

Abstract
The potent transcriptional activity of p53 (TP53) must be kept in check for normal cell growth and survival. Tumors, which drastically deviate from these parameters, have evolved multiple mechanisms to inactivate TP53, the most prevalent of which is the emergence of TP53 missense mutations, some of which have gain-of function activities. Another important mechanism by which tumors bypass TP53 functions is via increased levels of two TP53 inhibitors, MDM2 and MDM4. Studies in humans and in mice reveal the complexity of TP53 regulation and the exquisite sensitivity of this pathway to small changes in regulation. Here we summarize the factors that impinge on TP53 activity and thus, cell death/arrest or tumor development.
Keywords: Mdm2, Mdm4, mouse models, p53, TCGA
Introduction
Cellular distress signals activate p53 (MIM#191170), and cells respond to this activation by ceasing proliferation, senescing, differentiating, or dying. Cellular proteins such as Mdm2 (MIM#164785) and Mdm4 (MIM#602704) can dampen p53 activity and allow the cell to live and propagate. The relationship between p53 and its inhibitors is dynamic and is usurped by tumor cells. This review focuses on the mechanisms that regulate p53 activity in mouse and human cancers, with particular attention to Mdm2 and Mdm4.
Inactivation of the p53 tumor suppressor pathway by multiple mechanisms
p53 is a key transcriptional regulator that controls multiple signals by activating genes with roles in cell cycle arrest, senescence, apoptosis, metabolism, differentiation, and survival (Vousden and Prives, 2009). Cell cycle arrest, senescence, and apoptosis are tumor suppressive mechanisms, whereas changes in metabolism often fuel tumor cell growth and survival. In addition, the ability of p53 to inhibit its own activity through Mdm2 regulates survival and allows cells no longer stressed to survive. Clearly, these activities underscore the importance of p53 in regulating cell fate decisions. Therefore, mutations that deplete TP53 activities and deletions of TP53 (GenBank accession number NM000546) are common events in the development of most human cancers. In particular, missense mutations account for approximately 75% of all TP53 mutations in cancer (Hamroun, et al., 2006; Olivier, et al., 2010)
The most common mechanism of altering the p53 pathway is through mutation of the TP53 gene itself. A single base change can alter an amino acid crucial for TP53’s most important biological function as a transcriptional regulator. Modification of any of five arginine residues that are hotspots for TP53 mutations in human cancers disables TP53 sequence-specific DNA binding (Cho, et al., 1994). Moreover, signature mutations in the DNA binding domain (some of which occur at these same hotspots) can arise through exposure to common insults such as ultraviolet light, smoking, and exposure to aflatoxin or other chemical carcinogens (Vogelstein and Kinzler, 1992).
TP53 missense mutations clearly predominate over frameshift and nonsense mutations in cancer cells (Olivier, et al., 2002). Thus, besides eliminating wild type TP53 transcriptional activity, another reason missense mutations are common is that these mutations lead not only to the loss of TP53 tumor suppressive functions but also to the acquisition of other activities known as gain of function (GOF) (Sigal and Rotter, 2000). Early experiments indicated that immortalized cells lacking TP53 gained additional transforming properties upon expression of mutant TP53 proteins (Dittmer, et al., 1993). Cells expressing the most common TP53 mutants, in contrast to cells lacking TP53, show increased metastatic potential and invasiveness (Crook and Vousden, 1992; Hsiao, et al., 1994). In Li-Fraumeni syndrome (LFS), individuals with TP53 missense mutations show a higher cancer incidence and an earlier age of tumor onset than individuals with truncating or splicing mutations (Birch, et al., 1998). These data are supported by studies in mice that inherit a single amino acid change in the DNA binding domain of p53 (GenBank accession number NM011640) that mimics a mutation hotspot found in human cancers (Lozano, 2007). Mutant p53-heterozygous mice develop many metastatic tumors that are rare in mice lacking p53 (Lang, et al., 2004; Olive, et al., 2004). Additionally, differences in the tumor spectrum were apparent when comparing two different hotspot mutations in the 129Sv strain of mice, (Olive, et al., 2004). Thus, cells expressing mutant p53 appear to have a survival and/or growth advantage over cells lacking p53. These in vitro and in vivo data support the gain-of-function phenotype of mutant p53 in tumorigenesis.
Although deletion or mutation of TP53 itself accounts for approximately 50% of the alterations in TP53, other modifications also derail the p53 pathway (Figure 1) (Vogelstein, et al., 2000). The best example is the overexpression of the TP53 inhibitors MDM2 and/or MDM4. MDM2 is highly expressed in numerous types of human tumors, and the majority of these retain wild-type TP53 (Rayburn, et al., 2005; Wade, et al., 2013). Mdm2 is an E3 ubiquitin ligase that ubiquitylates and targets p53 for degradation (Haupt, et al., 1997; Honda, et al., 1997; Kubbutat, et al., 1997). Under some circumstances, Mdm2 can also ubiquitylate itself and Mdm4, which leads to the opposite phenotype: p53 stability and activity (Linke, et al., 2008). The critical function of Mdm2 as a negative regulator of p53 was discovered in mouse models. Loss of Mdm2 leads to cell death by apoptosis and embryonic lethality at the blastocyst stage, and this phenotype is completely rescued by concomitant deletion of p53 (Chavez-Reyes, et al., 2003; Jones, et al., 1995; Montes de Oca Luna, et al., 1995).
Figure 1. A model showing some of the effectors of the p53 pathway.

Dotted lines denote tumor suppressors and solid lines denote activated or over produced proteins that contribute to tumor development. The shaded box denotes p53-independent functions of Mdm2. Mdm2 binds and inhibits Nbs1, a member of the MRN complex responsible for DNA break repair.
The Mdm2-interacting protein Mdm4 (also known as MdmX) also negatively regulates the tumor suppressor function of p53 by binding and masking the amino terminal transcriptional activation domain of p53 (Shvarts, et al., 1996). Mice lacking Mdm4 exhibit embryonic lethal phenotypes that are also completely rescued by p53 deletion (Finch, et al., 2002; Migliorini, et al., 2002; Parant, et al., 2001). As with MDM2, MDM4 is overexpressed in many human cancers (Danovi, et al., 2004; Gembarska, et al., 2012; Han, et al., 2007; Leventaki, et al., 2012; Ramos, et al., 2001; Riemenschneider, et al., 1999; Valentin-Vega, et al., 2007; Wade, et al., 2013). Recent data indicate the MDM4 gene is amplified in 65% of human retinoblastoma, a tumor of the eye that often results from inherited mutations in the RB1 gene (MIM#614041) (Laurie, et al., 2006). In the same study, the recapitulation of RB1 loss and MDM4 overexpression in primary human retina explants yielded a retinoblastoma phenotype. Retinoblastoma was once thought to be an example of a cancer that develops without the need to inactivate TP53, because TP53 mutations are very rare in this cancer type (Kato, et al., 1996); however, the discovery of MDM4 amplification in the majority of retinoblastomas proved this assumption incorrect. Melanoma is another tumor in which TP53 is rarely mutated, but approximately 65% of these tumors also have high levels of MDM4 (Gembarska, et al., 2012). These data confirm an important role of MDM4 in tumorigenesis. Thus, tumors that rarely acquire TP53 mutations have developed other mechanisms to inactivate the p53 pathway.
Both Mdm2 and Mdm4 contain RING domains. However, only the RING domain of Mdm2 encodes a functional E3-ubiquitin ligase that promotes p53 degradation (Linke, et al., 2008). Mdm2 and Mdm4 also interact with each other through their respective RING domains, and this interaction is essential for inhibition of p53 (Figure 1). Genetic deletion or mutation of the Mdm4 RING domain, which disrupts interaction with Mdm2, leads to p53-dependent embryonic lethal phenotypes, which indicates the importance of the Mdm2/Mdm4 heterodimer in regulating p53 activity (Huang, et al., 2011; Pant, et al., 2011). These data indicate that Mdm4 is a key cofactor of Mdm2 and that the Mdm2-Mdm4 complex is the most efficient inhibitor of p53 activity.
Other regulators of the p53 pathway
Numerous other proteins bind Mdm2 and/or p53 and affect p53 levels and activities (Riley and Lozano, 2012). Here, we highlight the interactions that are altered in tumors that lead to inhibition of p53 activity (directly or indirectly through Mdm2) and thus, contribute to tumor development. The discovery of the alternative reading frame of the Ink4a locus (ARF; CDKN2A; MIM#600160) and its association with Mdm2 significantly increased our understanding of the p53 pathway and its regulation (Kamijo, et al., 1998; Kamijo, et al., 1997; Quelle, et al., 1995; Zhang, et al., 1998). ARF is normally expressed at very low levels in cells until there is cellular distress, such as deregulation of an oncogene. Oncogenic activity transcriptionally up regulates and post-translationally stabilizes ARF, which directly binds Mdm2 and holds Mdm2 away from p53 either in the nucleolus or the nucleus (Figure 1) (Chen, et al., 2010; Eischen, et al., 1999; Llanos, et al., 2001; Weber, et al., 1999; Zhang, et al., 1998; Zindy, et al., 1998). This allows p53 activation and induction of apoptosis, cell cycle arrest, or senescence. There are conflicting reports as to whether ARF directly binds and regulates Mdm4 (Jackson, et al., 2001; Wang, et al., 2001). However, if ARF does not directly bind Mdm4, it may still regulate Mdm4 indirectly through its interaction with Mdm2 if Mdm4 is bound to Mdm2 at the time (Li, et al., 2012). Additional experiments are needed to test the influence of ARF on Mdm4, and Mdm4:Mdm2 complexes.
Studies in mice show that Arf does not regulate Mdm2 (and consequently p53) in proliferating tissues during development and under homeostatic conditions except germ cells (Li, et al., 2013; O’Leary, et al., 2004). However, under stressful conditions, such as when oncogenes are activated, loss of Arf results in deregulated Mdm2 and the inability of a cell to effectively activate p53 (Eischen, et al., 1999; Schmitt, et al., 1999; Zindy, et al., 1998). ARF is inactivated in human cancers primarily through two mechanisms. First, tumors frequently delete the entire INK4A locus (CDKN2A), which includes p14ARF and p16; thus, concurrently inactivating the RB pathway, since p16 inhibits Cyclin D:CDK4/6-mediated phosphorylation of RB1 and activation of the E2Fs. The ARF promoter can also be methylated in malignancies, which prevents ARF expression, but ARF is rarely mutated in human cancers (Esteller, et al., 2000; Kim and Sharpless, 2006). Arf proved to be a classic tumor suppressor in knockout mouse studies. Deletion of one or two alleles of Arf in mice leads to tumorigenesis (primarily lymphomas and sarcomas). Tumors that develop in Arf heterozygous mice show loss of heterozygosity of the second allele of Arf (Kamijo, et al., 1999). Therefore, ARF has an essential role in preventing tumor development through regulation of Mdm2, but this function appears unnecessary in most tissues during development.
