

Abstract
Oscillations of cytosolic Ca2+ concentration are a widespread mode of signalling. Oscillatory spikes rely on repetitive exchanges of Ca2+ between the endoplasmic reticulum (ER) and the cytosol, due to the regulation of inositol 1,4,5-trisphosphate receptors. Mitochondria also sequester and release Ca2+, thus affecting Ca2+ signalling. Mitochondrial Ca2+ activates key enzymes involved in ATP synthesis. We propose a new integrative model for Ca2+ signalling and mitochondrial metabolism in electrically non-excitable cells. The model accounts for (1) the phase relationship of the Ca2+ changes in the cytosol, the ER and mitochondria, (2) the dynamics of mitochondrial metabolites in response to cytosolic Ca2+ changes, and (3) the impacts of cytosol/mitochondria Ca2+ exchanges and of mitochondrial metabolism on Ca2+ oscillations. Simulations predict that as expected, oscillations are slowed down by decreasing the rate of Ca2+ efflux from mitochondria, but also by decreasing the rate of Ca2+ influx through the mitochondrial Ca2+ uniporter (MCU). These predictions were experimentally validated by inhibiting MCU expression. Despite the highly non-linear character of Ca2+ dynamics and mitochondrial metabolism, bioenergetics were found to be robust with respect to changes in frequency and amplitude of Ca2+ oscillations.
In most organisms, mitochondria play an important role in ATP production and act as Ca2+ stores, both functions of these organelles being tightly connected. Mitochondria sequester and release Ca2+, thereby affecting the shape, the frequency and the amplitude of the Ca2+ spikes in the cytosol1,2,3. In turn, increased mitochondrial Ca2+ ([Ca2+]m) linked to the transfer of Ca2+ from the cytosol to mitochondria during [Ca2+]c signals stimulates mitochondrial metabolism and allows the coupling of ATP supply with energy demand4,5,6,7.
At rest, [Ca2+]m and [Ca2+]c are similar, in the 100 nM range8. Upon cell stimulation by an agonist, inositol 1,4,5-trisphosphate (IP3) is produced and triggers cytosolic Ca2+ oscillations9. In non-excitable cells, these oscillations are due to a cyclical exchange of Ca2+ between the cytosol and the endoplasmic reticulum (ER), through the biphasic regulation of the IP3 receptor (IP3R) by cytosolic Ca2+. Fast activation and slow inhibition of the opening of the IP3R by Ca2+ indeed suffice to generate either Ca2+ oscillations in classical deterministic models or repetitive spiking, if noise is considered to play a predominant role in cellular Ca2+ dynamics10,11,12. Ca2+ entry into mitochondria occurs through a multistep mechanism. By extruding protons out of mitochondria, the respiratory chain creates a large inside-negative potential difference across the inner mitochondrial membrane. This ΔΨ, which is harnessed by the ATP synthase for the production of ATP, allows the Mitochondrial Calcium Uniporter (MCU) to transport Ca2+ inside mitochondria13,14. Ca2+ entry then depolarizes the mitochondria, thus reducing its own driving force. When [Ca2+]c returns to its basal value, extrusion of Ca2+ out of mitochondria occurs through both a Na+-Ca2+ exchanger (NCX) and a H+-Ca2+ exchanger, possibly identified as LETM1, although the contribution of this channel to mitochondrial Ca2+ transport is not yet firmly established15,16,17.
Uptake by mitochondria of pyruvate, the end product of cytosolic glycolysis, is at the onset of the oxidative phosphorylation cascade. A pyruvate dehydrogenase transforms substrates into acetyl-CoA which enters the Krebs cycle, also called the acid citric cycle or tricarboxylic acid cycle (TCA). This 9-step cycle converts the chemical energy of pyruvate into the reducing power of NADH. In addition, the activity of the malate-aspartate shuttle (MAS) also increases mitochondrial NADH18. NADH then feeds the oxidative phosphorylation pathway in which electrons are transferred and finally used to extrude protons and establish a proton gradient between the intermembrane space and the interior of mitochondria. This electrochemical source of energy is then harnessed by the F1Fo-ATPase to phosphorylate ADP into ATP. An increase in [Ca2+]m activates metabolism as pyruvate dehydrogenase and two rate-limiting enzymes of the TCA cycle, isocitrate dehydrogenase and α-ketoglutarate dehydrogenase, are up-regulated by Ca2+ 19. Thus, upon stimulation, the transfer of Ca2+ from the cytosol into mitochondria allows for the enhancement of mitochondrial ATP production by the F1Fo-ATPase. Cytosolic Ca2+ also directly influences mitochondrial metabolism as a component of the MAS, the aspartate-glutamate carrier (AGC), is stimulated by modest increases in cytosolic Ca2+ 18. Given the complexity and highly non-linear character of Ca2+ and mitochondrial dynamics, it is useful to resort to computational modelling to clarify their interplay. Several models have been proposed. Many of them focus on mitochondrial metabolism, but less on the effect of this metabolism on Ca2+ signalling20,21,22,23,24,25. Other models in contrast, extend computational descriptions of ER-cytosolic Ca2+ exchanges to incorporate Ca2+ handling by mitochondria. These models shed new light on important specific questions, such as the effect of mitochondria on the amplitude of the Ca2+ spikes26, the effect of glucose on the frequency of Ca2+ oscillations27, the modulation of NADH metabolism in pancreatic β-cells28, the mechanism responsible for Ca2+ wave propagation in mitochondrial suspensions29, or the importance of the distance between the ER membranes and mitochondria for their cross talk30.
Here we extend these previous studies to propose a model accounting for a variety of already published experimental observations centred on Ca2+ in non-excitable cells. These observations concern the effect of Ca2+ on mitochondrial metabolism, as well as changes in cytosolic Ca2+ dynamics occurring when the kinetic properties of the mitochondria are modified. The model is mainly built from the combination of previously published kinetic expressions. One of our aims is to clarify this complex issue by using simple expressions when possible and by motivating the resort to complex mathematical expressions when necessary. This is for example the case for the kinetics of the MCU that we analysed in details. We also found that a reversible Ca2+ leak between the cytosol and the mitochondria, which could correspond to the low conductance mode of the mitochondrial permeability transition pore, was necessary to account for a number of experimental observations. The phase relationship between Ca2+ peaks in the cytosol, the ER, and the mitochondria31 was investigated in details. The effect of modifying the activity of the MCU or of the NCX was next analysed in the simulations, with respect to previous experimental observations in HeLa cells32,33.
We found that increasing the activity of the MCU first increases and then decreases the frequency of Ca2+ oscillations. We tested this counter-intuitive prediction by silencing the MCU in HeLa cells. We then switched to the validation of the model concerning mitochondrial metabolism by comparing computationally obtained time series with the evolution of NADH4, voltage difference across the inner mitochondrial membrane (ΔΨ)34, and ATP in response to a single Ca2+ spike34 or in response to sustained Ca2+ oscillations4,35. This validated model could then be used to investigate the sensitivity of mitochondrial metabolism to changes in the frequency and amplitude of cytosolic Ca2+ spikes. Besides a strong robustness of mitochondrial metabolism with respect to the characteristics of the cytosolic Ca2+ changes, the model predicted that ATP synthesis by mitochondria is most efficient for frequencies and amplitudes of Ca2+ spikes usually observed experimentally.
Methods
Model
The processes considered in the model are schematized in Fig. 1. Fluxes and reactions are described by ordinary differential equations. As we neglect spatial aspects, we do not consider Ca2+ microdomains. More specifically, we do not explicitly incorporate MAM (mitochondria-associated ER membranes) in the model36 and focus on cellular average behaviour. All concentrations (including Michaelis-Menten constants) are thus averages on the volume of a given intracellular compartment: cytosol (c), endoplasmic reticulum (ER) or mitochondria (m). Thus, the fluxes are scaled by the appropriate volume ratio when necessary (see the legend of Table 1 for a more accurate description).
Figure 1. Schematic representation of the model for Ca2+ dynamics and mitochondrial metabolism.

Non standard abbreviations: Jo: rate of NADH oxidation; JPDH: rate of NADH production by the pyruvate dehydrogenase, followed by the Krebs cycle; JANT: rate of the ATP/ADP translocator; Jx: bidirectional Ca2+ leak between the cytosol and mitochondria (model hypothesis); JIPR: Ca2+ flux through the IP3 receptor. JAGC: rate of NADH production induced by the MAS NADH shuttle. See text.
