

Abstract
Bromodomain-containing proteins have emerged as desirable targets for anti-neoplastic and anti-inflammatory drug discovery. Toward the development of selective inhibitors of the BET family of bromodomains, we optimized bead-based assays to detect interactions between bromodomains and poly-acetylated histone peptides. Donor and acceptor beads bound to target and ligand are brought into proximity by this protein-protein interaction. After laser illumination, singlet oxygen evolved from donor beads travels to the spatially close acceptor beads, resulting in chemiluminesence. This AlphaScreen assay has proven amendable to high-throughput screening, secondary validation, and specificity profiling during lead discovery and optimization. Here we report our protocol for assay development to measure inhibition of ligand binding to bromodomain containing protein 4 (BRD4). We discuss the discovery of an appropriate probe, optimization of bead, probe, and protein concentrations, and the derivation of protein-probe inhibition curves. Finally, we explore the implementation of this technology for high-throughput screening of potential BRD4 inhibitors.
Keywords: Bromodomain, BRD4, JQ1, AlphaScreen, High-throughput screening, Protein-protein interaction
Introduction
AlphaScreen (Amplified Luminescent Proximity Homogeneous Assay Screen) technology was first developed under the name LOCI (Luminescent Oxygen Channeling Immunoassay) by Dade Behring, Inc. Originally published in 1994, PerkinElmer obtained the rights to manufacture this bead technology in 1999 under the name AlphaScreen for drug discovery purposes (Eglen et al., 2007; Ullman et al., 1994; Ullman et al., 1996). Since then, this commercial technology has found a wide range of application in monitoring molecular interactions between targets and their ligands in labs around the world (Eglen et al., 2007).
As the name implies, AlphaScreen is a homogenous bead-based proximity assay amendable to high-throughput screening (HTS) for monitoring biomolecular interactions in a multiwell microplate format. AlphaScreen assays can be adapted to monitor interactions between proteins, peptides, lipids, and nucleic acids, and their potential ligands, which can also be proteins, peptides, lipids, nucleic acids or small molecules. In short, donor and acceptor beads that have been bound to target molecules and potential ligands, respectively, are brought into close proximity by a specific molecular interaction. When illuminated by a light source at 680 nm, reactive singlet state oxygen is produced by the donor bead and diffuses from the donor to the acceptor bead, initiating a cascade of reactions that result in a luminescent signal at 520-620 nm (Figure 1). If the donor bead is not in close proximity to the acceptor bead (>200 nm), the singlet oxygen decays to ground state and no signal is produced. Thus, AlphaScreen technology can be used to assay for the presence of molecular interactions, but it is also amenable to monitoring the disruption of molecular interactions. Compounds that disrupt the interaction between target and ligand will separate the two beads, and result in signal loss—in this manner potential inhibitors can be identified.
Figure 1. Schematic depiction of a competitive AlphaScreen assay.
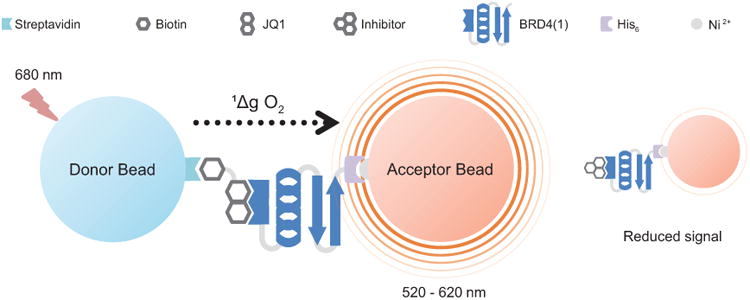
When His6-tagged recombinant protein (BRD4(1)) binds to biotinylated probe (Bio-JQ1), streptavidin-coated donor beads and nickel-coated acceptor beads are brought into close proximity (<200 nm). Upon illumination at 680 nm, the photosensitized donor bead transfers singlet state oxygen (1Δg O2) to the acceptor bead, initializing a series of reactions resulting in a fluorescent signal at 520-620 nm. Incubation with an inhibitor can disrupt the protein-probe complex, resulting in loss of signal.
We and others have deployed bead-based nanomaterial assays to study diverse protein-protein and protein-small molecule interactions (Filippakopoulos et al., 2010; McKeown et al., 2014; Yi et al., 2014). Here, we illustrate our approach to optimize and deploy this technology to screen for inhibitors of the bromodomain and extra-terminal (BET) family member, bromodomain containing protein 4 (BRD4) (Eglen et al., 2007; Quinn et al., 2010). Bromodomains mediate context-specific recognition of acetyl-lysine residues on histone proteins and specific transcription factors (Zeng and Zhou, 2002). Molecular recognition of acetylated histones occurs in regulatory regions of euchromatin, typically at active promoter and enhancer elements (Chapuy et al., 2013). Recently, BET bromodomains have been identified as desirable targets for anti-cancer drug development owing to the rearrangement of BRD4 and BRD3 as proto-oncogenes in an aggressive form of lung and head & neck cancer (French, 2012) and as critical co-activators of MYC expression (Delmore et al., 2011; Zuber et al., 2011). BRD4 contains two bromodomains (BRD4(1) and BRD4(2)); while the first bromodomain recognizes acetylated lysine, there have been several reports that BRD4(2) is involved in coactivation of P-TEFb through binding of triacetylated cyclin T1 (Huang et al., 2009; Yang et al., 2005). Previously, our group in collaboration with Prof. Stefan Knapp developed first direct-acting inhibitors of BET bromodomains, including the prototypical chemical probe JQ1(Filippakopoulos et al., 2010). This research established the druggability of human bromodomains, and encouraged the development of chemically diverse BET inhibitors by our group and others (Filippakopoulos and Knapp, 2014).
