

Abstract
Background:
Nonsyndromic hearing loss (NSHL) is highly heterogeneous, in which more than 90 causative genes have currently been identified. DFNA5 is one of the deafness genes that known to cause autosomal dominant NSHL. Until date, only five DFNA5 mutations have been described in eight families worldwide. In this study, we reported the identification of a novel pathogenic mutation causing DFNA5 deafness in a five-generation Chinese family.
Methods:
After detailed clinical evaluations of this family, the genomic DNA of three affected individuals was selected for targeted exome sequencing of 101 known deafness genes, as well as mitochondrial DNA and microRNA regions. Co-segregation analysis between the hearing loss and the candidate variant was confirmed in available family members by direct polymerase chain reaction (PCR)-Sanger sequencing. Real-time PCR (RT-PCR) was performed to investigate the potential effect of the pathogenic mutation on messenger RNA splicing.
Results:
Clinical evaluations revealed a similar deafness phenotype in this family to that of previously reported DFNA5 families with autosomal dominant, late-onset hearing loss. Molecular analysis identified a novel splice site mutation in DFNA5 intron 8 (IVS8+1 delG). The mutation segregated with the hearing loss of the family and was absent in 120 unrelated control DNA samples of Chinese origin. RT-PCR showed skipping of exon 8 in the mutant transcript.
Conclusions:
We identified a novel DFNA5 mutation IVS8+1 delG in a Chinese family which led to skipping of exon 8. This is the sixth DFNA5 mutation relates to hearing loss and the second one in DFNA5 intron 8. Our findings provide further support to the hypothesis that the DFNA5-associated hearing loss represents a mechanism of gain-of-function.
Keywords: DFNA5, Hearing Loss, Mutation, Targeted Genomic Capture
INTRODUCTION
Hearing loss is one of the most genetically heterogeneous disorders, present in one of every 500 newborns. During the past two decades, tremendous progress has been made in the identification of causative genes that cause hereditary nonsyndromic hearing loss (NSHL). Up to now, approximately 30 dominant (DFNA), 55 recessive (DFNB), 4 X-linked (DFNX), and 2 mitochondrial genes have been identified (http://hereditaryhearingloss.org/). These genes involved in many functions in the ear crucial for normal hearing, such as cochlear fluid homeostasis, ionic channels, stereocilia morphology and function, synaptic transmission, gene regulation, and others.[1] However, the function of some of these genes remains a mystery.
DFNA5 is one of the deafness genes with currently unknown function, which was mapped to chromosome 7p15 in 1995 and identified in 1998 in an extended Dutch family.[2,3] Since then, only eight families with DFNA5 mutations segregating hereditary NSHL have been described in the literature.[3,4,5,6,7,8,9] The hearing loss in these families is strikingly similar, being progressive, nonsyndromic, and sensorineural predominantly affecting the high frequencies, although the age of onset differs slightly. Each reported mutation causes skipping of exon 8 at the messenger (mRNA) level, resulting in a frame-shift and produces a prematurely truncated DFNA5 protein. In this study, we reported the identification of a novel DFNA5 mutation, IVS8+1 delG, in a Chinese family which again led to exon 8 skipping and subsequently to nonsyndromic sensorineural hearing impairment. To the best of our knowledge, this is the second description of a DFNA5 mutation in intron 8.
METHODS
Pedigree and clinical evaluation
A five-generation Chinese family, designated as JSNY-052, was ascertained from the Department of Otolaryngology, First Affiliated Hospital of Nanjing Medical University [Figure 1]. Eighteen family members, including 12 presumably affected and 6 presumably unaffected individuals, participated in this study. Ethical approval was obtained from the Ethical Committee of Nanjing Medical University for human studies and written informed consent was obtained from all participants. Medical histories, including the degree of hearing loss, the age of onset, the progression of hearing impairment, use of aminoglycosides, noise exposure and other relevant clinical manifestations, were collected. Nongenetic causes of hearing loss were excluded. All individuals included in this study underwent otological examination, with particular attention paid to the presence of any syndromic findings. Pure-tone audiometry was performed in 8 affected and 6 unaffected family members. Immittance, auditory brainstem response (ABR) and distortion product otoacoustic emissions (DPOAEs) were performed in two affected subjects only. The computerized tomography of the temporal bone was done in the proband (IV-12).
