

Abstract
Cerebral ischemia is among the leading causes of death worldwide. It is characterized by a lack of blood flow to the brain that results in cell death and damage, ultimately causing motor, sensory, and cognitive impairments. Today, clinical treatment of cerebral ischemia, mostly stroke and cardiac arrest, is limited and new neuroprotective therapies are desperately needed. The Sirtuin family of oxidized nicotinamide adenine dinucleotide (NAD+)-dependent deacylases has been shown to govern several processes within the central nervous system as well as to possess neuroprotective properties in a variety of pathological conditions such as Alzheimer’s Disease, Parkinson’s Disease, and Huntington’s Disease, among others. Recently, Sirt1 in particular has been identified as a mediator of cerebral ischemia, with potential as a possible therapeutic target. To gather studies relevant to this topic, we used PubMed and previous reviews to locate, select, and resynthesize the lines of evidence presented here. In this review, we will first describe some functions of Sirt1 in the brain, mainly neurodevelopment, learning and memory, and metabolic regulation. Second, we will discuss the experimental evidence that has implicated Sirt1 as a key protein in the regulation of cerebral ischemia as well as a potential target for the induction of ischemic tolerance.
Keywords: Cerebral ischemia, neuroprotection, Sirt1
Introduction
Cerebral ischemia, most notably in the form of stroke and cardiac arrest, is an ever-growing burden on both the US and world populations.[1] It can be defined as a reduction in blood flow to the brain that fails to meet the metabolic demand of this tissue, in turn causing cell death or what is known as infarct. During ischemia, failure to meet the metabolic demand results in mostly necrotic cell death as a result of excitotoxicity, severe disruption of ion homeostasis, and calcium dysregulation. Following ischemia, reperfusion of the brain marks the onset of hyperoxygenation that leads to increased and adverse free radical production, cellular swelling, and tissue edema due to ionic imbalances and, ultimately, apoptosis via numerous mechanisms. In the days, weeks, and months after an ischemic insult, the infarcted area can continue to grow as the neuroinflammatory response takes hold.
Currently, clinical treatment is limited; for example, there is only one clinically approved treatment for stroke, recombinant tissue plasminogen activator (a thrombolytic agent),[2] which is administered to very few stroke patients. In an attempt to combat this limitation, researchers have identified various molecular targets shown to modulate ischemic cell death. In the last decade, a body of evidence demonstrating the role of Sirtuins, and in particular Sirt1, in neuroprotection from ischemia has emerged.[3]
The history of mammalian Sirtuins research can be traced back to roughly 20 years ago when silent information regulator 4 (Sir4) in yeast was found to extend the lifespan.[4] Initially, this was thought to occur through interaction with its associated Sir2 and Sir3 proteins, but it was not until a few years later that this phenomenon was attributed fully to Sir2.[5] Much excitement surrounded this discovery, eventually leading to the identification of a human Sir2 homologue [silent information regulator two 1 (Sirt1)][6] and the rest of the mammalian “Sirtuins,” as they are now referred to.
There are seven proteins in the Sirtuin family (Sirt1–7),[7] and while they share a conserved catalytic core domain to a certain degree, they differ at their N- and C-termini. Perhaps these differences account for some of their versatility in terms of enzymatic character. Originally classified as strictly deacetylases, Sirtuins are now known collectively as deacylases. Sirt1–4 and Sirt1–7 demonstrate the strongest deacetylase activity, whereas Sirt5 and 6 are primarily deacylases.[8] In terms of their subcellular localization, generally Sirt1, Sirt6, and Sirt7 are found in the nucleus, Sirt3, Sirt4, and Sirt5 in the mitochondrion, and Sirt2 in the cytoplasm. However, recent evidence demonstrates the ability of many Sirtuins to translocate to or shuttle between subcellular compartments, adding to the versatility of their function. They all share the requirement for oxidized nicotinamide adenine dinucleotide (NAD+) as a cofactor of their enzymatic activity (hence their classification as class III histone deacetylases).[9] This requirement gives rise to the energy-sensing functions of the Sirtuins and makes them unique as metabolic regulators within many organ systems.[10]
The Sirtuins are expressed throughout the body, in tissues such as the heart, liver, kidney, muscle, adipose, endothelium, and the brain, among others.[11] In response to changes in cellular energy state, they coordinate the regulation of various metabolic pathways and processes such as glycolysis, fatty acid synthesis, gluconeogenesis, the cell cycle, DNA repair, cell survival, and mitochondrial biogenesis, to name a few.[12] This is achieved through epigenetic targeting of histone and nonhistone proteins in the nucleus to globally modulate gene expression as well as downstream of transcription by targeting specific protein substrates within the mitochondria and cytoplasm.[11] In this review, we will look briefly at the physiological roles of the most extensively studied Sirtuin, Sirt1, specifically in the brain, and discuss the evidence surrounding Sirt1 in the context of ischemic neuroprotection.
Functions of Sirt1 in the Brain
Sirt1 expression in the adult brain is widespread, with high levels in the hippocampus, cerebellum, cortex, and hypothalamus.[13] Most of this expression is neuronally localized;[13–15] however, Sirt1 is also found in neural stem cells (NSCs), neural progenitor cells (NPCs),[16] astrocytes and microglia in culture[17,18] as well as glial cells in the human brain.[19,20] The distribution of Sirt1 in the highly specialized areas mentioned above already provides great insight into its physiological roles in the brain. Here we will discuss the brain-specific functions of Sirt1. For general background material on Sirt1 functions, the reader is directed to a study by Rahman et al.[21]
Neurodevelopment
Sirt1 plays a role in neurodevelopment, where peak Sirt1 expression can be found at E4.5 in the spinal cord and dorsal root ganglion.[15] Concerning neurons specifically, Sirt1 influences their structure at several levels, including axon growth through Akt and GSK3,[22] neurite outgrowth via mTOR,[23] and dendritic arborization[24] by way of Rho-associated kinase (ROCK). In the adult brain, differentiation between NSCs and NPCs depends upon Sirt1. Proliferation and self-renewal of hippocampal NSCs is enhanced in Sirt1−/− mice.[25] The opposite effect was seen when agonizing Sirt1 with resveratrol in neurosphere cultures from wild-type mice. Additionally, another group showed that resveratrol reduced the proliferation of cultured multipotent NPCs as well as the survival of hippocampal NPCs in the dentate gyrus in vivo.[26] Furthermore, inhibition of Sirt1 with nicotinamide enhanced the differentiation of induced pluripotent stem cells (iPSCs) into NSCs in vitro.[27] Moreover, brain-specific inactivation of Sirt1 was shown to increase proliferating NSCs and oligodendrocyte progenitor cells (OPCs).[28] Here, oligodendrocyte expansion was mediated to some extent by signaling downstream of Platelet-derived growth factor receptor α (PDGFRα), mainly Akt and p38, and implicated Sirt1 in demyelinating disorders. These studies highlight the role of Sirt1 in modulating both neurogenesis and gliogenesis.
Learning and memory
Sirt1 is also implicated in learning and memory. Sirt1−/− mice exhibit a decrease in the branching of dendrites, the length of branching, and complexity of dendritic arbors. [24,29–31] In a similar line of evidence, brain-specific Sirt1−/− mice display reductions in the number of functional synapses and dendritic spine density, supporting the role for Sirt1 in neuronal development and suggesting a role in synaptic plasticity.[24,31] Indeed, long-term potentiation (LTP) in hippocampal CA1 neurons is impaired in these two Sirt1 knockout mouse models.[24,31] In both cases, basal synaptic activity was unaffected, demonstrating that deficiencies in LTP were not caused by impaired synaptic transmission. Overexpression of Sirt1 was comparable to normal LTP responses, whereas activation of Sirt1 actually enhanced LTP. In line with LTP abrogation, Sirt1−/− mice show deficits in associative memory (both contextual and tone-dependent fear-conditioning), spatial memory (Morris water maze, Barnes maze), and immediate memory (Y maze, novel object recognition test).[24,31]
Further study into the molecular mechanism of Sirt1’s involvement in learning and memory has elucidated a pathway surrounding miR-134.[31] MiR-134 is a microRNA species that when unrepressed acts to downregulate two important proteins for LTP induction, cAMP response element-binding protein (CREB) and brain-derived neurotrophic factor (BDNF). Sirt1 represses miR-134 expression via a complex with a transcription factor Yin Yang 1 (YY1), keeping it in check. In the case of Sirt1 deficiency, miR-134 limits CREB and BDNF, resulting in impaired synaptic plasticity. These data establish the involvement of Sirt1 in learning and memory at the behavioral and molecular levels.