Our development of acetyl-lysine competitive BET bromodomain inhibitors was supported by a series of orthogonal biochemical and biophysical assays. Here, we describe the foundational assay utilized in high-throughput screening and follow-up chemistry. We describe our experiences with developing an AlphaScreen assay for inhibitors of BRD4(1), but the approach outlined may be adapted for other protein-protein or protein-ligand interactions. In Basic Protocol 1, we describe how to optimize target and probe concentrations using recombinant His6-tagged BRD4(1), the biotinylated form of the BRD4 inhibitor (+)-JQ1, and PerkinElmer's AlphaScreen Histidine (Nickel Chelate) Detection Kit. Compound screening in dose-response format is detailed in Basic Protocol 2. And finally, in Basic Protocol 3, we describe how to develop a high-throughput screening strategy utilizing large chemical libraries and how to distinguish true inhibitors from false positive results.
Strategic Planning
Bead Choice and Design
PerkinElmer offers several different types of AlphaScreen Donor and Acceptor beads for screening assays and bead selection is an important consideration. The choice of bead is mainly influenced by the biomolecules to be studied and how they are available (tagged or untagged). Histidine-tagged affinity nickel chelate beads, streptavidin-coated beads, antibody-coated beads, and as yet unconjugated beads are commercially available as both donor and acceptor beads. Additionally, two different types of acceptor beads are available with various coatings: the AlphaScreen and AlphaLISA beads. These acceptor beads differ in the fluorophores used to generate signal. AlphaScreen acceptor beads use rubene, which emits light in the 520-620 nm range, whereas the AlphaLISA beads use a europium chelate that fluoresces in a much narrower range at 615 nm. This renders the AlphaLISA Acceptor bead less prone to interference from buffer components (e.g. serum, plasma) or complex biological samples that may contain components that absorb light between 520 and 600 nm (e.g. heme). Interference is often not a concern with simple buffers, as used here for BRD4(1). However, the more sensitive AlphaLISA beads may still prove advantageous in a compound library screen, as some compounds in the screen may interfere with the absorbance and luminescence across 520-600 nm, resulting in false positive readings. False positive readings can also arise from compounds that compete with the protein or probe for binding to their respective Alpha bead (e.g. biotin mimetics when using streptavidin coated beads). Due to the possibility of false positive readings when screening a library of compounds with unestablished structure activity relationships (SAR) for BRD4, it is important to test the compound library against a control assay (see Basic Protocol 3).
Probe and Target Design
The design of the competitive binding assay starts with finding an appropriate probe, often a small molecule or peptide, with high affinity (Kd < 1 uM) for the specific protein domain being interrogated. As bromodomains bind to acetylated lysine, one option is to synthesize a probe using a chemically-tagged acetylated peptide. In particular, BRD4 binds tightly to human histone H4 tetra-acetylated peptides (Dey et al., 2003; Jung et al., 2014), which can be synthesized in many academic labs or purchased commercially. Biotinylated tetra-acetylated peptides are also available commercially (Epigentek).
To establish a highly sensitive assay, we developed an affinity reagent biased for BET binding by appending JQ1 to biotin with a PEG linker positioned at the site least likely to impair molecular recognition of BRD4 (Bio-JQ1;(Anders et al., 2013). As available, crystal structures of protein and ligand can help in identifying a solvent-exposed position at which to chemically attach a retrievable chemical handle. Ensuring that biotinylated probe molecules bind to targets with similar affinity (Kd) as the unmodified probe is essential before beginning the AlphaScreen assay. Additionally, modification of the protein target with the addition of a tag (e.g. His6 or GST) should not significantly alter binding to the probe. To this end, most tags should be attached to the N- or C-termini of peptides to prevent interfering with binding of acetylated lysine by the bromodomain.
Determining the binding affinity of a biotinylated probe to its modified target protein can be accomplished using several methods such as isothermal titration calorimetry (ITC) or surface plasmon resonance. Commercial services are available to determine binding affinity or potency of inhibitors of BRD4 (Reaction Biology, DiscoveRx).
Basic Protocol 1 – Ligand, target, and bead concentration optimization for enhanced AlphaScreen Signal
After selecting beads, probe and target protein for the AlphaScreen assay (see Strategic Planning, above), the next step is to systematically vary the probe, protein, and bead concentrations for optimized signal. Each bead has a certain binding capacity for probe and protein determined by several factors such as size and affinity of the biomolecule associating with the bead. For instance, a bead's binding capacity for small biomolecules will be greater than that of larger biomolecules with comparable affinity. Affinity also affects this binding capacity. Anti-His6 antibody-coated acceptor beads and nickel chelate beads are both available to bind to His-tagged targets and probes; however, the affinity of anti-His6 antibodies is often greater than the affinity of nickel for His6 tags. This means that anti-His6 antibody-coated beads may reach saturation more quickly than nickel chelate beads, because for a given concentration of protein, more material may be associated with the anti-His6 beads. Signal increases as more and more protein or probe is added until the bead binding capacity becomes saturated. Further increases in protein or probe in solution can bind to their respective targets, without binding to a bead—effectively acting as an inhibitor of bead-bead association and resulting in loss of signal intensity. This so-called “hook effect” is expected in assay development, and facilitates selection of final reagent concentrations (Figure 2). Beyond varying ligand and protein concentrations, bead concentration may be varied to optimize signal, and minimize background and cost. Assay signal is strongly influenced by bead concentration (Figure 2). Changing bead inputs may also influence concentrations where the hook effect is observed, frequently requiring iterative optimization of assay parameters.
Figure 2. Ligand and protein concentration optimization.

Graphs show the change in Alpha signal (counts) with varying protein (BRD4(1)) and ligand (Bio-JQ1) concentrations for (A) 10 μg/mL and (B) 1 μg/mL of AlphaScreen beads. A 3D surface was mapped onto the average of triplicate data points. While the general curves are the same, 1 μg/mL of AlphaScreen beads results in a much lower signal. The hook point in (A) is at approximately 10-6.5 M (316 nM) and in (B) at 10-7.5 M (31.6 nM) ligand.