Table 1. List of parameter values.
| Parameter | Definition | Value | Ref. |
|---|---|---|---|
| a1 | Scaling factor between NADH consumption and change in membrane voltage | 20 | This work |
| a2 | Scaling factor between ATP production by ATPase and change in membrane voltage | 3.43 | 28 |
| α | Volumic ratio between the endoplasmic reticulum and the cytosol | 0.1 | 40 |
| αc | Factor taking cytosolic ADP and ATP buffering into account | 0.111 | 27 |
| αm | Factor taking mitochondrial ADP and ATP buffering into account | 0.139 | 27 |
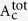 |
Total concentration in cytosolic adenine nucleotides | 2500 μM | 73 |
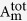 |
Total concentration in mitochondrial adenine nucleotides | 15000 μM | 28 |
| b | Ca2+ leak from the endoplasmic reticulum | 0.01 | This work |
| Cp | Mitochondrial inner membrane capacitance divided by F | 1.8 μM.mV−1 | 28 |
| δ | Volumic ratio between the mitochondria and the cytosol | 0.0733 | 28 |
| F | Faraday constant | 96480 C.mol−1 | |
| fc | Fraction of free over buffer-bound Ca2+ in the cytosol | 0.01 | 27 |
| fER | Fraction of free over buffer-bound Ca2+ in the ER | 0.01 | 27 |
| fm | Fraction of free over buffer-bound Ca2+ in mitochondria | 0.0003 | 27 |
| k1 | Rate constant of the Ca2+ flux through IP3R | 30 s−1 | This work |
| K1 | Dissociation constant for Ca2+ translocation by the MCU | 6 μM | This work |
| K2 | Dissociation constant for MCU activation by Ca2+ | 0.38 μM | 20 |
| Ka | Dissociation constant of Ca2+ from the activating site of the IP3R | 0.3 μM | 74 |
| KAGC | Dissociation constant of Ca2+ from AGC | 0.14 μM | 50 |
| Ke | Dissociation constant of ATP from SERCA pumps | 0.05 μM | 41 |
| kGLY | Velocity of glycolysis (empirical) | 450 μM.s−1 | 28 |
| Kh | Michaelis-Menten constant for ATP hydrolysis | 1000 μM | This work |
| kHYD | Maximal rate of ATP hydrolysis | 100 μM.s−1 | This work |
| Ki | Dissociation constant of IP3 binding from its receptor | 1 μM | 54 |
| k− | Rate constant of Ca2+ dissociation from the inactivating site of the IP3 receptor | 0.02 s−1 | 74 |
| ko | Rate constant of NADH oxidation by ETC | 600 μM.s−1 | 28 |
| Kp | Dissociation constant of Ca2+ from SERCA | 0.35 μM | 54 |
| k+ | Rate constant of Ca2+ binding to the inhibiting site of the IP3R | 20 μM−4.s−1 | This work |
| kx | Rate constant of bidirectional Ca2+ leak from mitochondria | 0.008 s−1 | This work |
| L | Allosteric equilibrium constant for uniporter conformations | 50 | 21 |
| na | Hill coefficient of Ca2+ binding to the activating site of the IP3R | 3 | 74 |
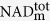 |
Total concentration of mitochondrial pyridine nucleotides | 250 μM | This work |
| ni | Hill coefficient of Ca2+ binding to the inhibiting site of the IP3R | 4 | 74 |
| p1 | Voltage dependence coefficient of MCU activity | 0.1 mV−1 | This work |
| p2 | Voltage dependence coefficient of NCX activity | 0.016 mV−1 | This work |
| p3 | Voltage dependence coefficient of calcium leak | 0.05 mV−1 | This work |
| p4 | Voltage dependence coefficient of AGC activity | 0.01 mV−1 | This work |
| q1 | Michaelis-Menten-like constant for NAD+ consumption by the Krebs cycle | 1 | 28 |
| q2 | S0.5 value for activation the Krebs cycle by Ca2+ | 0.1 μM | This work |
| S0.5 value for indirect inhibition of the AGC by cytosolic Ca2+ | 0.1 μM | This work | |
| q3 | Michaelis-Menten constant for NADH consumption by the ETC | 100 μM | 28 |
| q4 | Voltage dependence coefficient 1 of ETC activity | 177 mV | 28 |
| q5 | Voltage dependence coefficient 2 of ETC activity | 5 mV | 28 |
| q6 | Inhibition constant of ATPase activity by ATP | 10000 μM | 28 |
| q7 | Voltage dependence coefficient of ATPase activity | 190 mV | 28 |
| q8 | Voltage dependence coefficient of ATPase activity | 8.5 mV | 28 |
| q9 | Voltage dependence of the proton leak | 2 μM.s−1.mV−1 | 28 |
| q10 | Rate constant of the voltage-independent proton leak | −30 μM.s−1 | 28 |
| R | Perfect gas constant | 8315 mJ.mol−1.K−1 | |
| T | Temperature | 310.16 K | |
| VANT | Rate constant of the adenine nucleotide translocator | 5000 μM.s−1 | 28 |
| VAGC | Rate constant of NADH production via malate-aspartate shuttle | 25 μM.s−1 | This work |
| VF1FO | Rate constant of the F1FO ATPase | 35000 μM.s−1 | 28 |
| VMCU | Rate constant of the MCU | 0.0006 μM.s−1 | This work |
| VNCX | Rate constant of the NCX | 0.35 μM.s−1 | This work |
| Vp | Rate constant of the SERCA pumps | 120 μM.s−1 | 54 |
Fluxes are defined with respect to the volumes of the cytoplasm, the ER or the mitochondria as indicated in the text. Thus, as described in Fall and Keizer27, the values of the rate constants obtained experimentally and expressed in nmol.(mg.s)−1 are multiplied by the total protein amounts for each compartment and divided by the volume of the compartment.
We adopt a fully deterministic approach, which is appropriate to obtain predictions about the average effect of the different individual fluxes on Ca2+ dynamics37 and mitochondrial metabolism. As described in previous approaches27,28, there is no distinction between the inter-membrane mitochondrial space and the cytosol, as the outer membrane is highly permeable to Ca2+ and H+. The model is defined by 7 evolution equations and 4 conservation relations given here below (Eqs. (1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11)). The fluxes appearing in these equations are defined by Eqs. (12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23).
Evolution equations:
Cytosolic Ca2+ concentration
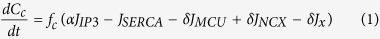
Using the framework of rapid buffering approximation38, fc is the Ca2+ buffering capacity of the cytosol. The different volumes of the compartments are taken into account via α = VER/Vc and δ = Vm/Vc. Jx is a bidirectional Ca2+ leak (see the ‘Results’ section for the rationale of this term). Ca2+ exchanges with the extracellular medium are not taken into account since they do not play a major role in the interplay between cytosolic and mitochondrial Ca2+ during the early spikes following stimulation31. Also, we do not consider Ca2+ fluxes through the mitochondrial Ca2+/H+ exchanger. Although this is not firmly established17, the Ca2+/H+ exchanger is thought to correspond to the LETM1 protein39 and the levels of expression of LETM1 appear to be inconsequential for mitochondrial Ca2+ export during physiological stimulation of HeLa cells17.
Mitochondrial Ca2+ concentration
 fm is the Ca2+ buffering capacity of mitochondria.
fm is the Ca2+ buffering capacity of mitochondria.Fraction of inactivated IP3 receptors
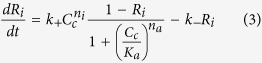 This equation reflects the biphasic regulation of the IP3 receptors by Ca2+
10,40.
This equation reflects the biphasic regulation of the IP3 receptors by Ca2+
10,40.Mitochondrial NADH concentration
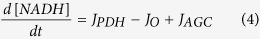
Mitochondrial ADP concentration
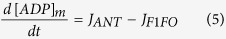
Cytosolic ADP concentration
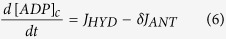
Voltage difference across the inner mitochondrial membrane (positive corresponding to an excess of positive charges in the cytosol)
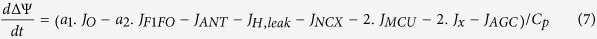 Cp is a constant that includes both the membrane capacitance and the Faraday constant.
Cp is a constant that includes both the membrane capacitance and the Faraday constant.Conservation of intracellular Ca2+
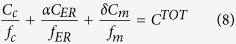 fER is the Ca2+ buffering capacity of the ER.
fER is the Ca2+ buffering capacity of the ER.Conservation of total (oxidized and reduced) NADH
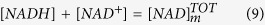
Conservation of di- and trisphosphorylated adenine nucleotides in mitochondria
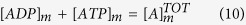
Conservation of di- and trisphosphorylated adenine nucleotides in the cytosol
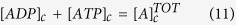 |
The conservation relations described by Eqs. (10) and (11) do not always hold as adenine nucleotide/Mg2+ transporters, known as SCaMCs, mediate net transfer of ADP and/or ATP to the mitochondrial matrix. However, on the short time scales investigated in this study, we expect this transporter to have a minor role since SCaMCs are much slower than the adenine nucleotide translocases (ANT) and have a low affinity for cytoplasmic Ca2+ 18.
