

Abstract
In the past two decades, the study of cancer therapy has gradually advanced to the “Nano” era. Numerous novel nanomaterials armed with unique physical properties have been introduced into biomedical research. At the same time, functional nucleic acid molecules, especially aptamers, have aroused broad attention from the biomedical community. Benefiting from the advancement of molecular engineering strategies, it is now feasible to combine the cancer specific recognition capability of aptamers with various other special functions of nanomaterials to develop cancer specific drugs at the nanoscale. Nanodrugs are now offering an unprecedented opportunity to achieve the goal of efficient targeted delivery as well as controlled release. This review highlights some achievements of multiple aptamer-based nanodrug systems which have emerged in recent years, including studies in the infant stage of “proof-of-concept”.
Keywords: aptamer, controlled release, drug delivery, nanodrug, nanotechnology
1. Introduction
It has been over a quarter century since scientists proposed that nucleic acids could serve as recognition probes for any target of interest.[1] Functional single-strand oligonucleotides, called “aptamers”, bind with specificity and affinity rivaling the capability of antibodies. Nucleic acids molecules are much easier to synthesize, manipulate, and functionalize, and, compared to antibodies, aptamers perform better in terms of stability and immunogenicity.
Formation of an aptamer follows a relatively simple protocol: 1) chemical synthesis of a randomized nucleic acid library, usually composed of 20–40 nucleotides (nt) flanked by primer binding sections; 2) exposure of the target to the library pool and PCR amplification of the binding sequences; 3) repetition of the screening process for 8–20 rounds until the library is significantly enriched; 4) binding pool sequencing followed by characterization. Termed Systematic Evolution of Ligands by EXponential enrichment (SELEX), this method has led scientists to generate aptamers towards a variety of small molecules like ATP[2] and cocaine,[3] to macromolecules like proteins[4] and carbohydrates.[5] Some aptamers have entered the clinic, albeit in modified forms.[6]
In 2006, the Tan group first reported the cell-SELEX technology,[7] which uses whole cells as the target without prior knowledge of the membrane binding sites. The generated aptamers can recognize the target cells in the presence of the surrounding non-target cells (e.g. cancer cells in the presence of normal cells), and can be applied for drug delivery or cellular detection.[8] Aptamer sgc8, one of our most studied as well as the best known aptamer generated against leukemia cells, has been widely exploited for developing novel drug delivery systems.[9] Aptamers can also serve as tools for cancer-related biomarker discovery.[10] As aptamers bind to sites specific on cancer cell membranes and not present on normal cells, it is possible to characterize the site as a biomarker for cancer.
As researchers understand more deeply about the nature of nucleic acid molecules, a variety of DNA specific reactions and molecular engineering methods have emerged, such as hybridization chain reaction (HCR),[11] rolling circle amplification (RCA),[12] strand displacement reaction (SDR),[13] and even DNA origami technologies.[14] These developments have made it possible to build DNA-based nanostructures with a variety of functions. Used in conjunction with other nanomaterials, like gold nanoparticles and magnetic nanoparticles, DNA-based nanostructures have lifted the platform of drug delivery study to an unprecedented high level.[9]
Generally, the ultimate goals for cancer-related drug delivery are clear: targeted delivery, stimuli-responsive release, and logic-based cell destruction. Combining the recognition capability of aptamers, the advantages of a variety of nanostructures for drug loading and stimulus response, and the information coding ability of DNA molecules, these three goals can now be achieved. This review describes recent research in the development of aptamer-based nanodrugs by the Tan group, as well as by other investigators.
2. Methods of DNA-drug conjugation
Once an effective cancer cell-targeting aptamer has been identified, the next challenge is finding a way to conjugate it to drug molecules. The direct drug-DNA conjugation also serves as a basic unit for further assembly of much more complex nanodrugs. DNA aptamers and drugs have been conjugated by non-covalent intercalation[15] or by covalent bonding via a chemical linker, either through solid-phase synthesis[16] or post-synthesis modification.[17] Different applications of these approaches are described in greater detail below.
2.1. Non-covalent conjugates
Doxorubicin (Dox) is an anthracycline chemotherapeutic agent that has been extensively used in the treatment of various cancers. The cytotoxicity of Dox is due to its intercalation into genomic DNA, which undermines replication and transcription, and therefore induces cell death. This mechanism can in turn be adopted for Dox delivery, in which Dox is conjugated to DNA sequences via non-covalent interactions.
Efforts in this area include the initial work by Bagalkot and his colleagues, who intercalated Dox into the A10 2′-fluoropyrimidine RNA aptamer with a long duplex tail for prostate-specific membrane antigen (PSMA) targeted therapy,[18] recent work by Hu et al. that employed MA3 aptamer to target MUC1-expressing tumor cells,[15a] and a similar study that used DNA aptamer HB5 (specific for HER2) for targeting breast cancer.[15b] In 2012, the Tan group modified liver cancer LH86 specific aptamer TLS11a with a long repeated GC tail to form a dimer structure and carry Dox at a ratio of up to 28 Dox per tail (Figure 1A).[19] In addition to anthracycline chemotherapeutic agents, photosensitizers can also be delivered into target cells through simple intercalation with aptamers. As described by Shieh et al., a photosensitizing agent (TMPyP4) was intercalated into a guanine-rich AS1411 DNA aptamer to selectively target nucleolin overexpressed by MCF-7 breast cancer cells. This intercalation was formed by the aromatic and cationic quadruplex ligand of TMPyP4 binding to the G-quadruplex of AS1411 aptamer.[15c] A similar strategy was exploited in the Tan group, but the more cancer cell-specific aptamer sgc8 was used.[20] (Figure 1B).
Figure 1.

A) Intercalation of Dox into GC modified aptamer TLS11a to form physical conjugate. Reprinted by permission from reference.[19] Copyright 2012 Meng et al. B) G-quadruplex-aptamer as a drug carrier. The design utilizes the G-quadruplex as the drug carrier and the aptamer as the targeting molecule to deliver TMPyP4, which is known to bind and stabilize different types of quadruplexes. The helical strand represents the aptamer. Reprinted by permission from reference.[20] Copyright 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. C) (Top) Synthesis of ApDCs from phosphoramidites A, T, C, G, and the phosphoramidite containing a drug moiety with a photocleavable linker (denoted as phosphoramdite D). (Bottom) Structural features of phosphoramidite D. Reprinted by permission from reference.[16] Copyright 2014 American Chemical Society.
Despite the effectiveness of intercalation, several concerns need to be addressed, including the instability of the aptamer-drug conjugates due to the reversibility of the interaction, the limited numbers of drugs that are feasible for intercalation, the disruption of aptamer structure and function upon intercalation,[21] and the limited drug payload capacity.
