCC-specific deletion of Ptch1 reduces ENCC proliferation. Reichenbach et al. (2008) similarly found Ptc1 expressed by Phox2b-expressing ENCCs in zebrafish. By contrast, however, reporter mice expressing β-galactosidase driven by Ptc1, Gli1 or Gli2 showed no lacZ expression in ENCCs (Washington Smoak et al., 2005; Kolterud et al., 2009), suggesting that these Hh pathway genes are not expressed. We performed Ptc1 and Ptc2 in situ hybridization in embryonic avian hindgut and also in cultured ENCCs and find that both receptors are expressed by the mesenchymal compartment of the gut, but not by the ENCCs. We found a similar expression pattern of Ptc1 in the mesenchyme, but not the ENS, in embryonic mouse hindgut. Importantly, we observed an analogous role for Shh in the intestines of both species. Although we cannot explain the contradictory results obtained in these studies, our results suggest that interspecies differences, at least between avian and rodent, do not exist.
The lack of Shh receptor expression leaves two possibilities to explain the observed Shh-mediated effects on ENS development: (1) Shh acts directly on ENCCs via a Ptc-independent pathway; or (2) the effect of Shh on the ENS is indirect via modification of the ENCC environment. To determine whether Shh can directly influence ENCCs, we treated intestinal explants with Gdnf, which promotes the robust migration of ENCCs out of the gut and onto a fibronectin-coated surface. Twenty-four hours later, after the cells had emigrated out of the gut wall, Shh protein was added to the culture medium, yet this did not affect continued ENCC migration. By contrast, when Shh and Gdnf were both added prior to the onset of cell migration, no migration occurs, consistent with previous results in embryonic mouse gut (Fu et al., 2004). This indicates that Shh does not directly inhibit ENCC migration, but rather must do so indirectly, presumably by altering the gut microenvironment and thus preventing their ability to respond to exogenous Gdnf.
We tested the effect of Shh overexpression and inhibition ex vivo and in ovo and observed that modulating Shh activity leads to dramatic changes in the expression of ECM proteins that are known regulators of neural crest cell migration (Payette et al., 1988; Newgreen and Hartley, 1995; Nagy et al., 2009). Whereas laminin, fibronectin and collagen I are permissive to neural crest cell migration, the CSPG family, including versican and collagen IX, are inhibitory (Newgreen and Thiery, 1980; Bronner-Fraser, 1986; Oakley et al., 1994; Perris et al., 1996; Dutt et al., 2006). We found that Shh strongly induces the expression of versican, collagen IX and CS-56 in the intestine, whereas cyclopamine reduces their expression. Furthermore, both the gut epithelium and a Shh-soaked bead are able to induce ectopic expression of collagen IX in the bursa of Fabricius, which normally does not express this protein. A similar induction of versican expression by Shh has been demonstrated in the developing trigeminal ganglia (Fedtsova et al., 2003). The induction of these factors that inhibit neural crest cell migration could account for the aganglionosis associated with Shh overexpression in both our cultured intestine and our in ovo model. We hypothesize that the gradient of epithelial-derived Shh protein in the lamina propria thus leads to the creation of an inhibitory ECM environment that prevents ENCCs from migrating too close to the epithelium, counteracting the chemoattractive effect of netrin (Jiang et al., 2003) and thus contributing to patterning the submucosal plexus.
In addition to its effect on ECM composition, Shh also decreases the expression of Gdnf, a mesenchymal factor important for ENS development (Goldstein et al., 2013). Gdnf promotes ENCC proliferation and migration via a chemoattractive effect (Young et al., 2001; Nagy and Goldstein, 2006a,b; Mwizerwa et al., 2011), and loss of Gdnf in rodents and avians leads to aganglionosis (Moore et al., 1996; Mwizerwa et al., 2011). Gdnf downregulation provides another example of Shh indirectly inhibiting ENS development. We noted that the addition of exogenous Gdnf to cultured intestine does not alter versican expression. This suggests that Shh primarily acts to alter ECM patterning, which we hypothesize secondarily decreases Gdnf expression. Shh has previously been shown to directly induce versican in cranial mesenchyme (Fedtsova et al., 2003) and collagen IX in somitic mesenchyme (Cairns et al., 2008), consistent with our observation of Shh-induced collagen IX in bursa mesenchyme. Further studies are needed to understand the mechanisms by which the microenvironment influences ENS development in normal and pathological development.