Figure 1.
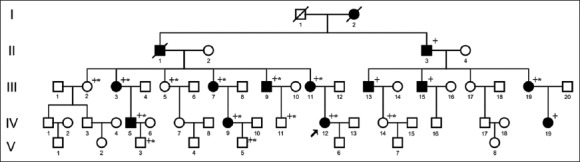
Pedigree of family JSNY-052. ↗: Proband; +: Mutation analysis; *: Pure-tone audiometry performed.
Targeted genomic capture and variation analysis
Peripheral blood samples were obtained from all the participants, and genomic DNA was extracted using a blood genomic DNA extraction kit (TianGen, Beijing, China). The coding exons and flanking regions of 101 known deafness genes, as well as 3 mitochondrial DNA and 3 microRNA regions, were target-enriched as previously described.[10] A minimum of 3 μg DNA was used to generate indexed Illumina libraries, and the final library size was 300–400 base pairs. Captured DNA fragments were sequenced on an Illumina HiSeq2000 Analyzer with paired-end reads of 100 base pairs. After that, high-quality reads were retrieved from raw reads by filtering out the low-quality reads and adaptor sequences using the SolexaQA and the Cutadapt (http://code.google.com/p/cutadapt/). Variations were first selected if they appeared in the 1000 Genome Project database with a MAF >0.05, then were selected if they appeared in the 300 local Asian Genome database. Reads were aligned to the reference genome (NCBI37/hg19) using the BWA software (Solectek Corporation). Single nucleotide polymorphism (SNPs) and InDels were identified using the SOAPsnp and GATK programs, respectively. Finally, nonsynonymous variant information was collected and predicted for pathogenicity using four algorithms, PolyPhen, SIFT, PANTHER, and PMut.
Co-segregation analysis between the hearing loss and the candidate pathogenic variants were performed for all available family members by direct polymerase chain reaction (PCR)-Sanger sequencing. Genomic DNA was amplified with the primers 5’- GCTGCGGGTCGTACTTTCCTC-3’ and 5’- TAGGGAGGCTTGTGCTGGATG-3’. As for controls, DNA samples from 120 unrelated normal hearing individuals were also collected.
Real-time polymerase chain reaction analysis
Total RNA was isolated from peripheral blood of the family members using Trizol reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's guidelines. One-step real-time PCRs (RT-PCRs) were performed using a Prime Script RT reagent kit (Takara, Dalian, China). For initial testing of skipping of exon 8, the cDNA was amplified using a forward primer from exon 7, 5’-AACAGACAGCTTTGAGTGACA-3’, and a reverse primer from exon 10, 5’- ATCCCAAACCTTTCTGTATCT-3’. For the detection of alternative splicing in other parts of the mRNA in DFNA5, the primers were selected as previously described.[5] PCR reactions were performed in a standard time/temperature profile, and the amplified fragments were sequenced on an automated sequencer (ABI 3730, Applied Biosystems, Foster City, CA, USA).
RESULTS
Clinical findings
The pedigree of the family [Figure 1] comprises 53 members in five-generations and shows autosomal dominant inheritance. Fourteen individuals were diagnosed as having a hereditary hearing loss by audiological evaluation and medical history taking. Of whom, 12 were still alive and available for this study. The hearing loss was first reported at ages from 8 to 30 years and manifested itself as progressive and nonsyndromic. Pure-tone audiograms of eight patients showed bilateral moderate to profound sensorineural hearing loss [Table 1]; both ears were similarly affected except III-9, who suffered from chronic suppurative otitis media in the left ear and demonstrated an asymmetric hearing loss [Figure 2]. In most cases, the hearing loss was severest at the high frequencies, making a down-sloping audiometric configuration. Other audiometric configurations included the mid-frequency U-shaped form in two patients (III-11, IV-12), and the flat form in one patient (III-9). Extended audiological examinations, including immittance, ABR and DPOAEs in the 2 affected individuals revealed a cochlear involvement. Aside from hearing loss, the patients were phenotypically normal. None of them complained of vestibular symptoms. An imaging study of the temporal bone in the proband was normal.
Table 1.