2.2. Covalent linkage
Covalent linkage is an alternative to improve conjugate stability and widen the scope of applicable drug molecules. In an early attempt, Huang et al. covalently bonded Dox to aptamer sgc8 via an acid-labile hydrazone linker for intracellular delivery.[17b] The binding affinity and capability for internalization of sgc8[22] were preserved upon conjugation with Dox, while the cytotoxicity potency was comparable to that of nude Dox on target cells but minimal on control cells. However, when a bifunctional linker was used for a multiple-Dox-aptamer conjugate (sgc8c-3Dox), the killing efficiency was compromised, probably due to diminished binding affinity of the aptamers. Very recently, another attempt to prepare sgc8-Dox conjugates with high payload capacity was reported by Zhu et al.[17c] Here, formaldehyde acted as a reducing and crosslinking agent that specifically reacted with deoxyguanosine of DNA to form the DNA-drug adduct. Not only was the pay load capacity of Dox significantly increased, but also the DNA-drug adduct was nuclease-resistant.
Covalent linkage to DNA has also been reported for other drug molecules. For example, the photosensitizer chlorin e6 was linked to aptamers targeting tumor-associated mucin markers such as MUC1 peptides and Tn antigens.[17a] And Yang et al. incorporated multiple aptamers into a cytotoxic polymer, to achieve selective targeted therapy, which was effective even on drug-resistant cancer cells.[15e]
Aside from post-synthesis DNA modification, Wang et al. designed and synthesized a phosphoramidite containing a drug moiety with a photocleavable linker for solid phase synthesis of DNA aptamers (Figure 1C). This module allowed multiple drugs to be efficiently incorporated into aptamers at controlled designated positions. The aptamer-drug conjugates could be manipulated for developing an effective chemotherapy with minimum side effects.[16]
3. Programmable DNA based nanostructures
In the past two decades, the study of DNA has advanced to a new level, as a result of scientists’ realization that the ability of nucleic acids to encode vast amounts of information via canonical Watson-Crick base-pairing interactions could be manipulated.[23] Accordingly, innumerable DNA-based nanostructures have emerged, many of them armed with therapeutic agents for drug delivery. This series of researches is significant, not only due to the programmability of DNA, making it possible to build intricate structures, load larger amounts of drugs and achieve logic-based delivery, but also because of the nature of nucleic acids as biopolymers with excellent biocompatibility.
3.1. Aptamer-based self-assembly of DNA nanostructures
The Tan group has extensively explored the possibility to combine the strengths of DNA aptamers and the advantages of self-assembled DNA nanostructures for targeted drug delivery. In one attempt, Zhu et al. built self-assembled DNA nanostructures, called aptamer-tethered DNA nanotrains (aptNTrs), to load Dox for targeted delivery (Figure 2).[15d] When exposed to the sgc8 trigger, the two hairpin monomers self-assembled into an elongated duplex DNA chain via a hybridization chain reaction (HCR). This created many addressable loading sites (“boxcars”) in one single aptNTr, resulting in a payload molar ratio of drug/sgc8 aptNTr as high as 50/1. The aptamer acts as both a trigger for the HCR, as well as a “locomotive” to guide the direction (targeting) and provide the driving force to facilitate the endocytosis. Several further studies showed that the aptNTr had many unique properties, such as selective recognition abilities, selective drug transport, and applicability to a variety of target cell types and drugs.
Figure 2.

Schematics of aptamer-tethered DNA nanotrains (aptNTrs) for theranostic applications. A) Self-assembly of aptNTrs from short DNA building blocks initiated by a chimeric aptamer-tethered trigger probe. The tethered aptamers work as locomotives with multiple repetitive “boxcars” to be loaded with molecular drugs. B) The drugs are specifically transported to target cancer cells via aptNTrs. Reprinted by permission from reference.[15d] Copyright 2013 Zhu et al.
In another example, Zhu and Hu et al. employed the rolling circle amplification (RCA) method to self-assemble DNA Nanoflowers (NFs).[24] Densely compacted multifunctional DNA nanostructures were made of elongated non-nicked building blocks that contained a small number of only a few DNA strands. This work overcame the disadvantages of conventional DNA nanostructures, which rely on Watson-Crick base-pairing between short DNA building blocks, necessitating complicated design procedures, large amounts of DNA for bulky preparation, and intrinsic nicks that affected biostability. As facile and biologically stable DNA nanostructures, NFs have presented a number of biological applications, including therapeutics delivery and intracellular imaging.[24]
3.2. Programmable DNA nanodevices for logic-based controlled release
Cancer targeting and therapy have also been based on DNA logic gates. Cancer cells are different from normal cells in several ways, including expression levels of the cell membrane components. Recognition of multiple biomarkers on the cell surface provides information to accurately differentiate cancer cells from normal cells. For example, reliability increases if multiple targets on the cell membrane can be targeted simultaneously. For this reason, nanodevices based on logic gates are essential for accurate controlled release. In 2012, the technology of DNA origami was adopted by Douglas and his coworkers to develop a DNA nanorobot with a 3-dimensional hexagonal barrel-shaped nanostructure.[25] The nanorobot can intelligently release the drug payload when combinations of cell surface biomarkers match its aptamer pattern.
In year 2013, Han et al. engineered an aptamer-based logic circuit targeting cancer cells to enable photodynamic cancer therapy (PDT) by activating Chlorin e6 (Ce6) photosensitizer.[26] In the presence of target cells, the circuit triggers a cascade reaction to release the quencher-labeled strand and activate Ce6, which generates singlet oxygen upon irradiation at 404 nm. This method avoids the common disadvantages of Ce6, including short lifetime and short diffusion distance, as well as the broad spectrum of cytotoxicity of singlet oxygen.
As more and more cell targeted aptamers become available, it is possible to build more complex nanodevices to sense multiple biomarkers on the cell surface and generate a more comprehensive disease profile and yield a more accurate intervention. You et al. realized this AND logic gate system by designing a “nano-claw” for autonomous analysis of cell surface biomarker profiling to yield a diagnostic signal or targeted PDT response (Figure 3).[27] The nanodevice was designed in either a trivalent “Y”-shape or a three-input tetravalent “X”-shape for testing different numbers of biomarkers. The scaffold backbone was composed of a duplex oligonucleotide, and the “capture/effector toes” were connected through complementary sticky ends. Switchable aptamers act as “capture toes”, which are in charge of targeting multiple biomarkers on the cell surface and subsequently releasing the “barcode strands”. Only if all “barcode strands” from all “capture toes” are released and interact with the corresponding strands on the “effector toe” can the therapeutic agent Ce6 be activated.
Figure 3.