MATERIALS AND METHODS
Animals
Fertilized White Leghorn chicken (Gallus gallus) and quail (Coturnix coturnix japonica) eggs were incubated at 37°C in a humidified incubator. Embryos were staged according to Hamburger and Hamilton (HH) (Hamburger and Hamilton, 1992) or the number of embryonic days (E). All studies were approved by the Animal Ethical Committee at Semmelweis University and by the Institutional Subcommittee on Research Animal Care at Massachusetts General Hospital.
In vitro epithelium-mesenchyme recombination
Epithelium from E6 chick hindgut or E9 chick bursa was recombined with aganglionic E5 quail hindgut mesenchyme as described (Nagy and Oláh, 2010). E6 chick hindgut epithelium was also recombined with E9 chick bursa mesenchyme. To separate epithelium from mesenchyme, tissue was incubated in DMEM (Sigma) containing 0.03% collagenase (Sigma) for 15 min. Tissue recombinants were embedded in a three-dimensional collagen gel overnight (BD Biosciences) as described (Nagy and Goldstein, 2006a), then harvested from the gel and implanted into E3 chick coelom (Nagy et al., 2005; Nagy and Goldstein, 2006b) (Fig. S3). After 9 days, grafts were removed and immunohistochemistry performed. A total of 31 chimeric experiments were performed.
In the recombinants, measurements included gut wall thickness (from epithelial basement membrane to outer edge of intestine) and muscle wall thickness (from inner to outer layer of muscularis propria). Both measurements were made at the bottom of the epithelial folds and included three measurements per gut and three guts per group. Additional measurements included submucosal ganglion diameter in the radial direction and distance of the inner edge of the submucosal enteric ganglia from the epithelial basement membrane (three guts per group).
Intestinal organ culture assay
Gut was removed from E5 chick and embedded in collagen gel with recombinant mouse Shh protein (2 μg/ml; R&D Systems), monoclonal Shh-blocking antibody 5E1 (50 µg/ml; DSHB), normal mouse IgG (50 µg/ml; Invitrogen, 10400C), or cyclopamine (2 μM; Toronto Research Chemicals) for 48-72 h. For migration assays, midgut was removed from E8 chick embryos and cultured in collagen gel or fibronectin-coated dishes with Gdnf (10 ng/ml; R&D Systems) or Shh protein (2 μg/ml). Mouse intestine was dissected from C57BL/6 mice at E12.5 (Jackson Laboratory), pinned to a silicone-coated Petri dish, and cultured for 48 h with no additive, Shh protein (2 μg/ml) or cyclopamine (2 μM).
In ovo Shh-RCAS viral misexpression
Shh misexpression was achieved with the replication-competent retroviral vector RCAS. DF-1 chicken fibroblast cells (ATCC) were transduced with RCAS-Shh construct (gift of Cliff Tabin; Riddle et al., 1993). Viral harvesting, concentration and titering were performed as described (Logan and Tabin, 1998). For in ovo infection, embryos were incubated until E2 (HH10-12) and, using a Hamilton syringe, 1 μl virus was injected into the presumptive distal gut mesoderm, based on chick fate maps (Matsushita, 1995). Eggs were harvested 4-6 days later. Controls were uninjected or injected with empty RCAS vector.
Chorioallantoic membrane transplants
Hindgut, including ceca and cloaca, was dissected from E5 embryos and transplanted on the CAM of E9 chick. For retroviral experiments, the nerve of Remak was removed from E5 intestine and RCAS-Shh injected into the hindgut wall, which was cultured on the CAM for 9 days.
Shh bead implantation
Affi-Gel Blue agarose beads (70-150 μm diameter; BioRad) were soaked in 100 μg/ml Shh protein at 37°C for 2 h (Tiecke and Tickle, 2007). Beads were inserted into the E9 bursa mesenchyme, which was cultured on the CAM for 7 days. PBS-soaked beads served as controls.