Kinetic expressions for fluxes and reaction rates
 |
where 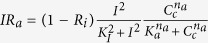
This is a classical expression for the Ca2+ flux through the IP3 receptor that is rapidly activated at low cytosolic Ca2+ and more slowly inhibited at high cytosolic Ca2+ 40. It is defined with respect to the volume of the ER. I stands for the concentration of IP3 that is taken as a parameter whose value directly reflects the level of cell stimulation.
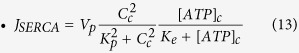 |
It represents the Ca2+ flux through an unidirectional SERCA ATPase, that transports Ca2+ from the cytosol into the ER, against the concentration gradient, using the energy provided by the hydrolysis of ATP41. It is defined with respect to the volume of the cytosol.
We now describe the kinetic expressions for mitochondrial Ca2+ fluxes and metabolism. Most of these processes have been already individually described by complex models quantitatively accounting for energetic-redox mechanisms under various (patho) physiological conditions. Assembling these models would lead to an extremely complex network. Our aim here is to build an intelligible model, centred around Ca2+ signalling and its interplay with mitochondrial metabolism. The kinetic expressions used are thus a trade-off between complexity and appropriate conceptualization.
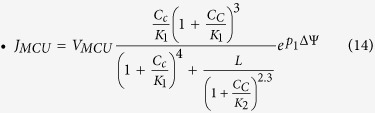 |
JMCU stands for the flux of Ca2+ from the cytosol into mitochondria that occurs through the MCU, defined with respect to the mitochondrial volume. This flux is cooperatively stimulated by cytosolic Ca2+ through the MICU1 subunit42,43. We based this kinetic equation on that proposed by Magnus and Keizer20. Although this expression was proposed before the discovery44 of the molecular identity of the MCU (previously called the Ca2+ uniporter), it is in agreement with the observed properties of this channel. Supplementary Fig. S1 shows the flux through the MCU as a function of cytosolic Ca2+ concentration (black curve). This curve, entirely based on the expression and the parameter values proposed by Magnus and Keizer20 is in very good agreement with the results of Csordás et al. (2013) on the MCU42. If the activation by cytosolic Ca2+ is removed (K2 → ∞), the curve (in red) becomes less sigmoidal and is shifted to the left, as observed for the MICU1 knock-down. As shown by the blue curve, the simplified expression proposed by Bertram et al.28 does not accurately reproduce the original expression if [Ca2+] > 1 μM. Eq. (14) is based on the Monod-Wyman-Changeux formalism for allosteric enzymes45 and further assumes that Ca2+ cannot bind to the transporter when subunits are in the so-called “tense” form. Concerning the dependence of this rate on the mitochondrial potential, we have simplified the expression used by Magnus and Keizer and simply used Eyring’s theory. Thus, the rate constant of Ca2+ transport increases with the potential difference across the mitochondrial membrane in an exponential manner. As explained in the ‘Results’ section, this change was moreover required to account for the observation that energising mitochondria leads to a decreased frequency of Ca2+ oscillations1,46.
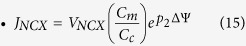 |
This is the expression proposed by Bertram et al.28 for the rate of Ca2+ extrusion out of mitochondria mediated by the Na+/Ca2+ exchanger, defined with respect to the mitochondrial volume. This channel exchanges 1 Ca2+ for 3 Na+ and is thus electrogenic. It is assumed that the cytosolic Na+ concentration remains constant and that channel activity is favoured by a large ratio of Ca2+ concentrations between the cytosol and mitochondria. As reported previously47, we did not consider a possible reverse mode (Cc towards Cm). The dependency on the potential has the same form as for the MCU.
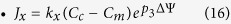 |
This flux, defined with respect to the mitochondrial volume, was not considered in previous models. Its molecular nature remains to be fully identified (see ‘Results’). We found necessary to consider this flux to account for many experimental results and, in particular, for the fact that Cm does not drop in MCU-knocked down cells42. The existence of this flux also accounts for the observation that mitochondria take up Ca2+ from the cytosol at nanomolar concentrations, at which the MCU is inactive15. It is chosen bidirectional to allow for the existence of sustained Cc oscillations when the NCX is totally inhibited33. If the flux was unidirectional, NCX inhibition would otherwise lead to Ca2+ accumulation in mitochondria and arrest of oscillations. As explained in the Results section, we hypothesize that this flux may reflect the low conductance state of the mitochondrial Permeability Transition Pore (mPTP)48.
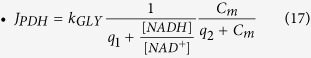 |
This expression is taken from Bertram et al.28, who proposed a simplified mathematical expression showing the same behaviour as that derived by Magnus and Keizer (1998)21 and taking into account the activation of this enzyme through Ca2+-sensitive reversible phosphorylation. This rate not only accounts for the Pyruvate DeHydrogenase(PDH)-catalysed reaction but gathers the glycolytic pathway (kGLY) and the Krebs cycle. The Krebs cycle reduces NAD+ into NADH, hence the dependency on the adenine nicotinamide ratio. [NAD+] is computed from the conservation relationship (Eq. (9)). We have modified the value of 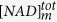 with respect to Bertram et al.28 to account for experimental observations in HeLa cells49. The last factor in Eq. (17) reflects the activation of both the PDH dehydrogenase and the Krebs cycle by Ca2+, the latter one occurring at the levels of both the isocitrate- and the α-ketoglutarate dehydrogenases. In agreement with experimental data19, the KD value for activation by Ca2+ (q2) is taken equal to 0.1 μM.
with respect to Bertram et al.28 to account for experimental observations in HeLa cells49. The last factor in Eq. (17) reflects the activation of both the PDH dehydrogenase and the Krebs cycle by Ca2+, the latter one occurring at the levels of both the isocitrate- and the α-ketoglutarate dehydrogenases. In agreement with experimental data19, the KD value for activation by Ca2+ (q2) is taken equal to 0.1 μM.
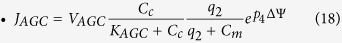 |
The aspartate-glutamate carrier is part of the MAS NADH shuttle. The two mammalian carriers, aralar and citrin, are activated by moderate cytosolic Ca2+ increases (100 nM < S0.5 < 350 nM)18,50. This is taken into account by the second factor in Eq. (18). In many cases, the activation of the MAS pathway is not maintained at Ca2+ levels at which the MCU becomes active, although the cross-talk between the Ca2+ activation of MAS and the mitochondrial dehydrogenases may vary from tissue to tissue. By activating the Krebs cycle, mitochondrial Ca2+ increase would lead to a decrease in the amount of α-ketoglutarate, a key-metabolite of the MAS18,50. To take this limiting step into account in the model, we considered in Eq. (18) that the activity of AGC is inhibited by mitochondrial Ca2+ with a threshold value q2 that is the same as the threshold for activation of the Krebs cycle by mitochondrial Ca2+. As the AGC is electrogenic, JAGC also appears in Eq. (7).
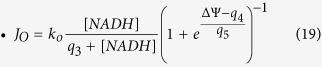 |
Downstream the Krebs cycle, NADH is then oxidized in the Electron Transport Chain (ETC) to extrude protons from mitochondria. JO thus represents both the rate at which NADH is oxidized (Eq. (4)) and the rate at which H+ are extruded. In the rate expression for JO (Eq. (19)), the change of rate with variation in the proton concentration gradient is not considered as in Magnus and Keizer (1997) and Bertram et al. (2006)20,28. Although protons are not considered explicitly in the model, they appear in the evolution equation of the membrane potential (protons extrusion leads to an increase in voltage). In Eq. (7), JO is multiplied by a1 to scale the NADH production into a change in voltage due to the proton flux. Eq. (19) is that proposed by Bertram et al.28 as a simplification of that initially proposed by Magnus and Keizer (1998)21. The exponential factor contains the dependency on both the membrane voltage and the proton gradient.
 |
This rather complex expression20 describes the activity of a translocator that needs to bind either ATP or ADP on both sides for a conformational change of the carrier to occur. This change allows for a ligand exchange. Thus, 4 possible combinations of ligands are possible (ATPc-ATPm; ATPc-ADPm; ADPc-ATPm; ADPc-ADPm). The α’s stand for the fact that only a fraction of nucleotides has access to the transporter. ADP and ATP are negatively charged (3- and 4-, respectively), which explains the potential dependence of this flux. It is defined with respect to the mitochondrial volume. This equation has been erroneously transcribed and simplified in previous publications51. We refer the readers to this reference for a discussion about the usefulness of keeping the original expression.