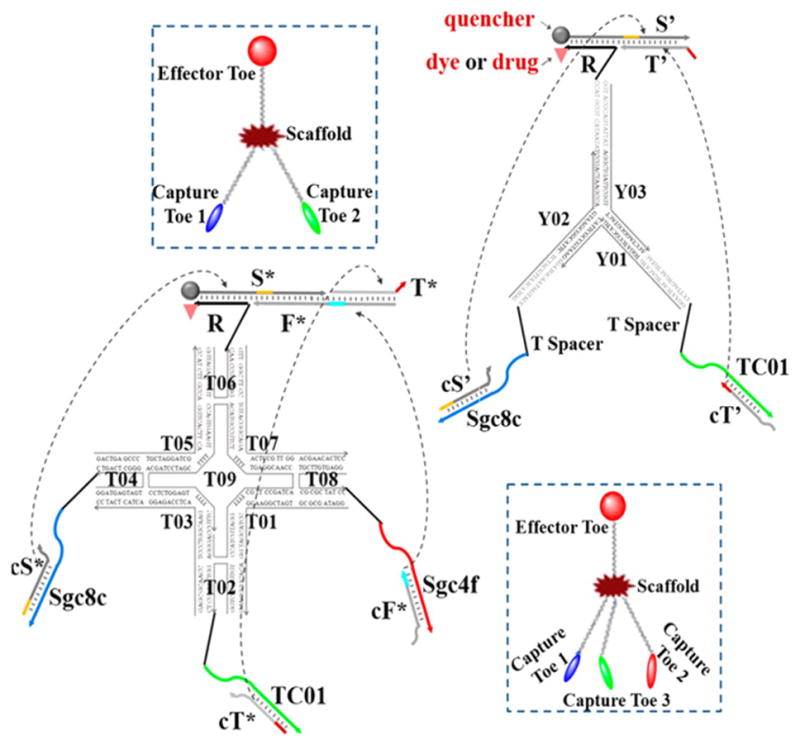
Construction of DNA-based “nano-claw”: two-input trivalent “Y”-shaped nano-claw and three-input tetravalent “X”-shaped nano-claw. Reprinted by permission from reference.[27] Copyright 2014 American Chemical Society
4. DNA molecular engineering
Aptamers can be readily chemically modified, and this is an important advantage over antibodies. Different functional groups or linker molecules can be incorporated into DNA strands and be used to construct more structurally complex nanomaterials, providing a large variety of properties for the drug delivery system. Among these structures, DNA micelles, DNA liposomes and DNA nanogels have aroused broad attention.
4.1. DNA micelles
One type of modification is addition of a hydrophobic tail to the hydrophilic DNA strands, thereby creating an amphiphilic molecule that can self-assemble into a spherical micelle structure above the critical micelle concentration (CMC). Compared to bare aptamer DNA strands, aptamer micelles exhibit the multivalent effect, which increases the binding affinity to the target. The resistance to enzymatic degradation is also increased due to steric hindrance from the close structure of this micelle assembly. Moreover, the hydrophobic core of the micelle can load hydrophobic drugs, and the oligonucleotide corona has the potential to be utilized for gene therapy.[28]
With these intrinsic advantages, the micelle structure has been extensively explored as a drug delivery system. For example, the Tan group synthesized an amphiphilic micelle monomer by attaching a diacyllipid to the end of an aptamer via a PEG linker (Figure 4A).[29] This aptamer micelle was size tunable and exhibited enhanced cell permeability. Selective binding of micelles formed with aptamer TDO5 demonstrated 80-fold enhanced permeability to Ramos cells compared to bare TDO5 aptamers at 37°C. A hydrophobic fluorescent dye was loaded into the micelle as an illustration of the delivery ability. After 2 h incubation, there was strong fluorescence from the target cells, while the free dye produced a very weak signal, indicating hydrophobic drug delivery ability of aptamer micelles.
Figure 4.
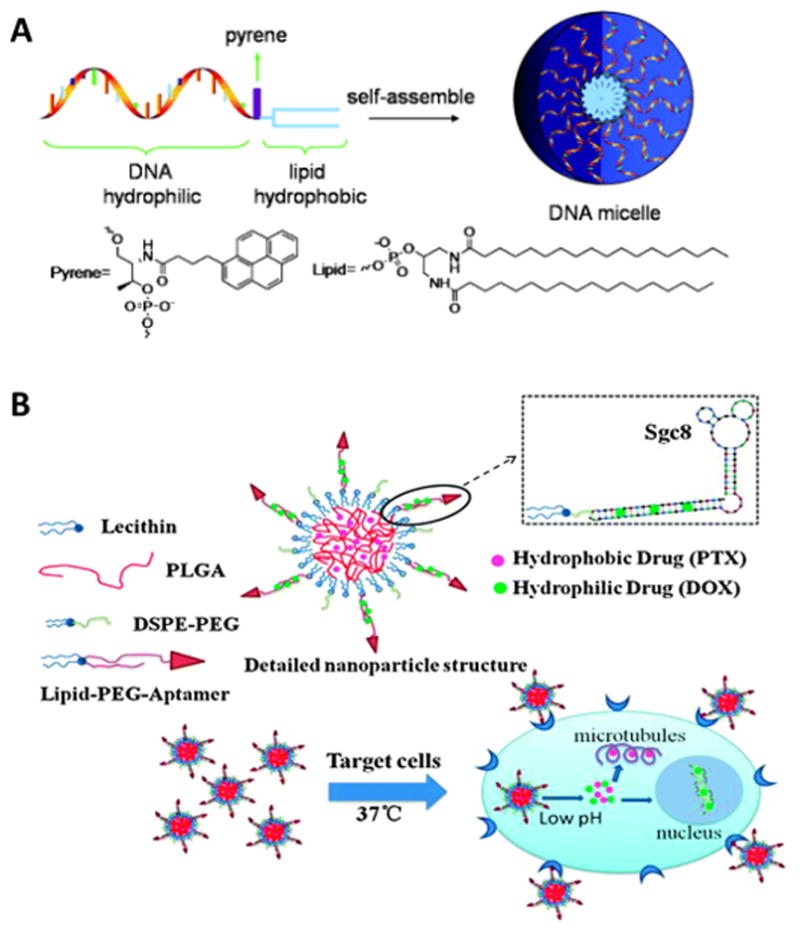
A) Schematic of the design and assembly of DNA micelles. (Top) Oligonucleotide micelles contain a DNA corona, a pyrene unit (fluorescence reporter), and a lipid core. (Bottom) Molecular structure of pyrene and lipid unit.[29b] Copyright 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim B) Structure of aptamer-coated PLGA hybrid nanoparticles and co-delivery of two drugs to cancer cells. PTX is encapsulated in the PLGA core and DOX is loaded in the lipid-PEG-aptamer shell. Lecithin and DSPE-PEG are used to stabilize the structure. Reprinted by permission from reference.[30] Copyright 2015 Royal Society of Chemistry.