Immunohistochemistry
For cryosections, tissue was fixed in 4% formaldehyde for 1 h, then infiltrated with 15% sucrose overnight, followed by 7.5% gelatin in 15% sucrose for 1-2 h, then rapidly frozen at −60°C in isopentane (Sigma). For Shh immunostaining, guts were fixed in Histochoice (EMS). Cryosections and wholemounts were stained using the primary antibodies listed in Table S1 as described (Nagy et al., 2007). Fluorescent secondary antibodies included Alexa Fluor 594 (goat anti-mouse IgG, IgM and IgG1), Alexa Fluor 488 goat anti-mouse IgG2a, and Alexa Fluor 546 goat anti-rabbit IgG (all Molecular Probes; 1:1000).
BrdU labeling and apoptosis detection
For cell proliferation, guts were incubated for 3 h in BrdU (5 mg/ml; Roche). To detect apoptosis, sections were examined with anti-activated caspase 3 (Cell Signaling). Nuclei were stained with DAPI (Molecular Probes).
In situ hybridization
In situ hybridization was performed for chick Shh, Ptc1 and Ptc2 on paraffin sections using digoxigenin-labeled riboprobes [kindly provided by Cliff Tabin (Riddle et al., 1993; Roberts et al., 1995)] according to standard protocols (Riddle et al., 1993; Acloque et al., 2008).
Quantitative PCR
Total mRNA was extracted using the RNeasy Mini Kit (Qiagen). cDNA was synthesized with the Superscript III Reverse Transcription Kit (Invitrogen). Gene expression levels were measured by quantitative PCR (qPCR) with Gapdh as the internal standard. Primers included (5′-3′, forward and reverse): Edn3, TGCGTCTACTACTGCCACCTC and CCAAACAAGAGCACCGAAAT; Gdnf, TCTCCACCTCACCCCTG and CTGGGTAATCCTCTGGCATATTAG; Gapdh, GTGCTAAGCGTGTTATCATCTC and GACAACTTTGGCATTGTGGA. Relative expression was calculated by 2−ΔCt.
Statistical analysis
Data are expressed as mean±s.d. Paired t-tests were used to evaluate statistical significance between groups. When more than two groups were compared, means were statistically compared by one-way ANOVA with Tukey's post-hoc multiple comparison tests of differences. P<0.05 was considered statistically significant.
Acknowledgements
Several antibodies were obtained from the Developmental Studies Hybridoma Bank (DSHB) developed under the auspices of the NICHD and maintained by University of Iowa, Department of Biological Sciences, Iowa City, IA 52242, USA.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
N.N. and A.M.G. designed the experiments, wrote and edited the manuscript. N.N., C.B., H.K.G., R.H., L.S.C. and N.F. assisted with experiments and analyzed the data. All authors reviewed and approved the final manuscript.
Funding
This work was supported by the National Institutes of Health (NIH)-NIDDK [R01DK080914] and the American Pediatric Surgical Association Foundation. N.N. was supported by a Bolyai Fellowship of the Hungarian Academy of Sciences. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.128132/-/DC1
References
- Acloque H., Wilkinson D. G. and Nieto M. A. (2008). In situ hybridization analysis of chick embryos in whole-mount and tissue sections. Methods Cell Biol. 87, 169-185. 10.1016/S0091-679X(08)00209-4 [DOI] [PubMed] [Google Scholar]
- Akbareian S. E., Nagy N., Steiger C. E., Mably J. D., Miller S. A., Hotta R., Molnar D. and Goldstein A. M. (2013). Enteric neural crest-derived cells promote their migration by modifying their microenvironment through tenascin-C production. Dev. Biol. 382, 446-456. 10.1016/j.ydbio.2013.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biau S., Jin S. and Fan C.-M. (2013). Gastrointestinal defects of the Gas1 mutant involve dysregulated Hedgehog and Ret signaling. Biol. Open 2, 144-155. 10.1242/bio.20123186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breau M. A., Dahmani A., Broders-Bondon F., Thiery J.-P. and Dufour S. (2009). Beta1 integrins are required for the invasion of the caecum and proximal hindgut by enteric neural crest cells. Development 136, 2791-2801. 10.1242/dev.031419 [DOI] [PubMed] [Google Scholar]