Summary of clinical data for eight affected family members
| Subjects | Gender | Age (years) | PTA (dB HL)* | Degree of hearing loss | Audiogram shape | ||
|---|---|---|---|---|---|---|---|
| At testing | At onset | Left | Right | ||||
| III-3 | Female | 64 | 18 | 87.5 | 96.25 | Severe | Down-slopping |
| III-7 | Female | 60 | 10 | 98.75 | >100 | Profound | Down-slopping |
| III-9 | Male | 55 | 30 | >100 | 92.5 | Profound | Flat |
| III-11 | Female | 48 | 12 | 91.25 | 92.5 | Profound | U-shaped |
| III-19 | Female | 55 | 12 | >101.25 | >110 | Profound | Down-slopping |
| IV-5 | Male | 33 | 15 | 88.75 | 95 | Severe | Down-slopping |
| IV-9 | Female | 33 | 8 | 101.25 | 97.5 | Profound | Down-slopping |
| IV-12 | Female | 24 | 10 | 66.25 | 70 | Moderate | U-shaped |
*The PTA was calculated from audiometric thresholds at 500, 1000, 2000, and 4000 Hz. The severity of HL was categorized as follows: Mild (PTA ≤40 dB), moderate (40 dB < PTA ≤70 dB), severe (70 dB < PTA ≤90 dB), and profound (PTA >90 dB). PTA: Pure-tone average.
Figure 2.

Pur-tone audiograms of patient III-9 and proband IV-12.
Identification of DFNA5 IVS8+1 delG mutation
The genomic DNA of three affected individuals was subjected to targeted exome sequencing. More than 96.50% of targeted regions were covered for each sample with an average sequencing depth of 142.8. The coverage of the targeted exons ranged from 95.20% to 96.10% for the >×10 reads and from 91.60% to 93.40% for the >×20 reads, respectively. All mitochondrial DNA and microRNA regions were sequenced at a depth of over × 100. Sequence quality of all known deafness genes or regions was measured. Finally, more than 140 variants were detected for each sample. To give priority to the identification of deleterious mutations (missense, nonsense and splice variants), a series of filtering strategies, including HapMap 28, SNP databases and multiple algorithms (PolyPhen, SIFT, PANTHER and PMut) were adopted. Following this step-by-step filtering protocol, only one novel DFNA5 splice site mutation, IVS8+1 delG, was likely to be pathogenic according to this criterion [Figures 3 and 4a]. Sanger sequencing of all available family members showed that this mutation faithfully co-segregated with the disease phenotype in this family. However, the mutation was absent in the 120 control participants.
Figure 3.
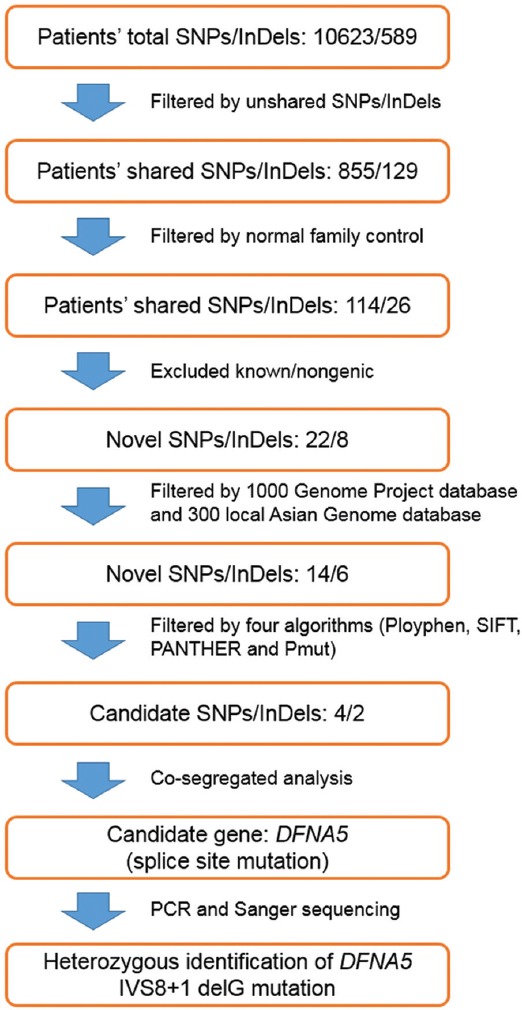
The filtering protocol of variants identified by targeted genomic capture.
Figure 4.