Metabolic regulation
The hypothalamus is responsible for carrying out many crucial brain functions related to systemic physiology and metabolic regulation. Sirt1 mediates many of these functions: For example, in the dorsomedial (DMH) and lateral hypothalamic nuclei (LH) Sirt1 deacetylates Nk2 homeobox 1 (Nkx2-1).[32] This in turn drives transcription of another protein, orexin receptor type 2 (Ox2r), leading to neuronal activation that regulates physiological parameters such as physical activity, O2 consumption, and body temperature.[32] Additionally, mitochondrial morphology and function in skeletal muscle, and perhaps other tissues, may feedback to the hypothalamus for another level of regulation. Interestingly, mice overexpressing Sirt1 specifically in the brain show significant life-span extension due to the Sirt1 interactions mentioned above and their downstream effects on the regulation of systemic metabolism.[32]
Another example of systemic metabolic regulation by Sirt1 can be seen in proopiomelanocortin (POMC) neurons of the arcuate nucleus.[33,34] The lack of Sirt1 in these neurons reduces energy expenditure and sensitizes mice to diet-induced obesity. Here, leptin was no longer able to properly remodel a particular region of white adipose tissue, as phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) signaling was disrupted. Additionally, impairment in visceral fat deposition was attributed to a reduction in sympathetic nerve activity. Collectively, these studies demonstrate the significant role Sirt1 plays in regulating metabolism at the organismal level from within the brain.
Regulation of circadian rhythms is another hypothalamic function of Sirt1.[35,36] In the suprachiasmatic nucleus (SCN), Sirt1 and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) work in concert to control the regulation of two circadian genes, aryl hydrocarbon receptor nuclear translocator-like protein 1 (BMAL1) and circadian locomotor output cycles kaput (CLOCK). BMAL1 and CLOCK have transcriptional control of the circadian machinery, which includes nicotinamide phosphoribosyltransferase (NAMPT) among other proteins.[37,38] When NAMPT enhances NAD+ for Sirt1 activity (discussed in more detail below), the feedback loop responsible for fluctuations in the circadian rhythm is complete and is regulated in this manner. Many of these pathways are deregulated in aging, where Sirt1 declines and adaptation of the circadian clock to the environment is impaired.[38] This decline in Sirt1 has also been linked to the quality of sleep,[39] which is also affected in the aging process. Taken together, these studies indicate a crucial role for Sirt1 in the regulation of the circadian clock.
Sirt1 and Ischemic Neuroprotection
There is a substantial amount of work demonstrating the neuroprotective properties of Sirt1 in an array of neurological disorders, such as Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease, among others.[40] This work has prompted the investigation of Sirt1 in cerebral ischemia, which is not as well established as yet.[40] Studies outlined in the next few sections highlight key experiments and pieces of evidence that support a neuroprotective role for Sirt1 in cerebral ischemia. We will begin by examining genetic and pharmacological employment of Sirt1 and then discuss several Sirt1-mediated signaling pathways that lead to ischemic tolerance.
Sirt1 expression
Genetic manipulation of Sirt1 expression has effects on ischemic injury outcome. Sirt1−/− mice displayed larger infarct volumes following permanent middle cerebral artery occlusion (pMCAo) compared to their wild-type counterparts.[41] In contrast, Sirt1-Tg mice that overexpress Sirt1 showed less hippocampal damage following bilateral common carotid artery occlusion (BCAo) than wild-type mice.[42] A similar protective effect was seen with Sirt1-Tg mice in a bilateral common carotid artery stenosis (BCAS) model of hypoperfusion injury,[43] a common pathological occurrence following stroke.
The physiological levels of Sirt1 are modulated by ischemic injury as well. For example, Sirt1 was upregulated in the peri-infarct area up to 7 days following pMCAo in mice.[41] On the other hand, in rats subjected to transient focal ischemia with varying periods of reperfusion, Sirt1 was found to be downregulated 6 h after reperfusion, compared with nonreperfused animals.[44] The discrepancy between up or downregulation could be due to the different species and ischemia models used in the two studies. However, what is clear from these studies is that Sirt1 expression is inherently regulated by and could modulate the outcome of ischemic injury.
Pharmacological manipulation of Sirt1
In another line of evidence, activation or inhibition of Sirt1 with pharmacological agents modulates cerebral ischemic outcome. Treatment with the Sirt1 activator Activator 3 at 10 min, 24 h, and 40 h following pMCAo reduced infarct volume, while treatment with the Sirtuin inhibitor Sirtinol increased infarct volume from vehicle levels.[41] The same holds true for treatments administered prior to ischemia, such as in preconditioning. Our laboratory has shown that both ischemic preconditioning (IPC) and resveratrol preconditioning (RPC) protect against oxygen and glucose deprivation (OGD, in vitro ischemia) in organotypic hippocampal slice cultures when administered 2 days prior to injury.[45] IPC increased Sirt1 activity 48 h later, whereas RPC increased activity after just 30 min. In both paradigms, Sirtinol blocked protection. Furthermore, we have confirmed these results in vivo, where IPC and RPC 2 days before asphyxia cardiac arrest (global ischemia) decreased hippocampal cell death.[46] Moreover, we showed that Sirt1 was a mediator of this protection, as Sirtinol was able to block these protective effects in the hippocampus.
Several other endogenous or exogenous compounds shown to induce ischemic tolerance were also linked to activation or upregulation of Sirt1. 2,3,5,4′-Tetrahy droxystilbene-2-O-ß-D-glucoside (TSG) is a naturally occurring compound with similarities to resveratrol that protects against OGD in vitro and MCAo in vivo.[47] Cultured cells exposed to TSG for 3 days showed increased Sirt1 protein levels as well as a partial loss of TSG-induced protection in the presence of the Sirt1 inhibitor nicotinamide. Another naturally occurring compound, alpha-lipoic acid (ALA) given 30 min prior to pMCAo reduced neurological deficit, infarct, and edema in 24 h following injury, where an increase in Sirt1 expression was observed.[48] Likewise, treatment with melatonin (once immediately following induction of MCAo, and then again at the onset of reperfusion) reduced neurological deficit, infarct, and edema 24 h after injury.[49] This coincided with an increase in Sirt1 expression linked to enhanced mitochondrial function that was blocked by the specific Sirt1 inhibitor 6-Chloro-2,3,4,9-tetrahydro-1H-carbazole-1-carboxamide (EX-527). Furthermore, icariin treatment (icariin is another naturally occurring compound of the flavonoid variety) each day for 7 days following reperfusion after MCAo reduced infarct volume and brain edema as well as improved neurological score at 7 days after injury.[50] Again, Sirt1 expression was increased at this 7-day time point. The authors go on to show that in neuronal culture, Sirt1 inhibitor III ablates icariin-induced protection from OGD. These pharmacological experiments provide further supporting evidence for Sirt1 as a key mediator in ischemic injury and as a possible target for ischemic neuroprotection.
It should be noted that increases in Sirt1 expression may not always translate into increased enzymatic activity. For instance, no changes in histone 3 and 4 acetylation were seen with Sirt1 activation or genetic ablation.[41] The difficulty of observing specific changes in acetylation of Sirt1 substrates in vivo may account for some of this discrepancy. Interestingly, the enzymatic activity of Sirt1 may only be partially responsible for its protective effects, as one study demonstrated that Sirt1-mediated neuroprotection can be independent of its deacetylase activity.[51] These are important faculties to consider in current and future studies.
Sirt1-dependent pathways
Mechanistically, several lines of evidence pinpoint different signaling pathways, all regulated at some level by Sirt1, that lead to neuroprotection from ischemia. The most important ones with potential roles in ischemic neuroprotection are discussed here and illustrated below in Figure 1.
Figure 1.