- Bronner-Fraser M. (1986). An antibody to a receptor for fibronectin and laminin perturbs cranial neural crest development in vivo. Dev. Biol. 117, 528-536. 10.1016/0012-1606(86)90320-9 [DOI] [PubMed] [Google Scholar]
- Burns A. J. and Le Douarin N. M. (1998). The sacral neural crest contributes neurons and glia to the post-umbilical gut: spatiotemporal analysis of the development of the enteric nervous system. Development 125, 4335-4347. [DOI] [PubMed] [Google Scholar]
- Burns A. J. and Le Douarin N. M. (2001). Enteric nervous system development: analysis of the selective developmental potentialities of vagal and sacral neural crest cells using quail-chick chimeras. Anat. Rec. 262, 16-28. [DOI] [PubMed] [Google Scholar]
- Cairns D. M., Sato M. E., Lee P. G., Lassar A. B. and Zeng L. (2008). A gradient of Shh establishes mutually repressing somitic cell fates induced by Nkx3.2 and Pax3. Dev. Biol. 323, 152-165. 10.1016/j.ydbio.2008.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druckenbrod N. R. and Epstein M. L. (2009). Age-dependent changes in the gut environment restrict the invasion of the hindgut by enteric neural progenitors. Development 136, 3195-3203. 10.1242/dev.031302 [DOI] [PubMed] [Google Scholar]
- Dutt S., Kleber M., Matasci M., Sommer L. and Zimmermann D. R. (2006). Versican V0 and V1 guide migratory neural crest cells. J. Biol. Chem. 281, 12123-12131. 10.1074/jbc.M510834200 [DOI] [PubMed] [Google Scholar]
- Dutt S., Cassoly E., Dours-Zimmermann M. T., Matasci M., Stoeckli E. T. and Zimmermann D. R. (2011). Versican V0 and V1 direct the growth of peripheral axons in the developing chick hindlimb. J. Neurosci. 31, 5262-5270. 10.1523/JNEUROSCI.4897-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericson J., Morton S., Kawakami A., Roelink H. and Jessell T. M. (1996). Two critical periods of Sonic Hedgehog signaling required for the specification of motor neuron identity. Cell 87, 661-673. 10.1016/S0092-8674(00)81386-0 [DOI] [PubMed] [Google Scholar]
- Fedtsova N., Perris R. and Turner E. E. (2003). Sonic hedgehog regulates the position of the trigeminal ganglia. Dev. Biol. 261, 456-469. 10.1016/S0012-1606(03)00316-6 [DOI] [PubMed] [Google Scholar]
- Fu M., Lui V. C., Sham M. H., Pachnis V. and Tam P. K. (2004). Sonic hedgehog regulates the proliferation, differentiation, and migration of enteric neural crest cells in gut. J. Cell Biol. 166, 673-684. 10.1083/jcb.200401077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein A. M. and Nagy N. (2008). A bird's eye view of enteric nervous system development: lessons from the avian embryo. Pediatr. Res. 64, 326-333. 10.1203/PDR.0b013e31818535e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein A. M., Brewer K. C., Doyle A. M., Nagy N. and Roberts D. J. (2005). BMP signaling is necessary for neural crest cell migration and ganglion formation in the enteric nervous system. Mech. Dev. 122, 821-833. 10.1016/j.mod.2005.03.003 [DOI] [PubMed] [Google Scholar]
- Goldstein A. M., Hofstra R. M. W. and Burns A. J. (2013). Building a brain in the gut: development of the enteric nervous system. Clin. Genet. 83, 307-316. 10.1111/cge.12054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburger V. and Hamilton H. L. (1992). A series of normal stages in the development of the chick embryo. 1951. Dev. Dyn. 195, 231-272. 10.1002/aja.1001950404 [DOI] [PubMed] [Google Scholar]