Many tumors in mice and humans inactivate either p53 or ARF, leading to the conclusion that these two proteins have an epistatic relationship (Sherr, 2006). Moreover, genetic studies have demonstrated functionally incompetent p53 in oncogene-activated tumor cells that have lost Arf or overexpress Mdm2. Specifically, deletion of Arf or overexpression of Mdm2 accelerated Myc (MIM#190080) oncogene-induced B cell lymphoma development at a rate similar to that caused by Myc overexpression and p53 deficiency. Moreover, loss of Arf or overexpression of Mdm2 significantly reduced the selection for p53 mutations that normally arise during Myc-induced B cell lymphomagenesis (Eischen, et al., 1999; Wang, et al., 2008).
In contrast, multiple aggressive tumors have been found to have alterations in both ARF and TP53 (Rozenblum, et al., 1997; Sanchez-Cespedes, et al., 1999; Smeds, et al., 2002). For example, approximately 40% of pancreatic ductal adenocarcinomas have mutated TP53 and deleted ARF (Heinmoller, et al., 2000; Hustinx, et al., 2005; Maitra, et al., 2003). However, it is unclear whether these two alterations coexist in a single cell or that a tumor has multiple clones with separate mutations. Alternatively, gain-of-function TP53 mutants may cooperate with alterations in ARF in the development of these tumors. Furthermore, deletion of Arf does not accelerate tumorigenesis in p53-null mice, except in a stable mixed genetic background, suggesting again that alterations of Arf and p53 may not be mutually exclusive under some conditions (Eischen and Boyd, 2012; Weber, et al., 2000). For example, we evaluated ARF and TP53 status in the data from The Cancer Genome Atlas (TCGA) and determined that malignancies such as squamous cell carcinoma of the lung or head and neck co-inactivate ARF and TP53 in approximately 20% of these tumors (Table 1; (Cerami, et al., 2012; Gao, et al., 2013). In addition, some tumors in mice and humans contain mutant p53 and overexpress Mdm2, indicating that Mdm2 may have functions independent of p53 (Alt, et al., 2003; Cordon-Cardo, et al., 1994; Eischen, et al., 1999; Lu, et al., 2002; Peng, et al., 2001; Sigalas, et al., 1996). Moreover, Mdm2 transgenic mice that lacked Arf had an accelerated tumor development compared to mice that lacked just Arf (Moore, et al., 2003). Arf has known functions independent of Mdm2 and p53 (Abida and Gu, 2008; Chen, et al., 2009; Sherr, 2006; Weber, et al., 2000), so it is possible for Arf and/or p53 to be inactivated and Mdm2 to be overexpressed in the same cancer. Identification and characterization of the functions of Arf that are independent of Mdm2 and p53 are needed and will further increase our understanding of the contributions of these proteins to human cancer (p53-independent functions of Mdm2 are discussed below).
Table 1.
Frequency of co-inactivation of TP53 and CDKN2A in human cancera
| Cancer | Samples | TP53 mut/delb | CDKN2A delc | Co-inactivationd |
|---|---|---|---|---|
| Acute Myeloid Leukemia | 187 | 15 (8.0%) | 2 (1.1%) | 0 |
| Adenoid Cystic Carcinoma | 60 | 2 (3.3%) | 2 (3.3%) | 1 (1.7%) |
| Bladder Carcinoma | 97 | 24 (24.7%) | 14 (14.4%) | 7 (7.2%) |
| Breast, Invasive Carcinoma | 482 | 169 (35.1%) | 8 (1.7%) | 11 (2.3%) |
| Colon and Rectum Adenocarcinoma | 212 | 108 (50.9%) | 1 (0.47%) | 1 (0.47%) |
| Glioblastoma Multiforme | 236 | 45 (19.1%) | 126 (53.4%) | 27 (11.4%) |
| Glioma, lower grade | 215 | 106 (49.3%) | 18 (8.4%) | 6 (2.8%) |
| Head and Neck Squamous Cell Carcinoma | 302 | 92 (30.5%) | 23 (7.6%) | 60 (19.9%) |
| Lung Adenocarcinoma | 182 | 76 (41.8%) | 6 (3.3%) | 12 (6.6%) |
| Lung, Squamous Cell Carcinoma | 178 | 91 (51.1%) | 5 (2.8%) | 42 (23.6%) |
| Melanoma (Cutaneous) | 225 | 22 (9.8%) | 64 (28.4%) | 3 (1.3%) |
| Ovarian Serous Cystadenocarcinoma | 316 | 292 (92.4%) | 1 (0.32%) | 6 (1.9%) |
| Prostate Adenocarcinoma | 103 | 5 (4.9%) | 3 (2.9%) | 0 |
| Renal (Kidney) Clear Cell Carcinoma | 418 | 7 (1.7%) | 12 (2.9%) | 1 (0.24%) |
| Soft Tissue Sarcoma | 201 | 25 (12.4%) | 9 (4.5%) | 3 (1.5%) |
| Stomach Adenocarcinoma | 115 | 43 (37.4%) | 8 (7.0%) | 6 (5.2%) |
| Thyroid Carcinoma | 318 | 3 (0.94%) | 0 | 0 |
| Uterine Corpus Endometrioid Carcinoma | 240 | 68 (28.3%) | 0 | 0 |
Data were obtained July 2013 through the cBioPortal for Cancer Genomics (Cerami, et al., 2012; Gao, et al., 2013)
Samples that only deleted or mutated TP53 with wild-type CDKN2A
Samples that only deleted CDKN2A with wild-type TP53 (mutated CDKN2A samples not included)
Samples that had both a TP53 mutation or deletion together with CDKN2A deletion
Because Mdm2 ubiquitination affects p53 levels and activity, it is not surprising to discover deubiquitinating enzymes that also regulate the p53 pathway. The herpesvirus-associated ubiquitin-specific protease Hausp (also known as USP7; MIM#602519) is a deubiquitinating enzyme first identified as a p53-interacting protein (Li, et al., 2002). However, subsequent data indicate that Hausp is a more important regulator of Mdm2 function and indirectly impinges on p53 activity. Hausp deubiquitinates Mdm2, which allows Mdm2 to functionally keep p53 levels low. Hausp loss by homologous recombination in human cell lines expressing wild-type p53 is incompatible with proliferation (Cummins, et al., 2004). The development of a conditional loss-of-function allele of Hausp in mice also leads to cell death in a p53-dependent manner in the developing neural system of the mouse (Kon, et al., 2011). However, p53 loss does not completely rescue the Hausp-null phenotype, which suggests that Hausp has additional functions. Thus, the physiological consequence of Hausp loss is destabilization of Mdm2, which leads to increased p53 levels and activity (Cummins, et al., 2004; Li, et al., 2004). More recent data suggest that Hausp also deubiquitinates Mdm4, another inhibitor of p53 activity (Meulmeester, et al., 2005). Thus, in normal cells, Hausp levels appear to finely tune p53 stability and activity, whereas in tumor cells, increased Hausp activity may be sufficient to keep Mdm2 and Mdm4 stable and consequently, p53 levels low. Our evaluation of TCGA data revealed that USP7 is rarely amplified in human cancer (Cerami, et al., 2012; Gao, et al., 2013). However, 5.3% (40 of 760) of breast adenocarcinomas contained amplified USP7 and of those, 75% maintained wild-type TP53 allele status, which implies that USP7 may have other functions related to oncogenesis. Studies examining the levels and locations of TP53, MDM2, and Hausp/USP7 in a single tumor type are needed. In addition, the generation of Hausp transgenic mice would further our understanding of these relationships in tumorigenesis.
Mdm2/4 gene dosage and haploinsufficiency
Small changes in the levels of Mdm2 and Mdm4 also significantly contribute to developmental and tumor phenotypes. Although Mdm2 or Mdm4 heterozygosity is sufficient for homeostasis, p53 activity cannot be effectively regulated by these proteins under conditions of stress. Consequently, gamma radiation or overexpression of the Myc oncogene leads to cell death when cells are haploinsufficient for Mdm2 (Alt, et al., 2003; O’Leary, et al., 2004; Terzian, et al., 2007). Moreover, mice carrying a hypomorphic and a null allele of Mdm2, which express about 30% of wild-type Mdm2 levels, have increased p53 activity and are small, lymphopenic, and radiosensitive (Mendrysa, et al., 2003). Recent results also indicate that mice lacking the p53-Mdm2 autoregulatory feedback loop are normal but succumb to hematopoietic failure upon exposure to ionizing radiation due to increased p53 activity (Pant, et al., 2013). Similar results have been observed for Mdm4 heterozygous mice with Mdm4 haploinsufficiency resulting in increased sensitivity to gamma radiation (Terzian, et al., 2007). Notably, heterozygosity of both Mdm2 and Mdm4 results in lethality, with 30% of mice dying in utero and the rest of hematopoietic failure within 21 days after birth. The observation that these phenotypes can be rescued with deletion of a single p53 allele indicates how precisely Mdm2 and Mdm4 regulate p53 levels (Terzian, et al., 2007). With regards to tumorigenesis, heterozygosity at the Mdm2 or Mdm4 locus delays tumor onset in mice carrying an Eμ-myc transgene (Alt, et al., 2003; Terzian, et al., 2007), and decreased Mdm2 levels delay the appearance of intestinal tumors in Apcmin/+ mice (Mendrysa, et al., 2006). Mdm4 heterozygous mice also show a lower tumor burden in an Rb+/− background (Fang, et al., 2013). Thus, a small difference (2-fold in these experiments) in Mdm2 or Mdm4 levels has significant in vivo effects on tumor phenotypes. These studies emphasize the important relationship between the Mdm proteins and p53 activity in development and tumorigenesis.
The p53 pathway is exquisitely sensitive not only to levels of Mdm2 and Mdm4, but also to the regulator of these regulators. For example, loss of just one allele of Arf can fully rescue the effects of Mdm2 haploinsufficiency and reduce the rate of Myc-induced lymphoma development to wild-type levels (Eischen, et al., 2004). However, deletion of Arf did not rescue the radiosensitivity of Mdm2 hypomorphic mice (O’Leary, et al., 2004), suggesting the p53-inducing stimuli dictate the requirements for or the effects of Arf. In the absence of oncogene activation, when Arf is deleted, Mdm2 can still regulate p53 effectively. Specifically, loss of one allele of Mdm2 significantly inhibits tumor development in mice lacking both alleles of Arf, which suggests that Mdm2 expression facilitates tumor development caused by the absence of Arf (Eischen and Boyd, 2012; Wang, et al., 2006). Mdm2 heterozygosity can also significantly delay tumor development in Arf −/− p53+/− and Arf +/− p53+/− mice, but not in mice lacking both alleles of p53 regardless of their Arf status (Eischen and Boyd, 2012). These results indicate that the effects of Mdm2 haploinsufficiency are dependent on p53. Therefore, depending on the stimulus that results in p53 activation, Arf levels may or may not be involved in the regulation of p53 (Figure 1).