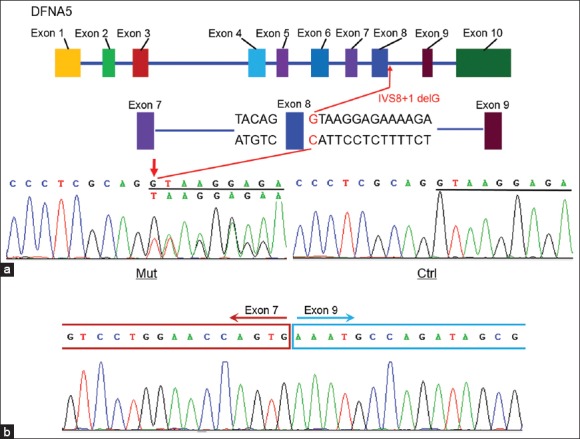
DNFA5 IVS8+1 delG mutation in family JSNY-052. (a) The genomic structure of DFNA5 and chromatograms of IVS8+1 delG in patients and controls. (b) Sequence chromatogram of the cDNA fragment amplified by real-time polymerase chain reaction showing the skipping of exon 8.
Validation of skipping of exon 8 in DFNA5
To investigate the potential effect of the IVS8+1 delG mutation on mRNA splicing, we designed primers from exon 7 and exon 10 to amplify the cDNA fragment from peripheral blood mRNA. This yielded a cDNA fragment of 424-bp in controls and an additional 231-bp fragment in the affected individuals. Sequence analysis of aberrant RT-PCR product showed skipping of exon 8 in the mutant transcript, which resulted in a direct connection of exon 7 to exon 9 [Figure 4b]. Skipping of exon 8 led to a frame-shift in coding sequences with a stop codon at position 372,[3] which resulted in changes of 41 amino acids (residuals 331–371) and in-frame deletion of 126 amino acids in the reading frame. No mutations in any part of the mRNA were detected by RT-PCR with other primers.
DISCUSSION
In the present study, we described the ninth DFNA5 family with autosomal dominant hearing impairment. The affected individuals exhibited a very similar clinical phenotype to that of previously reported families with the DFNA5 mutations.[3,4,5,6,7,8,9] The hearing loss is late-onset, nonsyndromic, progressive, bilaterally symmetric (except patient III-9), and sensorineural that usually started at high frequencies. None of the patients complained of vestibular symptoms although the vestibular function was not tested. In this family, targeted next-generation sequencing combined with co-segregation analysis led to the identification of a novel DFNA5 mutation, IVS8+1 delG, which is located in the splice site of intron 8. The mutation abolishes the known splice donor site of DFNA5 intron 8, leading to the abnormal splicing reaction between the splice acceptor site in intron 8 and the donor site in intron 7. As a consequence, this mutation would result in skipping of exon 8 at the mRNA level. The IVS8+1 delG was present in all affected individuals, and was not detected in other family members and 120 unrelated control DNA samples of Chinese origin, indicating that this mutation is the molecular basis for the hearing loss.
In addition to the genetic factors, hearing loss is also affected by the environmental factors. In this study, we cannot rule out the possibility that environmental factors modify the clinical phenotype of DFNA5 mutation, as the patient III-9 who suffered from chronic suppurative otitis media in the left ear clearly demonstrated a different severity of hearing loss for both ears, while the other seven patients without ear disease exhibited a symmetric hearing loss [Figure 2]. This finding indicates that otitis media may also contribute to the phenotypic expression of the deafness-associated DFNA5 mutation.
Until date, a total of six deafness-causing mutations in the DFNA5 gene have been reported in nine families [Table 2]. Apart from two Dutch families described by Van Laer et al.[3] and Bischoff et al.,[5] the others were all pedigrees of East Asian origin. In addition, of all reported DFNA5 mutations, a 3-bp deletion mutation (c. 991-15_991-13del) was confirmed in a Chinese, a Korean and two Japanese families with autosomal dominant NSHL.[4,7,8] After comparison of the mutation-linked haplotypes of these families, a single origin of this mutation was suggested.[7,8] These findings indicate the possibility of ethnic specificity for DFNA5 mutations, and the c. 991-15_991-13del mutation may be a frequent cause of hearing the loss in the East Asian populations. It is thus reasonable to perform a regular diagnostic test for the DFNA5 mutations in these populations with autosomal dominant NSHL.
Table 2.