In another work inspired by DNA micelles. Huang et al. coated diacyllipid conjugated aptamers to hydrophobic poly (lactide-co-glycolic acid) (PLGA) nanoparticles to form a hybrid core-shell structure (Figure 4B).[30] This hybrid assembly enabled co-delivery of two drugs that are synergistically effective: the hydrophilic drug DOX is loaded with DNA in the outer shell and the hydrophobic drug Paclitaxel (PTX) is encapsulated in the PLGA core. The system showed specific binding towards target cells with little leakage into control cells. The cytotoxicity test showed that co-delivery of two drugs selectively reduces cell viability more than single drug systems.
4.2. DNA liposomes
A liposome is another soft spherical vesicle composed of amphiphilic monomers, constructed like a lipid bilayer to resemble the structure of a cell membrane. Instead of having a hydrophobic core and hydrophilic corona as in micelles, liposomes contain a hydrophilic core and outer layer, as well as a hydrophobic environment between two layers. Liposomes are widely used as vehicles for drug delivery, and more than ten kinds of liposomal delivery systems have been approved for clinical use. Aptamers engineered with modification tails can be incorporated into liposomes by conjugation or hydrophobic interaction.
The Tan group has modified liposomes with aptamers via EDC/NHS coupling between a thiol group on the aptamer and a maleimide group on the liposome surface (Figure 5A).[31] FITC-Dextran was encapsulated in the liposome to show the delivery ability. With aptamer conjugation, highly specific and effective delivery toward positive cells rather than negative cells was proven by flow cytometry as well as confocal images. Moreover, the Kim group constructed an RNA aptamer-conjugated liposome, termed aptamosome, by a post-insertion method.[32] (Figure 5B) First they modified a DNA micelle with a tethering linker by EDC-NHS reaction. Then RNA aptamer with extended sequence complementary to linker DNA was connected to the liposome by annealing. By incubating the aptamer-conjugated micelle and liposome together, micelles were fused and inserted into the liposomes to form aptamosomes. Dox was encapsulated for in vivo study and the targeted antitumor efficacy was significant.
Figure 5.

A) Conjugation of aptamer to a liposome shell. Reprinted by permission from reference.[31] Copyright 2010 Royal Society of Chemistry B) Incorporation of aptamer on a liposome by fusing aptamer-connected micelle to liposome. Reprinted by permission from reference.[32] Copyright 2015 Elsevier B.V.
Besides direct conjugation, the DNA end can be modified with a hydrophobic moiety, which can intercalate into the phospholipid layer of the liposome by hydrophobic interactions, while the hydrophilic oligonucleotide strands stay outside the sphere.[33] The Mirkin group demonstrated the increased stability of the liposome as well as the reduction of particle-particle fusion by incorporation of DNA strands.
4.3. DNA-based nanogels
Hydrogels have been widely used as drug delivery systems because of their excellent biocompatibility by retaining large amounts of water, making them as flexible as human tissue. However as a delivery system, one common issue for hydrogels is that cargo release is usually very rapid. Sustained release is needed, because it increases the therapeutic efficacy of drugs and decreases side effects. An aptamer can be incorporated to achieve this goal by holding the protein drug inside the hydrogel. The release kinetics can be controlled by changing aptamers with different affinity to the protein,[34] or modifying the length and the sequence of the aptamer’s complementary DNA strand.[35]
The Wang group combined multiple protein drugs with an aptamer-functionalized hydrogel. They functionalized polystyrene microparticles with aptamers for vascular endothelial growth factor (VEGF) and platelet-derived growth factor BB (PDGF-BB).[34] Then the particles were physically incorporated into the hydrogel. In this way, they used different complementary sequences (CS) to trigger the release of different proteins. Since the release rate is dependent on the CS length and the time to release can be predetermined, this system has the potential for controllable release of different drugs based on the stages of the disease and treatment.
They also developed a superporous hydrogel synthesis using the method of free radical polymerization combined with gas foaming[35a] thereby avoiding the drawbacks of integration of the cargo loading process with hydrogel formation. First, the polymer synthesis always involves harsh conditions or organic solvents which may influence the structure and function of the proteins loaded in the hydrogel. Second, washing is necessary but may cause denaturation or loss of loaded proteins. Moreover, the ligands that do not bind to the hydrogel would cause an initial significant release. By taking advantage of the superporous gel to load the proteins after gel formation and washing, they eliminated these problems. In addition, these gels can be stored stable for a reasonable time and loading can be performed just before use.
Hydrogels entirely based on branched DNA have been developed since 2006.[35b] To better utilize this biocompatible and biodegradable material as a drug delivery system, the Tan group designed an aptamer-based DNA nanoassembly for targeted delivery of drugs and antisense oligonucleotides (Figure 6A).[36] Building units were first created by linking three Y-shaped DNA functional domains to an X-shaped connector. By modifying the ends of functional domains with acrydite, the building units can be photo-cross-linked to form a size-controllable nanostructure. Many different kinds of functional moieties can be added to the Y-shaped domain. Aptamers were connected to provide targeting ability; small molecule drugs such as Dox were intercalated between DNA base pairs, and antisense oligonucleotide MDR1-AS was combined to overcome the obstacle of multidrug resistance. This drug delivery system was easily manipulated, highly programmable, biostable and biocompatible with integrated multifunctionality.
Figure 6.

A) Building units and formation of multifunctional self-assembled nanoassembly. Reprinted by permission from reference.[36] Copyright 2013 American Chemical Society. B) Components and formation of DNA nano-hydrogel and the mechanism of cell internalization as well as stimulus-responsive degradation. Reprinted by permission from reference.[37] Copyright 2015 American Chemical Society.
A stimulus-responsive DNA nanogel was designed by the same group.[37] (Figure 6B) The building units of the hydrogel included a Y-shaped monomer with three sticky ends, a linker DNA with two sticky ends and the blocking and targeting unit with one sticky end and an aptamer. In addition, they incorporated disulfide linkages in the building units, which are reduced and cleaved by GSH in the cancer cell. The size is controllable by tuning the relative amounts of the building Y-shaped monomer and the blocking Y-shaped monomer.
5. DNA-nanoparticle conjugates
Nanotechnology has thrived in the past three decades, and the combination of functional nucleic acid probes like aptamers with nanomaterials has been broadly explored for advanced drug delivery methods. It is impossible to review all these advances within limited pages. Here we will focus on recent achievements from the Tan group, mainly conjugation of gold nanoparticles and magnetic nanoparticles with aptamers for the purpose of nanodrug development.
