

Abstract
The use of antibacterial drug combinations with synergistic effects is increasingly seen as a critical strategy to combat multi-drug resistant bacteria such as methicillin-resistant Staphylococcus aureus (MRSA). In this work, the proteome responses in MRSA under the stress of a sub-inhibitory dose of a synergistic drug combination of a novel erythromycin derivative, SIPI-8294, and oxacillin, were studied by label-free quantitative proteomics. Several control treatment groups were designed to isolate proteome responses potentially related to the synergy: (1) the non-synergistic drug combination of erythromycin and oxacillin, (2) SIPI-8294 only, (3) oxacillin only and (4) erythromycin only. Results showed that 200 proteins were differentially expressed in SIPI-8294/oxacillin-treated cells. Among these proteins, the level of penicillin binding protein 2a, the protein mainly responsible for oxacillin resistance in MRSA, was four times lower in the SIPI-8294/oxacillin group than in the erythromycin/oxacillin group, suggesting that SIPI-8294 may interfere with this known oxacillin resistance mechanism. Moreover, hierarchical clustering analysis of differentially expressed proteins under different treatments revealed that SIPI-8294/oxacillin elicits very different responses than the individual drugs or the non-synergistic erythromycin/oxacillin combination. Bioinformatic analysis indicated that the synergistic effect can be further traced to a disruption in oxidation-reduction homeostasis and cell wall biosynthesis.
Staphylococcus aureus (S. aureus) can cause various kinds of infections including skin abscesses, necrotizing pneumonia, joint infections, and endocarditis1. Methicillin-resistant S. aureus (MRSA) accounts for 60–70% of S. aureus infections in hospitals and causes the highest number of invasive infections among all antibiotic-resistant bacteria2. According to the U.S. Centers for Disease Control and Prevention (CDC), invasive infection of MRSA has a 14% fatality rate in 20113. While the majority of MRSA cases are acquired in hospitals and other healthcare settings, community-acquired MRSA infection has seen a big increase in prevalence, posing greater danger to the public4,5,6. Unfortunately, new antibiotic development has not kept pace with the emergence of resistance over the past few decades7. Although several newer antibiotics introduced after 2000, such as linezolid and daptomycin, remain largely effective against MRSA, strains resistant to those antibiotics have already been reported8,9. Therefore, the use of antimicrobial drug combinations is increasingly seen as a critical strategy to combat multi-drug resistant pathogens such as MRSA.
Generally, drug combinations can have synergistic, additive, and antagonistic effects, depending on whether the effect of combination is bigger than, equal to or smaller than the predicted sum of the effects of individual drugs10. Synergistic effects are the most desirable, because lower dose can be used, which not only reduce cost and toxicity, but also slow down the development of antibiotic resistance. However, elucidation of the mechanism underlying such effects has been difficult, partly because traditional reductionist approaches mainly focused on the immediate drug targets and the addition or modification of individual cellular components that underlie the resistance11,12. This is unlikely to reveal the long chain of interactions that are likely to be responsible for synergistic effects caused by multiple drugs. Instead, systems biology approaches are more suitable to unearth the key players in the biological network which are involved13. In this systems view, bacteria cells respond to antibiotic damage by regulating its metabolic pathways globally to compensate for that damage14. Bacteria that survive the antibiotic treatment will develop persistent adaptive responses, which make it possible to develop resistance over time15,16.
Although the antimicrobial and resistance mechanisms of antibiotics have been studied for decades, much of the biology remains unknown beyond the immediate drug targets, even for the most studied pathogens. Indeed, more and more studies support the notion of a global response to antibiotic stress17,18,19. In a very recent study, Cho et al. reported that β-lactam antibiotics not only inhibit their targets, namely the penicillin binding proteins (PBPs), in Escherichia coli as is commonly believed, but also induce a toxic malfunctioning of the biosynthetic machinery, thereby bolstering their bactericidal activity. The cells under antibiotic stress undergo futile cycles of cell wall synthesis and degradation, which deplete cellular resources20. Kohanski et al. has demonstrated that many distinct classes of antibiotics commonly accelerate the electron transport chain via tricarboxylic acid (TCA) cycle and damage the iron-sulfur cluster, which leads to increased oxidative stress. It was thus conjectured that oxidative stress is a common antimicrobial mechanism of antibiotics21. Recently, proteomics has been employed as a tool to study the adaption or global response of the bacteria to the environment, including to antibiotic stress14,22,23,24. The majority of studies showed that bacterial responses to antibiotic stress are not limited to a few molecular targets directly related to the known antimicrobial and resistance mechanisms, but appear to be global. A large number of proteins involved in various pathways were differentially regulated in the presence of antibiotics. Such studies suggested that proteomics and other systems approaches to biology can potentially provide a more comprehensive picture of bacterial responses to antibiotics, complementing the traditional reductionist approach.
SIPI-8294 (Chinese patent CN201010273264 and CN201410131277) is a new derivative of erythromycin synthesized by the Shanghai Institute of Pharmaceutical Industry25. The chemical structures of SIPI-8294 and erythromycin are shown in Fig. 1. SIPI-8294 retains partial of the erythromycin structures, 14-membered macrolactone ring and 5-position desosamine sugar, but is more hydrophobic than erythromycin due to several different functional groups (shown in Fig. 1 with pink color). It has been revealed recently that SIPI-8294 has remarkable synergistic effect with oxacillin against MRSA in vitro25. It was shown that in the presence of a low dose of SIPI-8294, the minimum inhibitory concentration (MIC) of oxacillin was vastly reduced, as if the susceptibility of MRSA against oxacillin has been restored. Specifically, SIPI-8294 could reduce the MIC of 18 out of 21 clinically used β-lactam antibiotics against MRSA (ATCC43300) by 4 to 128 times and had synergistic effects with oxacillin against 12 out of 16 clinical isolated MRSA strains. No synergistic effects were observed against methicillin-susceptible strains. Interestingly, SIPI-8294 has no apparent bactericidal effect on MRSA, nor has its parent compound, erythromycin. Unlike SIPI-8294, however, erythromycin and other macrolides antibiotics did not exhibit any synergistic effects with the β-lactams25. The mechanism for the synergistic effect of SIPI-8294 and oxacillin is not known yet. This study therefore aimed at revealing the global cellular responses of MRSA against sub-MIC dose of SIPI-8294 and oxacillin, and thereby obtaining new insights into the synergistic mechanism of the drug combination.
Figure 1. Chemical structures for SIPI-8294 and erythromycin.