- Huang H., Cotton J. L., Wang Y., Rajurkar M., Zhu L. J., Lewis B. C. and Mao J. (2013). Specific requirement of Gli transcription factors in Hedgehog-mediated intestinal development. J. Biol. Chem. 288, 17589-17596. 10.1074/jbc.M113.467498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y., Liu M.-T. and Gershon M. D. (2003). Netrins and DCC in the guidance of migrating neural crest-derived cells in the developing bowel and pancreas. Dev. Biol. 258, 364-384. 10.1016/S0012-1606(03)00136-2 [DOI] [PubMed] [Google Scholar]
- Jin S., Martinelli D. C., Zheng X., Tessier-Lavigne M. and Fan C.-M. (2015). Gas1 is a receptor for sonic hedgehog to repel enteric axons. Proc. Natl. Acad. Sci. USA 112, E73-E80. 10.1073/pnas.1418629112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur R. P. (2000). Developmental disorders of the enteric nervous system. Gut 47, Suppl. 4, iv81-iv83. 10.1136/gut.47.suppl_4.iv81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolterud Å., Grosse A. S., Zacharias W. J., Walton K. D., Kretovich K. E., Madison B. B., Waghray M., Ferris J. E., Hu C., Merchant J. L. et al. (2009). Paracrine Hedgehog signaling in stomach and intestine: new roles for hedgehog in gastrointestinal patterning. Gastroenterology 137, 618-628. 10.1053/j.gastro.2009.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake J. I. and Heuckeroth R. O. (2013). Enteric nervous system development: migration, differentiation, and disease. Am. J. Physiol. Gastrointest. Liver Physiol. 305, G1-G24. 10.1152/ajpgi.00452.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landolt R. M., Vaughan L., Winterhalter K. H. and Zimmermann D. R. (1995). Versican is selectively expressed in embryonic tissues that act as barriers to neural crest cell migration and axon outgrowth. Development 121, 2303-2312. [DOI] [PubMed] [Google Scholar]
- Liu H.-X., MacCallum D. K., Edwards C., Gaffield W. and Mistretta C. M. (2004). Sonic hedgehog exerts distinct, stage-specific effects on tongue and taste papilla development. Dev. Biol. 276, 280-300. 10.1016/j.ydbio.2004.07.042 [DOI] [PubMed] [Google Scholar]
- Logan M. and Tabin C. (1998). Targeted gene misexpression in chick limb buds using avian replication-competent retroviruses. Methods 14, 407-420. 10.1006/meth.1998.0595 [DOI] [PubMed] [Google Scholar]
- Mao J., Kim B. M., Rajurkar M., Shivdasani R. A. and McMahon A. P. (2010). Hedgehog signaling controls mesenchymal growth in the developing mammalian digestive tract. Development 137, 1721-1729. 10.1242/dev.044586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita S. (1995). Fate mapping study of the splanchnopleural mesoderm of the 1.5-day-old chick embryo. Roux's Arch. Dev. Biol. 204, 392-399. 10.1007/BF00360484 [DOI] [PubMed] [Google Scholar]
- Moore M. W., Klein R. D., Fariñas I., Sauer H., Armanini M., Phillips H., Reichardt L. F., Ryan A. M., Carver-Moore K. and Rosenthal A. (1996). Renal and neuronal abnormalities in mice lacking GDNF. Nature 382, 76-79. 10.1038/382076a0 [DOI] [PubMed] [Google Scholar]
- Mwizerwa O., Das P., Nagy N., Akbareian S. E., Mably J. D. and Goldstein A. M. (2011). Gdnf is mitogenic, neurotrophic, and chemoattractive to enteric neural crest cells in the embryonic colon. Dev. Dyn. 240, 1402-1411. 10.1002/dvdy.22630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy N. and Goldstein A. M. (2006a). Endothelin-3 regulates neural crest cell proliferation and differentiation in the hindgut enteric nervous system. Dev. Biol. 293, 203-217. 10.1016/j.ydbio.2006.01.032 [DOI] [PubMed] [Google Scholar]
- Nagy N. and Goldstein A. M. (2006b). Intestinal coelomic transplants: a novel method for studying enteric nervous system development. Cell Tissue Res. 326, 43-55. 10.1007/s00441-006-0207-3 [DOI] [PubMed] [Google Scholar]