5.1. Aptamer-conjugated gold nanoparticles for targeted cancer therapy
The extensive attention for gold nanoparticles (AuNPs) as cancer therapy platforms is attributed to their unique optical, chemical and biological properties.[9, 38] AuNPs can be precisely tuned with different sizes and shapes (e.g., gold nanospheres, nanorods, nanoshells and nanocages) through a variety of synthetic methods.[39] The as-prepared nanostructures exhibit unusual and tunable optical properties due to their localized surface plasmon resonance (SPR) properties. AuNPs can support plasmon resonance with remarkably high absorption cross sections at near-infrared (NIR) frequencies, which can achieve deep-tissue penetration. Moreover, photon energies absorbed by AuNPs can be converted into heat efficiently via a series of non-radiative processes. As a result, AuNPs are considered potent therapeutic agents in photothermal therapy (PTT).[40] AuNP surface modification through Au:S ligand exchange processes has become a high capacity, easy-to-handle and low-cost approach, owing to the large nanoparticle surface area and strong interaction between gold and the thiol group, including thiol-modified DNA aptamers.
Effective hyperthermia is considered as a relatively noninvasive and benign alternative for cancer treatment. In an early application, Huang et al.[41] developed an sgc8 aptamer-based nanoplatform, which combined the high absorption efficiency of Au-Ag nanorods (NRs) with multiple sgc8c aptamers through simple thiol covalent linkages. The results indicated that the Au-Ag nanorod combination offers selective and efficient photothermal killing of targeted tumor cells. In another work, Wang et al.[42] modified AuNRs with two different aptamers (aptamer CSC1 targeting DU145 prostate cancer cells and CSC13 targeting the subpopulation of DU145 cancer stem cells) to target and kill cancer cells and cancer stem cells simultaneously by NIR laser irradiation.
In recent years, there have been many studies that use various approaches for utilizing nanoparticles for PDT. They categorized nanoparticles into passive and active nanoparticles, either depending on the absence or presence of any target on their surface, or based on their involvement in the excitation of PS.[50, 51]
As this line of research continued to develop, the Tan group investigated the possibility of combining PTT with chemotherapy or photodynamic cancer therapy (PDT). Kang et al.[43] (Figure 7A) constructed a NIR light-responsive drug delivery nanogel based on Au-Ag nanorods (Au-Ag NRs) coated with DNA cross-linked polymeric shells. The Au-Ag NR-based nanogel can be functionalized with aptamers for targeted drug delivery. When illuminated with NIR light, the temperature of the surrounding gel increases rapidly because of the thermal effect generated from Au-Ag NRs, resulting in rapid release of the loaded drug with high controllability. In vitro studies demonstrated that aptamer-modified nanogels can be developed as targeted drug delivery carriers with high controllability by NIR irradiation.
Figure 7.

A) Schematic diagram of the formation of aptamer-functionalized Au-Ag nanorods for drug delivery. Reprinted by permission from reference.[43] Copyright 2011 American Chemical Society. B) Schematic diagram illustrating a cell-targeted photocontrolled nuclear uptake nanodrug delivery system for cancer therapy. Reprinted by permission from reference.[44] Copyright 2015 American Chemical Society. C) Schematic diagram illustrating ASP-photosensiter-AuNRs for PTT and PDT. Reprinted by permission from reference.[45] Copyright 2012 American Chemical Society.
In addition, Qiu et al.[44] (Figure 7B) used self-assembled DNA to develop a nuclear-uptake nanodrug system carried by a cell-targeted NIR-responsive nanotruck for drug-resistant cancer chemotherapy. Via DNA hybridization, small drug-loaded gold nanoparticles self-assemble onto the side face of a silver-gold nanorod (NR, termed nanotruck) whose end faces were modified with a cell type-specific internalizing aptamer. This DNA-based nanoplatform selectively transports across the cell membrane, and subsequent NIR irradiation triggers the release of nanodrugs from the nanotruck via the photothermal effect. The released small nanodrugs diffuse into the nuclei where they sustainably release Dox to induce cancer cell apoptosis. By using this size-photocontrollable nanodrug delivery system, anticancer drugs can be efficiently accumulated in the nuclei to effectively kill the cancer cells. This design greatly enhances therapeutic efficacy against drug-resistant cancer cells by effectively bypassing P-glycoprotein (P-gp), which can actively efflux various anticancer drugs from the cell, a major mechanism of multidrug resistance (MDR).
In combined photothermal and photodynamic therapy (PTT/PDT), the AuNRs also serve as ultra-efficient energy quenchers because of the strong surface plasmon absorption in the NIR region. In this application, PDT can be effectively controlled by manipulating the quenching and recovery of photosensitizer (PS) fluorescence, which is, in turn, controlled according to the distance between the quencher and PS. For example, Wang et al.[45] (Figure 7C) developed an aptamer switch probe (ASP) linking the PS molecule, Ce6, to the surface of AuNRs. In the presence of target cancer cells, a conformational change drives Ce6 away from the gold surface, resulting in production of singlet oxygen for PDT upon light irradiation. At the same time, AuNRs can also destroy cells by the hyperthermia effect. Combined with the high selectivity and specificity of ASP, this PDT/PTT multimodal strategy promises to be a more efficient therapeutic regimen against cancer cells than either PDT or PTT alone.
5.2. Aptamer-conjugated magnetic nanoparticles for targeted cancer therapy
In recent years, various types of magnetic nanoparticles (MNPs) have been developed for cancer therapy under an external magnetic field. Aptamer-functionalized MNPs (Apt-MNPs) have been formulated to meet the specific requirements of imaging reagents and drug carriers for targeted cancer therapy.
In an attempt to improve drug-carrying capacity, Zheng et al.[46] (Figure 8A) designed a superparamagnetic iron oxide nanoparticle (SPION)-based spherical nucleic acid (SPION-SNA) to achieve higher drug-loading capacity. In this study, a novel multifunctional SNA platform was engineered by a SPION-tethered initiator, two partially complementary hairpin monomers and an aptamer probe. Each copy of the initiator on the SPION can trigger a cascade of hybridization chain reactions (HCR), and multiple copies of AS1411 aptamer can be hybridized with the long DNA biopolymer on the surface of SPION. Moreover, Dox can intercalate in the double-helix sequence as a drug-loading module. Therefore, a multifunctional nanoplatform can be achieved with high drug loading efficacy and high specific binding capacity.
Figure 8.