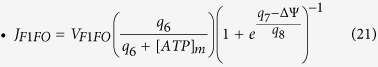 |
This is the rate of ATP synthesis by the F1Fo-ATPase. This expression, proposed by Bertram et al.28 accounts for a weak decrease in rate with increasing ATPm (i.e. with decreasing ADPm), but for a steep sigmoidal dependency on the mitochondrial potential. ATP synthesis is driven by the proton flux from the cytosol into the mitochondria, which depolarizes the membrane. Thus JF1FO also enters the evolution equation for ΔΨ (Eq. (7)), with the scaling factor a2.
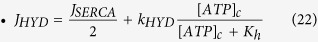 |
This term represents the rate of ATP consumption in the cytosol. In contrast with previous models, we incorporate the link between Ca2+ activity and ATP consumption in the cytoplasm. SERCA pumps (Eq. (13)) are Ca2+-ATPases transporting 2 Ca2+ ions for one molecule of ATP hydrolysed52. The second term of Eq. (22)) encompasses the other ATP-consuming processes in the cytoplasm.
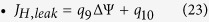 |
This simplified expression for the ohmic mitochondrial proton leak is taken from Bertram et al.28
Parameter values are listed in Table 1. These values are the same as those proposed in previous studies when we used kinetic expressions previously published. For new expressions, values were fitted manually to get reasonable agreement with available experimental data. To remain as coherent as possible, we chose data obtained in HeLa cells when available. The full system of equations is simulated using the software package XPPAUT developed by Bard Ermentrout53. Bifurcation diagrams are obtained numerically, by solving the differential equations with the MATLAB solver ode23, or ode23tb when using pulses.
Experiments
Cell lines and silencing MCU expression
HeLa cells were from ATCC and were grown in RPMI medium containing 5% fetal calf serum in a 5% CO2 incubator. Cells were transfected with a non-targeting siRNA (scrambled) or with two pooled siRNA sequences against the MCU44 using the lipofectamine RNAi Max (Invitrogen) following the manufacturer’s instructions. For all experiments MCU mRNA silencing (>70%) was confirmed at 48 h post-siRNA. The rabbit polyclonal antibody against the MCU and Cyclosporin A were from Sigma and siRNAs against the MCU were from Eurogentec.
Western-Blot analysis
The equivalent of 4 × 105 cells were scraped in Laemmli-loading buffer. Samples were subjected to Western-blot analysis using the indicated primary antibody followed by anti-rabbit HRP-conjugated antibody. Detection was performed using ECL plus reagent (GE Healthcare Biosciences).
Calcium fluorescence microscopy imaging
Analysis of Ca2+ variations was performed by loading cells with 3 μM Fluo-4-AM (Invitrogen) in EM buffer containing 120 mM NaCl, 7 mM KCl, 1.8 mM CaCl2, 0.8 mM MgCl2, 5 mM glucose, 25 mM HEPES pH 7.3, as described previously54. Samples were analysed at 33 °C on an inverted Nikon fluorescence microscope, and images were captured every 3 s, using an EM-CCD camera (Hamamatsu), digitized and integrated in real time by an image processor (Compix). All images were corrected for background fluorescence.
Results
Ca2+ dynamics
Ca2+ oscillations occur in the cytosol, the ER and the mitochondria with well-defined phase relationships31. We first compared the behaviour of the model defined by Eqs. (1) to (7), with respect to this relationship. Figure 2A shows the evolution of the free Ca2+ concentrations in the cytosol (black), the ER (red) and the mitochondria (blue) in the presence of first a relatively high (from 0 to 200 s), followed by a low IP3 concentration. The period of oscillations is of the order of 30 s, while their half-width is ~1/5 of the period. A detailed view of one peak is shown in Fig. 2B, where time 0 corresponds to the minimum of Ca2+ in the cytosol, during sustained oscillations. From this starting point, Ca2+ slowly increases both in the cytosol and in the ER, while Cm is still decreasing. Thus, release of mitochondrial Ca2+ is responsible for the increase in Cc, which itself allows the replenishment of the ER. The rise in Cc then stimulates the IP3R and CER starts decreasing fast. Cm only starts increasing during the fast rising phase of the cytosolic Ca2+ peak. Then, Cc reaches its maximum, slightly before CER gets to its minimum value (vertical lines in Fig. 2B). As long as Cc is large, the ER refills fast. A change in slope occurs when the rate of CER increase is imposed by the rate of Ca2+ release from the mitochondria into the cytosol. Concerning Cm, it keeps accumulating quite late after the peak in Cc. Cm finally decreases until the onset of the new cytosolic Ca2+ spike, but, interestingly, Cm does not recover to basal values during the interspike period. This mechanism is in agreement with the observations of Ishii et al.31 who observed the same sequence. Thus, mitochondria play an important role in triggering the cytosolic Ca2+ spike as they continuously release Ca2+ during the silent phase of cytosolic Ca2+ oscillations. This effect of mitochondria on Ca2+ dynamics is confirmed in the model by the fact that in the absence of mitochondria (JMCU = JNCX = Jx = 0), the frequency of cytosolic Ca2+ oscillations is lower, but the amplitude is higher. In addition to these modifications, mitochondria also alter the shape of the cytosolic Ca2+ peak. Indeed, when JMCU = JNCX = Jx = 0, the rate of cytosolic Ca2+ decrease is significantly lower (see Supplementary Fig. S2). This is due to the fact that mitochondria slowly release the Ca2+ accumulated during the rising part of the oscillation. This more asymmetric shape of the spike is in agreement with experimental observations in HeLa cells31.
Figure 2. Dynamics of Ca2+ exchanges between cytosol, ER and mitochondria.

Curves show the simulated changes in Ca2+ concentrations in the cytosol (black), the mitochondria (blue) and the ER (red). (A) Sustained oscillations triggered by 1 μM IP3, followed by the return to a non-stimulated situation (IP3 = 0.1 μM). (B) Detail of one Ca2+ peak occurring when IP3 = 1 μM allowing a detailed comparison of the phase relationships in the model and in the experiments of Ishii et al. (2006)31. Parameter values are listed in Table 1.
In response to a sudden decrease in IP3 (Fig. 2A), oscillations rapidly stop in the cytosol, while ER Ca2+ evolves towards a high steady value and mitochondrial Ca2+ slowly decreases towards its resting state, close to the basal cytosolic Ca2+ concentration. This slow decrease in mitochondrial Ca2+ down to a basal concentration of about 100 nM is in agreement with observations in HeLa cells17,31 and chromaffin cells8. Reported concentrations of mitochondrial Ca2+ during oscillations are highly variable, depending on the study: some experiments report levels in the μM range8,55 while others describe peaks in Cm reaching several tens of μM2,44. It is highly plausible that results are influenced by the nature of the probe used to monitor Ca2+ changes in the mitochondria56. However, intercellular and subcellular heterogeneity in mitochondrial Ca2+ signalling has been observed using the CEPIA probe in HeLa cells57 suggesting that specific characteristics of mitochondria could also explain this difference. In agreement with this hypothesis, we found that the value taken for the Ca2+ buffering capacity of mitochondria (fm) has a very strong influence on the values of Cm associated with changes in Cc (Fig. 3A). As shown in Fig. 3B, this factor does not much affect Ca2+ changes in the cytoplasm. Thus, different Ca2+ buffering capacities in mitochondria due either to intrinsic cell variability or to different concentrations of probes may explain, at least in part, the large range of reported Cm.
Figure 3. The Ca2+ buffering capacity of mitochondria modifies the amplitude of Ca2+ oscillations.

(A) Effect on mitochondrial Ca2+. (B) Effect on cytosolic Ca2+. Curves indicate the minima and maxima reached during oscillations. Parameter values are listed in Table 1. IP3 = 1 μM.
In a next step, we altered the rates of the individual Ca2+ fluxes between mitochondria and the cytosol. We first analysed the effect of changing the maximal rate of the NCX. A complete inhibition of the activity of the NCX leads to a decrease in the frequency of the Ca2+ spikes (Supplementary Fig. S3). This inhibition indeed leads to a slower Ca2+ release from mitochondria and hence to a delayed priming of the IP3R to generate the cytosolic Ca2+ spike. This decrease in frequency has been observed in HeLa cells stimulated by histamine in the presence of the inhibitor of the mitochondrial Na+-Ca2+ exchanger CGP3715733. The inhibition of the exchanger practically does not change the bifurcation diagram as a function of IP3, thus confirming that Ca2+ release from mitochondria modulates, but does not cause or inhibit, cytosolic Ca2+ oscillations.