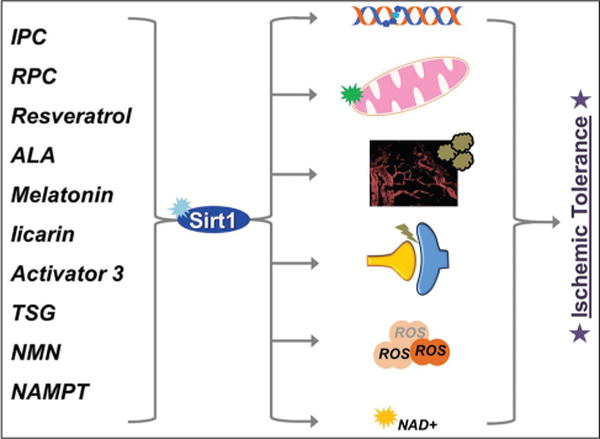
Overview of Sirt1 mediated ischemic tolerance
Left: IPC = Ischemic preconditioning, RPC = Resveratrol preconditioning, ALA = Alpha-lipoic acid, TSG = 2,3,5,4′-Tetrahydroxystilbene-2-O-ß-D-glucoside, NMN = Nicotinamide mononucleotide, NAMPT = Nicotinamide phosphoribosyltransferase
Right: DNA repair, Mitochondrial function, Blood flow and neuroinflammation, Synaptic function, Antioxidation, NAD+ metabolism
Mitochondrial function and antioxidation
PGC1α
It is well established that Sirt1 regulates antioxidant defenses and mitochondrial function through the activation of transcriptional coactivator PGC1α.[52] Sirt1 deacetylates and activates PGC1α. When activated, PGC1α interacts with other transcriptional coactivators, such as peroxisome proliferator-activated receptor gamma (PPARγ), inducing transcription of genes involved in antioxidation and mitochondrial biogenesis. Several studies have connected this pathway to ischemic neuroprotection. In the ALA and icariin paradigms mentioned above, increased expression of Sirt1 was congruent with the same effect on PGC1α. Knockdown of PGC1α with siRNA reversed the protection seen with icariin treatment in culture.[50] As for ALA, the authors witnessed an increase in superoxide dismutase (SOD) activity that was associated with enhanced PGC1α.[48] Additionally, another group demonstrated that PGC1α is upregulated following transient global ischemia, where they also observed an increase in mitochondrial uncoupling protein 2 (UCP2) and SOD2.[53] Antisense oligodeoxynucleotide-induced knockdown of PGC1α exacerbated oxidative damage following ischemia, perhaps due to the loss of UCP2 and SOD2 that was observed.
UCP2
Mentioned briefly above, UCP2 is another Sirt1-regulated protein contributing to the cellular redox state. Sirt1 binds directly to the UCP2 promoter, repressing its transcription. This allows for proper production of adenosine triphosphate (ATP) in response to glucose stimulation and subsequently insulin secretion in pancreatic β cells.[54] In the context of ischemia, varying lines of evidence suggest that both up-and downregulation of UCP2 can produce an ischemic protective effect. For example, one group showed that UCP2−/− mice were more susceptible to transient focal ischemia than wild-type mice,[55] however another group also showed that the same UCP2−/− mice were less susceptible than wild-type mice to permanent focal ischemia.[56] In support of the former, two studies add that mice overexpressing human UCP2 are protected from focal and global ischemic injuries.[57,58] In preconditioning studies, both up- and downregulation of UCP2 have been associated with improved ischemic outcome. IPC upregulated UCP2 in the rat (both in vitro and in vivo), an effect that was associated with ischemic protection.[58] On the other hand, our laboratory has shown that RPC reduces UCP2 levels, which corresponds to protection from global ischemia in vivo.[46] In all of these studies described above, UCP2-mediated protection, whether up- or downregulated, was attributed to changes in mitochondrial function, reactive oxygen species (ROS) production, and/or antioxidant defenses.
When debating the effects of UCP2 on mitochondrial physiology, one can argue that upregulation of UCP2 would result in beneficial uncoupling of mitochondria, dropping ROS production and, in turn, oxidative damage. Conversely, evidence also supports the argument that a reduction in UCP2 would couple mitochondria (raise the membrane potential), making ATP synthesis more efficient, as fewer wasted protons leak back across the inner mitochondrial matrix.[59] This would also stimulate ROS production, as the oxidative phosphorylation complexes ramp up their activity in response to a higher membrane potential. While this may seem like a deleterious effect, in fact studies have shown that an increase in ROS, such as in exercise,[60] can induce beneficial redox signaling pathways that culminate in the upregulation of antioxidant defenses. The studies mentioned in this section provide evidence in support of both of these mechanisms. Further studies are needed to sort out the discrepancies surrounding UCP2 in cerebral ischemia.
DNA repair
As a result of ischemia reperfusion injury, an increase in free radical production and subsequent oxidative damage can severely damage DNA and contribute to loss of cell viability.[61] Ku70 is a nuclear protein involved in repair of double stand breaks that is downregulated following ischemia[62] but upregulated following IPC.[63] Sirt1 increases the DNA repair activity of Ku70 by deacetylating two lysine residues at its C-terminus.[64] This was shown to play a protective role following cellular exposure to radiation in vitro.[65] Another repair enzyme, Poly [ADP-ribose] polymerase 1 (PARP1), is what one might call a double-edged sword in terms of its activity. On one hand, PARP1 helps repair single-strand breaks in DNA by adenosine diphosphate ribose (ADP)-ribosylation. On the other, ADP-ribosylation requires the ADP-ribose moiety of NAD+ that PARP1 consumes during this process.[66] PARP1 is a major culprit for the decline of NAD+ in aging[67] and acute depletion leading to cell death in ischemia.[68] PARP1 is overactive in response to substantial DNA damage resulting from ischemia, and inhibition of PARP1 restores NAD+ levels and protects against ischemic injury.[69] Sirt1 binds to and deacetylates PARP1, reducing its activity and promoting cell survival.[70,71]
Sirt1 also increases the activity of several other DNA repair pathways. For instance, human AP endonuclease (APE1) is an enzyme that cleaves apurinic/apyrimidinic sites in the base excision repair pathway.[72,73] Several lysine residues of APE1 are acetylated, a modification associated with decreased activity.[74] Sirt1 overexpression or resveratrol treatment induces deacetylation of APE1, increasing its activity.[75] Alternatively, knockdown of Sirt1 results in an increase of deleterious DNA base modifications and makes cells more susceptible to death after oxidative or genotoxic stress.[75] Additionally, APE1 has been shown to contribute to DNA repair after IPC,[76] again where Sirt1 is enhanced. Furthermore, NAD+ treatment has been shown to protect neurons against ischemia in vitro through upregulation of the base excision repair pathway,[77] where APE1 is prominent. In addition to APE1, other Sirt1 DNA repair targets include DNA repair protein complementing XP-A cells (XPA)[78] in nucleotide excision repair, and nibrin (NBS1)[79] in double-strand breaks, although they have yet to be directly implicated in ischemic injury. These results indicate that Sirt1-mediated DNA provides another level of protection following ischemic injury.
NAD+ metabolism
NAD+ is a versatile metabolite not only responsible for Sirtuin activation but also for steps in glycolysis, complex activity in oxidative phosphorylation, and the replenishing of antioxidants. Nicotinamide is converted to nicotinamide mononucleotide (NMN) by NAMPT.[80] Subsequently, NMNAT converts NMN to NAD+. Despite the fact that it is actually NMNAT that produces NAD+, NAMPT has been determined as the rate-limiting enzyme in this pathway of NAD+ biosynthesis and is accountable for increasing NAD+ levels. Given that Sirtuins are activated by NAD+, it is logical to hypothesize that boosting this metabolite will have ischemic neuroprotective effects. Such is indeed the case. Intranasal NAD+[81] or NMN administration[82] protected mice against focal cerebral ischemia. Additionally, inhibition of NAMPT worsens outcome following ischemia, whereas overexpression of NAMPT enhances NAD+, activates Sirt1, and lessens ischemic injury.[82] In Sirt1-null mice, these protective effects of NAMPT were lost. Moreover, our group has shown that preconditioning with IPC, RPC, or the PKCɛ activator pseudo-epsilon RACK (ΨɛRACK) increased mitochondrial NAMPT levels and enhanced the NAD+/reduced nicotinamide adenine dinucleotide (NADH) ratio in this compartment.[83] Furthermore, following IPC, NAD+ is increased in hippocampal slices,[84] where Sirt1 activity is increased and protection is lost upon its inhibition.