- Nagy N. and Oláh I. (2010). Experimental evidence for the ectodermal origin of the epithelial anlage of the chicken bursa of Fabricius. Development 137, 3019-3023. 10.1242/dev.055194 [DOI] [PubMed] [Google Scholar]
- Nagy N., Bíró E., Takács A., Pólos M., Magyar A. and Oláh I. (2005). Peripheral blood fibrocytes contribute to the formation of the avian spleen. Dev. Dyn. 232, 55-66. 10.1002/dvdy.20212 [DOI] [PubMed] [Google Scholar]
- Nagy N., Brewer K. C., Mwizerwa O. and Goldstein A. M. (2007). Pelvic plexus contributes ganglion cells to the hindgut enteric nervous system. Dev. Dyn. 236, 73-83. 10.1002/dvdy.20933 [DOI] [PubMed] [Google Scholar]
- Nagy N., Mwizerwa O., Yaniv K., Carmel L., Pieretti-Vanmarcke R., Weinstein B. M. and Goldstein A. M. (2009). Endothelial cells promote migration and proliferation of enteric neural crest cells via beta1 integrin signaling. Dev. Biol. 330, 263-272. 10.1016/j.ydbio.2009.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy N., Burns A. J. and Goldstein A. M. (2012). Immunophenotypic characterization of enteric neural crest cells in the developing avian colorectum. Dev. Dyn. 241, 842-851. 10.1002/dvdy.23767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newgreen D. F. and Hartley L. (1995). Extracellular matrix and adhesive molecules in the early development of the gut and its innervation in normal and spotting lethal rat embryos. Acta Anat. 154, 243-260. 10.1159/000147776 [DOI] [PubMed] [Google Scholar]
- Newgreen D. F. and Thiery J. P. (1980). Fibronectin in early avian embryos: synthesis and distribution along the migration pathways of neural crest cells. Cell Tissue Res. 211, 269-291. 10.1007/BF00236449 [DOI] [PubMed] [Google Scholar]
- Ngan E. S.-W., Garcia-Barceló M.-M., Yip B. H.-K., Poon H.-C., Lau S.-T., Kwok C. K.-M., Sat E., Sham M.-H., Wong K. K.-Y., Wainwright B. J. et al. (2011). Hedgehog/Notch-induced premature gliogenesis represents a new disease mechanism for Hirschsprung disease in mice and humans. J. Clin. Invest. 121, 3467-3478. 10.1172/JCI43737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley R. A., Lasky C. J., Erickson C. A. and Tosney K. W. (1994). Glycoconjugates mark a transient barrier to neural crest migration in the chicken embryo. Development 120, 103-114. [DOI] [PubMed] [Google Scholar]
- Payette R. F., Tennyson V. M., Pomeranz H. D., Pham T. D., Rothman T. P. and Gershon M. D. (1988). Accumulation of components of basal laminae: association with the failure of neural crest cells to colonize the presumptive aganglionic bowel of ls/ls mutant mice. Dev. Biol. 125, 341-360. 10.1016/0012-1606(88)90217-5 [DOI] [PubMed] [Google Scholar]
- Perris R., Perissinotto D., Pettway Z., Bronner-Fraser M., Morgelin M. and Kimata K. (1996). Inhibitory effects of PG-H/aggrecan and PG-M/versican on avian neural crest cell migration. FASEB J. 10, 293-301. [DOI] [PubMed] [Google Scholar]
- Potts W. M., Olsen M., Boettiger D. and Vogt V. M. (1987). Epitope mapping of monoclonal antibodies to gag protein p19 of avian sarcoma and leukaemia viruses. J. Gen. Virol. 68, 3177-3182. 10.1099/0022-1317-68-12-3177 [DOI] [PubMed] [Google Scholar]
- Raghavan S., Gilmont R. R. and Bitar K. N. (2013). Neuroglial differentiation of adult enteric neuronal progenitor cells as a function of extracellular matrix composition. Biomaterials 34, 6649-6658. 10.1016/j.biomaterials.2013.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalho-Santos M., Melton D. A. and McMahon A. P. (2000). Hedgehog signals regulate multiple aspects of gastrointestinal development. Development 127, 2763-2772. [DOI] [PubMed] [Google Scholar]