Summary of all reported DFNA5 mutations leading to hearing loss
| Families (n) | Mutation | Location | Effect of mutation | Authors (year) |
|---|---|---|---|---|
| Dutch (1) | c. 990+503_990+1691delins 132 | Intron 7 | Skipping of exon 8 | Van Laer et al. (1998)[3] |
| Chinese (1) | c. 991-15_991-13del | Intron 7 | Skipping of exon 8 | Yu et al. (2003)[4] |
| Dutch (1) | c. 991-6 C>G | Intron 7 | Skipping of exon 8 | Bischoff et al. (2004)[5] |
| Chinese (1) | c. 1183+4 A>G | Intron 8 | Skipping of exon 8 | Cheng et al. (2007)[6] |
| Korean (1) | c. 991-15_991-13del | Intron 7 | Skipping of exon 8 | Park et al. (2010)[7] |
| Japanese (2) | c. 991-15_991-13del | Intron 7 | Skipping of exon 8 | Nishio et al. (2014)[8] |
| Chinese (1) | c. 991_2A>G | Intron 7 | Skipping of exon 8 | Chai et al. (2014)[9] |
| Chinese (1) | IVS8+1 delG | Intron 8 | Skipping of exon 8 | This study |
The molecular pathogenic mechanism of NSHL associated with DFNA5 mutations remains unclear. As shown in Table 2, all the currently known DFNA5 mutations are located in introns 7 or 8, and are expected to lead to skipping of exon 8 in the mutant transcript although these mutations may differ at the genomic DNA level.[4,5] Skipping of exon 8 results in a shift in the reading frame and thereby causes premature truncation of the encoded protein. While a truncating mutation, c. 640insC, in exon 5 of DFNA5 did not segregate with the clinical phenotype in an Iranian family with the autosomal dominant hearing loss.[11] This mutation did not lead to exon 8 skipping, but truncated the protein in the fifth exon and led to a loss of function. Furthermore, a Dfna5 knockout mice generated through the deletion of exon 8 had no significant hearing loss, even though morphological studies demonstrated significant differences in the number of fourth row outer hair cells between Dfna5-/- mice and their wild-type littermates.[12] Van Laer et al.[12] believed that the differences in the type of mutation rather than the genetic backgrounds best explain the phenotypic difference between the human and the mouse mutant. These observations suggest that only skipping of exon 8 but not other mutations in other parts of the DFNA5 gene results in hearing loss, and the hearing loss associated with DFNA5 might represent a mechanism of gain-of-function rather than haplo-insufficiency. The description of two patients harboring a genomic deletion in the chromosomal region encompassing DFNA5 but showing no signs of hearing loss[13] further strengthens the hypothesis. Other evidence supporting the hypothesis of a deleterious new function for mutant DFNA5 derived from two experimental studies.[4,5] It was demonstrated that transfections of human mutant DFNA5 exerted toxic effects on both yeast[14] and mammalian cells.[15]
In conclusion, we described here a five-generation Chinese family with autosomal dominant late-onset NSHL. In this family, a novel DFNA5 mutation IVS8+1 delG leading to skipping of exon 8 was identified through targeted gnomic sequencing and co-segregation analysis. This is the sixth DFNA5 mutation relates to hearing loss and the second one in DFNA5 intron 8. Our results provide further support to the hypothesis that the DFNA5-associated hearing loss is caused by a very special gain-of-function mutation.
Financial support and sponsorship
This study was supported by a grant from the Jiangsu Health Administration (No. LJ201120), by a research grant award from the National Natural Science Foundation of China (No. 31171217), and by a grant from the Research Special Fund for Public Welfare Industry of Health, Ministry of Health of China (No. 201202005).