Another level of NAD+ regulation is added by adenosine monophosphate (AMP)-activated kinase (AMPK). AMPK is another metabolic regulator that, in response to an increase in the AMP/ATP ratio, inhibits cellular processes that consume energy and stimulates those that generate it.[85] Moreover, AMPK has been shown to enhance mitochondrial function and the oxidative stress response under certain stressful conditions. Aside from the AMP/ATP ratio, AMPK is activated by a kinase called liver kinase B1 (LKB1). LKB1 is deacetylated and in turn activated by Sirt1. NAMPT-induced neuroprotection from OGD was concomitant with LKB1 deacetylation and AMPK activation, where the effects on LKB1 and AMPK as well as protection were lost in Sirt1 knockout neurons.[82] Moreover, in a recurrent transient MCAo model, resveratrol-induced protection was associated with increased AMPK activity, NAD+/NADH ratio, and Sirt1 activity.[86] Here, inhibition or knockdown of AMPK or Sirt1 abolished resveratrol-induced protection in vitro.
It has also become apparent that AMPK can also be upstream of NAMPT. In skeletal muscle, two studies show that AMPK activation leads to increased NAMPT expression[87] and in turn modulation of the NAD+/NADH ratio.[88] In our group’s work, the preconditioning induced increase in mitochondrial NAMPT, and NAD+/NADH ratio was shown to be regulated in an AMPK-dependent fashion.[83] These studies demonstrate positive feedback for Sirt1 and NAD+ through NAMPT and AMPK. Together, these data implicate NAD+ as a neuroprotective metabolite in cerebral ischemia, at least in part by maintaining energy and substrates for Sirt1 activity.
Synaptic function
As described above, a connection has been established between Sirt1 and BDNF with regard to learning and memory. There is also evidence supporting beneficial effects of Sirt1-mediated BDNF expression in the context of neuroprotection from ischemia. Sirt1 regulates BDNF expression by several mechanisms; as indicated above, Sirt1 enhances BDNF pertaining to synaptic plasticity through miR-134. Another level of regulation exists at the BDNF promoter, where Sirt1 deacetylates the BDNF repressor MeCP2,[89] disrupting its binding in this region and promoting BNDF transcription. Moreover, new evidence also shows regulation of BDNF by Sirt1 through the binding of Sirt1 directly to the BDNF promoter region. This was shown by chromatin immunoprecipitation-sequencing in the mouse cerebellum.[90]
Previously, our laboratory has demonstrated that IPC results in the induction of the BDNF gene in th rat hippocampus.[91] An increase in BDNF protein levels was confirmed in vitro 24–48 h following IPC.[92] In the same study, IPC-induced tolerance against OGD was abolished when the BDNF receptor, tropomyosin receptor kinase B (TrkB) was blocked. Additionally, knockdown of miR-134 alleviates ischemic injury in vitro,[93] perhaps through Sirt1-mediated BDNF expression, as in the synaptic plasticity pathway. Many groups have demonstrated that direct BDNF administration reduces infarct volume and enhances function recovery following ischemic injury. This protection has been attributed to several signaling pathways and endpoint functional changes, such as a reduction in neurotoxicity, suppression of neuroinflammation, and promotion of neuronal regeneration, among others.[94] The enhancement of synaptic function via a Sirt1 BDNF pathway could have profound effects on the synapse during/following ischemia; however, additional studies are needed to confirm a direct relationship that is evident in the experiments highlighted here.
Blood flow and neuroinflammation
Given that disruptions in blood flow are ultimately the root cause of ischemia as well as a major culprit of reperfusion injury, it is no surprise that modulation of blood flow can have a significant impact on anatomical and functional outcome following ischemia. Several studies demonstrate that blood flow is modulated by Sirt1 and that this modulation leads to ischemic tolerance. Sirt1-Tg mice (overexpressing Sirt1) showed preserved cerebral blood flow (CBF) during BCAo, which was 45–50% of baseline, while only 20–25% of baseline CBF was observed in wild-type littermates.[42] These differences were seen at 5 min and 10 min of BCAo and came back to comparable levels at 2 h. This correlated with an improvement of histological outcome in the hippocampus. It is important to note that baseline CBF did not differ between Sirt1-Tg and wild-type mice. Additionally, the same group showed that 2 h and 24 h following BCAS, wild-type mice had a reduction in CBF to roughly 72–74% of baseline, whereas this reduction was not seen in Sirt1-Tg mice who had CBF values of 91–94% of baseline at the same time points.[43] Again, it should be noted that baseline CBF between the two groups was not different and the diameter of basal arteries did not differ, suggesting that the retention of CBF in Sirt1-Tg mice after BCAS is not due to changes in collateral brain circulation. The authors go on to reveal that irregularities in the vascular endothelium seen with wild-type mice after BCAS were not observed in Sirt1-Tg mice.
Prior to this study, it had been shown that Sirt1 activates endothelial nitric oxide synthase (eNOS) by deacetylation.[95] Acetylated eNOS was only observed in wild-type mice following BCAS but not Sirt1-Tg mice, suggesting a preservation of CBF through Sirt1 stimulated nitric oxide (NO) production by eNOS. This was validated by the fact that treatment with the eNOS inactivator cavtratin reduced CBF in Sirt1-Tg mice after BCAS and that Sirtinol abolished CBF retention and protection from BCAS. Furthermore, another group demonstrated that resveratrol increased eNOS and plasma NO following MCAo, and that this effect and ischemic protection were lost with application of the eNOS inhibitor N5-(1-iminoethyl)-L-ornithine, dihydrochloride (L-NIO).[96]
Modulation of blood flow is also linked to neuroinflammation, a damaging response (if not managed properly) that is initiated immediately following ischemia.[97] The main transcription factor that promotes pro-inflammatory mediators is nuclear factor-kappaB (NF-κB).[98] Groups have shown that activation of NF-κB exacerbates neuronal injury following cerebral ischemia.[99] Sirt1 deacetylates NF-κB at Lys310 of its p65 subunit, decreasing its transcriptional activity.[100] Resveratrol was shown to promote the inhibition of NF-κB signaling through the deacetyaltion of RelA/p65 Lys310 as a result of increased Sirt1 activity.[101] Additionally, TSG also inhibited NF-κB through increased Sirt1 and protected against ischemia.[47]
The link between neuroinflammation and blood flow comes by way of inducible NOS (iNOS), which is another form of NOS involved in vasodilation during the neuroinflammatory response and is predominantly refereed by NF-κB.[18] As mentioned above, NF-κB is deacetylated by Sirt1 decreasing its transcriptional drive. This links the activation of Sirt1 to the inhibition of iNOS expression, and in contrast to eNOS, reductions in iNOS have been connected to ischemic protection. While resveratrol increased eNOS, it decreased iNOS where protection was observed following MCAo.[96] TSG also inhibited iNOS expression induced by in vitro ischemia, an effect that was blocked by the Sirtuin inhibitor nicotinamide.[47] This suggests that the Sirt1 inhibition of NF-κB and subsequent iNOS signaling is an important pathway to possibly dampen the detrimental immune response following ischemic injury.
Another important aspect of blood flow modulation besides vascular tone is the formation of new blood vessels from preexisting ones, or angiogenesis. This process has been shown to improve outcome following ischemic injury,[102] possibly through Sirt1. Sirt1 has been shown to mediate several angiogenic processes, such as migration and sprouting of endothelial cells[103] as well as erythropoietin production. Under conditions of hypoxia as well as ischemia, Sirt1 activation results in the deacetylation of hypoxia-inducible factor-2alpha (HIF-2α), increasing its transcription.[104] This results in increased expression of erythropoietin, an important modulator of red blood cell production in adaptation to low oxygen. Moreover, erythropoietin is increased following IPC[105] and is sufficient by itself to confer a protective preconditioning response in rat hippocampal neurons.[106] Additionally, Sirt1 also stabilizes HIF-1α,[107] another hypoxia-inducible factor that drives the response to a low-oxygen environment to initiate cellular and molecular compensatory mechanisms pertaining to oxygen transport, glycolysis, cell survival, and angiogenesis. For example, resveratrol increased the proangiogenic factors matrix metalloproteinase-2 and vascular endothelial growth factor (VEGF) in the delayed injury stage following focal ischemia,[108] possibly through Sirt1 action upon HIF-1α. These results demonstrate the ability of Sirt1 to regulate blood flow through several avenues to promote neuroprotection from ischemia.
