

Abstract
Background
Primary hepatocellular carcinoma is one of the most common malignant tumors in China and its mortality rate shows no sign at present of ceasing to rise. In our previous study, we found that the mRNA level of Dynamin3 (DNM3), a member of the Dynamin family, is significantly lower in hepatocellular carcinoma tissues than in non-tumor tissues. The aim of this study was to investigate the expression pattern and potential function of DNM3 in hepatocellular carcinoma.
Material/Methods
First, we determined the expression ofDNM3 in human hepatocellular carcinoma tissues and cell lines. We then studied the biological function of DNM3 on hepatocellular carcinoma cells by proliferation assay and colony formation assay. Flow cytometry was used to study the effect of DNM3 on cell cycle and apoptosis.
Results
Expression of DNM3 was significantly downregulated in hepatocellular carcinoma tissues and was associated with vein invasion and tumor metastasis. In addition, upregulation of DNM3 reduced hepatocellular carcinoma cell proliferation and colony formation, induced hepatocellular carcinoma cell G0/G1 phase arrest, and stimulated hepatocellular carcinoma cell apoptosis. We also found that DNM3 may exert its anti-proliferative effect through upregulating p53.
Conclusions
Our findings suggest that DNM3 attenuates the proliferation and induces apoptosis of gastric cancer cells. Modulation of DNM3 may prove to be an efficient method of hepatocellular carcinoma treatment.
MeSH Keywords: Carcinoma, Hepatocellular; Cell Proliferation; Dynamin III; Genes, p53
Background
Primary liver cancer is one of the most common malignant tumors that do great harm to human health, and we have witnessed an increasing incidence rate in recent years. Among those, approximate half of the cases and deaths were estimated to occur in China. Hepatocellular carcinoma (HCC) represents the major histological subtype of primary liver cancer, accounting for 70% to 85% of the total hepatocellular carcinoma burden worldwide. The most influential risk factor for HCC is chronic hepatitis, caused by hepatotropic viruses (HBV and HCV), but the molecular mechanisms leading to HCC have not been well characterized [1]. Surgical resection remains the most effective treatment for hepatocellular carcinoma patients, but only a small fraction of patients are eligible for radical resection due to the sophisticated tumorigenesis and development process of HCC. As a result, the majority of the patients have not had a significant improvement in survival time and quality. With the development of molecular biotechnology, myriads of dysregulated genes, due to genetic and epigenetic alterations or environmental factors, have been discovered to participate in tumorigenesis of hepatocellular carcinoma. It is of great importance, therefore, to further investigate the mechanism and function of the dysregulated genes to develop better diagnosis and treatment of hepatocellular carcinoma.
DNM3, a member of Dynamin family, is a GTPase that is essential in endocytosis and possessing mechanochemical properties of tabulating and severing membranes [2]. It had seldom been related to malignant diseases until Shen et al. first reported that DNM3 is methylated in tumor tissues compared to adjacent normal tissues in 62 HCC patients with a genome-wide DNA methylation profile [3]. Later, a study conducted by Inokawa et al. revealed that methylaton of DNM3 promoter downregulates its expression and is correlated with worse prognosis for hepatocellular carcinoma patients, thus suggesting that DNM3 may be a candidate novel tumor suppressive gene [2]. Although DNM3 seems to be important in hepatocellular carcinoma, there are hardly any reports discribing the biological function of DNM3 in hepatocellular carcinoma and, of course, its underlying mechanisms remain largely unknown. We therefore aimed to elucidate the specific role of DNM3 in hepatocellular carcinoma and explain the mechanisms by a series of in vitro experiments.
Material and Methods
Clinical specimens
Human hepatocellular carcinoma and matched adjacent normal tissues (n=68) were obtained from patients at the Second Affiliated Hospital of Anhui Medical University. All patients provided written informed consent. The study protocol was approved by the Ethics Committee of Anhui Medical University.