This protocol describes a method for determining optimal protein and ligand concentrations using His6-tagged recombinant BRD4(1) and Bio-JQ1 with a bead concentration of 10 μg/mL. In this titration experiment, we monitor luminescence while varying both ligand and protein concentration simultaneously in a matrix format in a 384-well microtiter plate. A similar procedure can be used for other probes and bead designs, such as biotinylated tetra-acetylated peptides (Matzuk et al., 2012).
Materials
AlphaScreen buffer (see recipe)
Protein stock solution: 10 μM BRD4(1)-His6 in AlphaScreen buffer solution
Tagged probe stock solution: 100 μM Bio-JQ1 in AlphaScreen buffer (Anders et al., 2013; Filippakopoulos et al., 2010)
AlphaScreen 384-well microplates (PerkinElmer, cat. no. 6005350)
AlphaScreen Histidine (Nickel Chelate) Detection Kit (PerkinElmer, cat. no. 6760619R)
Alpha bead stock solution: 10 μg/mL each of donor and acceptor beads in AlphaScreen buffer
Plate seals (e.g. Corning, 6570)
Plastic reservoir (e.g. Corning, 4870)
AlphaScreen capable reader (e.g. Envision, Enspire)
Graphing software (e.g. PRISM GraphPad)
Protocol
Prepare protein and probe concentration matrix in triplicate
- Aliquot 1500 μL of protein stock solution into a microcentrifuge tube, and make a series of 15 three-fold dilutions into AlphaScreen buffer by sequentially mixing 1000 μL of buffer with 500 μL of the previous protein solution in a new microcentrifuge tube.It is preferable to start with as high a concentration of stock protein as possible in order uncover the ‘hook’ point. Of course, this may be limited by supply of protein and protein solubility.
Aliquot 1500 μL of probe stock solution into a microcentrifurge tube, and make a series of 15 three-fold dilutions into AlphaScreen buffer by sequentially mixing 1000 μL of buffer with 500 μL of the previous probe solution in a new microcentrifuge tube.
Allow sample to equilibrate to room temperature for approximately 15 minutes before addition to plates.
Transfer 5 μL of each of the 15 protein solutions to fill individual rows of a 384-well AlphaScreen microplate using a multi-channel pipette. Protein concentrations should decrease from top to bottom across the plate. Repeat this for three plates.
Centrifuge each plate for 1 minute at 500 × g, room temperature, to bring the contents of the plate to the bottom of each well.
Transfer 5 μL of each of the 15 probe solutions to fill individual columns of a 384-well AlphaScreen microplate using a multi-channel pipette. Probe concentrations should decrease from left to right across the plate. Repeat this for three plates.
Centrifuge each plate for 1 minute at 500 × g, room temperature.
- Add 10 μL of Alpha bead stock solution to each well of each plate using a multi-channel pipette, and centrifuge each plate for 1 minute at 500 × g, room temperature.Be sure to work with Alpha beads in low light conditions (<100 lux, similar to hallway lighting), as the donor beads are particularly light sensitive. Exposure to bright lights can result in premature release of singlet state oxygen which can react with surface streptavidin and decrease its binding efficiency.
Seal plates to prevent evaporation and exposure to light and incubate for 1 hour at room temperature before reading. Signal is temperature dependent, so it is important to keep incubation temperatures the same.
Obtain Alpha signal readings and analyze data
10. Load the plate into the plate reader and follow the manufacturer's protocol to determine the correct plate dimensions and measurement height. Appropriate reader mirrors and filters are sold by PerkinElmer (AlphaScreen 680 excitation / 570 emission filter set). Be sure that AlphaScreen mode is selected on the instrument.
11. Read the plate and obtain measurements of AlphaScreen fluorescence for each sample.
12. Export and average the data for each triplicate measurement.
13. Calculate the log base 10 of the protein and ligand concentrations.
14. Plot the average of the triplicate data for each well as a function of the log transformed protein concentration for each concentration of ligand. Also plot the data as a function of the log transformed ligand concentration for each concentration of protein.
15. Determine the optimal ligand and protein concentrations. The optimal concentrations should be within the linear range of signal increase and below the hook point at the lowest concentration that still gives a good signal to noise ratio. Most AlphaScreen assays will utilize a final biotinylated probe concentration in the low nanomolar range and a protein binding partner concentration between the low to mid-nanomolar range, depending on binding efficiency and bead concentration.
Basic Protocol 2 – Measurement of competitive binding to BRD4 using AlphaScreen
After determining the optimal concentrations of target protein and probe, the BRD4 AlphaScreen assay can be used to discover potential acetyl-lysine competitive binders of BRD4. As the concentration of competitive inhibitor increases, it will displace Bio-JQ1 from BRD4, separating donor and acceptor beads and resulting in signal loss (Figure 1). In order to establish assay robustness it is helpful to have a positive control compound – an inhibitor of binding that can reduce the fluorescent output signal as its concentration is increased in the assay. The standard for this control is the non-biotinylated version of the probe, in this case, JQ1. When plotted on a logarithmic scale, the displacement curve should have a characteristic sigmoidal shape readily fit by non-linear logistic regression to interpolate the concentration inhibiting 50% of the original signal (IC50).
The protocol below describes assaying potential inhibitory compounds of BRD4(1) and generating a competitive binding curve. Optimized concentrations of both probe and protein obtained from Basic Protocol 1 should be used here [e.g. 20 nM of both BRD4(1) and Bio-JQ1 (Figure 2)]. With the ultimate goal of developing this assay for high-throughput screening, the use of automated compound transfer to the assay plate is described (e.g. robotic pin transfer), but serial dilutions of compounds using a pipette may also be implemented. Compound stocks are dissolved in DMSO, thus, DMSO is used as a negative control in this assay and all data will be normalized to multiple vehicle-only wells. A typical transfer volume with a pinning tool is 100 nL, therefore compounds are prepared in the compound stock plate at 200× (for a final assay volume of 20 μL/well). Starting at a concentration of 10 mM in the compound stock plate (50 μM final concentration in the assay plate) and serially diluting compounds in a 10 point dilution series by a dilution factor of √10 at each step should provide a concentration range with well-spaced data points with which to fit the data and determine an IC50. A well-established binding assay will give a characteristic sigmoidal inhibition curve for the control compound with minimal error bars (Figure 3) and consistent IC50 values when repeated.