Potentially detrimental effects of Sirt1
Besides its cofactor function toward enzymatic activity, NAD+ and the NAD+/NADH ratio functions in glycolysis, oxidative phosphorylation and antioxidation. Preservation of NAD+ and its redox ratio to carry out these functions is vital to cell survival both in normal physiology and especially pathological stress such as ischemia.[109] More specifically, marked reductions in NAD+ (and disruption of its relative NADH) following ischemia are concomitant with energy depletion leading to cell death. Maintenance of NAD+ is pivotal for a better outcome, evidenced by the fact that supplying NAD+ both prior to and following OGD significantly reduces ischemic injury.[77] Repair of DNA damage can also deplete NAD+ by way of PARP, as mentioned above. Sirt1, also utilizing NAD+ for enzymatic activity, may contribute to diminishing levels. Mice overexpressing Sirt1 did not show any significant protection from ischemia.[110] One possible explanation is that a chronic increase in Sirt1 expression dwindles NAD+ and actually hinders its beneficial effects. This has been substantiated in vitro by the fact that nicotinamide (a Sirt1 inhibitor) preserved NAD+ levels and protected neurons from excitotoxitcy-induced cell death (a hallmark of ischemic injury).[111] Concordantly, inhibition of Sirt1 with Sirtinol also protected against excitotoxicity in vitro. It may be that when Sirt1 activation is sustained, one needs to compensate for NAD+ expenditure and increase its availability. This compensatory mechanism can be seen in IPC, which increases NADH oxidation,[84,112] possibly preventing loss of NAD+ levels during sustained Sirt1 activity. Further studies are needed to unravel the contribution of Sirt1 activity to NAD+ depletion in the context of ischemia.
Conclusions
Neuroprotective therapies for cerebral ischemia in the clinic are desperately needed. The evidence gathered here demonstrates a potential therapy in the activation of Sirt1. It is clear that Sirt1 has many physiological roles in the brain as well as roles in response to pathological conditions of metabolic stress, such as ischemia. Several pharmacological agents that activate Sirt1 and are detailed above show potential for translation into the clinic. In the current state, more studies are warranted to tease apart the Sirt1-dependent pathways that foster neuroprotection in the context of ischemia. However, it appears that Sirt1 is a promising candidate for further investigation.
Acknowledgments
Financial support and sponsorship:
Nil
Footnotes
For reprints contact: reprints@medknow.com
Conflicts of interest
There are no conflicts of interest.
References
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, et al. American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Executive summary: Heart disease and stroke statistics-2014 update: A report from the American Heart Association. Circulation. 2014;129:399–410. doi: 10.1161/01.cir.0000442015.53336.12. [DOI] [PubMed] [Google Scholar]
- 2.Wardlaw JM, Murray V, Berge E, del Zoppo G, Sandercock P, Lindley RL, et al. Recombinant tissue plasminogen activator for acute ischaemic stroke: An updated systematic review and meta-analysis. Lancet. 2012;379:2364–72. doi: 10.1016/S0140-6736(12)60738-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang Y, Duan W, Li Y, Yan J, Yi W, Liang Z, et al. New role of silent information regulator 1 in cerebral ischemia. Neurobiol Aging. 2013;34:2879–88. doi: 10.1016/j.neurobiolaging.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 4.Kennedy BK, Austriaco NR, Jr, Zhang J, Guarente L. Mutation in the silencing gene SIR4 can delay aging in S. cerevisiae. Cell. 1995;80:485–96. doi: 10.1016/0092-8674(95)90499-9. [DOI] [PubMed] [Google Scholar]
- 5.Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–80. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–59. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 7.Anderson KA, Green MF, Huynh FK, Wagner GR, Hirschey MD. SnapShot: Mammalian sirtuins. Cell. 2014;159:956.e1. doi: 10.1016/j.cell.2014.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakagawa T, Guarente L. SnapShot: Sirtuins, NAD, and aging. Cell Metab. 2014;20:192.e1. doi: 10.1016/j.cmet.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 9.Schemies J, Uciechowska U, Sippl W, Jung M. NAD(+) −dependent histone deacetylases (sirtuins) as novel therapeutic targets. Med Res Rev. 2010;30:861–89. doi: 10.1002/med.20178. [DOI] [PubMed] [Google Scholar]
- 10.Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol. 2012;13:225–38. doi: 10.1038/nrm3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dali-Youcef N, Lagouge M, Froelich S, Koehl C, Schoonjans K, Auwerx J. Sirtuins: The ‘magnificent seven’, function, metabolism and longevity. Ann Med. 2007;39:335–45. doi: 10.1080/07853890701408194. [DOI] [PubMed] [Google Scholar]
- 12.Chang HC, Guarente L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol Metab. 2014;25:138–45. doi: 10.1016/j.tem.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramadori G, Lee CE, Bookout AL, Lee S, Williams KW, Anderson J, et al. Brain SIRT1: Anatomical distribution and regulation by energy availability. J Neurosci. 2008;28:9989–96. doi: 10.1523/JNEUROSCI.3257-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hisahara S, Chiba S, Matsumoto H, Tanno M, Yagi H, Shimohama S, et al. Histone deacetylase SIRT1 modulates neuronal differentiation by its nuclear translocation. Proc Natl Acad Sci U S A. 2008;105:15599–604. doi: 10.1073/pnas.0800612105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sakamoto J, Miura T, Shimamoto K, Horio Y. Predominant expression of Sir2alpha, an NAD-dependent histone deacetylase, in the embryonic mouse heart and brain. FEBS Lett. 2004;556:281–6. doi: 10.1016/s0014-5793(03)01444-3. [DOI] [PubMed] [Google Scholar]
- 16.Herskovits AZ, Guarente L. SIRT1 in neurodevelopment and brain senescence. Neuron. 2014;81:471–83. doi: 10.1016/j.neuron.2014.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thompson JW, Dave KR, Saul I, Narayanan SV, Perez-Pinzon MA. Epsilon PKC increases brain mitochondrial SIRT1 protein levels via heat shock protein 90 following ischemic preconditioning in rats. PloS One. 2013;8:e75753. doi: 10.1371/journal.pone.0075753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen J, Zhou Y, Mueller-Steiner S, Chen LF, Kwon H, Yi S, et al. SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-kappaB signaling. J Biol Chem. 2005;280:40364–74. doi: 10.1074/jbc.M509329200. [DOI] [PubMed] [Google Scholar]
- 19.Körner S, Böselt S, Thau N, Rath KJ, Dengler R, Petri S. Differential sirtuin expression patterns in amyotrophic lateral sclerosis (ALS) postmortem tissue: Neuroprotective or neurotoxic properties of sirtuins in ALS? Neurodegener Dis. 2013;11:141–52. doi: 10.1159/000338048. [DOI] [PubMed] [Google Scholar]
- 20.Lutz MI, Milenkovic I, Regelsberger G, Kovacs GG. Distinct patterns of sirtuin expression during progression of Alzheimer〉s disease. Neuromolecular Med. 2014;16:405–14. doi: 10.1007/s12017-014-8288-8. [DOI] [PubMed] [Google Scholar]
- 21.Rahman S, Islam R. Mammalian Sirt1: Insights on its biological functions. Cell Commun Signal. 2011;9:11. doi: 10.1186/1478-811X-9-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li XH, Chen C, Tu Y, Sun HT, Zhao ML, Cheng SX, et al. Sirt1 promotes axonogenesis by deacetylation of Akt and inactivation of GSK3. Mol Neurobiol. 2013;48:490–9. doi: 10.1007/s12035-013-8437-3. [DOI] [PubMed] [Google Scholar]