More relevant to human malignancies is the finding that many tumors produce elevated levels of MDM2 and/or MDM4 (Onel and Cordon-Cardo, 2004; Wade, et al., 2013). Mdm2 transgenic mice, in which the Mdm2 transgene was driven from its normal promoter to create an approximately 4-fold elevation in Mdm2 expression, have an increased incidence of cancer, but not until later in life (Jones, et al., 1998). Another study found that elevated levels of Mdm2 induce an increase in genome instability early in life in these mice, but again, cancer did not develop until well into adulthood (Lushnikova, et al., 2011). Increased levels of Mdm2 cooperated with the overexpression of Myc to accelerate lymphoma development in mice (Wang, et al., 2008). Similarly, overexpression of Mdm4 together with mutant oncogenic Ras transforms cells in culture (Danovi, et al., 2004), suggesting that Mdm4 is oncogenic. To determine whether Mdm4 is oncogenic in vivo, transgenic mice were generated by two different labs. Increased expression of Mdm4 in the mice in which Mdm4 was driven from the chicken β-actin promoter led to an increase in tumor development (Xiong, et al., 2010). In contrast, Myc-tagged Mdm4 expression from an artificial promoter in the Rosa26 locus did not result in tumor development (De Clercq, et al., 2010). The differences in cancer incidence between the two experiments may be due to differences in Mdm4 levels. Alternatively, the Myc-tagged Mdm4 transgene may alter an Mdm4 activity essential for tumorigenesis. Thus, Mdm2 or Mdm4 overexpression in mice promotes tumor development, and reduces overall survival. Because Mdm2 and Mdm4 work together to inhibit p53, it will be necessary to accurately measure the expression of both in human malignancies and to determine how their ratios contribute to inactivation of p53 and ultimately cancer.
In humans, MDM2 and MDM4 contribute to tumor development and impact patient survival (Lenos, et al., 2012; Onel and Cordon-Cardo, 2004; Rayburn, et al., 2005). Humans with cancers that contain MDM4 or MDM2 amplification have lower survival rates. For example, analysis of TCGA data (Cerami, et al., 2012; Gao, et al., 2013) showed that 6.6% (24 of 363) of uterine carcinomas had amplified MDM4, and patients with tumors with MDM4 amplification had lower survival rates (Figure 2A). In comparison, Mdm2 amplification rarely occurs in uterine carcinomas (0.28%; 1 in 363). TCGA data (Cerami, et al., 2012; Gao, et al., 2013) also showed a trend toward lower survival rates in patients with glioblastoma that have amplified MDM4 (9 of 206), but because of the low numbers of cases with MDM4 amplification (4.4%), statistical significance was not reached. However, reduced survival rates were observed for glioblastoma patients with MDM2 amplification, which was detected in 10.7% (22 of 206) of cases (Figure 2B). In glioblastoma, only 1 of 206 tumors amplified both MDM2 and MDM4 suggesting that such co-amplification is rare in this cancer. Thus, there may be a preference for overexpression of one or the other MDM family member in specific tumor types or the selection of only one (regardless of which one), and this can lead to shorter survival. Clearly, much additional research is needed to identify why a particular cancer, such as retinoblastoma (Laurie, et al., 2006), would have a bias toward MDM4 overexpression over MDM2 overexpression when other cancers have an equal distribution of expression of these genes.
Figure 2. Amplification of MDM4 or MDM2 genes in specific cancers can result in decreased patient survival.

Gene copy number alterations and survival data were obtained from the cBioPortal for Cancer Genomics (http://www.cbiopor2tal.org/public-portal/) July 2013 (Cerami, et al., 2012; Gao, et al., 2013). Kaplan-Meier survival curves of patients with (A) uterine carcinoma with and without MDM4 amplification and (B) glioblastoma with and without MDM2 amplification. The number (n) of samples of each is indicated, and p values were calculated by log-rank tests comparing survival between groups.
In some human cancers, MDM2 and MDM4 overexpression occurs through gene amplification, but high MDM2 levels can also be achieved through increased transcription and translation (Capoulade, et al., 1998; Landers, et al., 1994). Whether MDM4 expression is increased due to mechanisms other than gene amplification is unknown, but likely. Additional research is needed to determine when and how MDM2 and MDM4 mRNA and protein are overexpressed during tumorigenesis. Moreover, a thorough analysis of the levels of both proteins in human cancers is needed. These studies have been hampered by technical issues with MDM2 antibodies and the numerous post-translational modifications that can occur on MDM2, which inhibit binding of some antibodies (Cheng and Chen, 2011; Eischen, 2011; Maya and Oren, 2000; Meek and Hupp, 2010; Zhang and Prives, 2001). Consequently, the frequency of elevated MDM2 protein levels in human cancers is likely significantly underestimated. Additionally, what constitutes MDM2 overexpression also needs to be re-evaluated. It is now clear that small (2- to 4-fold) increases in MDM2 protein levels, such as those that occur with MDM2SNP309 (see below), can significantly increase tumor susceptibility (Bond, et al., 2004; Post, et al., 2010).
The gene dosage of Mdm2 also dictates the type and the number of primary tumors that develop. Over a decade of Mdm2 research suggests that both increased and decreased levels of Mdm2 can alter the tumor type that emerges in various mouse models of cancer. Specifically, overexpression of Mdm2 causes more sarcomas and fewer lymphomas to develop in p53-null mice (Jones, et al., 1998). Mdm2 heterozygosity in the absence of p53 also results in a higher frequency of sarcomas than that seen in p53-null mice (Eischen and Boyd, 2012; McDonnell, et al., 1999). Moreover, a third of Mdm2+/− Arf−/− mice develop two primary malignancies, whereas Arf−/− mice develop a single cancer (Wang, et al., 2006). Alterations in Mdm2 concomitant with deficiencies in both Arf and p53 also reveal additional effects of Mdm2 on tumorigenesis. Specifically, two primary malignancies develop in half of the Mdm2−/− Arf−/− p53−/− mice compared to a third of the Arf−/− p53−/− mice, and three primary cancers are only observed in Mdm2−/− Arf−/− p53−/− mice (Weber, et al., 2000). Mdm2+/− Arf−/− p53−/− and Mdm2+/− Arf+/− p53−/− mice have a significantly higher frequency of lymphoma and lower frequency of sarcoma compared to their Mdm2 wild-type counterparts (Eischen and Boyd, 2012). In addition, Mdm2+/− Arf+/− p53+/− mice had a lower frequency of sarcoma and a higher frequency of carcinoma than Mdm2+/+ Arf+/− p53+/− mice (Eischen and Boyd, 2012). Thus, Mdm2 levels can dramatically influence the rate and type of tumors that develop. Future studies will be needed to determine the contribution of Mdm4 to these phenotypes. These data suggest that the role of the p53 pathway in tumor-initiating cells may differ depending on cell type, and that these differences may dictate the tumor type and the rate at which the tumor develops.
Mdm2 regulation of mutant p53
The generation of mice with p53 missense mutations has also yielded some surprising findings regarding the stability of wild-type and mutant p53. Not all tumors that develop in p53-mutant mice had stable mutant p53 (Lang, et al., 2004; Olive, et al., 2004). A detailed characterization of mice with one of these mutations, p53R172H (the amino acid numbering system is based on the initiation codon as codon 1), indicates that homozygous mutant mice had undetectable levels of mutant p53 in normal cells, suggesting that tumor-specific alterations result in stable mutant p53 in some, but not all, cells (Terzian, et al., 2008). Because tissue culture experiments suggested that Mdm2 could modulate the levels of mutant p53 as well as wild-type p53 (Midgley and Lane, 1997), the role of Mdm2 in mutant p53 stability in vivo was examined. Loss of Mdm2 results in mutant p53 stability in many (but not all) cells, an increase in tumor incidence, and the development of a gain-of-function metastatic phenotype (Terzian, et al., 2008). These results suggest that endogenous levels of Mdm2 are sufficient to keep mutant p53 levels low. The data also suggest that alterations in upstream pathways that impinge on p53 activity also contribute to the tumor phenotype. Because p16INK4a loss is a common event in human tumorigenesis via the Rb pathway, the role of p16 in regulating mutant p53 stability was also examined. Loss of p16 resulted in mutant p53 stability that was associated with increased tumor incidence and a metastatic gain-of-function phenotype (Terzian, et al., 2008). Thus, this study indicates that tumors with p53 missense mutations that are not stable resemble tumors with loss of p53, and the stabilization of mutant p53 is a prerequisite for its gain-of-function phenotype. Additional studies clearly show that other signals that stabilize wild-type p53, such as ionizing radiation, reactive oxygen species, and K-Ras activation, also stabilize mutant p53 in vivo (Suh, et al., 2011).
Modifiers of the p53 pathway
A recently discovered polymorphism in the MDM2 gene (SNP309) affects the expression levels of MDM2 (Bond, et al., 2004). The presence of a G nucleotide at SNP309 in the MDM2 promoter creates a better binding site for the SP1 transcription factor, and thus, results in elevated levels of MDM2. In Li-Fraumeni syndrome patients with germline TP53 mutations in one allele, the presence of MDM2 SNP309G contributes to an earlier onset of the tumor phenotype (Bond, et al., 2004; Bougeard, et al., 2006; Ruijs, et al., 2007). The MDM2 SNP309G allele also associates with increased risk of breast and ovarian cancer in patients with inherited BRCA1 and BRCA2 mutations (Yarden, et al., 2008). In contrast, several studies, some of them with thousands of patient samples, show no association between SNP309G and increased tumorigenesis (Schmidt, et al., 2007; Wilkening, et al., 2007). In another study, SNP309G was associated with increased risk of lung cancer in never or light smokers (Liu, et al., 2008). Gender and estrogen status also cooperate with SNP309 and may account for some of the discrepancies between data sets (Bond and Levine, 2007). An adjacent SNP (SNP285) was recently found to offset the impact of SNP309 (Knappskog, et al., 2011). SNP285C strongly reduces binding by the SP1 transcription factor to the MDM2 promoter, which results in lower levels of MDM2, and therefore, higher levels of TP53. SNP285C is associated with a reduction in the risk of breast and ovarian cancers (Knappskog, et al., 2011). Therefore, SNPs in MDM2 can alter cancer risk in opposing ways. Since MDM4 also inhibits TP53 function, it will be important in the future to also evaluate SNPs in MDM4 for their contribution to cancer.