Cell lines and culture conditions
Human HCC cells (HepG2, Hep3B, HLE, Huh7, and PLC/PRF/5) cell lines and the human immortalized liver cell line HL-7702 were purchased from the Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, CA, USA) supplemented with 10% fetal bovine serum (FBS) (Biowest, France), 100 IU/ml penicillin, and 100 IU/ml streptomycin at 37°C under a humidified atmosphere in a 5% CO2 incubator.
Total RNA isolation and quantitative realtime polymerase chain reaction (qRT-PCR)
Total RNA was extracted from 1×105 cells by Trizol reagent (Invitrogen, Carlsbad, CA, USA). One microgram of RNA was converted into cDNA using the Reverse Transcription System (Promega) with oligo dT. QRT-PCR was carried out with a set of primers for DNM3 were 5′-AGTTCGCCTTGAGATTGAAGC-3′ (F), 5′-CGTGTGGGGAATAGACTCGTAAA-3′ (R), GAPDH were 5′-G GACCTGACCTGCCGTCTAG-3′ (F), 5′-GTAGCCCAGGATGCCCTTGA-3′ (R), respectively. The PCR reactions were carried out at 95°C for 2 min, then a 3-step cycle procedure was used (denaturation at 95°C for 10 s, annealing at 61°C for 20 s, and elongation at 72°C for 40 s) for 40 cycles, with a final extension at 72°C for 10 min. GAPDH served as the constitutive control. PCRs of each sample were conducted in triplicate. The relative expression levels were determined by the following equation:
Protein extraction and Western blot
Whole-cell proteins were extracted from cells using RIPA buffer containing Protease Inhibitor Cocktail (Pierce, Rockford, IL, USA) and protein quantification was performed by BCA Protein Assay kit (Pierce, Rockford, IL, USA). Cell extracts (80 ug) were separated by 12.5% sodium dodecyl sulfate polyacrylamide gel electrophoresis, and transferred onto polyvinylidene fluoride membranes. After blocking with 5% nonfat milk in TBST for 2 h at room temperature, the membrane was hybridized with rabbit anti-DNM3 (1:500; Ab3458, Abcam), rabbit anti-GAPDH (1:5000; Ab9485, Abcam), and mouse anti-P53 (1:500; Ab26, Abcam) and visualized by an enhanced chemiluminescence detection system (Amersham Bioscience, Piscataway, NJ, USA) following the manufacturer’s protocol.
Plasmid and transfection
DNM3 full-length cDNA was cloned from HL-7702 cDNA and then inserted into the pIRES2-EGFP vector. One day before transfection, HepG2 and Hep3B cells were plated onto 6-well plates. After 24 h, when the cell confluence was about 60%, transfection was performed with Lipofectamine 2000 (Invitrogen) following the manufacturer’s instructions. Cells that had been transfected with the constructed plasmid were then selected by antibiotic resistance in cell culture medium containing 1200 ug/ml G418 to obtain the cell strains with stable expression of DNM3.
Cell proliferation assay
The Cell Counting Kit-8 reagent (CCK-8, Dojindo, Japan) was used to assess the proliferation ability of HCC cells. Cells were seeded into 96-well plates (1.5×103 cells/well) and incubated for various timepoints (24, 48, 72, 96, and 120 h). Growth medium was then removed and replaced with 200 μl of DMEM medium containing 10% FBS and 20 μl of CCK-8 and the plates were incubated at 37°C for 2 h. The absorbance was read at 450 nm using a microplate reader (BIO-TEK, USA) according to the manufacturer’s instructions.
Colony formation assay
Cells were seeded into 6-well plates (500 cells/well) and culture medium was replaced every 3–4 days. After 14 days of culture in DMEM containing fetal calf serum, cell colonies were fixed with 4% paraformaldehyde for 10 min, stained with 0.1% crystal violet for 20 min, and photographed. The number of cell colonies with more than 50 cells was counted in each dish.