We next modified the rate constant of the MCU, which is the mitochondrial Ca2+ flux opposite to the NCX. As shown in Fig. 4A (black curve), the model predicts a biphasic effect: increasing the activity of the MCU first increases then decreases the frequency of oscillations. At large rates, raising VMCU slightly slows down the oscillations (Fig. 4A, black curve) because mitochondria buffer Ca2+ changes in the cytosol. This slows down the ER-cytosol Ca2+ exchanges. Surprisingly, at moderate rates the frequency of oscillations decreases when decreasing the rate of Ca2+ entry into mitochondria, as also exemplified in Fig. 4B. Indeed, in the range of low values of VMCU, Ca2+ accumulation in the mitochondria becomes so limited that the subsequent release of Ca2+ through the NCX is too weak to boost the IP3 receptor. In other words, the amount of Ca2+ entering via the MCU becomes the limiting factor in mitochondrial Ca2+ handling. In agreement with this explanation, the frequency of Ca2+ oscillations is a monotonous increasing function of VMCU when the rate constant of the NCX is increased (red curve in Fig. 4A); in these conditions indeed, mitochondria never slow down the cytosol-ER Ca2+ exchanges as the Ca2+ sequestered by mitochondria is always rapidly released into the cytosol. This complex relationship between the frequency of Ca2+ oscillations and the activity of the uniporter accounts for the observation that activators of the MCU (PPT and kaempferol) stimulate and inhibit the oscillatory behaviour in HeLa cells and in fibroblasts32. Interestingly, it is reported in the same studies that the inhibitory effect (i.e. decrease in frequency) is more pronounced if the activity of the NCX is reduced.
Figure 4. The rate of Ca2+ entry into mitochondria alters cytosolic Ca2+ oscillations.

(A) Relationship between the frequency of oscillations and the rate constant of the MCU. The rate constant of the NCX is the default value (black curve, VNCX = 0.35 μM.s−1) or is increased (red curve, VNCX = 1 μM.s−1). (B) Effect of the rate constant of the MCU on cytosolic Ca2+ oscillations, as predicted by the model. The black curve shows oscillations for the default value (VMCU = 0.0006 μM.s−1) given in Table 1, while the red curve shows oscillations obtained when VMCU = 0. (C,D) Measurement of Ca2+ variations in control (C) or MCU-silenced HeLa cells (D). Cells loaded with Fluo4 were perfused with 3 μM histamine for the time shown by the black line. Experiments are representative of more than five trials. See also Supplementary Fig. S4.
That decreasing the rate of Ca2+ entry into mitochondria through the MCU slows down Ca2+ oscillations appeared as a rather counter-intuitive prediction of the model. We tested this prediction experimentally using HeLa cells transiently transfected with scrambled siRNA or siRNA against MCU (see Supplementary Fig. S4). Indeed, it has been shown that inhibition of the MCU allows to selectively modify the Ca2+ uptake capacity of mitochondria without interfering with bioenergetic properties or organelle structure44,58. The results of a typical experiment are shown in Fig. 4C (control) and D (siRNA anti-MCU). In qualitative agreement with the model, the average period of Ca2+ oscillations in response to stimulation by 3 μM histamine increases from 31 s (±19 s, n = 63) to 97 s (±39 s, n = 28) in the cells that do not express the MCU. Experiments thus confirmed that MCU inhibition leads to a decrease in the frequency of Ca2+ oscillations in HeLa cells. Our interpretation of this observation is that, in the absence of MCU, the very low level of mitochondrial Ca2+ does not allow the release of mitochondrial Ca2+ between two ER-generated spikes, release that is responsible for the ‘pacemaker-like’ Ca2+ trigger of the IP3 receptor occurring in control cells.
The increase in period observed in the experiments is, however, much larger than that predicted by the model. We thus investigated if the MCU siRNA treatment shifts the effective dose response to histamine. Supplementary Table S1 shows that this was indeed the case: at 0.3 μM histamine, only 3% of MCU siRNA treated cells displayed a Ca2+ response while 39% of control cells oscillated. However, this shift is not the only reason for the observed decrease in period in the cells inhibited for the MCU. Indeed, the period of Ca2+ oscillations at the highest histamine dose in anti-MCU siRNA treated cells (10 μM histamine; 98s) is still larger than that seen at the lowest histamine dose in Scrbl siRNA treated cells (0.1 μM histamine; 50 s). Thus, oscillations in the absence of MCU at maximal stimulation, which corresponds to the smallest possible period, are still slower than those obtained at low levels of stimulation in the presence of MCU, pointing to a specific role of the MCU in the control of the frequency of oscillations.
Possible nature of the bidirectional Ca2+ flux between the mitochondria and the cytosol
In the model, there is another flux allowing a Ca2+ exchange between the mitochondria and the cytosol (Jx, see Eqs. (2) and (16)). This flux was initially incorporated in the model to account for the observation that mitochondria can still take up Ca2+ when the MCU is inactive42. We found, however, that assuming a reversible flux, the direction of which depending on the electrochemical gradient, led to a better agreement with experimental results as oscillations are maintained when the NCX is inhibited33, indicative that Ca2+ is extruded from mitochondria by another pathway. For both directions, best agreement is obtained when assuming a low conductance, proportional to the Ca2+ concentration gradient. A plausible candidate for this flux is the low conductance mode of the mitochondrial permeability transition pore (PTP)59. We analysed the impact of this flux on the simulated Ca2+ oscillations. As this flux is bidirectional, it could either boost or slow down Ca2+ oscillations. Figure 5A shows that its suppression always decreases the frequency of Ca2+ oscillations, as for the NCX. Interestingly, in hepatocytes, an inhibition of the PTP by cyclosporin A (CSA) results in an increase in the interspike interval60, in agreement with the behaviour of the model. Experiments also reveal a rise in the mitochondrial membrane potential60, which is also observed in the model provided that the flux of protons is reduced, reflecting the fact that the PTP is also permeable to protons.
Figure 5. Analysis of the bidirectional Ca2+ flux between the cytosol and the mitochondria, Jx.

(A) Effect of altering Jx on the simulated Ca2+ oscillations. The black curve shows oscillations for the default value (kx = 0.008 s−1) given in Table 1, while the red curve shows oscillations obtained when kx = 0. In the latter case, the period of oscillations is slightly increased. IP3 = 1 μM. (B) Experimental investigation of the effect of inhibiting the mPTP with CSA (1 μM) on the period of Ca2+ oscillations in Hela cells. n = 64, 18, 16, 14 for control cells (0.3 and 10 μM histamine) and CSA-treated cells (0.3 and 10 μM histamine), respectively. Two groups were compared with an unpaired student’s t-test and two-tail p-value. Results were considered statistically significant when p < 0.05 (*p < 0.05 and **p < 0.01).
To further challenge the prediction that the low conductance mode of the mPTP could be involved during cytosolic Ca2+ oscillations in HeLa cells, we stimulated CSA-treated cells with histamine. As shown in Fig. 5B, inhibition of the mPTP indeed increased the period of Ca2+ oscillations in response to both 0.3 and 1 μM histamine. It should be noted that CSA has been reported to stimulate SERCA pumps in addition to its effect on the mPTP60. An increased rate of Ca2+ pumping back to ER might thus also participate in the effect shown in Fig. 5B. As in hepatocytes the effect of CSA on the period of Ca2+ oscillations was eliminated in the presence of mitochondrial inhibition60, we conclude from our observations that the mPTP is a realistic candidate for the bidirectional flux Jx.
Mitochondrial variables
We next investigated the dynamics of the mitochondrial variables in response to a prototypic Ca2+ peak in the cytosol. The Ca2+ peak simulated in Fig. 6 is a square wave pulse of 10 s duration and 1.5 μM amplitude. We chose this type of stimulation to optimise the comparison with experimental data. Figure 6A shows the massive and long-lasting (>200 s) increase in NADH resulting from the cytosolic Ca2+ spike. It is in agreement with observations in phenylephrine-stimulated hepatocytes4. This accumulation of NADH stimulates the Krebs cycle (Eq. (19)) and increases the mitochondrial potential. However, this increase is preceded by a transient decrease in ΔΨ due to the entry of Ca2+ from the cytosol into mitochondria (Fig. 6B). Such dynamics for ΔΨ has been observed in HeLa cells stimulated by histamine34. The biphasic change in potential induces a similar dynamics of ATP (Fig. 6C) synthesis as the F1Fo-ATPase is highly sensitive to the mitochondrial voltage (Eq. (21)). The initial drop in mitochondrial ATP is also observed experimentally35,61. In one of these studies35, the authors suggested that the drop is a consequence of the initial activation of cytosolic processes, such as those involving ion pumps or contractile proteins, which consume ATP. The drop in mitochondrial ATP would result from its rapid transfer to the cytosol in supply for energy demand. As the increase in NADH occurs later than the rise in Ca2+, ATP synthesis is expected to occur later. The model however disagrees with this interpretation. A rise in ATP consumption by the SERCA pumps is indeed observed in the cytosol (Fig. 6D). However, the flux of the translocator (Eq. (20)) is outward for ATP, wich suggests that the initial decrease in ATP can be ascribed to the decrease in ΔΨ.