- 23.Guo W, Qian L, Zhang J, Zhang W, Morrison A, Hayes P, et al. Sirt1 overexpression in neurons promotes neurite outgrowth and cell survival through inhibition of the mTOR signaling. J Neurosci Res. 2011;89:1723–36. doi: 10.1002/jnr.22725. [DOI] [PubMed] [Google Scholar]
- 24.Michan S, Li Y, Chou MM, Parrella E, Ge H, Long JM, et al. SIRT1 is essential for normal cognitive function and synaptic plasticity. J Neurosci. 2010;30:9695–707. doi: 10.1523/JNEUROSCI.0027-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma CY, Yao MJ, Zhai QW, Jiao JW, Yuan XB, Poo MM. SIRT1 suppresses self-renewal of adult hippocampal neural stem cells. Development. 2014;141:4697–709. doi: 10.1242/dev.117937. [DOI] [PubMed] [Google Scholar]
- 26.Park HR, Kong KH, Yu BP, Mattson MP, Lee J. Resveratrol inhibits the proliferation of neural progenitor cells and hippocampal neurogenesis. J Biol Chem. 2012;287:42588–600. doi: 10.1074/jbc.M112.406413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu B, Guo Y, Chen C, Li Q, Niu X, Guo S, et al. Repression of SIRT1 promotes the differentiation of mouse induced pluripotent stem cells into neural stem cells. Cell Mol Neurobiol. 2014;34:905–12. doi: 10.1007/s10571-014-0071-8. [DOI] [PubMed] [Google Scholar]
- 28.Rafalski VA, Ho PP, Brett JO, Ucar D, Dugas JC, Pollina EA, et al. Expansion of oligodendrocyte progenitor cells following SIRT1 inactivation in the adult brain. Nat Cell Biol. 2013;15:614–24. doi: 10.1038/ncb2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dietrich MO, Antunes C, Geliang G, Liu ZW, Borok E, Nie Y, et al. Agrp neurons mediate Sirt1’s action on the melanocortin system and energy balance: Roles for Sirt1 in neuronal firing and synaptic plasticity. J Neurosci. 2010;30:11815–25. doi: 10.1523/JNEUROSCI.2234-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferrante RJ, Browne SE, Shinobu LA, Bowling AC, Baik MJ, MacGarvey U, et al. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J Neurochem. 1997;69:2064–74. doi: 10.1046/j.1471-4159.1997.69052064.x. [DOI] [PubMed] [Google Scholar]
- 31.Gao J, Wang WY, Mao YW, Gräff J, Guan JS, Pan L, et al. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature. 2010;466:1105–9. doi: 10.1038/nature09271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Satoh A, Brace CS, Rensing N, Cliften P, Wozniak DF, Herzog ED, et al. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 2013;18:416–30. doi: 10.1016/j.cmet.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramadori G, Fujikawa T, Fukuda M, Anderson J, Morgan DA, Mostoslavsky R, et al. SIRT1 deacetylase in POMC neurons is required for homeostatic defenses against diet-induced obesity. Cell Metab. 2010;12:78–87. doi: 10.1016/j.cmet.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramadori G, Fujikawa T, Anderson J, Berglund ED, Frazao R, Michán S, et al. SIRT1 deacetylase in SF1 neurons protects against metabolic imbalance. Cell Metab. 2011;14:301–12. doi: 10.1016/j.cmet.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, Kreppel F, et al. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell. 2008;134:317–28. doi: 10.1016/j.cell.2008.06.050. [DOI] [PubMed] [Google Scholar]
- 36.Nakahata Y, Kaluzova M, Grimaldi B, Sahar S, Hirayama J, Chen D, et al. The NAD+ –dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell. 2008;134:329–40. doi: 10.1016/j.cell.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Musiek ES, Lim MM, Yang G, Bauer AQ, Qi L, Lee Y, et al. Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. J Clin Invest. 2013;123:5389–400. doi: 10.1172/JCI70317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang HC, Guarente L. SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell. 2013;153:1448–60. doi: 10.1016/j.cell.2013.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Panossian L, Fenik P, Zhu Y, Zhan G, McBurney MW, Veasey S. SIRT1 regulation of wakefulness and senescence-like phenotype in wake neurons. J Neurosci. 2011;31:4025–36. doi: 10.1523/JNEUROSCI.5166-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang F, Wang S, Gan L, Vosler PS, Gao Y, Zigmond MJ, et al. Protective effects and mechanisms of sirtuins in the nervous system. Prog Neurobiol. 2011;95:373–95. doi: 10.1016/j.pneurobio.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hernández-Jiménez M, Hurtado O, Cuartero MI, Ballesteros I, Moraga A, Pradillo JM, et al. Silent information regulator 1 protects the brain against cerebral ischemic damage. Stroke. 2013;44:2333–7. doi: 10.1161/STROKEAHA.113.001715. [DOI] [PubMed] [Google Scholar]
- 42.Hattori Y, Okamoto Y, Nagatsuka K, Takahashi R, Kalaria RN, Kinoshita M, et al. SIRT1 attenuates severe ischemic damage by preserving cerebral blood flow. Neuroreport. 2015;26:113–7. doi: 10.1097/WNR.0000000000000308. [DOI] [PubMed] [Google Scholar]
- 43.Hattori Y, Okamoto Y, Maki T, Yamamoto Y, Oishi N, Yamahara K, et al. Silent information regulator 2 homolog 1 counters cerebral hypoperfusion injury by deacetylating endothelial nitric oxide synthase. Stroke. 2014;45:3403–11. doi: 10.1161/STROKEAHA.114.006265. [DOI] [PubMed] [Google Scholar]
- 44.Kalaivani P, Ganesh M, Sathiya S, Ranju V, Gayathiri V, Saravana Babu C. Alteration in bioenergetic regulators, SirT1 and Parp1 expression precedes oxidative stress in rats subjected to transient cerebral focal ischemia: Molecular and histopathologic evidences. J Stroke Cerebrovasc Dis. 2014;23:2753–66. doi: 10.1016/j.jstrokecerebrovasdis.2014.06.026. [DOI] [PubMed] [Google Scholar]
- 45.Raval AP, Dave KR, Pérez-Pinzón MA. Resveratrol mimics ischemic preconditioning in the brain. J Cereb Blood Flow Metab. 2006;26:1141–7. doi: 10.1038/sj.jcbfm.9600262. [DOI] [PubMed] [Google Scholar]
- 46.Della-Morte D, Dave KR, DeFazio RA, Bao YC, Raval AP, Perez-Pinzon MA. Resveratrol pretreatment protects rat brain from cerebral ischemic damage via a sirtuin 1-uncoupling protein 2 pathway. Neuroscience. 2009;159:993–1002. doi: 10.1016/j.neuroscience.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang T, Gu J, Wu PF, Wang F, Xiong Z, Yang YJ, et al. Protection by tetrahydroxystilbene glucoside against cerebral ischemia: Involvement of JNK, SIRT1, and NF-kappaB pathways and inhibition of intracellular ROS/RNS generation. Free Radic Biol Med. 2009;47:229–40. doi: 10.1016/j.freeradbiomed.2009.02.027. [DOI] [PubMed] [Google Scholar]
- 48.Fu B, Zhang J, Zhang X, Zhang C, Li Y, Zhang Y, et al. Alpha-lipoic acid upregulates SIRT1-dependent PGC-1α expression and protects mouse brain against focal ischemia. Neuroscience. 2014;281C:251–7. doi: 10.1016/j.neuroscience.2014.09.058. [DOI] [PubMed] [Google Scholar]
- 49.Yang Y, Jiang S, Dong Y, Fan C, Zhao L, Yang X, et al. Melatonin prevents cell death and mitochondrial dysfunction via a SIRT1-dependent mechanism during ischemic-stroke in mice. J Pineal Res. 2015;58:61–70. doi: 10.1111/jpi.12193. [DOI] [PubMed] [Google Scholar]
- 50.Zhu HR, Wang ZY, Zhu XL, Wu XX, Li EG, Xu Y. Icariin protects against brain injury by enhancing SIRT1-dependent PGC-1alpha expression in experimental stroke. Neuropharmacology. 2010;59:70–6. doi: 10.1016/j.neuropharm.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 51.Pfister JA, Ma C, Morrison BE, D’Mello SR. Opposing effects of sirtuins on neuronal survival: SIRT1-mediated neuroprotection is independent of its deacetylase activity. PloS One. 2008;3:e4090. doi: 10.1371/journal.pone.0004090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Higashida K, Kim SH, Jung SR, Asaka M, Holloszy JO, Han DH. Effects of resveratrol and SIRT1 on PGC-1α activity and mitochondrial biogenesis: A reevaluation. PLoS Biol. 2013;11:e1001603. doi: 10.1371/journal.pbio.1001603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen SD, Lin TK, Yang DI, Lee SY, Shaw FZ, Liou CW, et al. Protective effects of peroxisome proliferator-activated receptors gamma coactivator-1alpha against neuronal cell death in the hippocampal CA1 subfield after transient global ischemia. J Neurosci Res. 2010;88:605–13. doi: 10.1002/jnr.22225. [DOI] [PubMed] [Google Scholar]