Analysis of cell cycle and apoptosis
For flow cytometrical analysis of cell cycle distribution, cells were collected and fixed in 70% ethanol overnight at 4°C, washed with PBS, and then treated with RNase A (50 mg/ml) and stained with propidium iodide (PI) (50 mg/ml) for 30 min in the dark. The stained cells we reanalyzed by flow cytometry (FACSCalibur, Becton-Dickinson). For apoptosis analysis, the cells were harvested and fixed, PI and AnnexinV-APC double staining was performed, and flow cytometry (FACSCalibur, Becton-Dickinson) was used to detect the apoptosis of cells. Annexin V-APC-positive and PI-negative cells were defined as undergoing apoptosis. Three replicate tests were performed in each group, and the average values of 3 groups were calculated.
Statistical analysis
Data were expressed as the mean ± standard deviation (v±s). The t test was used to compare the difference between 2 groups, and one-way ANOVA was used among multiple groups. P<0.05 was set as statistical significance and P<0.01 was considered highly significant.
Results
DNM 3 expression is downregulated in hepatocellular carcinoma tissues
We first tested DNM3 expression at mRNA level in 68 hepatocellular carcinomas and their corresponding normal liver tissues, showing that DNM3 mRNA was significantly downregulated in hepatocellular carcinoma tissues (Figure 1). Due to its remarkably discrete expression pattern between cancerous and normal tissues, we investigated the relationship between DNM3 expression and clinicopathological features of hepatocellular carcinoma. As shown in Table 1, downregulation of DNM3 was closely associated with tumor metastasis (P=0.001) and vein invasion (P=0.047), but no significant correlation was found with other features, including sex, age, differentiation, and tumor size (Table 1).
Figure 1.
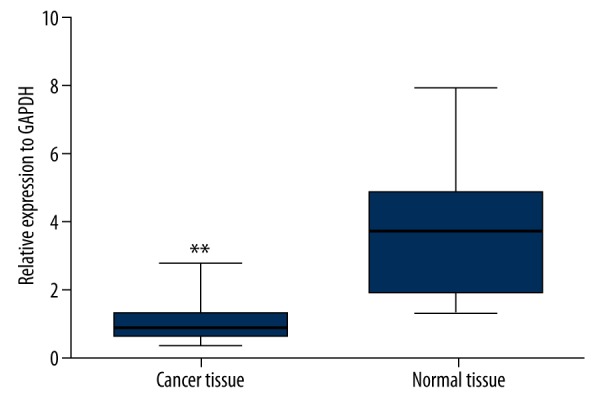
Expression of DNM3 in clinical hepatocellular carcinoma tissues. DNM3mRNA expression in hepatocellular carcinoma tissues and paired adjacent non-tumor tissues was examined by quantitative reverse transcription-polymerase chain reaction (qRT-PCR) and normalized to GAPDH. Bars represent the means of DNM3 relative expression in cancer tissues and non-tumor tissues, respectively.
Table 1.
Association between DNM3 expression and clinicopathological factors of hepatocellular carcinoma patients.