Figure 6. Dynamics of mitochondrial variables.

Curves show the simulated changes in the concentrations of the variables related to mitochondrial metabolism in response to a square-wave Ca2+ pulse in the cytosol (1.5 μM for 10 s). In all panels, the horizontal line indicates the time of the Ca2+ pulse. Mitochondrial Ca2+ is shown in blue and the variable indicated on the vertical left axis in red. Parameter values are listed in Table 1.
Figure 6 shows that the rate of decay of [NADH]m is much lower than that of Ca2+. Thus, when Cc displays sustained oscillations, [NADH]m does not decrease significantly between the spikes, and remains elevated as long as stimulation is maintained (Supplementary Fig. S5). This is due to the slow kinetics of NADH decrease and to the fact that mitochondrial Ca2+ does not recover to basal level during the interspike interval (Fig. 2A). The oscillations of NADH on an elevated level are observed in hepatocytes stimulated with high concentrations of phenylephrine4.
We next examined how mitochondrial metabolism affects Ca2+ oscillations. A classical result in this field is that the frequency of Ca2+ oscillations decreases with the amount of mitochondrial substrates46. We tested this in the model by decreasing kGLY (Eq. (17)) that represents the input of the glycolytic pathway. As shown in Fig. 7, this leads to a decrease in the period of Ca2+ oscillations, as observed experimentally. A less active glycolytic pathway indeed decreases the mitochondrial voltage and thereby reduces the activity of the MCU. In consequence, Ca2+ is less actively imported in mitochondria, leading to increased cytosolic Ca2+ between successive spikes. This higher levels of interspike cytosolic Ca2+ accelerate Ca2+ release through the IP3R and thus, the next Ca2+ spike. To get this result with the model, we needed to impose a rather steep dependence of the MCU on ΔΨ and, in particular, a sensitivity that is larger than that of the NCX (p1 > p2).
Figure 7. Decreasing the glycolytic input slows down the cytosolic Ca2+ oscillations.
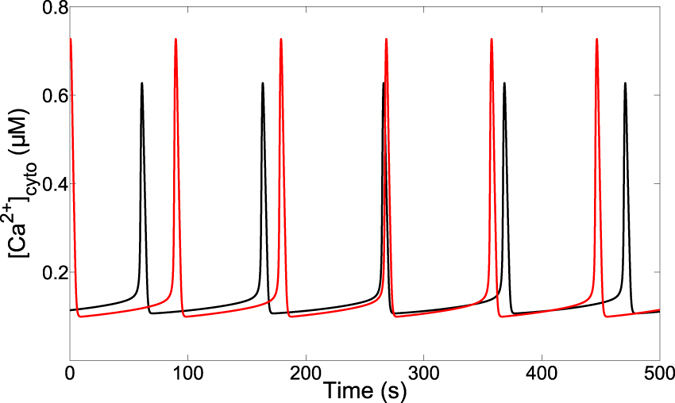
The curve in black shows cytosolic Ca2+ oscillations for the default values of the parameters given in Table 1, while the red curve shows the effect of decreasing the rate of the glycolytic pathway (kGLY) to 250 μM.s−1. IP3 = 0.7 μM.
Robustness of mitochondrial metabolism with respect to Ca2+ dynamics
Stimulus-induced Ca2+ rises in mitochondria have been suggested as essential for physiological cell bioenergetics62,63. For such an important function, one expects Ca2+ regulation of mitochondrial metabolism to be both versatile and robust. Given the complexity and the highly non-linear character of the regulation of mitochondrial metabolism, this question is best addressed by modelling. Thus, we simulated Ca2+ spikes of different frequencies and amplitudes and computed the resulting average values of mitochondrial NADH and ATP concentrations. The results shown in Fig. 8A,B were obtained when simulating artificial square-wave Ca2+ pulses lasting 10 s, with a 100 nM basal level of Ca2+. We found that maximal average values of NADHm and ATPm were obtained for frequencies and amplitudes of Ca2+ spiking that are typically observed in HeLa cells (~0.04 s−1 and ~800 nM). Interestingly, optimised metabolism is naturally obtained in the model for the parameter values listed in Table 1, which were independently adjusted to account for published observations on Ca2+ and mitochondrial metabolism.
Figure 8. Effect of changing the characteristics of Ca2+ spikes on mitochondrial metabolism.

The color code indicates the values of [NADH]m (A) and [ATP]m (B) averaged over one period of the Ca2+ repetitive spikes. Baseline Ca2+ is set to 100 nM and the duration of the spikes always equals 10 s.
Average [NADH]m is an increasing function of both the frequency and the amplitude of the Ca2+ spikes. As shown in Supplementary Fig. S6(A), at low frequencies the minimal values reached during oscillations much depend on the frequency. As NADH decrease is slow (Fig. 6A and Supplementary Fig. S5), [NADH]m does not regain its basal level between the Ca2+ spikes, the minimal value reached being fixed by the oscillations’ period. Moreover, AGC also influences this minimal value, as it is activated by low levels of cytosolic Ca2+. In consequence, one observes a frequency modulation of the average [NADH]m, which is more pronounced with a lower amplitude of the Ca2+ spikes. There is a similar amplitude modulation of the average [ATP]m, more pronounced with a lower frequency of the Ca2+ spikes (Supplementary Fig. S6(C)). In contrast to [NADH]m, average values of [ATP]m decrease with large frequency and/or amplitude of the Ca2+ spikes. This is due to the decrease in ΔΨ associated with Ca2+ entry in mitochondria, which leads to a decrease of the F1Fo ATPase activity. Consequently, an optimal amplitude and frequency of Ca2+ spiking, above which ATP synthesis decreases, can be observed (Fig. 6B and Supplementary Fig. S6 (B and D)).
Discussion
We are proposing a model for intracellular Ca2+ dynamics and mitochondrial metabolism accounting for previously published experimental observations about: (1) the effect of mitochondrial metabolism on Ca2+ cycling between ER, cytoplasm and mitochondria during oscillations induced by IP3-generating agonists, and (2) the effect of changes in cytosolic Ca2+ on mitochondrial metabolism. The model is built from a selection of previously proposed kinetic expressions for the various fluxes and reaction rates, except for the increase of mitochondrial NADH mediated by the malate-aspartate shuttle, which, to the best of our knowledge, has not been considered in previous models. We have modified the expressions for the MCU concerning its dependence with respect to the voltage, and for the SERCA pump, where the ATP requirement was included in the rate equation (Eq. (13)). In a previous study, Fall and Keizer27 also modified the original expression of Magnus and Keizer for the MCU21. However, these former authors included a dependence of the uptake rate on intra-mitochondrial Ca2+, which was not reported in later studies on the MCU42,44. Concerning the dependence of the SERCA pumps on [ATP], this was introduced in the model for internal coherence. We found that ATP consumption by this pump only slightly affects cytosolic ATP dynamics. Ca2+-stimulated production of mitochondrial ATP overtakes this effect by far, in contrast to what has been assumed35,64.
As compared with previous modelling approaches, we also considered the existence of one additional Ca2+ exchange flux between the cytosol and the mitochondria (Eq. (16)). As put forward in some experimental studies, a small influx into mitochondria was found necessary to account for the observed moderate increase in mitochondrial Ca2+ when the MCU is inactive15,42. Similarly, a small flux from mitochondria is required to account for the maintenance of cytosolic Ca2+ oscillations when the NCX is inhibited33. Given these requirements, a simple assumption is the existence of a passive small flux, whose direction is imposed by the electrochemical gradient. A plausible channel that could mediate this flux is the mitochondrial PTP in its low conductance state. This assumption is corroborated by the observed increase in the period of Ca2+ oscillations in CSA-treated HeLa cells (Fig. 5B). Accordingly, a MCU-independent Ca2+ influx pathway has been characterized in HeLa cells65. In disagreement with our simple expression for Jx (Eq. (16)), studies performed in neurons however report that the mPTP can only open in response to a rise in mitochondrial Ca2+ 66. On the other hand, the requirement for an efflux pathway other than the NCX has been proposed by Bernardi and Von Stockum67, who further assumed that this efflux should correspond to transient openings of the PTP.