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Li-Shao Guo
REFERENCES
- 1.Angeli S, Lin X, Liu XZ. Genetics of hearing and deafness. Anat Rec (Hoboken) 2012;295:1812–29. doi: 10.1002/ar.22579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Camp G, Coucke P, Balemans W, van Velzen D, van de Bilt C, van Laer L, et al. Localization of a gene for non-syndromic hearing loss (DFNA5) to chromosome 7p15. Hum Mol Genet. 1995;4:2159–63. doi: 10.1093/hmg/4.11.2159. [DOI] [PubMed] [Google Scholar]
- 3.Van Laer L, Huizing EH, Verstreken M, van Zuijlen D, Wauters JG, Bossuyt PJ, et al. Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nat Genet. 1998;20:194–7. doi: 10.1038/2503. [DOI] [PubMed] [Google Scholar]
- 4.Yu C, Meng X, Zhang S, Zhao G, Hu L, Kong X. A 3-nucleotide deletion in the polypyrimidine tract of intron 7 of the DFNA5 gene causes nonsyndromic hearing impairment in a Chinese family. Genomics. 2003;82:575–9. doi: 10.1016/s0888-7543(03)00175-7. [DOI] [PubMed] [Google Scholar]
- 5.Bischoff AM, Luijendijk MW, Huygen PL, van Duijnhoven G, De Leenheer EM, Oudesluijs GG, et al. A novel mutation identified in the DFNA5 gene in a Dutch family: A clinical and genetic evaluation. Audiol Neurootol. 2004;9:34–46. doi: 10.1159/000074185. [DOI] [PubMed] [Google Scholar]
- 6.Cheng J, Han DY, Dai P, Sun HJ, Tao R, Sun Q, et al. A novel DFNA5 mutation, IVS8 4 A>G, in the splice donor site of intron 8 causes late-onset non-syndromic hearing loss in a Chinese family. Clin Genet. 2007;72:471–7. doi: 10.1111/j.1399-0004.2007.00889.x. [DOI] [PubMed] [Google Scholar]
- 7.Park HJ, Cho HJ, Baek JI, Ben-Yosef T, Kwon TJ, Griffith AJ, et al. Evidence for a founder mutation causing DFNA5 hearing loss in East Asians. J Hum Genet. 2010;55:59–62. doi: 10.1038/jhg.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nishio A, Noguchi Y, Sato T, Naruse TK, Kimura A, Takagi A, et al. A DFNA5 mutation identified in Japanese families with autosomal dominant hereditary hearing loss. Ann Hum Genet. 2014;78:83–91. doi: 10.1111/ahg.12053. [DOI] [PubMed] [Google Scholar]
- 9.Chai Y, Chen D, Wang X, Wu H, Yang T. A novel splice site mutation in DFNA5 causes late-onset progressive non-syndromic hearing loss in a Chinese family. Int J Pediatr Otorhinolaryngol. 2014;78:1265–8. doi: 10.1016/j.ijporl.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 10.Wei Q, Zhu H, Qian X, Chen Z, Yao J, Lu Y, et al. Targeted genomic capture and massively parallel sequencing to identify novel variants causing Chinese hereditary hearing loss. J Transl Med. 2014;12:311. doi: 10.1186/s12967-014-0311-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Laer L, Meyer NC, Malekpour M, Riazalhosseini Y, Moghannibashi M, Kahrizi K, et al. A novel DFNA5 mutation does not cause hearing loss in an Iranian family. J Hum Genet. 2007;52:549–52. doi: 10.1007/s10038-007-0137-2. [DOI] [PubMed] [Google Scholar]
- 12.Van Laer L, Pfister M, Thys S, Vrijens K, Mueller M, Umans L, et al. Mice lacking Dfna5 show a diverging number of cochlear fourth row outer hair cells. Neurobiol Dis. 2005;19:386–99. doi: 10.1016/j.nbd.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 13.Dunø M, Hove H, Kirchhoff M, Devriendt K, Schwartz M. Mapping genomic deletions down to the base: A quantitative copy number scanning approach used to characterise and clone the breakpoints of a recurrent 7p14.2p15.3 deletion. Hum Genet. 2004;115:459–67. doi: 10.1007/s00439-004-1174-y. [DOI] [PubMed] [Google Scholar]
- 14.Gregan J, Van Laer L, Lieto LD, Van Camp G, Kearsey SE. A yeast model for the study of human DFNA5, a gene mutated in nonsyndromic hearing impairment. Biochim Biophys Acta. 2003;1638:179–86. doi: 10.1016/s0925-4439(03)00083-8. [DOI] [PubMed] [Google Scholar]
- 15.Van Laer L, Vrijens K, Thys S, Van Tendeloo VF, Smith RJ, Van Bockstaele DR, et al. DFNA5: Hearing impairment exon instead of hearing impairment gene? J Med Genet. 2004;41:401–6. doi: 10.1136/jmg.2003.015073. [DOI] [PMC free article] [PubMed] [Google Scholar]