Figure 3. Competitive inhibition assay plate and dose-response curves for inhibitors of BRD4(1).
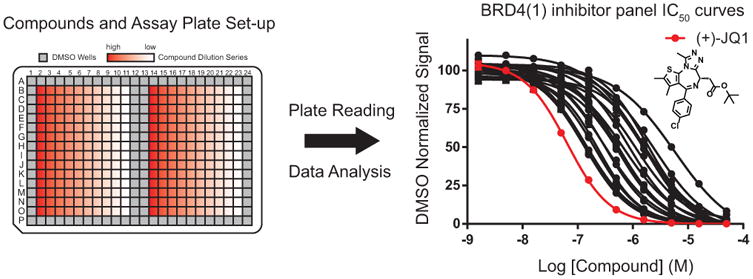
The left panel depicts the setup of a 384-well compound plate with serial dilutions of potential inhibitors. Samples are excluded from perimeter wells to prevent signal inconsistency from evaporation and light exposure at the edge of the plate. DMSO-containing wells in the center of the plate will be used to normalize signal. After plate reading and analysis, dose response curves and IC50 values for inhibitors of BRD4(1) are generated, as depicted in the right panel. (+)-JQ1 is included as a positive control (shown in red).
Materials
AlphaScreen buffer (see recipe)
AlphaScreen Histidine (Nickel Chelate) Detection Kit (PerkinElmer, cat. no. 6760619R)
AlphaScreen 384-well microplates (PerkinElmer, cat. no. 6005350)
Plate seals (e.g. Corning, cat. no. 6570)
384-well compound storage plates (e.g. Greiner Bio One, cat. no. 784201)
DMSO
Compound stock solutions: 10 mM in DMSO
Reagent Stock Solution: 40 nM BRD4(1)-His6, 40 nM Bio-JQ1, and 10 μg/mL nickel coated acceptor beads in AlphaScreen buffer.
Donor Bead Stock Solution: 10 μg/mL of streptavidin coated donor beads in AlphaScreen buffer
AlphaScreen capable reader (e.g. Envision, Enspire)
Graphing software (e.g. excel, PRISM GraphPad)
Plastic reservoir (e.g. Corning, 4870)
Compound pin transfer tool (e.g. V&P Scientific)
Protocol
Prepare compound source plate
- Create a 384-well compound plate with serial dilutions of potential inhibitors by first adding 11 μL of DMSO to each well in columns 1, and 3-12 (Figure 3).Wells on the perimeter of the assay plate will not be used, as signal variation in those wells is common due to evaporation and temperature fluctuations.
Add 16.1 μL each of up to 14 different 10 mM compound stock solutions to each well in column 2 (rows B-O). Be sure to add a positive control to every plate, in this case unlabeled (+)-JQ1.
- Serially dilute compounds by transferring 5.1 μL of solution from column 2 to column 3 and mix. Then take the same volume and transfer to row 4 and so on until row 11. The same procedure can be repeated for columns 13 – 24 if there are more compounds to test.The dilution factor given by these volumes (√10) provides a generally useful concentration range with data points close enough together to fit a competitive inhibition curve. Row 12 will contain DMSO to act as a negative control and for signal normalization.
Centrifuge each compound plate for 1 minute at 500 × g, room temperature.
Prepare the assay plate and transfer compound solutions
5. Dispense 10 μL of Reagent Stock Solution into each well of a 384-well AlphaScreen plate with a multichannel pipette. Centrifuge each plate for 1 minute at 500 × g, room temperature. Use enough plates in order to assay each compound/dilution in triplicate.
6. Pin 100 nL of compound from the compound source plate into each assay plate. The assay plate setup after being pinned is shown in Figure 3.
- 7. Seal each assay plate and incubate 30 minutes at room temperature.This incubation period depends on how long it takes potential inhibitors to reach equilibrium in displacing the probe. This can change depending on the compounds being used as well as the target, but the time provided here works in most cases.
8. Add 10 μL of Donor Bead Stock Solution into each well of the assay plate and centrifuge each plate for 1 minute at 500 × g, room temperature.
- 9. Seal each assay plate and incubate 1 hour at room temperature.It is advisable to run a time-series to determine when the Alpha signal becomes stable, and for just how long it is stable. This can change depending on assay design, probes, protein, library molecules, and other factors.
Obtain Alpha signal readings and analyze data
10. Using an appropriate plate reader, read the plate and obtain measurements of AlphaScreen fluorescence for each sample (as described in Basic Protocol 1).
11. Export the data and average the DMSO well signals from columns 12 and 13 to obtain a negative control signal.
12. Normalize the data by dividing the signal of each sample well by the DMSO negative control signal for that plate and multiply by 100 to generate the DMSO normalized signal as a percent.
13. Average the normalized values for each sample well across replicates.
14. Calculate the log base 10 of the compound concentrations.
15. Plot the average DMSO normalized signal as a function of the log transformed concentration of each compound. This should produce a characteristic sigmoidal curve for the positive control and potential inhibitors (Figure 3).
16. Fit the data using a 4-parameter logistic curve and determine the LogIC50 from the midpoint of that fit. Take the antilog to obtain an IC50 value. Statistical programs such as GraphPad PRISM can do this regression automatically (graphpad.com).
Basic Protocol 3 – Adaptation of the competitive BRD4 binding assay for HTS
After a competitive binding assay has been well established, it may be adapted for screening purposes to discover new molecules that inhibit the probe-target binding interaction (Figure 4A). High-throughput screening usually entails filling plates with solutions via an automated liquid-handling system, rather than using a pipette, and transferring library compounds (commercially available, or from other sources such as the US National Cancer Institute (NCI)) to the assay plate using a pin transfer robot or other precision instrument. Many diverse compounds can be profiled against the target very quickly in this manner. However, the assay must be sensitive and robust enough to remain reliable while testing these compounds at only one dose in either singlicate or duplicate and with automated steps. To determine assay quality for screening purposes, an often used statistic is the Z′ factor, (Zhang and Chung, 1999) given by:
Figure 4. Identification of true inhibitors of BRD4(1) and false positives in HTS.