Other modes of Ca2+ uptake into mitochondria, not considered in the model but reported to be active at low concentrations of cytosolic Ca2+, could also play a role. In particular, Ryanodine receptors have been shown to transport Ca2+ inside mitochondria in cardiomyocytes and neurons68. Also, a rapid mode of Ca2+ uptake by mitochondria (RaM) of yet poorly identified molecular nature has been reported in mitochondria isolated from liver and cardiac cells15,69. The present model could be easily extended to take these fluxes into account (see for example Bazil and Dash (2011))70.
The model is fully consistent with the reported effect of alterations in the rates of mitochondrial Ca2+ exchanges on the characteristics of Ca2+ oscillations. A surprising result of the simulations is that the frequency of oscillations can decrease when inhibiting the activity either of the NCX (Supplementary Fig. S3) or of the MCU (Fig. 4A) despite the fact that these fluxes have opposite directions. This behaviour of the model is confirmed experimentally33 (and Fig. 4). It emphasizes the fact that the slow release of Ca2+ from mitochondria that occurs between the cytosolic Ca2+ spikes paces the oscillations, as suggested by Ishii et al.31. The increase in the period of oscillations in the absence of MCU is however much larger in the experiments than in the model. This suggests that the absence of MCU alters processes distinct from the oscillatory mechanism and modifies the effective sensitivity to histamine (Supplementary Table S1). In any case, our results confirm that besides IP3, mitochondria play a role in determining the interspike interval in addition to other processes more often considered in models, as the time taken by the IP3 receptors to recover from Ca2+-induced inhibition71 or random opening of a sufficient number of channels11,12.
Altering mitochondrial metabolism in the model also accounts for corresponding published experimental observations about energising mitochondria for example (Fig. 7). Along these lines, we also found (not shown) that decreasing the activity of the ETC (ko in Eq. (19)) increases the frequency of Ca2+ oscillations by diminishing the rate of Ca2+ pumping into mitochondria, as we have previously reported for Hint2-knock down hepatocytes72. Besides, we found that the MAS NADH shuttle participates in the frequency- and amplitude- modulation of Ca2+-activated metabolism, by boosting metabolism at low levels of cytosolic Ca2+. Thus, the MAS NADH shuttle can increase the amplitude of NADHm oscillations. Interestingly if the effect of the shuttle is not limited when the activity of the MCU becomes significant, NADH oscillations tend to follow cytosolic Ca2+ variations and become spiky instead of saw tooth-like as observed experimentally. This in silico observation thus confirms the hypothesis that, upon massive activation of the Krebs cycle, α-ketoglutarate becomes limiting for the activity of the MAS NADH shuttle18.
The model finally predicted that mitochondrial metabolism remains relatively robust with respect to the amplitude and frequency of the stimulating Ca2+ oscillations. From a physiological point of view, this property ensures that mitochondrial metabolism filters out the randomness inherent to Ca2+ oscillations11 to stabilize ATP synthesis, which is a vital process. This was not expected a priori from a modeller’s point of view, given that mitochondrial metabolism is described by highly non-linear kinetic equations. The theoretically predicted robustness of mitochondrial metabolism with respect to changes in Ca2+ dynamics is in agreement with MCU knock-down experiments reporting no changes in mitochondrial respiration when MCU expression was reduced58. ATP production, however, modestly varies with the dynamical characteristics of the Ca2+ spikes, being at large values for periods around 25 s and amplitudes around 800 nM. These values are in the range of those usually seen in HeLa cells, on which our parameter values had been calibrated. Thus, in such a prototypic non-excitable cell, mitochondrial metabolism and Ca2+ dynamics are coordinated to optimise bioenergetics.
Additional Information
How to cite this article: Wacquier, B. et al. Interplay between intracellular Ca2+ oscillations and Ca2+-stimulated mitochondrial metabolism. Sci. Rep. 6, 19316; doi: 10.1038/srep19316 (2016).
Supplementary Material
Acknowledgments
B.W. and G.D. are, respectively, Research Fellow and Research Director at the Belgian F.R.S-F.N.R.S. This work was supported by the Fonds de la Recherche Scientifique-FNRS under Grant n° J0043.14, the ‘Fonds David and Alice Van Buuren’ and the french MITOPATHO ANR program. L.C., G.D. and G.T.V.N. benefit from a WBI-France exchange program (Wallonie-Bruxelles International, Fonds de la Recherche Scientifique, Ministère Français des Affaires étrangères et européennes, Ministère de l’Enseignement supérieur et de la Recherche dans le cadre des Partenariats Hubert Curien).
Footnotes
Author Contributions B.W., L.C., G.T.V.N. and G.D. conceived and designed the simulations and experiments. B.W., L.C. and G.T.V.N. performed the experiments. B.W. performed the simulations. B.W., L.C., G.T.V.N. and G.D. analysed the data. B.W., L.C., G.T.V.N. and G.D. wrote the manuscript.
References
- Zimmermann B. Control of InsP3-induced Ca2+ oscillations in permeabilized blowfly salivary gland cells: contribution of mitochondria. J Physiol 525, 707–719 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadkai G. & Duchen M. R. Mitochondria: the hub of cellular Ca2+ signaling. Physiology 23, 84–94 (2008). [DOI] [PubMed] [Google Scholar]
- Rizzuto R., De Stefani D., Raffaello A. & Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol 13, 566–78 (2012). [DOI] [PubMed] [Google Scholar]
- Gaspers L. D., Memin E. & Thomas A. P. Calcium-dependent physiologic and pathologic stimulus-metabolic response coupling in hepatocytes. Cell Calcium 52, 93–102 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov A. I. et al. Frequency-dependent mitochondrial Ca2+ accumulation regulates ATP synthesis in pancreatic β-cells. Pflugers Arch 465, 543–554 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glancy B. & Balaban R. S. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 51, 2959–2973 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen M. R. Contributions of mitochondria to animal physiology : from homeostatic sensor to calcium signalling and cell death. J Physiol 516, 1–17 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock D. F., Herrington J., Goodwin P. C., Park Y. B. & Hille B. Mitochondrial participation in the intracellular Ca2+ newtork. J Cell Biol 136, 833–44 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge M., Bootman M. & Roderick H. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4, 517–29 (2003). [DOI] [PubMed] [Google Scholar]
- Keener J. & Sneyd J. in Mathematical Physiology 2nd edn , Vol. 8 (Springer 2009). [Google Scholar]
- Skupin A., Kettenmann A. H. & Falcke M. Calcium signals driven by single channel noise. PLoS Comput Biol 5, e1000870 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thul R., Bellamy T., Roderick H., Bootman M. & Coombes S. Calcium oscillations. Adv Exp Med Biol 641, 1–27 (2008). [DOI] [PubMed] [Google Scholar]
- Raffaello A. et al. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J 32, 2362–76 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patron M. et al. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell 53, 726–37 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santo-Domingo J. & Demaurex C. Calcium uptake mechanisms of mitochondria. Biochim Biophys Acta . 1797, 907–912 (2010). [DOI] [PubMed] [Google Scholar]
- Jiang D., Zhao L. & Clapham D. E. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 326, 144–147 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Marchi U. et al. NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. J Biol Chem 289, 20377–20385 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satrústegui J., Pardo B. & Del Arco A. Mitochondrial transporters as novel targets for intracellular calcium signaling. Physiol Rev 87, 29–67 (2007). [DOI] [PubMed] [Google Scholar]
- Denton R. M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta 1787, 1309–16 (2009). [DOI] [PubMed] [Google Scholar]
- Magnus G. & Keizer J. Minimal model for calcium handling by mitochondria. Am. J. Physiol. 273, C717–C733 (1997). [DOI] [PubMed] [Google Scholar]
- Magnus G. & Keizer J. Model of β-cell mitochondrial calcium handling and electrical activity. [I]. cytoplasmic variables. Am J Physiol 274, C1158–73 (1998). [DOI] [PubMed] [Google Scholar]
- Magnus G. & Keizer J. Model of β-cell mitochondrial calcium handling and electrical activity. [II]. mitochondrial variables. Am J Physiol 274, C1174–84 (1998). [DOI] [PubMed] [Google Scholar]
- Cortassa S., Aon M. A., Marbán E., Winslow R. L. & O’Rourke B. An integrated model of cardiac mitochondrial energy metabolism and calcium dynamics. Biophys J 84, 2734–55 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter T. E. & Sheu S. S. Characteristics and possible functions of mitochondrial Ca2+ transport mechanisms. Biochim Biophys Acta 1787, 1291–308 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen M. H. & Jafri M. S. Mitochondrial calcium signaling and energy metabolism. Ann N Y Acad Sci 1047, 127–37 (2005). [DOI] [PubMed] [Google Scholar]