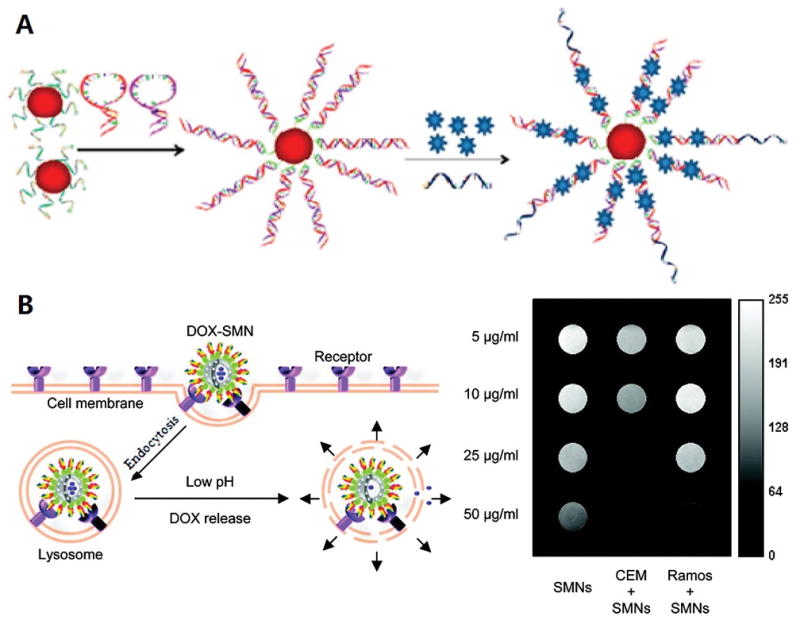
A) Schematic representation of multifunctional SPION-SNA platform for cancer therapy. Reprinted by permission from reference.[46] Copyright 2013 American Chemical Society. B) Schematic illustration of Apt-PHMNPs for cancer therapy. Reprinted by permission from reference.[48] Copyright 2011 American Chemical Society.
Porous hollow magnetic nanoparticles (PHMNPs) have been used for targeted chemotherapy and magnetic resonance imaging of cancer cells in vitro. The porous structure of PHMNPs facilitates high drug loading capacity. Moreover, the porous shell is relatively stable in neutral or basic physiological conditions. However, In an acidic environment, the pores are susceptible to acidic etching, resulting in rapid release of the drug cargo.[47] Chen et al.[48] (Figure 8B) modified PHMNPs with a heterobifunctional PEG ligand containing active carboxyl functional groups. Aptamers with amino groups were anchored onto the surface of PHMNPs (Apt-PHMNPs) by EDC/NHS crosslinker. Anticancer drug Dox can be loaded in the hollow interior of PHMNPs. Apt-PHMNPs specifically enter target CEM cells through receptor-mediated endocytosis and reside in the lysosome. The acidic environment results in pore size enlargement and release of Dox to kill the target cancer cells.
6. Perspective
In the battle against cancers, the specific molecular recognition capability of DNA aptamers selected against cancer cell membrane biomarkers has advanced cancer nanomedicine to a new level. From DNA aptamer-drug conjugates to aptamer-modified nanodrug carriers through the methodology of molecular engineering, aptamers have shown promising potential in cancer diagnostics and targeted therapeutics. Nine antagonistic aptamer-related drugs underwent phase I, II or III clinical trials in 2011, and this number has increased to a total of 26 aptamer-related clinical trials in 2014.[49] Although the powerful cell-SELEX allows identification of aptamers for any cancer cell type in vitro, the in vivo efficacy of these cancer-related aptamers still faces challenges, such as off-target recognition, as a result of the heterogeneity of tumors. The potential solution will be profiling of patient specific aptamers through rapid selection in a short SELEX period, as well as development of in vivo selection techniques with improved targeting efficacy. The other critical issue in targeted cancer therapeutics is biomarker identification of bound aptamers. In contrast to antibody-antigen interactions, many cancer cell membrane receptors that bind to aptamers remain unclear, making it difficult to distinguish highly similar cancer and non-cancer cells. Identification of different aptamer-bound receptors and design of multiple-aptamer targeting nanosystems will realize accurate diagnosis of cancer cells and tumors and finally facilitate cancer treatment. With the further generation of more aptamers with higher binding affinity and specificity through optimized selection techniques, it is certain that more sophisticated and effective aptamer-based nanosystems will be designed for the ultimate goal of cancer diagnosis and therapy.
Acknowledgments
The authors thank Dr Kathryn R. Williams for manuscript review. This work is supported by grants awarded by the National Institutes of Health (GM079359 and CA133086).
Biographies
Sena Cansiz obtained her BSc in Biology (2008) and MSc in Biotechnology (2010) from Middle East Technical University, Turkey. She is currently a PhD candidate in the Department of Chemistry at the University of Florida. Her research interests are focused on aptamer-target molecule interactions and using aptamers for cancer biomarker discovery.
Liqin Zhang received his BS (2009) and MS (2011) in Peking University Health Science Center, China. He is currently a PhD candidate in the Department of Chemistry at the University of Florida. His research interests center on incorporating artificial expanded genetic systems into nucleic acids to develop molecular probes and devices for molecular diagnosis and targeted tumor therapy.
Cuichen Wu received his bachelor’s degree in Chemistry and MS degree in Chemical Biology at Xiamen University in 2007 and 2010, respectively. He is currently a PhD candidate under the supervision of Professor Weihong Tan in the Department of Chemistry at the University of Florida. His research interests focus on the development of self-assembled nucleic acid nanostructures for bio-imaging, disease diagnosis and cancer therapy via molecular engineering.
Yuan Wu received her BS from Wuhan Institute of Technology in 2011. She is currently a PhD candidate under the supervision of Professor Weihong Tan in the Department of Chemistry at Hunan University. Her research interests focus on the application of functional nucleic acids in bio-imaging and cancer therapy.
Weihong Tan, Distinguished University Professor and V. T. and Louis Jackson Professor of Chemistry at the University of Florida, earned his PhD (1993) from the University of Michigan. Dr Tan’s research interests include Chemical Biology and Bioanalytical Chemistry. His group developed the cell-SELEX technique to screen nucleic acid aptamers for specific recognition of diseased living cells. They further used these aptamers for early disease diagnosis, targeted drug delivery, and cancer cell enrichment and separation. In addition, the Tan group has engineered a variety of DNA nanostructures to be used as drug carriers, biosensors and nanomotors.