| Variables | Number of cases | DNM3 expression | P | |
|---|---|---|---|---|
| Low (n=52) | High (n=16) | |||
| Gender | ||||
| Male | 44 | 34 | 10 | NS |
| Female | 24 | 18 | 6 | |
| Age (years) | ||||
| ≥60 | 42 | 30 | 12 | NS |
| <60 | 26 | 22 | 4 | |
| Tumor differentiation | ||||
| Well to moderate | 26 | 18 | 8 | NS |
| Poor | 42 | 34 | 8 | |
| Metastasis | ||||
| Positive | 20 | 10 | 10 | 0.001 |
| Negative | 48 | 42 | 6 | |
| Vein invasion | ||||
| Presence | 28 | 18 | 10 | 0.047 |
| Absence | 40 | 34 | 6 | |
| HBsAg | ||||
| (−) | 29 | 22 | 7 | NS |
| (+) | 39 | 30 | 9 | |
| Tumor size | ||||
| ≤5 cm | 34 | 23 | 11 | NS |
| >5 cm | 34 | 29 | 5 | |
| No. of tumor nodes | ||||
| Single | 42 | 32 | 10 | NS |
| Multiple ≥2 | 26 | 20 | 6 | |
| Capsular formation | ||||
| Presence | 21 | 17 | 4 | NS |
| Absence | 47 | 35 | 12 | |
| Cirrhosis | ||||
| Positive | 41 | 30 | 11 | NS |
| Negative | 27 | 22 | 5 | |
| AFP (ng/mL) | ||||
| ≤50 | 27 | 23 | 4 | NS |
| >50 | 41 | 29 | 12 | |
NS – no significant difference between groups.
Overexpression of DNM3 inhibits hepatocellular carcinoma cell proliferation and colony formation
We determined the mRNA and protein expression of DNM3 in the 5 hepatocellular carcinoma cell lines and the normal human liver cell line HL-7702, using qRT-PCR and Western blot analysis, respectively. Our results showed that the mRNA and protein levels of DNM3 were significantly lower in the hepatocellular carcinoma cell lines than in the normal human liver cell line HL-7702 (Figure 2). We chose HepG2 and Hep3B, whose DNM3 expression was relatively low, for further investigation. After introducing DNM3 cDNA by pIRES2-EGFP, we established 2 stable transfected cell lines, HepG2/DNM3 and Hep3B/DNM3, whose DNM3 expression were dramatically upregulated, confirmed by qRT-PCR and Western blot (Figure 3A, 3B). The growth rate of HepG2/DNM3 or Hep3B/DNM3 was significantly slower than that of the cells transfected with negative control vector (Figure 3C). Moreover, we found a striking reduction of colony formation in HepG2/DNM3 and Hep3B/DNM3 compared with the control cells (Figure 4A, 4B). Thus, we concluded that DNM3 can inhibit proliferation of hepatocellular carcinoma cells, at least in vitro.
Figure 2.

The expression ofDNM3 in human hepatocellular carcinoma cell lines. (A) qRT-PCR analysis of DNM3 mRNA in 6 hepatocellular carcinoma cell lines. (B) Western blotting analysis of DNM3 protein in 6 hepatocellular carcinoma cell lines.
Figure 3.

Up-regulation of DNM3in human hepatocellular carcinoma cells attenuated hepatocellular carcinoma cell proliferation. (A) qRT-PCR analysis of DNM3 mRNA in HepG2/DNM3, Hep3B/DNM3, and their control groups. (B) Western blotting analysis of DNM3 protein in whole-cell lysates and conditioned medium using anti-DNM3 and anti-GAPDH antibody. (C) Up-regulation of DNM3 inhibited HepG2 and Hep3B proliferation.* P<0.05; ** P<0.01.
Figure 4.