The transgenic Mdm models discussed above accurately mimic increases in Mdm2/4 levels that affect tumorigenesis, but only partially reproduce the actual genomic changes that occur in human cancers. The identification of a high-frequency single nucleotide polymorphism (SNP309) in the MDM2 promoter that increases MDM2 levels and is associated with an increased risk of spontaneous and inherited human cancers presented an opportunity to study the role of SNP309 in cancer (Bond, et al., 2004). Mice were, therefore, generated by introducing the human promoter sequences into the homologous murine locus to examine the direct effects of SNP309 on tumorigenesis (Post, et al., 2010). Mdm2SNP309G/G mice exhibit an increased risk of spontaneous tumors, and the Mdm2SNP309G allele potentiates the tumor phenotype and alters the tumor spectrum in mice inheriting a p53R172H hotspot mutation. These data provide causal evidence for increased cancer risk in humans with the MDM2 SNP309G allele. Since these mice did not contain the SNP285C sequence, future studies should investigate whether SNP285C can mitigate the effects of SNP309G in mice as well as humans.
The complexity and number of proteins that regulate p53 activity suggest that changes in any of these proteins may affect cancer risk. Although the inheritance of a TP53 mutation in Li-Fraumeni syndrome (LFS) patients provides a dramatic increase in cancer risk, smaller changes in TP53 levels via regulators of TP53 likely contribute to an individual’s risk of developing cancer, and may explain the wide variation in tumor onset even within a TP53 mutant lineage (Balmain, et al., 2003). In fact, different combinations of null and hypomorphic p53 alleles in mice show that as little as a 7% difference in p53 protein levels alters survival (Figure 3) (Wang, et al., 2011). Additionally, changes in p53 pathway activation could combine with other cancer-predisposing alterations (e.g., mutations in BRCA1 or BRCA2) or environmental carcinogens (e.g., smoking) to increase cancer risk. In fact, smoking contributes to an increased risk of cancer in LFS individuals (Hwang, et al., 2003). Thus, combinations of polymorphisms in several genes that lead to suppression of the p53 pathway are likely to modify cancer risk.
Figure 3. Small differences in p53 levels alter tumor development and survival.
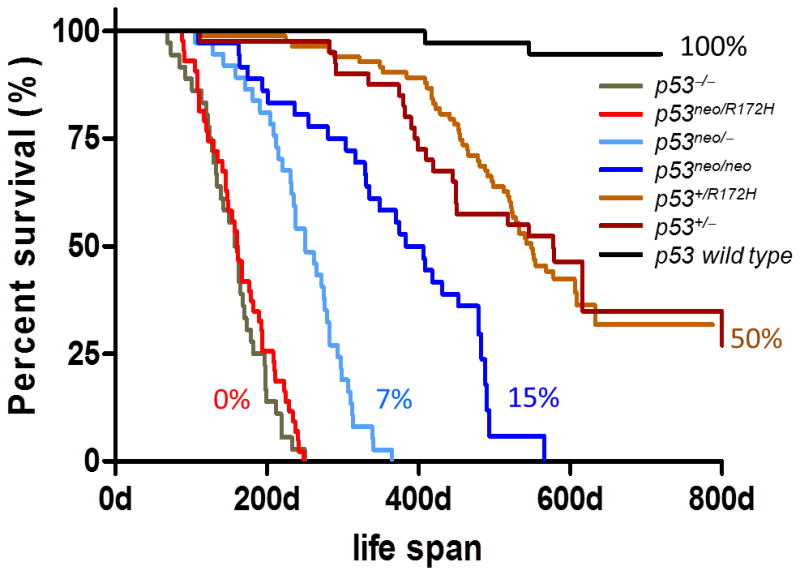
Survival curves of p53 mutant mice with various alleles. The p53neo allele is a hypomorphic allele that expresses approximately 7% of wild type p53 levels (Wang, et al., 2011). The percentages indicated equal the levels of p53 protein detected by western blot analysis compared to the wild type, which was set at 100%. From (Wang, et al., 2011) with permission.
p53 gene dosage
The most common alterations in TP53 as opposed to other tumor suppressors are missense mutations. Approximately 80% of TP53 mutations are missense. This observation alone suggests that TP53 mutants have additional transforming activities and mouse models with knockin alleles have provided clear evidence for this hypothesis (Lang, et al., 2004; Olive, et al., 2004). Often, a mutation of one allele of TP53 is followed by loss of the second allele and this process eliminates wild type TP53 transcriptional activity and unmasks GOF activities of mutant TP53. However, not all tumor cells with TP53 mutations lose their wild-type TP53 allele. In LFS patients with TP53 mutations, only 44% of tumors lose their wild-type TP53 gene (Varley, et al., 1997). These data are supported by studies in mice. Approximately 50% of p53 heterozygous mice that develop tumors under 18 months of age retain the wild-type p53 gene (Venkatachalam, et al., 1998). The retention of a wild-type p53 allele suggests that possibly a half dose of p53 is insufficient to maintain normality, an unlikely possibility given that p53 heterozygous patients and mice appear normal and only develop tumors with age, which suggests the need for additional mutations. Another possibility is that the p53 protein levels generated from one allele are sufficient under some stress conditions in many cells, but insufficient to deal with other kinds of stress signals. A third possibility is that a combination of molecular changes upstream or downstream in the p53 pathway could lead to the same end result: insufficient p53 levels or activity to effect apoptosis, senescence, or cell cycle arrest. The presence of abnormally high levels of the p53 inhibitors Mdm2 and Mdm4, for example, would preclude loss of the remaining wild-type allele. Thus, a cell with a TP53 mutation and with increased levels of TP53 inhibitors may not need to lose the remaining wild-type TP53 allele in the process of transformation.
The discussion above suggests that the number of modifications required to inactivate the p53 pathway may vary depending on circumstances. The nature of the p53 mutation may also contribute to the number of changes required for transformation. Obviously, loss of p53 predisposes a cell to tumor development. However, some TP53 mutations identified in human cancers are clearly partial loss-of-function mutations that retain the ability to activate some, but not all, TP53 targets (Resnick and Inga, 2003). For example, the rare TP53R175P mutation is able to activate the cell cycle arrest but not the apoptotic program of p53 (Ludwig, et al., 1996). Mice homozygous for the equivalent mutation develop tumors with longer latency than p53-null mice, which indicates that the ability to arrest the cell cycle is also a tumor-suppressive activity of p53. Further, these homozygous p53 mutant mice combined with loss of p21, a p53 target that encodes a cell cycle inhibitor, yields a tumor phenotype that more closely resembles that of p53-null mice (Barboza, et al., 2006). Thus, compounding weaker mutations may yield the same end result: inactivation of the p53 pathway. However, these combinations may be rare. Our evaluation of TCGA data in 13 different tumor types shows that p21 (CDKN1A; MIM#116899) is rarely deleted or mutated; only 0.84% (18 of 2148) tumors analyzed had p21 deletions or mutations (Cerami, et al., 2012; Gao, et al., 2013). Finally, other mutations, such as the gain-of-function mutations discussed above, would require even fewer changes. Splicing mutations may fall into either category depending on whether or not a truncated protein is made. Even a truncated p53 protein may have some gain-of-function activities.
It is important to note that different cancers alter the p53 pathway by different mechanisms. For example, 50% of lung tumors have TP53 mutations, while 30% of osteosarcomas have MDM2 amplification and 65% of retinoblastomas have MDM4 amplification (Laurie, et al., 2006; Oliner, et al., 1992)(and TCGA database). So why is MDM4 amplified in retinoblastoma while TP53 mutations are more common in lung cancers? How do chromatin modifications affect genetic alterations? Or does the accumulation of mutations in a cell depend on what mutation occurred first? Or does the cell of origin matter? New animal models will be invaluable in addressing these questions.
p53-independent functions of Mdm2 and Mdm4
Critics have used the fact that loss of p53 can rescue the embryonic lethality of Mdm2 or Mdm4 deletion (Jones, et al., 1995; Montes de Oca Luna, et al., 1995; Parant, et al., 2001) to conclude that the sole function of Mdm2 and Mdm4 is to regulate p53. However, functions in development only reveal part of the story and do not explain data generated over the past 20 years that indicate Mdm2 and Mdm4 have p53-independent functions that contribute to tumorigenesis (Melo and Eischen, 2012). For example, tumors that overexpress MDM2 and/or MDM4 have less TP53 activity, but some tumor cells contain both inactivated TP53 and overexpressed MDM2 and/or MDM4 (Cordon-Cardo, et al., 1994; Giglio, et al., 2005; Gunther, et al., 1997; Liu, et al., 2012; Lu, et al., 2002; Peng, et al., 2001; Ramos, et al., 2001; Sigalas, et al., 1996; Suda, et al., 2011; Wade, et al., 2013; Zou, et al., 1995). Our analysis of TCGA data showed that up to 6% of specific human malignancies have amplified MDM2 or MDM4 (and even more have increased MDM2 or MDM4 mRNA expression) with concomitant TP53 mutations (Cerami, et al., 2012; Gao, et al., 2013). These data have resulted in a line of investigation into the p53-independent functions of Mdm2 and Mdm4 that have revealed unexpected results.
Multiple independent investigations have shown that Mdm2 can contribute to tumorigenesis independent of p53. For example, deletion of Arf in a p53-null background does not alter the rate of tumor development, but deletion of Arf concurrently with Mdm2 overexpression accelerates tumorigenesis (Moore, et al., 2003). Mdm2 overexpression in p53-null mice results in an altered tumor spectrum compared to the tumor spectrum in p53-null mice (Jones, et al., 1998). There is increased anchorage-independent growth in cells lacking p53 and overexpressing Mdm2 compared to cells that only lack p53 (Dubs-Poterszman, et al., 1995). Furthermore, Mdm2 overexpression regardless of p53 status leads to genome instability in vivo and in vitro (Bouska, et al., 2008; Lushnikova, et al., 2011). Mice that lack both Mdm4 and p53 have an accelerated rate of tumor development, and cells from these mice have increased genome instability (Matijasevic, et al., 2008a; Matijasevic, et al., 2008b). Together, these data indicate that there are p53-independent functions of Mdm2 and Mdm4 in cancer that need to be considered when targeting these proteins. In addition, more research is needed to fully investigate the p53-independent functions of Mdm2 and Mdm4 and their contributions to cancer development.