- Marhl M., Schuster S. & Brumen M. Mitochondria as an important factor in the maintenance of constant amplitudes of cytosolic calcium oscillations. Biophys Chem 71, 125–32 (1998). [PubMed] [Google Scholar]
- Fall C. P. & Keizer J. E. Mitochondrial modulation of intracellular Ca2+ signaling. J Theor Biol 210, 151–165 (2001). [DOI] [PubMed] [Google Scholar]
- Bertram A., Pedersen M. G., Luciani D. S. & Sherman A. A simplified model for mitochondrial ATP production. J Theor Biol 243, 576–586 (2006). [DOI] [PubMed] [Google Scholar]
- Oster A. M., Thomas B., Terman D. & Fall C. P. The low conductance mitochondrial permeability transition pore confers excitability and CICR wave propagation in a computational model. J Theor Biol 273, 216–231 (2011). [DOI] [PubMed] [Google Scholar]
- Qi H., Li L. & Shuai J. Optimal microdomain crosstalkbetween endoplasmic reticulum and mitochondria for Ca2+ oscillations. Scientific Reports 5, 7984 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii K., Hirose K. & Iino M. Ca2+ shuttling beetween endoplasmic reticulum and mitochondria underlying Ca2+ oscillations. EMBO report 7, 390–396 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vay L. et al. Modulation of Ca2+ release and Ca2+ oscillations in HeLa cells and fibroblasts by mitochondrial Ca2+ uniporter stimulation. J Physiol 580, 33–49 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-SanMiguel E. et al. The mitochondrial Na+/Ca2+ exchanger plays a key role in the control of cytosolic Ca2+ oscillations. Cell Calcium 40, 53–61 (2006). [DOI] [PubMed] [Google Scholar]
- Jouaville L. S., Pinton P., Bastianutto C., Rutter G. A. & Rizzuto R. Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc Natl Acad Sci USA 96, 13807–13812 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths E. J. & Rutter G. A. Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochim Biophys Acta 1787, 1324–1333 (2009). [DOI] [PubMed] [Google Scholar]
- Hayashi T., Rizzuto R., Hajnoczky G. & Su T. P. MAM: more than just a housekeeper. Trends Cell Biol 19, 81–8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao P., Tan X., Donovan G., Sanderson M. J. & Sneyd J. A deterministic model predicts the properties of stochastic calcium oscillations in airway smooth muscle cells. PLoS Comput Biol 10, e1003783. (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith G. D., Wagner J. & Keizer J. Validity of the rapid buffering approximation near a point source of calcium ions. Biophys J 70, 2527–2539 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai M. F., Jiang D., Zhao L., Clapham D. & Miller C. Functional reconstitution of the mitochondrial Ca2+/H+ antiporter Letm1. J Gen Physiol 143, 67–73 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont G. & Croisier H. Spatiotemporal organization of Ca2+ dynamics: a modeling-based approach. HFSP Journal 4, 43–51 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytton J., Westlin M., Burk S. E., Shull G. E. & MacLennan D. H. Functional comparisons between isoforms of the sarcoplasmic or endoplasmic reticulum family of calcium pumps. J. Biol. Chem. 267, 14483–9 (1992). [PubMed] [Google Scholar]
- Csordás G. et al. MICU1 controls both the treshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metabolism 17, 976–987 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahuja M. & Muallem S. The gatekeepers of mitochondrial calcium influx: MICU1 and MICU2. EMBO Rep 15, 205–6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D., Raffaello A., Teardo E., Szaba I. & Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 19, 336–340 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J., Wyman J. & Changeux J. P. On the nature of allosteric transitions: a plausible model. J Mol Biol 12, 88–118 (1965). [DOI] [PubMed] [Google Scholar]
- Jouaville L. S., Ichas F., Holmuhamedov E. L., Camacho P. & Lechleiter J. D. Synchronization of calcium waves by mitochondrial substrates in Xenopus laevis oocytes. Nature 377, 438–441 (1995). [DOI] [PubMed] [Google Scholar]
- Pradhan R., Beard D. & Dash R. A biophysically based mathematical model for the kinetics of mitochondrial Na+-Ca2+ antiporter. Biophys J 98, 218–30 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. et al. Superoxide flashes in single mitochondria. Cell 134, 279–90 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson G. T. et al. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J Biol Chem 26, 13044–53 (2004). [DOI] [PubMed] [Google Scholar]
- Contreras L. et al. Ca2+ Activation Kinetics of the Two Aspartate-Glutamate Mitochondrial Carriers, Aralar and Citrin: Role in the Heart Malate-Aspartate NADH Shuttle. J Biol Chem 282, 7098–7106 (2007). [DOI] [PubMed] [Google Scholar]
- Saa A. & Siqueira K. M. Modeling the ATP production in mitochondria. Bull Math Biol 75, 1636–1651 (2013). [DOI] [PubMed] [Google Scholar]
- Tran K., Smith N. P., Loiselle D. S. & Crampin E. J. A thermodynamic model of the cardiac sarcoplasmic/endoplasmic Ca2+ (SERCA) pump. Biophys J 100, 2853 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermentrout G. B. in Simulating, analyzing, and animating dynamical system : a guide to XPPAUT for researchers and students (SIAM Books, Philadelphia, 2002). [Google Scholar]
- Tran Van Nhieu G. et al. Actin-based confinement of calcium repsonses during shigella invasion. Nat Commun 4, 1567 (2013). [DOI] [PubMed] [Google Scholar]
- Robb-Gaspers L. D. et al. Integrating cytosolic calcium signals into mitochondrial metabolic responses. EMBO J 17, 4987–5000 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer A. E. et al. Ca2+ indicators based on computationally redesigned calmodulin-peptide pairs. Chem Biol 13, 521–30 (2006). [DOI] [PubMed] [Google Scholar]
- Suzuki J. et al. Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat Commun 5, 4153 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman J. et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–5 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichas F. & Mazat J.-P. From calcium signaling to cell death: two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochim Biophys Acta 1366, 33–50 (1998). [DOI] [PubMed] [Google Scholar]
- Smaili S. S., Stellato K. A., Burnett P., Thomas A. P. & Gaspers L. D. Cyclosporin A inhibits inositol 1,4,5-trisphosphate-dependent Ca2+ signals by enhancing Ca2+ uptake into the endoplasmic reticulum and mitochondria. J Biol Chem 276, 23329–23340 (2001). [DOI] [PubMed] [Google Scholar]
- Nakano M., Imamura H., Nagai T. & Noji H. Ca2+ regulation of mitochondrial ATP synthesis visualized at the single cell level. ACS Chem Biol 6, 709–715 (2011). [DOI] [PubMed] [Google Scholar]
- Jouaville L. S., Ichas F. & Mazat J. P. Modulation of cell calcium signals by mitochondria. Mol Cell Biochem 184, 371–6 (1998). [PubMed] [Google Scholar]
- Cárdenas C. et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142, 270–83 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts M. & Sherman A. Modeling the pancreatic α-cell: dual mechanisms of glucose suppression of glucagon secretion. Biophys J 106, 741–751 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondarenko A., Jean-Qartier C., Malli R. & Graier W. Characterization of distinct single-channel properties of Ca2+ inward currents in mitochondria. Pflugers Arch 465, 997–1010 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsukova A. et al. Activation of the mitochondrial permeability transition pore modulates Ca2+ responses to physiological stimuli in adult neurons. Eur J Neurosci 33, 831–42 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P. & von Stockum S. The permeability transition pore as a Ca2+ release channel: new answers to an old question. Cell Calcium 52, 22–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob R. et al. Molecular and functional indentification of a mitochondrial ryanodine receptor in neurons. Neurosci Lett 575, 7–12 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buntinas L., Gunter K. K., Sparagna G. C. & Gunter T. E. The rapid mode of calcium uptake into heart mitochondria (RaM): comparison to RaM in liver mitochondria. Biochim Biophys Acta 1504, 248–61 (2001). [DOI] [PubMed] [Google Scholar]
- Bazil J. N. & Dash R. K. A minimal model for the mitochondrial rapid mode of Ca2+ uptake mechanism. PLoS One 6, e21324 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont G., Combettes L., Bird G. S. & Putney J. W. Calcium oscillations. Cold Spring Harb Perspect Biol 3, pii: a004226 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndiaye D. et al. Characterization of the effect of the mitochondrial protein Hint2 on intracellular Ca2+ dynamics. Biophys J 105, 1268–75 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beis I. & Newsholme E. A. The contents of adenine nucleotides, phosphagens and some glycolytic intermediates in resting muscles from vertebrates and invertebrates. Biochem J 152, 23–32 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swillens S., Combettes L. & Champeil P. Transient inositol 1,4,5-trisphosphate-induced Ca2+ release: a model based on regulatory Ca2+-binding sites along the permeation pathway. Proc Natl Acad Sci USA 91, 10074–10078 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.