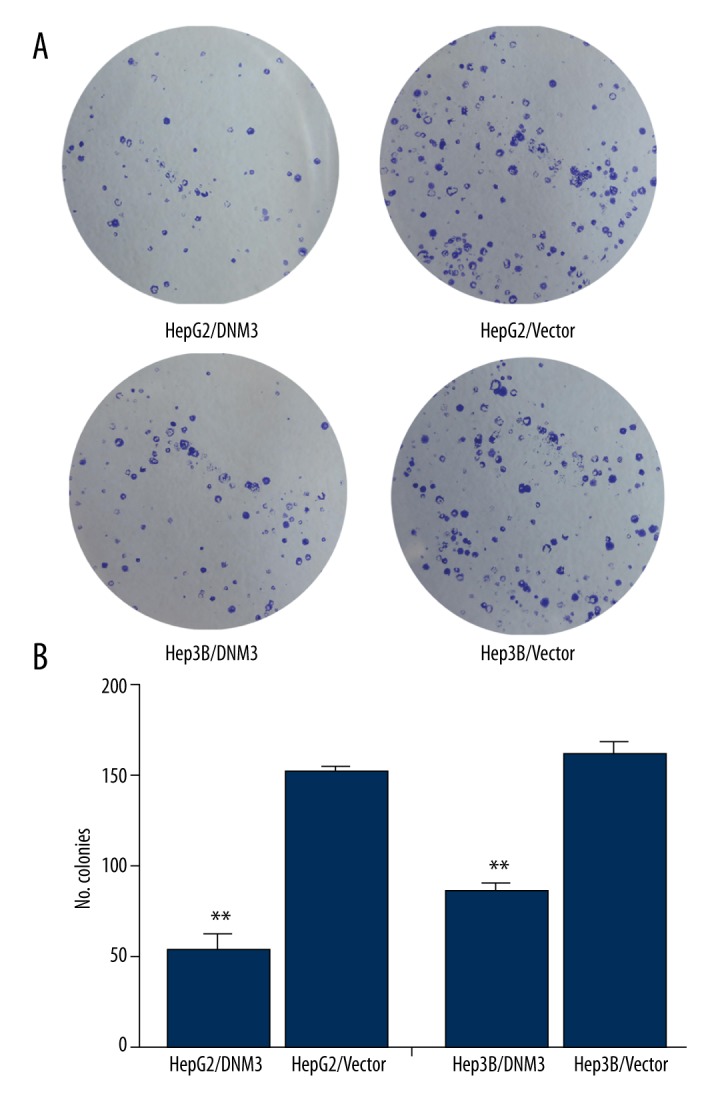
DNM3 inhibited the colony formation of hepatocellular carcinoma cells. (A, B) Representative images and quantification of colony formation assays in HepG2/DNM3, Hep3B/DNM3, and their control groups.* P<0.05; ** P<0.01.
Overexpression of DNM 3 induces G0/G1 cell cycle arrest and promotes apoptosis
In order to further demonstrate the mechanism underlying the anti-proliferative effect of DNM3, we tested its influence on cell cycle and apoptosis by flow cytometry. The results revealed that there was a higher proportion in G0/G1 phase and a correspondingly lower proportion in G2/M phase in HepG2/DNM3 or Hep3B/DNM3 compared with the control cells (Figure 5A, 5B). Similarly, HepG2/DNM3 and Hep3B/DNM3 also showed a higher rate of apoptosis (Figure 6A, 6B). Since p53 is a crucial regulator in both cell cycle and apoptosis pathway, we investigated whether p53 is involved in the anti-proliferative effect of DNM3. Western blot analysis showed that p53 was significantly upregulated in HepG2/DNM3 and Hep3B/DNM3 (Figure 6C); therefore, we concluded that DNM3 induces G0/G1 cell cycle arrest and promotes apoptosis though p53 signaling pathway.
Figure 5.

The effect of DNM3 on cell cycle distribution of hepatocellular carcinoma cells. (A) Proportion of cells in various phases of the cell cycle. (B) Representative histograms depicting cell cycle profiles of HepG2/DNM3, Hep3B/DNM3, HepG2/Vector, and Hep3B/Vector cells.* P<0.05; ** P<0.01.
Figure 6.

DNM3 induced hepatocellular carcinoma cell apoptosis. (A) Cells staining positive for Annexin V-APC and negative for propidium iodide (PI) were considered to have undergone apoptosis. (B) Representative histograms depicting apoptosis of each group of hepatocellular carcinoma cells. (C) P53 protein expression was evaluated by Western blot.* P<0.05; ** P<0.01.