Small increases in Mdm2 levels in normal cells are tolerated, but higher levels are incompatible with life in vivo and in vitro, except in some cases when p53 is also inactivated (Brown, et al., 1998; Carroll, et al., 1999; De Clercq, et al., 2010; Jones, et al., 1998; Kubbutat, et al., 1997). High levels of Mdm2 result in genome instability, and this occurs even in the absence of p53. Specifically, there was increased polyploidy in mammary epithelial cells in Mdm2 transgenic mice with or without p53 (Lundgren, et al., 1997). Elevated levels of Mdm2 result in increased chromatid and chromosome breaks as well as fusions (which are indicative of chromosome breaks), and these abnormalities have also been detected in cells overexpressing Mdm2 and lacking p53 (Bouska, et al., 2008; Lushnikova, et al., 2011). In recent years, it has been elucidated that Mdm2 directly binds to Nbs1 (MIM#602667) of the Mre11-Rad50-Nbs1 (MRN) DNA repair complex. The MRN complex was identified in an unbiased screen for novel proteins that interact with endogenous Mdm2. Binding between Mdm2 and Nbs1, but not between Mdm2 and p53, was required for Mdm2 to delay DNA break repair. This delay in DNA break repair resulted in increased genome instability and did not require the E3 ubiquitin ligase activity of Mdm2 (Alt, et al., 2005; Bouska, et al., 2008). Although the precise mechanism for the delay in DNA break repair caused by Mdm2 continues to be elucidated, multiple lines of evidence indicate that Mdm2 inhibits the early DNA damage signal that results in phosphorylation of multiple protein targets and marks broken DNA for repair (Bouska, et al., 2008; Melo and Eischen, 2012). We have also generated similar data for Mdm4 (Carrillo, et al., 2013), indicating the regulation of DNA break repair is a conserved function for the members of the Mdm family of proteins. Importantly, normal physiological levels of Mdm2 were shown to regulate Nbs1-mediated DNA break repair (Bouska, et al., 2008), which indicates that this function of Mdm2 is not aberrant and not a consequence of its overexpression in cancer. Therefore, when Mdm2 or Mdm4 is high in tumors, inhibition of both p53 and Nbs1-mediated DNA break repair would lead to increased genome instability. These important data provide new insight into another function of Mdm2 and Mdm4, whose deregulation most likely contribute to tumor development in multiple ways. In addition, this function of Mdm2 and Mdm4 also provides a therapeutic opportunity that could be capitalized on for the treatment of human malignancies that overexpress MDM2 or MDM4.
Conclusions
The p53 pathway is central in blocking proliferation and mediating apoptosis and is thus, often altered in tumor development by various mechanisms described here. MDM2 and MDM4 are critical inhibitors of TP53 that are frequently selected for overexpression in human malignancies as a mechanism of inactivating TP53 and also likely capitalizing on TP53-independent functions. Because tumors with high levels of MDM2 and MDM4 often have wild-type TP53, it is not surprising that drug companies want to activate TP53 in these cells by disrupting the interactions of MDM proteins with TP53. Consequently, numerous drugs have been designed to activate TP53 by interfering with MDM2 and/or MDM4 binding (Li and Lozano, 2013). Although these drugs do lead to TP53 activation, cancers have a unique ability to develop resistance to drugs that cause their destruction and subsequently acquired compensatory TP53 mutations (Aziz, et al., 2011; Michaelis, et al., 2011) that make the malignancy more difficult to treat. As noted in this review, MDM2 also regulates mutant TP53 stability, and these drugs would then stabilize mutant TP53, with adverse consequences. Frequent biopsies and monitoring of patients undergoing treatments is critically important. Moreover, drugs that bind MDM2 and/or MDM4 may have other unintended consequences, such as stabilizing these proteins and causing them to be present at high levels where their TP53-independent functions may predominate and negatively impact genome stability. Future studies of these drugs will need to take these confounding factors into consideration. With the large number of questions that remain unanswered and the mechanisms that are unresolved, researchers in this field should be quite busy for years to come.
Acknowledgments
We would like to thank Vinod Pant for review of this manuscript. GL is supported by NCI grant R01CA47296 and CME is supported by NCI grants R01CA117935, R01CA160432, and P30CA068485.
Footnotes
The authors have no conflict of interest to declare.
Contributor Information
Christine M. Eischen, Email: christine.eischen@vanderbilt.edu.
Guillermina Lozano, Email: gglozano@mdanderson.org.
Literature cited
- Abida WM, Gu W. p53-Dependent and p53-independent activation of autophagy by ARF. Cancer Res. 2008;68(2):352–7. doi: 10.1158/0008-5472.CAN-07-2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alt JR, Bouska A, Fernandez MR, Cerny RL, Xiao H, Eischen CM. Mdm2 binds to Nbs1 at sites of DNA damage and regulates double strand break repair. J Biol Chem. 2005;280(19):18771–81. doi: 10.1074/jbc.M413387200. [DOI] [PubMed] [Google Scholar]
- Alt JR, Greiner TC, Cleveland JL, Eischen CM. Mdm2 haplo-insufficiency profoundly inhibits Myc-induced lymphomagenesis. EMBO J. 2003;22(6):1442–50. doi: 10.1093/emboj/cdg133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz MH, Shen H, Maki CG. Acquisition of p53 mutations in response to the non-genotoxic p53 activator Nutlin-3. Oncogene. 2011;30(46):4678–86. doi: 10.1038/onc.2011.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmain A, Gray J, Ponder B. The genetics and genomics of cancer. Nat Genet. 2003;33(Suppl):238–44. doi: 10.1038/ng1107. [DOI] [PubMed] [Google Scholar]
- Barboza JA, Liu G, Ju Z, El-Naggar AK, Lozano G. p21 delays tumor onset by preservation of chromosomal stability. Proc Natl Acad Sci U S A. 2006;103(52):19842–7. doi: 10.1073/pnas.0606343104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birch JM, Blair V, Kelsey AM, Evans DG, Harris M, Tricker KJ, Varley JM. Cancer phenotype correlates with constitutional TP53 genotype in families with the Li-Fraumeni syndrome. Oncogene. 1998;17(9):1061–8. doi: 10.1038/sj.onc.1202033. [DOI] [PubMed] [Google Scholar]
- Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, Bargonetti J, Bartel F, Taubert H, Wuerl P, Onel K, Yip L, et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004;119(5):591–602. doi: 10.1016/j.cell.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Bond GL, Levine AJ. A single nucleotide polymorphism in the p53 pathway interacts with gender, environmental stresses and tumor genetics to influence cancer in humans. Oncogene. 2007;26(9):1317–23. doi: 10.1038/sj.onc.1210199. [DOI] [PubMed] [Google Scholar]
- Bougeard G, Baert-Desurmont S, Tournier I, Vasseur S, Martin C, Brugieres L, Chompret A, Bressac-de Paillerets B, Stoppa-Lyonnet D, Bonaiti-Pellie C, Frebourg T. Impact of the MDM2 SNP309 and p53 Arg72Pro polymorphism on age of tumour onset in Li-Fraumeni syndrome. J Med Genet. 2006;43(6):531–3. doi: 10.1136/jmg.2005.037952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouska A, Lushnikova T, Plaza S, Eischen CM. Mdm2 promotes genetic instability and transformation independent of p53. Mol Cell Biol. 2008;28(15):4862–74. doi: 10.1128/MCB.01584-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DR, Thomas CA, Deb SP. The human oncoprotein MDM2 arrests the cell cycle: elimination of its cell-cycle-inhibitory function induces tumorigenesis. EMBO J. 1998;17(9):2513–25. doi: 10.1093/emboj/17.9.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capoulade C, Bressac-de Paillerets B, Lefrere I, Ronsin M, Feunteun J, Tursz T, Wiels J. Overexpression of MDM2, due to enhanced translation, results in inactivation of wild-type p53 in Burkitt’s lymphoma cells. Oncogene. 1998;16(12):1603–10. doi: 10.1038/sj.onc.1201702. [DOI] [PubMed] [Google Scholar]
- Carrillo AM, Bouska A, Arrate MP, Eischen CM. Mdmx promotes genomic instability independent of p53 and Mdm2. Oncogene. 2013 doi: 10.1038/onc.2014.27. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll PE, Okuda M, Horn HF, Biddinger P, Stambrook PJ, Gleich LL, Li YQ, Tarapore P, Fukasawa K. Centrosome hyperamplification in human cancer: chromosome instability induced by p53 mutation and/or Mdm2 overexpression. Oncogene. 1999;18(11):1935–44. doi: 10.1038/sj.onc.1202515. [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez-Reyes A, Parant JM, Amelse LL, de Oca Luna RM, Korsmeyer SJ, Lozano G. Switching mechanisms of cell death in mdm2- and mdm4-null mice by deletion of p53 downstream targets. Cancer Res. 2003;63(24):8664–9. [PubMed] [Google Scholar]
- Chen D, Shan J, Zhu WG, Qin J, Gu W. Transcription-independent ARF regulation in oncogenic stress-mediated p53 responses. Nature. 2010;464(7288):624–7. doi: 10.1038/nature08820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Carracedo A, Lin HK, Koutcher JA, Behrendt N, Egia A, Alimonti A, Carver BS, Gerald W, Teruya-Feldstein J, Loda M, Pandolfi PP. Differential p53-independent outcomes of p19(Arf) loss in oncogenesis. Sci Signal. 2009;2(84):ra44. doi: 10.1126/scisignal.2000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q, Chen J. The phenotype of MDM2 auto-degradation after DNA damage is due to epitope masking by phosphorylation. Cell Cycle. 2011;10(7):1162–6. doi: 10.4161/cc.10.7.15249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994;265(5170):346–55. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- Cordon-Cardo C, Latres E, Drobnjak M, Oliva MR, Pollack D, Woodruff JM, Marechal V, Chen J, Brennan MF, Levine AJ. Molecular abnormalities of mdm2 and p53 genes in adult soft tissue sarcomas. Cancer Res. 1994;54(3):794–9. [PubMed] [Google Scholar]