- Reichenbach B., Delalande J.-M., Kolmogorova E., Prier A., Nguyen T., Smith C. M., Holzschuh J. and Shepherd I. T. (2008). Endoderm-derived Sonic hedgehog and mesoderm Hand2 expression are required for enteric nervous system development in zebrafish. Dev. Biol. 318, 52-64. 10.1016/j.ydbio.2008.02.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle R. D., Johnson R. L., Laufer E. and Tabin C. (1993). Sonic hedgehog mediates the polarizing activity of the ZPA. Cell 75, 1401-1416. 10.1016/0092-8674(93)90626-2 [DOI] [PubMed] [Google Scholar]
- Ring C., Hassell J. and Halfter W. (1996). Expression pattern of collagen IX and potential role in the segmentation of the peripheral nervous system. Dev. Biol. 180, 41-53. 10.1006/dbio.1996.0283 [DOI] [PubMed] [Google Scholar]
- Roberts D. J. (2000). Molecular mechanisms of development of the gastrointestinal tract. Dev. Dyn. 219, 109-120. [DOI] [PubMed] [Google Scholar]
- Roberts D. J., Johnson R. L., Burke A. C., Nelson C. E., Morgan B. A. and Tabin C. (1995). Sonic hedgehog is an endodermal signal inducing Bmp-4 and Hox genes during induction and regionalization of the chick hindgut. Development 121, 3163-3174. [DOI] [PubMed] [Google Scholar]
- Sukegawa A., Narita T., Kameda T., Saitoh K., Nohno T., Iba H., Yasugi S. and Fukuda K. (2000). The concentric structure of the developing gut is regulated by Sonic hedgehog derived from endodermal epithelium. Development 127, 1971-1978. [DOI] [PubMed] [Google Scholar]
- Tiecke E. and Tickle C. (2007). Application of sonic hedgehog to the developing chick limb. Methods Mol. Biol. 397, 23-33. 10.1007/978-1-59745-516-9_2 [DOI] [PubMed] [Google Scholar]
- Washington Smoak I., Byrd N. A., Abu-Issa R., Goddeeris M. M., Anderson R., Morris J., Yamamura K., Klingensmith J. and Meyers E. N. (2005). Sonic hedgehog is required for cardiac outflow tract and neural crest cell development. Dev. Biol. 283, 357-372. 10.1016/j.ydbio.2005.04.029 [DOI] [PubMed] [Google Scholar]
- Weskamp G. and Reichardt L. F. (1991). Evidence that biological activity of NGF is mediated through a novel subclass of high affinity receptors. Neuron 6, 649-663. 10.1016/0896-6273(91)90067-A [DOI] [PubMed] [Google Scholar]
- Wu J. J., Chen J. X., Rothman T. P. and Gershon M. D. (1999). Inhibition of in vitro enteric neuronal development by endothelin-3: mediation by endothelin B receptors. Development 126, 1161-1173. [DOI] [PubMed] [Google Scholar]
- Yang J. T., Liu C. Z., Villavicencio E. H., Yoon J. W., Walterhouse D. and Iannaccone P. M. (1997). Expression of human GLI in mice results in failure to thrive, early death, and patchy Hirschsprung-like gastrointestinal dilatation. Mol. Med. 3, 826-835. [PMC free article] [PubMed] [Google Scholar]
- Yntema C. L. and Hammond W. S. (1954). The origin of intrinsic ganglia of trunk viscera from vagal neural crest in the chick embryo. J. Comp. Neurol. 101, 515-541. 10.1002/cne.901010212 [DOI] [PubMed] [Google Scholar]
- Young H. M., Hearn C. J., Ciampoli D., Southwell B. R., Brunet J.-F. and Newgreen D. F. (1998). A single rostrocaudal colonization of the rodent intestine by enteric neuron precursors is revealed by the expression of Phox2b, Ret, and p75 and by explants grown under the kidney capsule or in organ culture. Dev. Biol. 202, 67-84. 10.1006/dbio.1998.8987 [DOI] [PubMed] [Google Scholar]
- Young H. M., Hearn C. J., Farlie P. G., Canty A. J., Thomas P. Q. and Newgreen D. F. (2001). GDNF is a chemoattractant for enteric neural cells. Dev. Biol. 229, 503-516. 10.1006/dbio.2000.0100 [DOI] [PubMed] [Google Scholar]
- Zanin M. K. B., Bundy J., Ernst H., Wessels A., Conway S. J. and Hoffman S. (1999). Distinct spatial and temporal distributions of aggrecan and versican in the embryonic chick heart. Anat. Rec. 256, 366-380. [DOI] [PubMed] [Google Scholar]