Discussion
The present study confirmed the previous result that DNM3 is indeed downregulated in hepatocellular carcinoma tissues compared to adjacent normal tissues, and we demonstrated the correlation between DNM3 downregulation and clinical features. Abnormal hypermethylation of DNM3 promoter is likely to lead to its remarkable down-expression, which indicates it may serve as a tumor suppressor in hepatocellular carcinoma. We, for the first time, proved that overexpression of DNM3 can inhibit hepatocellular carcinoma cell proliferation by cck-8 growth assay and colony formation assay. Furthermore, flow cytometry proved that DNM3 can hinder mitosis progression by inducing G0/G1 cell cycle arrest; on the other hand, DNM3 also promotes apoptosis to a significant degree. In addition to its apparent phenotype, we found that p53 protein level is upregulated in DNM3-overexpressed cell lines. Given its important role in both cell cycle and apoptosis signaling pathway, we speculated that DNM3 might exert its anti-proliferative effect through upregulating p53. Similar to their potent factors [4–6], DNM3 seems to be an influential molecule in the process of tumorigenesis.
DNM3 is a member of the DNM family, which consists of 3 typical DNM proteins that are prevalent throughout inferior and superior organisms. DNMs are recognized as modulators of membrane scission events; however, these proteins also facilitate specific signaling transduction by virtue of distinct protein interactions. In this regard, Cao et al. found that the proline-rich domain (PRD) of DNM2 is able to bind the FAD-binding region of the endothelial nitric-oxide synthase (eNOS) reductase domain, which consequently activates eNOS and enhances NO production [7]. It was recently found that all of these 3 dynamics can potentiate NO production in rat renal inner medullary collecting ducts via protein-protein interaction with NOS1, suggesting DNM’s anti-tumor function may be closely associated with NO regulation [8].
Due to its gaseous and lipid-soluble properties at body temperatures, NO is extremely diffusive and can penetrate the biomembrane to exert its biological function with very little effort. On the other hand, NO is a very unstable free radical with a half-time of merely 3–5 s; it transforms into nitrate and nitrite, making the total concentration of nitrate and nitrite an important measurement of NO production.
NO is closely related not only to tumorigenesis and development, but also to chemotherapy and immunotherapy of various malignancies. It is widely believed that NO, depending on its concentration, has dual effects on tumors. Persistent low concentrations of NO can promote tumor proliferation; on the contrary, high concentrations of NO induce apoptosis by its cytotoxicity.
NO is synthesized under the catalysis of nitric oxide synthase (NOS) in organisms, so the release of NO relies upon, to a large extent, the expression and activity of NOS. NOS in mammals can be divided into 3 classes: nervous NOS (nNOS, NOS1), inducible NOS (iNOS, NOS2), and endothelial NOS (eNOS, NOS3). Both NOS2 and NOS3 exist in hepatocellular carcinoma tissues, and, due to their low activity, produce low concentrations of NO, thus promoting tumorigenesis and development.[9] On the other hand, overexpressing NOS can inhibit tumor growth and metastasis as a result of high concentrations of NO [10].
High concentrations of NO are also involved in the anti-tumor effect of many chemotherapeutic drugs. Yang et al. proposed that the enhanced mRNA expression of iNOS and total NO content might be responsible for the apoptotic and anti-proliferative effects of simvastatin in K562 cells [11]. By increasing the effect of NO, p53 effects can be enhanced in various conditions, such as in gastrointestinal tumors; thereby, progression of late adenoma to early cancer can be canceled. Therefore, DNM3 may also have antitumor effect at various gastrointestinal tumors [12,13]. Similarly, N-acetyl cysteine (NAC), an ROS scavenger, was proved to enhance imatinib-induced apoptosis of Bcr-Abl cells by endothelial nitric oxide synthase-mediated production of nitric oxide [14]. Notas et al. concluded that the stilbene resveratrol exerts antiproliferative and proapoptotic actions on hepatocellular carcinoma cells by increasing iNOS and eNOS expression, NOS activity, and NO production [15].