- 54.Bordone L, Motta MC, Picard F, Robinson A, Jhala US, Apfeld J, et al. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol. 2006;4:e31. doi: 10.1371/journal.pbio.0040031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haines BA, Mehta SL, Pratt SM, Warden CH, Li PA. Deletion of mitochondrial uncoupling protein-2 increases ischemic brain damage after transient focal ischemia by altering gene expression patterns and enhancing inflammatory cytokines. J Cereb Blood Flow Metab. 2010;30:1825–33. doi: 10.1038/jcbfm.2010.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de Bilbao F, Arsenijevic D, Vallet P, Hjelle OP, Ottersen OP, Bouras C, et al. Resistance to cerebral ischemic injury in UCP2 knockout mice: Evidence for a role of UCP2 as a regulator of mitochondrial glutathione levels. J Neurochem. 2004;89:1283–92. doi: 10.1111/j.1471-4159.2004.02432.x. [DOI] [PubMed] [Google Scholar]
- 57.Haines B, Li PA. Overexpression of mitochondrial uncoupling protein 2 inhibits inflammatory cytokines and activates cell survival factors after cerebral ischemia. PloS One. 2012;7:e31739. doi: 10.1371/journal.pone.0031739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mattiasson G, Shamloo M, Gido G, Mathi K, Tomasevic G, Yi S, et al. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat Med. 2003;9:1062–8. doi: 10.1038/nm903. [DOI] [PubMed] [Google Scholar]
- 59.Andrews ZB, Diano S, Horvath TL. Mitochondrial uncoupling proteins in the CNS: In support of function and survival. Nat Rev Neurosci. 2005;6:829–40. doi: 10.1038/nrn1767. [DOI] [PubMed] [Google Scholar]
- 60.Peternelj TT, Coombes JS. Antioxidant supplementation during exercise training: Beneficial or detrimental? Sports Med. 2011;41:1043–69. doi: 10.2165/11594400-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 61.Li P, Hu X, Gan Y, Gao Y, Liang W, Chen J. Mechanistic insight into DNA damage and repair in ischemic stroke: Exploiting the base excision repair pathway as a model of neuroprotection. Antioxid Redox Signal. 2011;14:1905–18. doi: 10.1089/ars.2010.3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim GW, Noshita N, Sugawara T, Chan PH. Early decrease in dna repair proteins, Ku70 and Ku86, and subsequent DNA fragmentation after transient focal cerebral ischemia in mice. Stroke. 2001;32:1401–7. doi: 10.1161/01.str.32.6.1401. [DOI] [PubMed] [Google Scholar]
- 63.Sugawara T, Noshita N, Lewén A, Kim GW, Chan PH. Neuronal expression of the DNA repair protein Ku 70 after ischemic preconditioning corresponds to tolerance to global cerebral ischemia. Stroke. 2001;32:2388–93. doi: 10.1161/hs1001.097109. [DOI] [PubMed] [Google Scholar]
- 64.Cohen HY, Lavu S, Bitterman KJ, Hekking B, Imahiyerobo TA, Miller C, et al. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol Cell. 2004;13:627–38. doi: 10.1016/s1097-2765(04)00094-2. [DOI] [PubMed] [Google Scholar]
- 65.Jeong J, Juhn K, Lee H, Kim SH, Min BH, Lee KM, et al. SIRT1 promotes DNA repair activity and deacetylation of Ku70. Exp Mol Med. 2007;39:8–13. doi: 10.1038/emm.2007.2. [DOI] [PubMed] [Google Scholar]
- 66.Strosznajder RP, Czubowicz K, Jesko H, Strosznajder JB. Poly (ADP-ribose) metabolism in brain and its role in ischemia pathology. Mol Neurobiol. 2010;41:187–96. doi: 10.1007/s12035-010-8124-6. [DOI] [PubMed] [Google Scholar]
- 67.Bai P, Cantó C, Oudart H, Brunyánszki A, Cen Y, Thomas C, et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011;13:461–8. doi: 10.1016/j.cmet.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J Neurosci. 2010;30:2967–78. doi: 10.1523/JNEUROSCI.5552-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moroni F. Poly (ADP-ribose) polymerase 1 (PARP-1) and postischemic brain damage. Curr Opin Pharmacol. 2008;8:96–103. doi: 10.1016/j.coph.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 70.Kolthur-Seetharam U, Dantzer F, McBurney MW, de Murcia G, Sassone-Corsi P. Control of AIF-mediated cell death by the functional interplay of SIRT1 and PARP-1 in response to DNA damage. Cell Cycle. 2006;5:873–7. doi: 10.4161/cc.5.8.2690. [DOI] [PubMed] [Google Scholar]
- 71.Rajamohan SB, Pillai VB, Gupta M, Sundaresan NR, Birukov KG, Samant S, et al. SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly(ADP-ribose) polymerase 1. Mol Cell Biol. 2009;29:4116–29. doi: 10.1128/MCB.00121-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Parsons JL, Dianova II, Dianov GL. APE1 is the major 3′-phosphoglycolate activity in human cell extracts. Nucleic Acids Res. 2004;32:3531–6. doi: 10.1093/nar/gkh676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Srivastava DK, Berg BJ, Prasad R, Molina JT, Beard WA, Tomkinson AE, et al. Mammalian abasic site base excision repair. Identification of the reaction sequence and rate-determining steps. J Biol Chem. 1998;273:21203–9. doi: 10.1074/jbc.273.33.21203. [DOI] [PubMed] [Google Scholar]
- 74.Fantini D, Vascotto C, Marasco D, D’Ambrosio C, Romanello M, Vitagliano L, et al. Critical lysine residues within the overlooked N-terminal domain of human APE1 regulate its biological functions. Nucleic Acids Res. 2010;38:8239–56. doi: 10.1093/nar/gkq691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yamamori T, DeRicco J, Naqvi A, Hoffman TA, Mattagajasingh I, Kasuno K, et al. SIRT1 deacetylates APE1 and regulates cellular base excision repair. Nucleic Acids Res. 2010;38:832–45. doi: 10.1093/nar/gkp1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li W, Luo Y, Zhang F, Signore AP, Gobbel GT, Simon RP, et al. Ischemic preconditioning in the rat brain enhances the repair of endogenous oxidative DNA damage by activating the base-excision repair pathway. J Cereb Blood Flow Metab. 2006;26:181–98. doi: 10.1038/sj.jcbfm.9600180. [DOI] [PubMed] [Google Scholar]
- 77.Wang S, Xing Z, Vosler PS, Yin H, Li W, Zhang F, et al. Cellular NAD replenishment confers marked neuroprotection against ischemic cell death: Role of enhanced DNA repair. Stroke. 2008;39:2587–95. doi: 10.1161/STROKEAHA.107.509158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fan W, Luo J. SIRT1 regulates UV-induced DNA repair through deacetylating XPA. Mol Cell. 2010;39:247–58. doi: 10.1016/j.molcel.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 79.Yuan Z, Zhang X, Sengupta N, Lane WS, Seto E. SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol Cell. 2007;27:149–62. doi: 10.1016/j.molcel.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stein LR, Imai S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol Metab. 2012;23:420–8. doi: 10.1016/j.tem.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ying W, Wei G, Wang D, Wang Q, Tang X, Shi J, et al. Intranasal administration with NAD+ profoundly decreases brain injury in a rat model of transient focal ischemia. Front Biosci. 2007;12:2728–34. doi: 10.2741/2267. [DOI] [PubMed] [Google Scholar]
- 82.Wang P, Xu TY, Guan YF, Tian WW, Viollet B, Rui YC, et al. Nicotinamide phosphoribosyltransferase protects against ischemic stroke through SIRT1-dependent adenosine monophosphate-activated kinase pathway. Ann Neurol. 2011;69:360–74. doi: 10.1002/ana.22236. [DOI] [PubMed] [Google Scholar]