- Crook T, Vousden KH. Properties of p53 mutations detected in primary and secondary cervical cancers suggest mechanisms of metastasis and involvement of environmental carcinogens. EMBO J. 1992;11(11):3935–40. doi: 10.1002/j.1460-2075.1992.tb05487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins JM, Rago C, Kohli M, Kinzler KW, Lengauer C, Vogelstein B. Tumour suppression: disruption of HAUSP gene stabilizes p53. Nature. 2004;428(6982):1. doi: 10.1038/nature02501. p following 486. [DOI] [PubMed] [Google Scholar]
- Danovi D, Meulmeester E, Pasini D, Migliorini D, Capra M, Frenk R, de Graaf P, Francoz S, Gasparini P, Gobbi A, Helin K, Pelicci PG, et al. Amplification of Mdmx (or Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol Cell Biol. 2004;24(13):5835–43. doi: 10.1128/MCB.24.13.5835-5843.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq S, Gembarska A, Denecker G, Maetens M, Naessens M, Haigh K, Haigh JJ, Marine JC. Widespread overexpression of epitope-tagged Mdm4 does not accelerate tumor formation in vivo. Mol Cell Biol. 2010;30(22):5394–405. doi: 10.1128/MCB.00330-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmer D, Pati S, Zambetti G, Chu S, Teresky AK, Moore M, Finlay C, Levine AJ. Gain of function mutations in p53. Nat Genet. 1993;4(1):42–6. doi: 10.1038/ng0593-42. [DOI] [PubMed] [Google Scholar]
- Dubs-Poterszman MC, Tocque B, Wasylyk B. MDM2 transformation in the absence of p53 and abrogation of the p107 G1 cell-cycle arrest. Oncogene. 1995;11(11):2445–9. [PubMed] [Google Scholar]
- Eischen CM. Decreased Mdm2 levels after DNA damage: antibody masking or protein degradation? Cell Cycle. 2011;10(9):1347. doi: 10.4161/cc.10.9.15437. [DOI] [PubMed] [Google Scholar]
- Eischen CM, Alt JR, Wang P. Loss of one allele of ARF rescues Mdm2 haploinsufficiency effects on apoptosis and lymphoma development. Oncogene. 2004;23(55):8931–40. doi: 10.1038/sj.onc.1208052. [DOI] [PubMed] [Google Scholar]
- Eischen CM, Boyd K. Decreased Mdm2 expression inhibits tumor development and extends survival independent of Arf and dependent on p53. PLoS One. 2012;7(9):e46148. doi: 10.1371/journal.pone.0046148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999;13(20):2658–69. doi: 10.1101/gad.13.20.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M, Tortola S, Toyota M, Capella G, Peinado MA, Baylin SB, Herman JG. Hypermethylation-associated inactivation of p14(ARF) is independent of p16(INK4a) methylation and p53 mutational status. Cancer Res. 2000;60(1):129–33. [PubMed] [Google Scholar]
- Fang M, Simeonova I, Bardot B, Lejour V, Jaber S, Bouarich-Bourimi R, Morin A, Toledo F. Mdm4 loss in mice expressing a p53 hypomorph alters tumor spectrum without improving survival. Oncogene. 2013 doi: 10.1038/onc.2013.62. [DOI] [PubMed] [Google Scholar]
- Finch RA, Donoviel DB, Potter D, Shi M, Fan A, Freed DD, Wang CY, Zambrowicz BP, Ramirez-Solis R, Sands AT, Zhang N. mdmx is a negative regulator of p53 activity in vivo. Cancer Res. 2002;62(11):3221–5. [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gembarska A, Luciani F, Fedele C, Russell EA, Dewaele M, Villar S, Zwolinska A, Haupt S, de Lange J, Yip D, Goydos J, Haigh JJ, et al. MDM4 is a key therapeutic target in cutaneous melanoma. Nat Med. 2012;18(8):1239–47. doi: 10.1038/nm.2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giglio S, Mancini F, Gentiletti F, Sparaco G, Felicioni L, Barassi F, Martella C, Prodosmo A, Iacovelli S, Buttitta F, Farsetti A, Soddu S, et al. Identification of an aberrantly spliced form of HDMX in human tumors: a new mechanism for HDM2 stabilization. Cancer Res. 2005;65(21):9687–94. doi: 10.1158/0008-5472.CAN-05-0450. [DOI] [PubMed] [Google Scholar]
- Gunther T, Schneider-Stock R, Rys J, Niezabitowski A, Roessner A. p53 gene mutations and expression of p53 and mdm2 proteins in invasive breast carcinoma. A comparative analysis with clinico-pathological factors. J Cancer Res Clin Oncol. 1997;123(7):388–94. doi: 10.1007/BF01240122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamroun D, Kato S, Ishioka C, Claustres M, Beroud C, Soussi T. The UMD TP53 database and website: update and revisions. Hum Mutat. 2006;27(1):14–20. doi: 10.1002/humu.20269. [DOI] [PubMed] [Google Scholar]
- Han X, Garcia-Manero G, McDonnell TJ, Lozano G, Medeiros LJ, Xiao L, Rosner G, Nguyen M, Fernandez M, Valentin-Vega YA, Barboza J, Jones DM, et al. HDM4 (HDMX) is widely expressed in adult pre-B acute lymphoblastic leukemia and is a potential therapeutic target. Mod Pathol. 2007;20(1):54–62. doi: 10.1038/modpathol.3800727. [DOI] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387(6630):296–9. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- Heinmoller E, Dietmaier W, Zirngibl H, Heinmoller P, Scaringe W, Jauch KW, Hofstadter F, Ruschoff J. Molecular analysis of microdissected tumors and preneoplastic intraductal lesions in pancreatic carcinoma. Am J Pathol. 2000;157(1):83–92. doi: 10.1016/S0002-9440(10)64520-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420(1):25–7. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- Hsiao M, Low J, Dorn E, Ku D, Pattengale P, Yeargin J, Haas M. Gain-of-function mutations of the p53 gene induce lymphohematopoietic metastatic potential and tissue invasiveness. Am J Pathol. 1994;145(3):702–14. [PMC free article] [PubMed] [Google Scholar]
- Huang L, Yan Z, Liao X, Li Y, Yang J, Wang ZG, Zuo Y, Kawai H, Shadfan M, Ganapathy S, Yuan ZM. The p53 inhibitors MDM2/MDMX complex is required for control of p53 activity in vivo. Proc Natl Acad Sci U S A. 2011;108(29):12001–6. doi: 10.1073/pnas.1102309108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hustinx SR, Leoni LM, Yeo CJ, Brown PN, Goggins M, Kern SE, Hruban RH, Maitra A. Concordant loss of MTAP and p16/CDKN2A expression in pancreatic intraepithelial neoplasia: evidence of homozygous deletion in a noninvasive precursor lesion. Mod Pathol. 2005;18(7):959–63. doi: 10.1038/modpathol.3800377. [DOI] [PubMed] [Google Scholar]
- Hwang SJ, Cheng LS, Lozano G, Amos CI, Gu X, Strong LC. Lung cancer risk in germline p53 mutation carriers: association between an inherited cancer predisposition, cigarette smoking, and cancer risk. Hum Genet. 2003;113(3):238–43. doi: 10.1007/s00439-003-0968-7. [DOI] [PubMed] [Google Scholar]
- Jackson MW, Lindstrom MS, Berberich SJ. MdmX binding to ARF affects Mdm2 protein stability and p53 transactivation. J Biol Chem. 2001;276(27):25336–41. doi: 10.1074/jbc.M010685200. [DOI] [PubMed] [Google Scholar]
- Jones SN, Hancock AR, Vogel H, Donehower LA, Bradley A. Overexpression of Mdm2 in mice reveals a p53-independent role for Mdm2 in tumorigenesis. Proc Natl Acad Sci U S A. 1998;95(26):15608–12. doi: 10.1073/pnas.95.26.15608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378(6553):206–8. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- Kamijo T, Bodner S, van de Kamp E, Randle DH, Sherr CJ. Tumor spectrum in ARF-deficient mice. Cancer Res. 1999;59(9):2217–22. [PubMed] [Google Scholar]
- Kamijo T, Weber JD, Zambetti G, Zindy F, Roussel MF, Sherr CJ. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc Natl Acad Sci U S A. 1998;95(14):8292–7. doi: 10.1073/pnas.95.14.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, Grosveld G, Sherr CJ. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91(5):649–59. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- Kato MV, Shimizu T, Ishizaki K, Kaneko A, Yandell DW, Toguchida J, Sasaki MS. Loss of heterozygosity on chromosome 17 and mutation of the p53 gene in retinoblastoma. Cancer Lett. 1996;106(1):75–82. doi: 10.1016/0304-3835(96)04305-4. [DOI] [PubMed] [Google Scholar]
- Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127(2):265–75. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Knappskog S, Bjornslett M, Myklebust LM, Huijts PE, Vreeswijk MP, Edvardsen H, Guo Y, Zhang X, Yang M, Ylisaukko-Oja SK, Alhopuro P, Arola J, et al. The MDM2 promoter SNP285C/309G haplotype diminishes Sp1 transcription factor binding and reduces risk for breast and ovarian cancer in Caucasians. Cancer Cell. 2011;19(2):273–82. doi: 10.1016/j.ccr.2010.12.019. [DOI] [PubMed] [Google Scholar]
- Kon N, Zhong J, Kobayashi Y, Li M, Szabolcs M, Ludwig T, Canoll PD, Gu W. Roles of HAUSP-mediated p53 regulation in central nervous system development. Cell Death Differ. 2011;18(8):1366–75. doi: 10.1038/cdd.2011.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387(6630):299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- Landers JE, Haines DS, Strauss JF, 3rd, George DL. Enhanced translation: a novel mechanism of mdm2 oncogene overexpression identified in human tumor cells. Oncogene. 1994;9(9):2745–50. [PubMed] [Google Scholar]
- Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, El-Naggar AK, Lozano G. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119(6):861–72. doi: 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Laurie NA, Donovan SL, Shih CS, Zhang J, Mills N, Fuller C, Teunisse A, Lam S, Ramos Y, Mohan A, Johnson D, Wilson M, et al. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444(7115):61–6. doi: 10.1038/nature05194. [DOI] [PubMed] [Google Scholar]