High concentrations of NO may exert anti-tumor effects through multiple mechanisms. (a) There is an unpaired electron in NO’s track, and many oxygen free radicals are generated after NO binding superoxide anions in the cell. These oxygen free radicals induce apoptosis by various approaches [16,17]. (b) Normally, p53 is easily degradable, but degradation is obstructed when p53 is phosphorylated on various sites during DNA damage and other malfunction periods. Accumulated p53 upregulates p21 and causes G1 phase cell cycle arrest. On the other hand, p53 also induces apoptosis through activating Bax or inhibiting Bcl-2 [18–20]. (c) Inactivating crucial metabolic enzymes [21], potentiating macrophages [22], and inhibiting platelet aggregation [23] may also attribute to NO’s anti-tumor capability.
In the present study, we observed the potent inhibitive effect of DNM3 on hepatocellular carcinoma cell growth, and the involvement of p53 was also confirmed by Western blot. We thus propose that the possible mechanism may be that DNM3 directly activates NOS with its proline-rich domain, and, consequently, NOS gives rise to a high concentration of NO. As a result, p53, activated by NOS, induces cell cycle arrest and promotes apoptosis of hepatocellular carcinoma cells. Further studies need to done to substantiate the increase of NOS activity and NO content, as well as to investigate other possible pathways after p53 activation. We believe that modulation of DNM3 and NO may prove to be an efficient method of hepatocellular carcinoma treatment.
Conclusions
DNM3 is significantly downregulated in hepatocellular carcinoma and can cause attenuation of cell growThis also induces G0/G1 cell cycle arrest and promotes apoptosis via induction of P53 expression, which might provide a new strategy for hepatocellular carcinoma treatment.
Footnotes
Source of support: Departmental sources
References
- 1.Jemal A, Bray F, Center MM, et al. Global cancer statistics. Cancer J Clin. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Inokawa Y, Nomoto S, Hishida M, et al. Dynamin 3: A new candidate tumor suppressor gene in hepatocellular carcinoma detected by triple combination array analysis. Onco Targets Ther. 2013;6:1417–24. doi: 10.2147/OTT.S51913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shen J, Wang S, Zhang YJ, et al. Genome-wide DNA methylation profiles in hepatocellular carcinoma. Hepatology. 2012;55(6):1799–808. doi: 10.1002/hep.25569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang D, Xu C, Li Z, et al. Protective action of hepatocyte growth factor on transforming growth factor beta-1-induced alpha-smooth muscle actin and extracellular matrix in cultured human peritoneal fibroblasts. Med Sci Monit. 2010;16(8):BR250–54. [PubMed] [Google Scholar]
- 5.Shao Z, Ji W, Liu A, et al. TSG101 silencing suppresses hepatocellular carcinoma cell growth by inducing cell cycle arrest and autophagic cell death. Med Sci Monit. 2015;21:3371–79. doi: 10.12659/MSM.894447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang BF, Lin S, Bai MH, et al. Effects of SSd combined with radiation on inhibiting SMMC-7721 hepatoma cell growth. Med Sci Monit. 2014;20:1340–44. doi: 10.12659/MSM.891355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao S, Yao J, Shah V. The proline-rich domain of dynamin-2 is responsible for dynamin-dependent in vitro potentiation of endothelial nitric-oxide synthase activity via selective effects on reductase domain function. J Biol Chem. 2003;278(8):5894–901. doi: 10.1074/jbc.M212546200. [DOI] [PubMed] [Google Scholar]