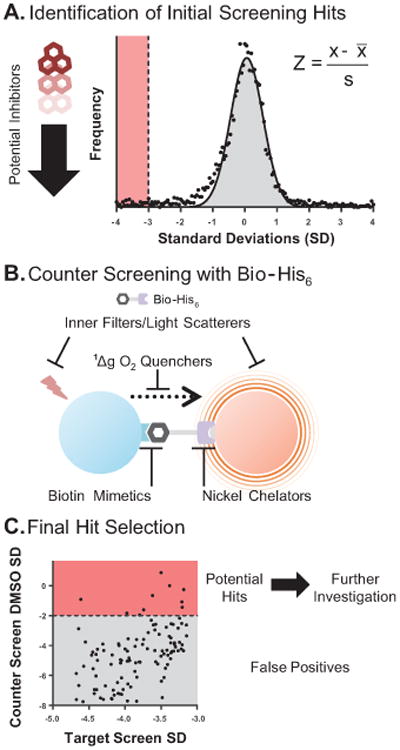
After compounds are tested in a high-throughput AlphaScreen, potential inhibitors are identified. (A.) Data can be transformed to a standard normal distribution by calculating each compound's Z-score. A typical cutoff for inhibitors is 3SD below the mean SD for all compounds (highlighted in red). (B.) Potential inhibitors are then tested in a counter screen assay to identify false positives. Here, Bio-His6 is used to bring streptavidin-coated donor beads and nickel chelate acceptor beads into close proximity (positive Alpha signal). Interference from compounds that act as inner filters, light scatterers, singlet oxygen quenchers, biotin mimetics, or nickel chelators will non-specifically reduce Bio-His6 binding or light absorbance/emission. (C.) Finally, compounds that non-specifically reduce Alpha signal in the counter screen assay are identified as false positives (gray), while compounds found to reduce Alpha signal in the initial screen and not in the counter screen are identified as true hits and investigated further (red).
| Equation 1 |
In equation 1, σ and μ represents the standard deviations (SD) and means of the positive and negative controls, respectively. The Z′ factor is most often calculated from a multi-well assay plate containing half positive and half negative controls. In our case, DMSO will serve as the negative control, and (+)-JQ1, at the lowest concentration to fully inhibit Alpha signal (determined in Basic Protocol 2) would be the positive control. Acceptable standards for high-throughput screening is a Z′ factor of at least 0.5, though Z′ values above 0.7 are characteristic of optimized homogeneous assays.
Critical to success in HTS is minimizing false-positive outcomes. A ‘false positive’ may arise by interference with the HTS assay in a non-specific manner unrelated to the engagement of the intended target. In this case, a false positive would be a compound that reduces the Alpha signal without actually disrupting the binding of BRD4(1) and Bio-JQ1. Eliminating false positives from a screen as early as possible saves time and effort after the screen is performed. The main categories of compound interference in an AlphaScreen assay are singlet oxygen quenchers, competitors for the probe or target protein with their respective bead, color quenchers/inner filters, and light scatterers (Figure 4B). Transition metals, anti-oxidants, and compounds with a thiophene-based structure are often strong quenchers of singlet oxygen and can reduce signal, resulting in false positives. In the streptavidin and nickel chelate system, biotin mimetics can disrupt probe binding to bead. Likewise, nickel chelators (e.g. imidazole based structures) can interfere with binding of His-tagged recombinant protein to the acceptor bead. Compounds that absorb in the same range as the excitation (680 ± 5 nm) or emission (520-620 nm) bands (usually blue/green in color) can act as inner filters and result in false positives. As mentioned in the strategic planning section, using AlphaLISA beads can reduce the window over which these inner filters might interfere (narrow emission window around 615 nm). Many compounds in compound libraries can precipitate out of solution when added to aqueous buffers, forming insoluble aggregates that scatter light at the excitation or emission wavelengths and reduce signal (McGovern et al., 2002). Problems arising from non-specific binding and aggregation can usually be solved with the addition of a detergent such as Tween-20 to the AlphaScreen buffer (Baell and Holloway, 2010; Ryan et al., 2003), primarily in HTS or secondarily as a confirmatory assay. In addition to the groups of “promiscuous” compounds mentioned here, there are many other typical compound classes described in the literature that can appear as false positives in high-throughput screening (Baell and Holloway, 2010; Johnston, 2011; Rishton, 1997). PerkinElmer sells several control kits for identification of false positives, such as their TruHits kit. They also sell a bioinylated-His6 (Bio-His6) molecule (included in the AlphaScreen Histidine (Nickel Chelate) Kit), which will bring streptavidin and nickel beads together without the presence of probe and target protein (Figure 4B). Potential inhibitors from a screen can be assayed in the presence of Bio-His6 and donor and acceptor beads. Any compounds that reduce Alpha signal more than 2SD below the DMSO average signal are potential false positives (Figure 4C). The hook point for Bio-His6 can be determined as described in Basic Protocol 1 however the protocol below uses a final concentration of 1 nM Bio-His6. Compounds found to reduce Alpha signal in the initial screen and not in the counter screen with Bio-His6 are identified as true inhibitors of BRD4(1)-JQ1 binding and can be further investigated for relative potency by performing a dose-response assays to determine an IC50 value (Basic Protocol 2).