- Lenos K, Grawenda AM, Lodder K, Kuijjer ML, Teunisse AF, Repapi E, Grochola LF, Bartel F, Hogendoorn PC, Wuerl P, Taubert H, Clinton-Jansen AM, et al. Alternate splicing of the p53 inhibitor HDMX offers a superior prognostic biomarker than p53 mutation in human cancer. Cancer Res. 2012;72(16):4074–84. doi: 10.1158/0008-5472.CAN-12-0215. [DOI] [PubMed] [Google Scholar]
- Leventaki V, Rodic V, Tripp SR, Bayerl MG, Perkins SL, Barnette P, Schiffman JD, Miles RR. TP53 pathway analysis in paediatric Burkitt lymphoma reveals increased MDM4 expression as the only TP53 pathway abnormality detected in a subset of cases. Br J Haematol. 2012;158(6):763–71. doi: 10.1111/j.1365-2141.2012.09243.x. [DOI] [PubMed] [Google Scholar]
- Li C, Finkelstein D, Sherr CJ. Arf tumor suppressor and miR-205 regulate cell adhesion and formation of extraembryonic endoderm from pluripotent stem cells. Proc Natl Acad Sci U S A. 2013;110(12):E1112–21. doi: 10.1073/pnas.1302184110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Brooks CL, Kon N, Gu W. A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol Cell. 2004;13(6):879–86. doi: 10.1016/s1097-2765(04)00157-1. [DOI] [PubMed] [Google Scholar]
- Li M, Chen D, Shiloh A, Luo J, Nikolaev AY, Qin J, Gu W. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature. 2002;416(6881):648–53. doi: 10.1038/nature737. [DOI] [PubMed] [Google Scholar]
- Li Q, Lozano G. Molecular pathways: targeting Mdm2 and Mdm4 in cancer therapy. Clin Cancer Res. 2013;19(1):34–41. doi: 10.1158/1078-0432.CCR-12-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Gilkes D, Li B, Cheng Q, Pernazza D, Lawrence H, Lawrence N, Chen J. Abnormal MDMX degradation in tumor cells due to ARF deficiency. Oncogene. 2012;31(32):3721–32. doi: 10.1038/onc.2011.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linke K, Mace PD, Smith CA, Vaux DL, Silke J, Day CL. Structure of the MDM2/MDMX RING domain heterodimer reveals dimerization is required for their ubiquitylation in trans. Cell Death Differ. 2008;15(5):841–8. doi: 10.1038/sj.cdd.4402309. [DOI] [PubMed] [Google Scholar]
- Liu G, Wheatley-Price P, Zhou W, Park S, Heist RS, Asomaning K, Wain JC, Lynch TJ, Su L, Christiani DC. Genetic polymorphisms of MDM2, cumulative cigarette smoking and nonsmall cell lung cancer risk. Int J Cancer. 2008;122(4):915–8. doi: 10.1002/ijc.23178. [DOI] [PubMed] [Google Scholar]
- Liu L, Fan L, Fang C, Zou ZJ, Yang S, Zhang LN, Li JY, Xu W. S-MDM4 mRNA overexpression indicates a poor prognosis and marks a potential therapeutic target in chronic lymphocytic leukemia. Cancer Sci. 2012;103(12):2056–63. doi: 10.1111/cas.12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llanos S, Clark PA, Rowe J, Peters G. Stabilization of p53 by p14ARF without relocation of MDM2 to the nucleolus. Nat Cell Biol. 2001;3(5):445–52. doi: 10.1038/35074506. [DOI] [PubMed] [Google Scholar]
- Lozano G. The oncogenic roles of p53 mutants in mouse models. Curr Opin Genet Dev. 2007;17(1):66–70. doi: 10.1016/j.gde.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Lu ML, Wikman F, Orntoft TF, Charytonowicz E, Rabbani F, Zhang Z, Dalbagni G, Pohar KS, Yu G, Cordon-Cardo C. Impact of alterations affecting the p53 pathway in bladder cancer on clinical outcome, assessed by conventional and array-based methods. Clin Cancer Res. 2002;8(1):171–9. [PubMed] [Google Scholar]
- Ludwig RL, Bates S, Vousden KH. Differential activation of target cellular promoters by p53 mutants with impaired apoptotic function. Mol Cell Biol. 1996;16(9):4952–60. doi: 10.1128/mcb.16.9.4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundgren K, Montes de Oca Luna R, McNeill YB, Emerick EP, Spencer B, Barfield CR, Lozano G, Rosenberg MP, Finlay CA. Targeted expression of MDM2 uncouples S phase from mitosis and inhibits mammary gland development independent of p53. Genes Dev. 1997;11(6):714–25. doi: 10.1101/gad.11.6.714. [DOI] [PubMed] [Google Scholar]
- Lushnikova T, Bouska A, Odvody J, Dupont WD, Eischen CM. Aging mice have increased chromosome instability that is exacerbated by elevated Mdm2 expression. Oncogene. 2011;30(46):4622–31. doi: 10.1038/onc.2011.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra A, Adsay NV, Argani P, Iacobuzio-Donahue C, De Marzo A, Cameron JL, Yeo CJ, Hruban RH. Multicomponent analysis of the pancreatic adenocarcinoma progression model using a pancreatic intraepithelial neoplasia tissue microarray. Mod Pathol. 2003;16(9):902–12. doi: 10.1097/01.MP.0000086072.56290.FB. [DOI] [PubMed] [Google Scholar]
- Matijasevic Z, Krzywicka-Racka A, Sluder G, Jones SN. MdmX regulates transformation and chromosomal stability in p53-deficient cells. Cell Cycle. 2008a;7(19):2967–73. doi: 10.4161/cc.7.19.6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matijasevic Z, Steinman HA, Hoover K, Jones SN. MdmX promotes bipolar mitosis to suppress transformation and tumorigenesis in p53-deficient cells and mice. Mol Cell Biol. 2008b;28(4):1265–73. doi: 10.1128/MCB.01108-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maya R, Oren M. Unmasking of phosphorylation-sensitive epitopes on p53 and Mdm2 by a simple Western-phosphatase procedure. Oncogene. 2000;19(28):3213–5. doi: 10.1038/sj.onc.1203658. [DOI] [PubMed] [Google Scholar]
- McDonnell TJ, Montes de Oca Luna R, Cho S, Amelse LL, Chavez-Reyes A, Lozano G. Loss of one but not two mdm2 null alleles alters the tumour spectrum in p53 null mice. J Pathol. 1999;188(3):322–8. doi: 10.1002/(SICI)1096-9896(199907)188:3<322::AID-PATH372>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Meek DW, Hupp TR. The regulation of MDM2 by multisite phosphorylation--opportunities for molecular-based intervention to target tumours? Semin Cancer Biol. 2010;20(1):19–28. doi: 10.1016/j.semcancer.2009.10.005. [DOI] [PubMed] [Google Scholar]
- Melo AN, Eischen CM. Protecting the genome from mdm2 and mdmx. Genes Cancer. 2012;3(3–4):283–90. doi: 10.1177/1947601912454139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendrysa SM, McElwee MK, Michalowski J, O’Leary KA, Young KM, Perry ME. mdm2 Is critical for inhibition of p53 during lymphopoiesis and the response to ionizing irradiation. Mol Cell Biol. 2003;23(2):462–72. doi: 10.1128/MCB.23.2.462-473.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendrysa SM, O’Leary KA, McElwee MK, Michalowski J, Eisenman RN, Powell DA, Perry ME. Tumor suppression and normal aging in mice with constitutively high p53 activity. Genes Dev. 2006;20(1):16–21. doi: 10.1101/gad.1378506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meulmeester E, Maurice MM, Boutell C, Teunisse AF, Ovaa H, Abraham TE, Dirks RW, Jochemsen AG. Loss of HAUSP-mediated deubiquitination contributes to DNA damage-induced destabilization of Hdmx and Hdm2. Mol Cell. 2005;18(5):565–76. doi: 10.1016/j.molcel.2005.04.024. [DOI] [PubMed] [Google Scholar]
- Michaelis M, Rothweiler F, Barth S, Cinatl J, van Rikxoort M, Loschmann N, Voges Y, Breitling R, von Deimling A, Rodel F, Weber K, Fehse B, et al. Adaptation of cancer cells from different entities to the MDM2 inhibitor nutlin-3 results in the emergence of p53-mutated multi-drug-resistant cancer cells. Cell Death Dis. 2011;2:e243. doi: 10.1038/cddis.2011.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midgley CA, Lane DP. p53 protein stability in tumour cells is not determined by mutation but is dependent on Mdm2 binding. Oncogene. 1997;15(10):1179–89. doi: 10.1038/sj.onc.1201459. [DOI] [PubMed] [Google Scholar]
- Migliorini D, Lazzerini Denchi E, Danovi D, Jochemsen A, Capillo M, Gobbi A, Helin K, Pelicci PG, Marine JC. Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol Cell Biol. 2002;22(15):5527–38. doi: 10.1128/MCB.22.15.5527-5538.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378(6553):203–6. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- Moore L, Venkatachalam S, Vogel H, Watt JC, Wu CL, Steinman H, Jones SN, Donehower LA. Cooperativity of p19ARF, Mdm2, and p53 in murine tumorigenesis. Oncogene. 2003;22(49):7831–7. doi: 10.1038/sj.onc.1206985. [DOI] [PubMed] [Google Scholar]
- O’Leary KA, Mendrysa SM, Vaccaro A, Perry ME. Mdm2 regulates p53 independently of p19(ARF) in homeostatic tissues. Mol Cell Biol. 2004;24(1):186–91. doi: 10.1128/MCB.24.1.186-191.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358(6381):80–3. doi: 10.1038/358080a0. [DOI] [PubMed] [Google Scholar]
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119(6):847–60. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum Mutat. 2002;19(6):607–14. doi: 10.1002/humu.10081. [DOI] [PubMed] [Google Scholar]
- Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2(1):a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onel K, Cordon-Cardo C. MDM2 and prognosis. Mol Cancer Res. 2004;2(1):1–8. [PubMed] [Google Scholar]
- Pant V, Xiong S, Iwakuma T, Quintas-Cardama A, Lozano G. Heterodimerization of Mdm2 and Mdm4 is critical for regulating p53 activity during embryogenesis but dispensable for p53 and Mdm2 stability. Proc Natl Acad Sci U S A. 2011;108(29):11995–2000. doi: 10.1073/pnas.1102241108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant V, Xiong S, Jackson JG, Post SM, Abbas HA, Quintas-Cardama A, Hamir AN, Lozano G. The p53-Mdm2 feedback loop protects against DNA damage by inhibiting p53 activity but is dispensable for p53 stability, development, and longevity. Genes Dev. 2013;27(17):1857–67. doi: 10.1101/gad.227249.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG, Lozano G. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet. 2001;29(1):92–5. doi: 10.1038/ng714. [DOI] [PubMed] [Google Scholar]