- 8.Hyndman KA, Musall JB, Xue J, Pollock JS. Dynamin activates NO production in rat renal inner medullary collecting ducts via protein-protein interaction with NOS1. Am J Physio Renal Physiol. 2011;301(1):F118–24. doi: 10.1152/ajprenal.00534.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jenkins DC, Charles IG, Thomsen LL, et al. Roles of nitric-oxide in tumor-growth. Proc Natl Acad Sci USA. 1995;92(10):4392–96. doi: 10.1073/pnas.92.10.4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le X, Wei D, Huang S, et al. Nitric oxide synthase II suppresses the growth and metastasis of human cancer regardless of its up-regulation of protumor factors. Proc Natl Acad Sci USA. 2005;102(24):8758–63. doi: 10.1073/pnas.0409581102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang YC, Xiao DW, Liu H, et al. Mechanism of simvastatin-induced K562 cell apoptosis. Pharmacology. 2009;84(4):191–95. doi: 10.1159/000235907. [DOI] [PubMed] [Google Scholar]
- 12.Isik A, Peker K, Firat D, et al. Importance of metastatic lymph node ratio in non-metastatic, lymph node-invaded colon cancer: A clinical trial. Med Sci Monit. 2014;20:1369–75. doi: 10.12659/MSM.890804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Isik A, Okan I, Firat D, et al. A new prognostic strategy for gastric carcinoma: Albumin level and metastatic lymph node ratio. Minerva Chir. 2014;69(3):147–53. [PubMed] [Google Scholar]
- 14.Rakshit S, Bagchi J, Mandal L, et al. N-acetyl cysteine enhances imatinib-induced apoptosis of Bcr-Abl(+) cells by endothelial nitric oxide synthase-mediated production of nitric oxide. Apoptosis. 2009;14(3):298–308. doi: 10.1007/s10495-008-0305-7. [DOI] [PubMed] [Google Scholar]
- 15.Notas G, Nifli AP, Kampa M, et al. Resveratrol exerts its antiproliferative effect on HepG2 hepatocellular carcinoma cells, by inducing cell cycle arrest, and NOS activation. Biochim Biophys Acta. 2006;1760(11):1657–66. doi: 10.1016/j.bbagen.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 16.Lala PK, Chakraborty C. Role of nitric oxide in carcinogenesis and tumour progression. Lancet Oncol. 2001;2(3):149–56. doi: 10.1016/S1470-2045(00)00256-4. [DOI] [PubMed] [Google Scholar]
- 17.Soliman MK, Mazzio E, Soliman KF. Levodopa modulating effects of inducible nitric oxide synthase and reactive oxygen species in glioma cells. Life Sci. 2002;72(2):185–98. doi: 10.1016/s0024-3205(02)02204-x. [DOI] [PubMed] [Google Scholar]
- 18.Lim S, Hung AC, Porter AG. Focused PCR screen reveals p53 dependence of nitric oxide-induced apoptosis and up-regulation of maspin and plasminogen activator inhibitor-1 in tumor cells. Mol Cancer Res. 2009;7(1):55–66. doi: 10.1158/1541-7786.MCR-08-0331. [DOI] [PubMed] [Google Scholar]
- 19.Gorospe M, Wang X, Holbrook NJ. Functional role of p21 during the cellular response to stress. Gene Expr. 1999;7(4–6):377–85. [PMC free article] [PubMed] [Google Scholar]
- 20.Amaral JD, Xavier JM, Steer CJ, Rodrigues CM. The role of p53 in apoptosis. Discov Med. 2010;9(45):145–52. [PubMed] [Google Scholar]
- 21.Gardner PR, Gardner AM, Brashear WT, et al. Hemoglobins dioxygenate nitric oxide with high fidelity. J Inorg Biochem. 2006;100(4):542–50. doi: 10.1016/j.jinorgbio.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 22.Vicetti Miguel RD, Cherpes TL, Watson LJ, McKenna KC. CTL induction of tumoricidal nitric oxide production by intratumoral macrophages is critical for tumor elimination. J Immunol. 2010;185(11):6706–18. doi: 10.4049/jimmunol.0903411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jurasz P, Sawicki G, Duszyk M, et al. Matrix metalloproteinase 2 in tumor cell-induced platelet aggregation: Regulation by nitric oxide. Cancer Res. 2001;61(1):376–82. [PubMed] [Google Scholar]