Materials
AlphaScreen buffer (see recipe)
AlphaScreen Histidine (Nickel Chelate) Detection Kit (PerkinElmer, cat. no. 6760619R)
AlphaScreen 384-well microplates (PerkinElmer, cat. no. 6005350)
Plate seals (e.g. Corning cat. no. 6570)
384-well compound storage plates (e.g. Greiner Bio One, cat. no. 784201)
DMSO
Pin transfer apparatus (e.g. JANUS automated liquid handling workstation, PerkinElmer)
Automated liquid dispenser for multiwell microplates (e.g. EL406, BioTek)
Commercial Compound Library
Reagent Stock Solution: 40 nM BRD4(1)-His6, 40 nM Bio-JQ1, 10 μg/mL nickel coated acceptor beads in AlphaScreen buffer
Donor Bead Stock Solution: 10 μg/mL of streptavidin coated donor beads in AlphaScreen buffer
Bio-His6 Stock Solution: 1 nM Bio-His6 and 10 μg/mL of nickel coated acceptor beads in AlphaScreen buffer
AlphaScreen capable reader (e.g. Envision, Enspire)
Graphing software (e.g. excel, PRISM GraphPad)
Plastic reservoir (e.g. Corning, cat. no. 4870)
Compound pin transfer tool (e.g. V&P Scientific)
Protocol
Prepare Z′ factor determination plate
Determine the minimal concentration of positive control (unlabeled (+)-JQ1) needed to completely inhibit binding based on the results from Basic Protocol 2 and make 3 mL of a 200× stock.
- To create a Z′ factor determination compound source plate, add 10 μL of positive control to half of the wells of a 384-well compound storage plate and 10 μL of negative control (DMSO) to the other half of wells.Compound library plates often have all of their wells full, so the Z′ plate should also have its perimeter wells full.
Dispense 10 μL of Reagent Stock Solution into each well of a 384-well AlphaScreen plate with an automated liquid handling system.
Pin 100 nL of compound from the Z′ determination compound source plate into the assay plate.
Seal the assay plate and incubate 30 minutes at room temperature.
Add 10 μL of Donor Bead Stock Solution into each well of the assay plate.
Seal the assay plate and incubate 1 hour at room temperature.
Read the plate and obtain measurements of Alpha assay signal for each sample (as described in Basic Protocol 1).
Calculate the Z′ factor for the plate using Equation 1. A Z′ factor >0.5 will ensure that the assay is robust enough for screening.
Library Screening
Dispense 10 μL of Reagent Stock Solution into each well of a 384-well AlphaScreen plate with a plate filler or an automated liquid handling system. Prepare enough plates to screen each compound library plate in duplicate.
Pin 100 nL of compound from the compound library plates into the assay plates in duplicate.
Seal each assay plate and incubate 30 minutes at room temperature.
Add 10 μL of Donor Bead Stock solution to each AlphaScreen plate as previously described.
Seal each assay plate and incubate 1 hour at room temperature.
Read the plates and obtain measurements of Alpha assay signal for each sample (as described in Basic Protocol 1).
Export the data and average the DMSO well signals on each plate to obtain a negative control signal.
Normalize the data by dividing the signal of each sample well by the DMSO negative control signal for that plate and multiply by 100 to generate the DMSO normalized signal as a percent.
Average the normalized values across replicates for each sample well.
Determine the mean and standard deviation of the normalized signals from all of the wells in the screen.
Calculate the Z-score for each compound by subtracting the screen mean from the normalized well signal and then divide by the screen standard deviation (see Figure 4A for equation).
- Identify compounds that inhibit BRD4(1) and Bio-JQ1 binding from the screen.For large unbiased chemical libraries the hit threshold can usually be set to 3SD below the mean (Figure 4A). In practice, sometimes it is easier to move the hit threshold more than 3SD below the mean to make the number of compounds to be tested in secondary assays more manageable.
Counter Screen with Bio-His6
13. Dispense 10 μL of Bio-His6 Stock Solution into each well of a 384-well AlphaScreen plate. Prepare enough plates to screen each compound library plate in duplicate.
14. Pin 100 nL of compound from the compound library plates into the respective AlphaScreen plates in duplicate.
15. Seal each assay plate and incubate 30 minutes at room temperature.
16. Add 10 μL of Donor Bead Stock Solution to each AlphaScreen plate as previously described.
17. Seal each assay plate and incubate 1 hour at room temperature.
18. Read plates and obtain measurements of Alpha assay signal for each sample.
19. Export the data and average the DMSO well signals on each plate to obtain a negative control signal.
20. Normalize the data by dividing the signal of each sample well by the DMSO negative control signal for that plate and multiply by 100 to generate the normalized signal as a percent.
21. Average the normalized values across replicates for each sample well.
22. Plot the normalized signal for each compound from the initial screen against the normalized counter screen signal.
23. Determine the SD of DMSO wells from the counter screen. Compounds that give a normalized signal below 2SD from the DMSO mean in the counter screen are potential false positives (Figure 4C).
Reagents and Solutions
All solutions should be made with Milli-Q grade water.
AlphaScreen Buffer solution
150 mM NaCl
50 mM HEPES
0.1% (w/v) BSA
0.01% (v/v) Tween 20
Adjust to pH 7.4 with NaOH and HCl, filter and store indefinitely at 4ᵒC
Add DTT fresh from 1M frozen stock to a final concentration of 1 mM
Once DTT is added, store up to 1 week at 4ᵒC
Biotinylated probe stock solution
Synthesize Bio-JQ1 as previously described and dilute in DMSO to a concentration of 100 μM (Anders et al., 2013; Filippakopoulos et al., 2010). Aliquot and store for 6 months at -20ᵒC to -80ᵒC. Store protected from light. Alternately, biotinylated tetra-acetylated peptide can be purchased commercially (Epigentek).
AlphaScreen Plates and Beads
All AlphaScreen materials can be obtained from PerkinElmer (Waltham, MA). Histidine (Nickel Chelate) Detection Kit cat. no. 6760619R; AlphaPlate-384 cat. no. 6005350
Commentary
Background information
We selected AlphaScreen technology as a primary BET inhibitor discovery assay for several reasons. Most important is its high sensitivity for signal detection, which was appealing as we considered the possibility that early screening positives could be of low potency. Second, the assay features a reverse Stokes shift, which may mitigate false-positives derived from compound auto-fluorescence. When samples are excited at 680 nm, there is a transfer of singlet state oxygen from a phthalocyanine photosensitizer in the donor bead to the acceptor bead. This singlet state oxygen reacts with a thioxene derivative in the donor bead which chemiluminesces at 370nm, subsequently activating other fluorophores (anthracene and rubrene) in the same bead that emit light at 520-620 nm. Usually the wavelength of an emitted photon is longer than that used to excite the fluorophore (called a Stokes shift). The assay sensitivity is enhanced by an anti-Stokes shift effect, in which the emitted light is a shorter wavelength than the absorbed light. Non-specific matrix fluorescence follows a typical Stokes shift to longer wavelengths, and is therefore not detected by the plate reader after passing through the 570 emission filter, thus reducing background interference (Ullman et al., 1996).