- Peng Y, Chen L, Li C, Lu W, Agrawal S, Chen J. Stabilization of the MDM2 oncoprotein by mutant p53. J Biol Chem. 2001;276(9):6874–8. doi: 10.1074/jbc.C000781200. [DOI] [PubMed] [Google Scholar]
- Post SM, Quintas-Cardama A, Pant V, Iwakuma T, Hamir A, Jackson JG, Maccio DR, Bond GL, Johnson DG, Levine AJ, Lozano G. A high-frequency regulatory polymorphism in the p53 pathway accelerates tumor development. Cancer Cell. 2010;18(3):220–30. doi: 10.1016/j.ccr.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83(6):993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- Ramos YF, Stad R, Attema J, Peltenburg LT, van der Eb AJ, Jochemsen AG. Aberrant expression of HDMX proteins in tumor cells correlates with wild-type p53. Cancer Res. 2001;61(5):1839–42. [PubMed] [Google Scholar]
- Rayburn E, Zhang R, He J, Wang H. MDM2 and human malignancies: expression, clinical pathology, prognostic markers, and implications for chemotherapy. Curr Cancer Drug Targets. 2005;5(1):27–41. doi: 10.2174/1568009053332636. [DOI] [PubMed] [Google Scholar]
- Resnick MA, Inga A. Functional mutants of the sequence-specific transcription factor p53 and implications for master genes of diversity. Proc Natl Acad Sci U S A. 2003;100(17):9934–9. doi: 10.1073/pnas.1633803100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemenschneider MJ, Buschges R, Wolter M, Reifenberger J, Bostrom J, Kraus JA, Schlegel U, Reifenberger G. Amplification and overexpression of the MDM4 (MDMX) gene from 1q32 in a subset of malignant gliomas without TP53 mutation or MDM2 amplification. Cancer Res. 1999;59(24):6091–6. [PubMed] [Google Scholar]
- Riley MF, Lozano G. The Many Faces of MDM2 Binding Partners. Genes Cancer. 2012;3(3–4):226–39. doi: 10.1177/1947601912455322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenblum E, Schutte M, Goggins M, Hahn SA, Panzer S, Zahurak M, Goodman SN, Sohn TA, Hruban RH, Yeo CJ, Kern SE. Tumor-suppressive pathways in pancreatic carcinoma. Cancer Res. 1997;57(9):1731–4. [PubMed] [Google Scholar]
- Ruijs MW, Schmidt MK, Nevanlinna H, Tommiska J, Aittomaki K, Pruntel R, Verhoef S, Van’t Veer LJ. The single-nucleotide polymorphism 309 in the MDM2 gene contributes to the Li-Fraumeni syndrome and related phenotypes. Eur J Hum Genet. 2007;15(1):110–4. doi: 10.1038/sj.ejhg.5201715. [DOI] [PubMed] [Google Scholar]
- Sanchez-Cespedes M, Reed AL, Buta M, Wu L, Westra WH, Herman JG, Yang SC, Jen J, Sidransky D. Inactivation of the INK4A/ARF locus frequently coexists with TP53 mutations in non-small cell lung cancer. Oncogene. 1999;18(43):5843–9. doi: 10.1038/sj.onc.1203003. [DOI] [PubMed] [Google Scholar]
- Schmidt MK, Reincke S, Broeks A, Braaf LM, Hogervorst FB, Tollenaar RA, Johnson N, Fletcher O, Peto J, Tommiska J, Blomqvist C, Nevanlinna HA, et al. Do MDM2 SNP309 and TP53 R72P interact in breast cancer susceptibility? A large pooled series from the breast cancer association consortium. Cancer Res. 2007;67(19):9584–90. doi: 10.1158/0008-5472.CAN-07-0738. [DOI] [PubMed] [Google Scholar]
- Schmitt CA, McCurrach ME, de Stanchina E, Wallace-Brodeur RR, Lowe SW. INK4a/ARF mutations accelerate lymphomagenesis and promote chemoresistance by disabling p53. Genes Dev. 1999;13(20):2670–7. doi: 10.1101/gad.13.20.2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer. 2006;6(9):663–73. doi: 10.1038/nrc1954. [DOI] [PubMed] [Google Scholar]
- Shvarts A, Steegenga WT, Riteco N, van Laar T, Dekker P, Bazuine M, van Ham RC, van der Houven van Oordt W, Hateboer G, van der Eb AJ, Jochemsen AG. MDMX: a novel p53-binding protein with some functional properties of MDM2. EMBO J. 1996;15(19):5349–57. [PMC free article] [PubMed] [Google Scholar]
- Sigal A, Rotter V. Oncogenic mutations of the p53 tumor suppressor: the demons of the guardian of the genome. Cancer Res. 2000;60(24):6788–93. [PubMed] [Google Scholar]
- Sigalas I, Calvert AH, Anderson JJ, Neal DE, Lunec J. Alternatively spliced mdm2 transcripts with loss of p53 binding domain sequences: transforming ability and frequent detection in human cancer. Nat Med. 1996;2(8):912–7. doi: 10.1038/nm0896-912. [DOI] [PubMed] [Google Scholar]
- Smeds J, Berggren P, Ma X, Xu Z, Hemminki K, Kumar R. Genetic status of cell cycle regulators in squamous cell carcinoma of the oesophagus: the CDKN2A (p16(INK4a) and p14(ARF) ) and p53 genes are major targets for inactivation. Carcinogenesis. 2002;23(4):645–55. doi: 10.1093/carcin/23.4.645. [DOI] [PubMed] [Google Scholar]
- Suda T, Yoshihara M, Nakamura Y, Sekiguchi H, Godai TI, Sugano N, Tsuchida K, Shiozawa M, Sakuma Y, Tsuchiya E, Kameda Y, Akaike M, et al. Rare MDM4 gene amplification in colorectal cancer: The principle of a mutually exclusive relationship between MDM alteration and TP53 inactivation is not applicable. Oncol Rep. 2011;26(1):49–54. doi: 10.3892/or.2011.1270. [DOI] [PubMed] [Google Scholar]
- Suh YA, Post SM, Elizondo-Fraire AC, Maccio DR, Jackson JG, El-Naggar AK, Van Pelt C, Terzian T, Lozano G. Multiple stress signals activate mutant p53 in vivo. Cancer Res. 2011;71(23):7168–75. doi: 10.1158/0008-5472.CAN-11-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzian T, Suh YA, Iwakuma T, Post SM, Neumann M, Lang GA, Van Pelt CS, Lozano G. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008;22(10):1337–44. doi: 10.1101/gad.1662908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzian T, Wang Y, Van Pelt CS, Box NF, Travis EL, Lozano G. Haploinsufficiency of Mdm2 and Mdm4 in tumorigenesis and development. Mol Cell Biol. 2007;27(15):5479–85. doi: 10.1128/MCB.00555-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentin-Vega YA, Barboza JA, Chau GP, El-Naggar AK, Lozano G. High levels of the p53 inhibitor MDM4 in head and neck squamous carcinomas. Hum Pathol. 2007;38(10):1553–62. doi: 10.1016/j.humpath.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varley JM, Thorncroft M, McGown G, Appleby J, Kelsey AM, Tricker KJ, Evans DG, Birch JM. A detailed study of loss of heterozygosity on chromosome 17 in tumours from Li-Fraumeni patients carrying a mutation to the TP53 gene. Oncogene. 1997;14(7):865–71. doi: 10.1038/sj.onc.1201041. [DOI] [PubMed] [Google Scholar]
- Venkatachalam S, Shi YP, Jones SN, Vogel H, Bradley A, Pinkel D, Donehower LA. Retention of wild-type p53 in tumors from p53 heterozygous mice: reduction of p53 dosage can promote cancer formation. EMBO J. 1998;17(16):4657–67. doi: 10.1093/emboj/17.16.4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Kinzler KW. Carcinogens leave fingerprints. Nature. 1992;355(6357):209–10. doi: 10.1038/355209a0. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408(6810):307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137(3):413–31. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013;13(2):83–96. doi: 10.1038/nrc3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Greiner TC, Lushnikova T, Eischen CM. Decreased Mdm2 expression inhibits tumor development induced by loss of ARF. Oncogene. 2006;25(26):3708–18. doi: 10.1038/sj.onc.1209411. [DOI] [PubMed] [Google Scholar]
- Wang P, Lushnikova T, Odvody J, Greiner TC, Jones SN, Eischen CM. Elevated Mdm2 expression induces chromosomal instability and confers a survival and growth advantage to B cells. Oncogene. 2008;27(11):1590–8. doi: 10.1038/sj.onc.1210788. [DOI] [PubMed] [Google Scholar]
- Wang X, Arooz T, Siu WY, Chiu CH, Lau A, Yamashita K, Poon RY. MDM2 and MDMX can interact differently with ARF and members of the p53 family. FEBS Lett. 2001;490(3):202–8. doi: 10.1016/s0014-5793(01)02124-x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Suh YA, Fuller MY, Jackson JG, Xiong S, Terzian T, Quintas-Cardama A, Bankson JA, El-Naggar AK, Lozano G. Restoring expression of wild-type p53 suppresses tumor growth but does not cause tumor regression in mice with a p53 missense mutation. J Clin Invest. 2011;121(3):893–904. doi: 10.1172/JCI44504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JD, Jeffers JR, Rehg JE, Randle DH, Lozano G, Roussel MF, Sherr CJ, Zambetti GP. p53-independent functions of the p19(ARF) tumor suppressor. Genes Dev. 2000;14(18):2358–65. doi: 10.1101/gad.827300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat Cell Biol. 1999;1(1):20–6. doi: 10.1038/8991. [DOI] [PubMed] [Google Scholar]
- Wilkening S, Bermejo JL, Hemminki K. MDM2 SNP309 and cancer risk: a combined analysis. Carcinogenesis. 2007;28(11):2262–7. doi: 10.1093/carcin/bgm191. [DOI] [PubMed] [Google Scholar]
- Xiong S, Pant V, Suh YA, Van Pelt CS, Wang Y, Valentin-Vega YA, Post SM, Lozano G. Spontaneous tumorigenesis in mice overexpressing the p53-negative regulator Mdm4. Cancer Res. 2010;70(18):7148–54. doi: 10.1158/0008-5472.CAN-10-1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarden RI, Friedman E, Metsuyanim S, Olender T, Ben-Asher E, Papa MZ. MDM2 SNP309 accelerates breast and ovarian carcinogenesis in BRCA1 and BRCA2 carriers of Jewish-Ashkenazi descent. Breast Cancer Res Treat. 2008;111(3):497–504. doi: 10.1007/s10549-007-9797-z. [DOI] [PubMed] [Google Scholar]
- Zhang T, Prives C. Cyclin a-CDK phosphorylation regulates MDM2 protein interactions. J Biol Chem. 2001;276(32):29702–10. doi: 10.1074/jbc.M011326200. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92(6):725–34. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998;12(15):2424–33. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou M, Shi Y, al-Sedairy S, Hussain SS, Farid NR. The expression of the MDM2 gene, a p53 binding protein, in thyroid carcinogenesis. Cancer. 1995;76(2):314–8. doi: 10.1002/1097-0142(19950715)76:2<314::aid-cncr2820760223>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]