References
- 1.Tuerk C, Gold L. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]; Ellington AD, Szostak JW. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 2.Sassanfar M, Szostak JW. Nature. 1993;364:550–553. doi: 10.1038/364550a0. [DOI] [PubMed] [Google Scholar]
- 3.Stojanovic MN, de Prada P, Landry DW. J Am Chem Soc. 2000;122:11547–11548. doi: 10.1021/ja0022223. [DOI] [PubMed] [Google Scholar]
- 4.Gopinath SCB. Anal Bioanal Chem. 2007;387:171–182. doi: 10.1007/s00216-006-0826-2. [DOI] [PubMed] [Google Scholar]
- 5.Sun W, Du LP, Li MY. Curr Pharm Des. 2010;16:2269–2278. doi: 10.2174/138161210791792877. [DOI] [PubMed] [Google Scholar]
- 6.Mitchell P, Annemans L, White R, Gallagher M, Thomas S. Pharmacoeconomics. 2011;29:107–131. doi: 10.2165/11585520-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 7.Shangguan D, Li Y, Tang Z, Cao ZC, Chen HW, Mallikaratchy P, Sefah K, Yang CJ, Tan W. Proc Natl Acad Sci USA. 2006;103:11838–11843. doi: 10.1073/pnas.0602615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan W, Donovan MJ, Jiang J. Chem Rev. 2013;113:2842–2862. doi: 10.1021/cr300468w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liang H, Zhang XB, Lv Y, Gong L, Wang R, Zhu X, Yang R, Tan W. Acc Chem Res. 2014;47:1891–1901. doi: 10.1021/ar500078f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shangguan D, Cao Z, Meng L, Mallikaratchy P, Sefah K, Wang H, Li Y, Tan W. J Proteome Res. 2008;7:2133–2139.7. doi: 10.1021/pr700894d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dirks RM, Pierce NA. Proc Natl Acad Sci USA. 2004;101:15275–15278. doi: 10.1073/pnas.0407024101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johne R, Muller H, Rector A, van Ranst M, Stevens H. Trends Microbiol. 2009;17:205–211. doi: 10.1016/j.tim.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 13.Zhang DY, Seelig G. Nat Chem. 2011;3:103–113. doi: 10.1038/nchem.957. [DOI] [PubMed] [Google Scholar]
- 14.Tørring T, Voigt NV, Nangreave J, Yan H, Gothelf KV. Chem Soc Rev. 2011;40:5636–5646. doi: 10.1039/c1cs15057j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu Y, Duan J, Zhan Q, Wang F, Lu X, Yang XD. PloS One. 2012;7:e31970. doi: 10.1371/journal.pone.0031970. [DOI] [PMC free article] [PubMed] [Google Scholar]; Liu Z, Duan JH, Song YM, Ma J, Wang FD, Lu X, Yang XD. J Transl Med. 2012;10:148. doi: 10.1186/1479-5876-10-148. [DOI] [PMC free article] [PubMed] [Google Scholar]; Shieh YA, Yang SJ, Wei MF, Shieh MJ. ACS nano. 2010;4:1433–1442. doi: 10.1021/nn901374b. [DOI] [PubMed] [Google Scholar]; Zhu G, Zheng J, Song E, Donovan M, Zhang K, Liu C, Tan W. Proc Natl Acad Sci USA. 2013;110:7998–8003. doi: 10.1073/pnas.1220817110. [DOI] [PMC free article] [PubMed] [Google Scholar]; Yang L, Meng L, Zhang X, Chen Y, Zhu G, Liu H, Xiong X, Sefah K, Tan W. J Am Chem Soc. 2011;133:13380–13386. doi: 10.1021/ja201285y. [DOI] [PMC free article] [PubMed] [Google Scholar]; Tan L, Gee Neoh K, Kang ET, Choe WS, Su X. J Pharm Sci. 2012;101:1672–1677. doi: 10.1002/jps.23101. [DOI] [PubMed] [Google Scholar]; Zhang Z, Ali MM, Eckert MA, Kang DK, Chen YY, Sender LS, Fruman DA, Zhao W. Biomaterials. 2013;34:9728–9735. doi: 10.1016/j.biomaterials.2013.08.079. [DOI] [PubMed] [Google Scholar]
- 16.Wang R, Zhu G, Mei L, Xie Y, Ma H, Ye M, Qing FL, Tan W. J Am Chem Soc. 2014;136:2731–2734. doi: 10.1021/ja4117395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferreira CS, Cheung MC, Missailidis S, Bisland S, Gariepy J. Nucleic Acids Res. 2009;37:866–876. doi: 10.1093/nar/gkn967. [DOI] [PMC free article] [PubMed] [Google Scholar]; Huang YF, Shangguan D, Liu H, Phillips JA, Zhang X, Chen Y, Tan W. ChemBioChem. 2009;10:862–868. doi: 10.1002/cbic.200800805. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zhu GC, Sena, You Mingxu, Qiu Liping, Han Da, Zhang Liqin, Mei Lei, Fu Ting, Chen Zhuo, Tan Weihong. NPG Asia Materials. 2015:7. [Google Scholar]
- 18.Bagalkot V, Farokhzad OC, Langer R, Jon S. Angew Chem Int Ed. 2006;45:8149–8152. doi: 10.1002/anie.200602251. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2006;118:8329–8332. [Google Scholar]
- 19.Meng L, Yang L, Zhao X, Zhang L, Zhu H, Liu C, Tan W. PloS One. 2012;7:e33434. doi: 10.1371/journal.pone.0033434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang K, You M, Chen Y, Han D, Zhu Z, Huang J, Williams K, Yang CJ, Tan W. Angew Chem Int Ed. 2011;50:6098–6101. doi: 10.1002/anie.201008053. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2011;123:6222–6225. [Google Scholar]
- 21.McGinely NL, Plumb JA, Wheate NJ. J Inorg Biochem. 2013;128:124–130. doi: 10.1016/j.jinorgbio.2013.07.021. [DOI] [PubMed] [Google Scholar]
- 22.Xiao Z, Shangguan D, Cao Z, Fang X, Tan W. Chem Eur J. 2008;14:1769–1775. doi: 10.1002/chem.200701330. [DOI] [PubMed] [Google Scholar]
- 23.Jones MR, Seeman NC, Mirkin CA. Science. 2015;347:1260901. doi: 10.1126/science.1260901. [DOI] [PubMed] [Google Scholar]
- 24.Zhu G, Hu R, Zhao Z, Chen Z, Zhang X, Tan W. J Am Chem Soc. 2013;135:16438–16445. doi: 10.1021/ja406115e. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hu R, Zhang X, Zhao Z, Zhu G, Chen T, Fu T, Tan W. Angew Chem Int Ed. 2014;53:5821–5826. doi: 10.1002/anie.201400323. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2014;126:5931–5936. [Google Scholar]