The singlet oxygen produced in this reaction can travel up to 200 nm during its 4 μs half-life before decaying to the stable triplet state, rendering this technology particularly useful for studying interactions between large complexes. Here, the technique has advantages over fluorescence resonance energy transfer (FRET) where signal is strongly influenced by close proximity (>10 nm) of acceptor and donor. Finally technical error may be minimized by the homogeneous nature of this sample addition assay, unlike flow cytometry, ELISA, and electrochemoluminescence-based assays that may require sample washing.
Critical Parameters
Alpha bead light and temperature sensitivity
The basis of AlphaScreen technology is the transfer of singlet state oxygen from a donor bead to an acceptor bead after laser excitation. However, prolonged light exposure can lead to premature evolution of singlet-state oxygen from donor beads, which can then react with streptavidin or other components on the surface of the bead, reducing efficiency. Users are instructed to always work with AlphaScreen beads under low light conditions. The manufacturer states on their website that beads should be handled under 100 lux, which is equivalent to an overcast day. As an extra reference, they cite office lighting at around 320-500 lux. Alternatively, green filters can be applied to light fixtures in the room where the assay is being conducted (Roscolux filters 389, Rosco). All incubations should be performed in the dark, covered by another opaque microplate, sheet of foil, or plate seal. It is also important to store beads at 4ᵒC to prevent signal decay due to bead degradation.
Confirming results from HTS
After initially screening compounds with a high-throughput AlphaScreen, counter screening to confirm that a compound is specifically disrupting BRD4(1)/Bio-JQ1 binding is essential. Like any screening assay based on light absorbance and emission, there is the potential for false positive results due to light interference (e.g. compounds that have precipitated out of solution may quench or scatter excitation and emission fluorescence). Additionally, there are compounds that can quench singlet oxygen or compete for binding of the probe or target to their respective beads. Thus, it is best to validate potential inhibitory compounds in an orthogonal, non-AlphaScreen assay in order to separate true from false results. In an ideal case, the compound library being used has already been screened in a similar AlphaScreen with a different probe/protein target combination. If a compound elicits a drop in Alpha signal when assayed with different sets of probes and target proteins, the reading can be considered false positive and additional screening is not necessary. In the case of our BRD4(1)/Bio-JQ1 AlphaScreen, we initially confirmed potential inhibitors in a counter screen using Bio-His6. Critically, orthogonal assays measuring target binding and competitive inhibition are needed to further assess assay positives. Here, we have utilized with success differential scanning fluorimetry (DSF) (Filippakopoulos et al., 2010). Other appropriate orthogonal assays may include fluorescence polarization (FP), surface plasmon resonance (SPR), and time-resolved fluorescence resonance energy transfer (TR-FRET).
Troubleshooting
Probe design
Probe (ligand) design is an essential part of early stage assay development. If the functionalized probe does not bind well to the target the assay will suffer from reduced (or no) signal when attempting to optimize the assay. If designing a new probe that has not been described in the literature already (such as Bio-JQ1 or Bio-tetraacetylated peptide for BRD4(1), there are a few steps that can help ensure success. First, knowledge about a natural substrate or small molecule that binds the target can be helpful. If there is a crystal structure of this ligand, a solvent exposed position at which to affix a chemical handle (e.g. biotin) that will not disrupt the binding interaction can be discerned. After synthesizing a modified ligand, services provided by a commercial company (ex. Reaction Biology, DiscoveRx) can be used to verify that the modified probe binds the target protein with equivalent affinity as the unmodified probe. Alternatively, methods such as ITC or differential scanning fluorimetry can be used to assess modified probe binding.
Buffer components
If fluorescent signal is low, it is possible that components of the buffer may be interfering with probe binding. It's important to avoid the use of singlet state oxygen quenchers such as azide and transition metals (Al2+, Fe2+, Fe3+, Cu2+, Ni2+ and Zn2+). Also avoid the use of components that absorb in the emission range of the AlphaScreen beads (520-680 nm). Alternatively, using AlphaLISA acceptor beads will reduce the emission window (narrow band around 615 nm) when analyzing complex biological samples such as serum. If the assay is suffering from a high background signal, increasing the concentrations of blocking agents and detergents such as BSA and Tween-20 may help to prevent non-specific interactions between assay components (Baell and Holloway, 2010; Ryan et al., 2003).
Temperature and incubation time
The amount of time required for the assay components and compounds in an AlphaScreen assay to reach equilibrium is dependent on temperature and individual protein and therefore, can vary. Colder room temperatures may extend the time required to reach equilibrium. It is best to keep room temperature consistent throughout screening and to determine the optimal incubation time by performing an incubation time series and monitoring the change in luminescence. This is also a useful way to determine signal stability. In addition, plates should be incubated at the same temperature as the plate reader to prevent fluctuations in signal upon reading.
Anticipated Results
Basic Protocol 1 will generate probe and target titration curves, allowing determination of the optimal concentrations to use for the BRD4 AlphaScreen assay based on signal and reagent availability. Basic Protocol 2 details testing the assay in a competitive dose response format to determine potential inhibitor IC50 values. Finally, in Basic Protocol 3, the assay is optimized for high-throughput screening by use of a Z′ factor determination plate to assess assay quality. After an appropriate Z′ value has been obtained (>0.5), the assay can be used to profile chemical libraries. False positives can be identified with the control provided by the manufacturer (Bio-His6) and hits can be further validated in the format described in Basic Protocol 2 and with other orthogonal (non-AlphaScreen) methods.