- 83.Morris-Blanco KC, Cohan CH, Neumann JT, Sick TJ, Perez-Pinzon MA. Protein kinase C epsilon regulates mitochondrial pools of Nampt and NAD following resveratrol and ischemic preconditioning in the rat cortex. J Cereb Blood Flow Metab. 2014;34:1024–32. doi: 10.1038/jcbfm.2014.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Centeno JM, Orti M, Salom JB, Sick TJ, Pérez-Pinzón MA. Nitric oxide is involved in anoxic preconditioning neuroprotection in rat hippocampal slices. Brain Res. 1999;836:62–9. doi: 10.1016/s0006-8993(99)01610-8. [DOI] [PubMed] [Google Scholar]
- 85.Ruderman NB, Xu XJ, Nelson L, Cacicedo JM, Saha AK, Lan F, et al. AMPK and SIRT1: A long-standing partnership? Am J Physiol Endocrinol Metab. 2010;298:E751–60. doi: 10.1152/ajpendo.00745.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang LM, Wang YJ, Cui M, Luo WJ, Wang XJ, Barber PA, et al. A dietary polyphenol resveratrol acts to provide neuroprotection in recurrent stroke models by regulating AMPK and SIRT1 signaling, thereby reducing energy requirements during ischemia. Eur J Neurosci. 2013;37:1669–81. doi: 10.1111/ejn.12162. [DOI] [PubMed] [Google Scholar]
- 87.Brandauer J, Vienberg SG, Andersen MA, Ringholm S, Risis S, Larsen PS, et al. AMP-activated protein kinase regulates nicotinamide phosphoribosyl transferase expression in skeletal muscle. J Physiol. 2013;591:5207–20. doi: 10.1113/jphysiol.2013.259515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fulco M, Cen Y, Zhao P, Hoffman EP, McBurney MW, Sauve AA, et al. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev Cell. 2008;14:661–73. doi: 10.1016/j.devcel.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zocchi L, Sassone-Corsi P. SIRT1-mediated deacetylation of MeCP2 contributes to BDNF expression. Epigenetics. 2012;7:695–700. doi: 10.4161/epi.20733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.MacIsaac KD, Lo KA, Gordon W, Motola S, Mazor T, Fraenkel E. A quantitative model of transcriptional regulation reveals the influence of binding location on expression. PLoS Comput Biol. 2010;6:e1000773. doi: 10.1371/journal.pcbi.1000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Truettner J, Busto R, Zhao W, Ginsberg MD, Pérez-Pinzón MA. Effect of ischemic preconditioning on the expression of putative neuroprotective genes in the rat brain. Brain Res Mol Brain Res. 2002;103:106–15. doi: 10.1016/s0169-328x(02)00191-2. [DOI] [PubMed] [Google Scholar]
- 92.Neumann JT, Thompson JW, Raval AP, Cohan CH, Koronowski KB, Perez-Pinzon MA. Increased BDNF protein expression after ischemic or PKC epsilon preconditioning promotes electrophysiologic changes that lead to neuroprotection. J Cereb Blood Flow Metab. 2015;35:121–30. doi: 10.1038/jcbfm.2014.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Huang W, Liu X, Cao J, Meng F, Li M, Chen B, et al. miR-134 Regulates ischemia/reperfusion injury-induced neuronal cell death by regulating CREB signaling. J Mol Neurosci. 2015;55:821–9. doi: 10.1007/s12031-014-0434-0. [DOI] [PubMed] [Google Scholar]
- 94.Chen A, Xiong LJ, Tong Y, Mao M. The neuroprotective roles of BDNF in hypoxic ischemic brain injury. Biomed Rep. 2013;1:167–76. doi: 10.3892/br.2012.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mattagajasingh I, Kim CS, Naqvi A, Yamamori T, Hoffman TA, Jung SB, et al. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 2007;104:14855–60. doi: 10.1073/pnas.0704329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tsai SK, Hung LM, Fu YT, Cheng H, Nien MW, Liu HY, et al. Resveratrol neuroprotective effects during focal cerebral ischemia injury via nitric oxide mechanism in rats. J Vasc Surg. 2007;46:346–53. doi: 10.1016/j.jvs.2007.04.044. [DOI] [PubMed] [Google Scholar]
- 97.Tobin MK, Bonds JA, Minshall RD, Pelligrino DA, Testai FD, Lazarov O. Neurogenesis and inflammation after ischemic stroke: What is known and where we go from here. J Cereb Blood Flow Metab. 2014;34:1573–84. doi: 10.1038/jcbfm.2014.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wong CH, Crack PJ. Modulation of neuro-inflammation and vascular response by oxidative stress following cerebral ischemia-reperfusion injury. Curr Med Chem. 2008;15:1–14. doi: 10.2174/092986708783330665. [DOI] [PubMed] [Google Scholar]
- 99.Pizzi M, Sarnico I, Lanzillotta A, Battistin L, Spano P. Post-ischemic brain damage: NF-kappaB dimer heterogeneity as a molecular determinant of neuron vulnerability. FEBS J. 2009;276:27–35. doi: 10.1111/j.1742-4658.2008.06767.x. [DOI] [PubMed] [Google Scholar]
- 100.Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–80. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–6. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 102.Fukuda S, Kaga S, Zhan L, Bagchi D, Das DK, Bertelli A, et al. Resveratrol ameliorates myocardial damage by inducing vascular endothelial growth factor-angiogenesis and tyrosine kinase receptor Flk-1. Cell Biochem Biophys. 2006;44:43–9. doi: 10.1385/CBB:44:1:043. [DOI] [PubMed] [Google Scholar]
- 103.Potente M, Ghaeni L, Baldessari D, Mostoslavsky R, Rossig L, Dequiedt F, et al. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev. 2007;21:2644–58. doi: 10.1101/gad.435107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dioum EM, Chen R, Alexander MS, Zhang Q, Hogg RT, Gerard RD, et al. Regulation of hypoxia-inducible factor 2alpha signaling by the stress-responsive deacetylase sirtuin 1. Science. 2009;324:1289–93. doi: 10.1126/science.1169956. [DOI] [PubMed] [Google Scholar]
- 105.Malhotra S, Savitz SI, Ocava L, Rosenbaum DM. Ischemic preconditioning is mediated by erythropoietin through PI-3 kinase signaling in an animal model of transient ischemic attack. J Neurosci Res. 2006;83:19–27. doi: 10.1002/jnr.20705. [DOI] [PubMed] [Google Scholar]
- 106.Zhang F, Signore AP, Zhou Z, Wang S, Cao G, Chen J. Erythropoietin protects CA1 neurons against global cerebral ischemia in rat: Potential signaling mechanisms. J Neurosci Res. 2006;83:1241–51. doi: 10.1002/jnr.20816. [DOI] [PubMed] [Google Scholar]
- 107.Rane S, He M, Sayed D, Vashistha H, Malhotra A, Sadoshima J, et al. Downregulation of miR-199a derepresses hypoxia-inducible factor-1alpha and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ Res. 2009;104:879–86. doi: 10.1161/CIRCRESAHA.108.193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dong W, Li N, Gao D, Zhen H, Zhang X, Li F. Resveratrol attenuates ischemic brain damage in the delayed phase after stroke and induces messenger RNA and protein express for angiogenic factors. J Vasc Surg. 2008;48:709–14. doi: 10.1016/j.jvs.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 109.Ying W, Xiong ZG. Oxidative stress and NAD+ in ischemic brain injury: Current advances and future perspectives. Curr Med Chem. 2010;17:2152–8. doi: 10.2174/092986710791299911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kakefuda K, Fujita Y, Oyagi A, Hyakkoku K, Kojima T, Umemura K, et al. Sirtuin 1 overexpression mice show a reference memory deficit, but not neuroprotection. Biochem Biophys Res Commun. 2009;387:784–8. doi: 10.1016/j.bbrc.2009.07.119. [DOI] [PubMed] [Google Scholar]
- 111.Liu D, Gharavi R, Pitta M, Gleichmann M, Mattson MP. Nicotinamide prevents NAD+ depletion and protects neurons against excitotoxicity and cerebral ischemia: NAD+ consumption by SIRT1 may endanger energetically compromised neurons. Neuromolecular Med. 2009;11:28–42. doi: 10.1007/s12017-009-8058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lin SJ, Ford E, Haigis M, Liszt G, Guarente L. Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. 2004;18:12–6. doi: 10.1101/gad.1164804. [DOI] [PMC free article] [PubMed] [Google Scholar]