- 25.Douglas SM, Bachelet I, Church GM. Science. 2012;335:831–834. doi: 10.1126/science.1214081. [DOI] [PubMed] [Google Scholar]
- 26.Han D, Zhu G, Wu C, Zhu Z, Chen T, Zhang X, Tan W. ACS Nano. 2013;7:2312–2319. doi: 10.1021/nn305484p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.You M, Peng L, Shao N, Zhang L, Qiu L, Cui C, Tan W. J Am Chem Soc. 2014;136:1256–1259. doi: 10.1021/ja4114903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen T, Wu CS, Jimenez E, Zhu Z, Dajac JG, You M, Han D, Zhang X, Tan W. Angew Chem Int Ed. 2013;52:2012–2016. doi: 10.1002/anie.201209440. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2013;125:2066–2070. [Google Scholar]
- 29.Wu Y, Sefah K, Liu H, Wang R, Tan W. Proc Natl Acad Sci USA. 2010;107:5–10. doi: 10.1073/pnas.0909611107. [DOI] [PMC free article] [PubMed] [Google Scholar]; Liu H, Zhu Z, Kang H, Wu Y, Sefan K, Tan W. Chem Eur J. 2010;16:3791–3797. doi: 10.1002/chem.200901546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang F, You M, Chen T, Zhu G, Liang H, Tan W. Chem Commun. 2014;50:3103–3105. doi: 10.1039/c3cc49003c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang H, O’Donoghue MB, Liu H, Tan W. Chem Commun. 2010;46:249–251. doi: 10.1039/b916911c. [DOI] [PubMed] [Google Scholar]
- 32.Baek SE, Lee KH, Park YS, Oh DK, Oh S, Kim KS, Kim DE. J Controlled Release. 2014;196:234–242. doi: 10.1016/j.jconrel.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 33.Banga RJ, Chernyak N, Narayan SP, Nguyen ST, Mirkin CA. J Am Chem Soc. 2014;136:9866–9869. doi: 10.1021/ja504845f. [DOI] [PMC free article] [PubMed] [Google Scholar]; Rodríguez-Pulido A, Kondrachuk AI, Prusty DK, Gao J, Loi MA, Herrmann A. Angew Chem Int Ed. 2013;52:1008–1012. doi: 10.1002/anie.201206783. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2013;125:1042–1046. [Google Scholar]
- 34.Battig MR, Huang Y, Chen N, Wang Y. Biomaterials. 2014;35:8040–8048. doi: 10.1016/j.biomaterials.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 35.Battig MR, Soontornworajit B, Wang Y. J Am Chem Soc. 2012;134:12410–12413. doi: 10.1021/ja305238a. [DOI] [PubMed] [Google Scholar]; Um SH, Lee JB, Park N, Kwon SY, Umbach CC, Luo D. Nat Mater. 2006;5:797–801. doi: 10.1038/nmat1741. [DOI] [PubMed] [Google Scholar]
- 36.Wu C, Han D, Chen T, Peng L, Zhu G, You M, Qiu L, Sefah K, Zhang X, Tan W. J Am Chem Soc. 2013;135:18644–18650. doi: 10.1021/ja4094617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li J, Zheng C, Cansiz S, Wu C, Xu J, Cui C, Liu Y, Hou W, Wang Y, Zhang L, Teng IT, Yang HH, Tan W. J Am Chem Soc. 2015;137:1412–1415. doi: 10.1021/ja512293f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daniel MC, Astruc D. Chem Rev. 2004;104:293–346. doi: 10.1021/cr030698+. [DOI] [PubMed] [Google Scholar]
- 39.Nikoobakht B, Wang X, Herzing A, Shi J. Chem Soc Rev. 2013;42:342–365. doi: 10.1039/c2cs35164a. [DOI] [PubMed] [Google Scholar]; Schwartzberg AM, Olson TY, Talley CE, Zhang JZ. J Phys Chem B. 2006;110:19935–19944. doi: 10.1021/jp062136a. [DOI] [PubMed] [Google Scholar]; Skrabalak SE, Chen J, Sun Y, Lu X, Au L, Cobley CM, Xia Y. Acc Chem Res. 2008;41:1587–1595. doi: 10.1021/ar800018v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu M, Chen J, Li ZY, Au L, Hartland GV, Li X, Marquez M, Xia Y. Chem Soc Rev. 2006;35:1084–1094. doi: 10.1039/b517615h. [DOI] [PubMed] [Google Scholar]
- 41.Huang YF, Sefah K, Bamrungsap S, Chang HT, Tan W. Langmuir. 2008;24:11860–11865. doi: 10.1021/la801969c. [DOI] [PubMed] [Google Scholar]
- 42.Wang J, Sefah K, Altman MB, Chen T, You M, Zhao Z, Huang CZ, Tan W. Chem Asian J. 2013;8:2417–2422. doi: 10.1002/asia.201300375. [DOI] [PubMed] [Google Scholar]
- 43.Kang H, Trondoli AC, Zhu G, Chen Y, Chang YJ, Liu H, Huang YF, Zhang X, Tan W. ACS Nano. 2011;5:5094–5099. doi: 10.1021/nn201171r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qiu L, Chen T, Ocsoy I, Yasun E, Wu C, Zhu G, You M, Han D, Jiang J, Yu R, Tan W. Nano Lett. 2015;15:457–463. doi: 10.1021/nl503777s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang J, Zhu G, You M, Song E, Shukoor MI, Zhang K, Altman MB, Chen Y, Zhu Z, Huang CZ, Tan W. ACS Nano. 2012;6:5070–5077. doi: 10.1021/nn300694v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zheng J, Zhu G, Li Y, Li C, You M, Chen T, Song E, Yang R, Tan W. ACS Nano. 2013;7:6545–6554. doi: 10.1021/nn402344v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng K, Peng S, Xu C, Sun S. J Am Chem Soc. 2009;131:10637–10644. doi: 10.1021/ja903300f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen T, Shukoor MI, Wang R, Zhao Z, Yuan Q, Bamrungsap S, Xiong X, Tan W. ACS Nano. 2011;5:7866–7873. doi: 10.1021/nn202073m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Esposito CL, Catuogno S, de Franciscis V, Cerchia L. Discovery Med. 2011;11:487–496. [PubMed] [Google Scholar]
- 50.Allemann E, Brasseur N, Benrezzak O, Rousseau J, Kudrevich SV, Boyle RW, Leroux JC, Gurny R, Van Lier JE. J Pharm Pharmacol. 1995;47:382. doi: 10.1111/j.2042-7158.1995.tb05815.x. [DOI] [PubMed] [Google Scholar]
- 51.Swarnalatha S, Lucky K, Zhang Y. Chem Rev. 2015;115:4. doi: 10.1021/cr5004198. [DOI] [PubMed] [Google Scholar]