Time Considerations
Once all of the necessary reagents and equipment are in hand, assay development can be performed in less than a day. However, unforeseen complications often arise requiring iterative rounds of assay optimization through buffer changes, probe re-design, repeated protein purification.
Acknowledgments
We thank members of the Bradner lab, particularly Alexander Federation, William Smith and Michael McKeown for contributing to BRD4 assay development and providing mentorship in deploying this technology, as well as Dennis Buckley and Jennifer Perry for critical reading of the manuscript. JEB is supported by the Burroughs-Wellcome Fund, the Damon-Runyon Cancer Research Foundation and the National Institutes of Health (RO1 CA176745).
Footnotes
Authors: In addition to updates in the protocols and other text to reflect new techniques and applications, please update references and supplier information.
Internet Resources: http://www.perkinelmer.com/
PerkinElmer homepage. All of the necessary reagents as well as background and FAQ's on Alpha technology and steps to creating a successful assay can be found here.
http://www.urmc.rochester.edu/hts/_source/AlphaScreenPracticalGuide.pdf
“A practical guide to working with AlphaScreen” posted by the University of Rochester's High Throughput Screening Core. This document gives a detailed background on working with the assay, including an exhaustive troubleshooting guide.
Literature Cited
- Anders L, Guenther MG, Qi J, Fan ZP. Genome-wide localization of small molecules. Nature Biotechnology. 2013 doi: 10.1038/nbt.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. Journal of medicinal chemistry. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Chapuy B, McKeown MR, Lin CY, Monti S. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013 doi: 10.1016/j.ccr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, Chesi M, Schinzel AC, McKeown MR, Heffernan TP, Vakoc CR, Bergsagel PL, Ghobrial IM, Richardson PG, Young RA, Hahn WC, Anderson KC, Kung AL, Bradner JE, Mitsiades CS. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey A, Chitsaz F, Abbasi A. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proceedings of the National Academy of Sciences. 2003 doi: 10.1073/pnas.1433065100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eglen RM, Reisine T, Roby P, Rouleau N, Illy C, Bossé R, Bielefeld M. The use of AlphaScreen technology in HTS: current status. Current chemical genomics. 2007;1:2–10. doi: 10.2174/1875397300801010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nature Reviews Drug Discovery. 2014;13:337–356. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French CA. Pathogenesis of NUT midline carcinoma. Annual Review of Pathology: Mechanisms of Disease. 2012 doi: 10.1146/annurev-pathol-011811-132438. [DOI] [PubMed] [Google Scholar]
- Huang B, Yang XDD, Zhou MMM, Ozato K, Chen LFF. Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Molecular and cellular biology. 2009;29:1375–1387. doi: 10.1128/MCB.01365-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston PA. Redox cycling compounds generate H2O2 in HTS buffers containing strong reducing reagents—real hits or promiscuous artifacts? Current opinion in chemical biology. 2011 doi: 10.1016/j.cbpa.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung M, Philpott M, Müller S, Schulze J. Affinity Map of Bromodomain Protein 4 (BRD4) Interactions with the Histone H4 Tail and the Small Molecule Inhibitor JQ1. Journal of Biological Chemistry. 2014 doi: 10.1074/jbc.M113.523019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzuk MM, McKeown MR, Filippakopoulos P, Li Q, Ma L, Agno JE, Lemieux ME, Picaud S, Yu RN, Qi J, Knapp S, Bradner JE. Small-molecule inhibition of BRDT for male contraception. Cell. 2012;150:673–684. doi: 10.1016/j.cell.2012.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGovern SL, Caselli E, Grigorieff N. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. Journal of medicinal chemistry. 2002 doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- McKeown MR, Shaw DL, Fu H, Liu S, Xu X, Marineau JJ, Huang Y, Zhang X, Buckley DL, Kadam A, Zhang Z, Blacklow SC, Qi J, Zhang W, Bradner JE. Biased multicomponent reactions to develop novel bromodomain inhibitors. Journal of medicinal chemistry. 2014 doi: 10.1021/jm501120z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn AM, Bedford MT, Espejo A, Spannhoff A, Austin CP, Oppermann U, Simeonov A. A homogeneous method for investigation of methylation-dependent protein–protein interactions in epigenetics. Nucleic Acids Research. 2010;38 doi: 10.1093/nar/gkp899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rishton GM. Reactive compounds and in vitro false positives in HTS. Drug Discovery Today 1997 [Google Scholar]
- Ryan AJ, Gray NM, Lowe P. Effect of detergent on “promiscuous” inhibitors. Journal of medicinal chemistry. 2003 doi: 10.1021/jm0340896. [DOI] [PubMed] [Google Scholar]
- Ullman EF, Kirakossian H, Singh S. Luminescent oxygen channeling immunoassay: measurement of particle binding kinetics by chemiluminescence. Proceedings of the National Academy of Sciences. 1994 doi: 10.1073/pnas.91.12.5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullman EF, Kirakossian H, Switchenko AC, Ishkanian J, Ericson M, Wartchow CA, Pirio M, Pease J, Irvin BR, Singh S, Singh R, Patel R, Dafforn A, Davalian D, Skold C, Kurn N, Wagner DB. Luminescent oxygen channeling assay (LOCI): sensitive, broadly applicable homogeneous immunoassay method. Clinical chemistry. 1996;42:1518–1526. [PubMed] [Google Scholar]
- Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Molecular cell. 2005;19:535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- Yi JS, Fderation AJ, Qi J, Dhe-Paganon S, Hadler M, Xu X, Pierre R, Varca AC, Wu L, Marineau JJ. Structure-Guided DOT1L Probe Optimization by Label-Free Ligand Displacement. ACS chemical biology. 2014 doi: 10.1021/cb500796d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Chung TDY. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. Journal of biomolecular screening. 1999 doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, Taylor MJ, Johns C, Chicas A, Mulloy JC, Kogan SC, Brown P, Valent P, Bradner JE, Lowe SW, Vakoc CR. RNAi screenidentifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]