The macrolactone ring is in red color and the 5-position disosamine sugar is in blue color. The different functional groups between SIPI-8294 and erythromycin are in pink color for SIPI-8294 and in black color for erythromycin.
Results and Discussion
Proteomic analysis
Spectral-counting based label-free quantitative proteomics was performed to investigate the synergistic effect of the combination of SIPI-8294 and oxacillin (SIPI-8294/Oxa). The workflow of sample preparation and data analysis is shown in Fig. 2. Besides the SIPI-8294/Oxa treatment group, four other treatment groups were also acquired: erythromycin/oxacillin (Ery/Oxa), oxacillin only (Oxa), SIPI-8294 only (SIPI-8294), and erythromycin only (Ery). Each treatment group was compared with the untreated control group respectively to identify differentially expressed proteins.
Figure 2. Experimental workflow for the sample preparation and data analysis.

Different drug treatment groups were compared with control (no drug treatment) and performed spectral-counting based label-free quantitation.
In all cases, sub-MIC doses of antibiotics and SIPI-8294 were applied to MRSA, so as to impose stress to the cells but not induce non-specific effects associated with cell death. In this circumstance, it is assumed that the cells will have sufficient time to change the allocation of the translational capacity to produce proteins needed to counteract the inflicted damage15. The growth curves for the treated cells under different conditions are shown in Supplementary Fig. S1. Under all treatment conditions including control group, MRSA continued to grow to the stationary phase and reach to the similar level after 24 hr incubation.
Figure 3 shows the total numbers of differentially expressed proteins in different treatment groups, relative to the untreated group. Figure 3a shows the volcano plot for the differentially expressed proteins in the SIPI-8294/Oxa and Ery/Oxa groups. The proteins with p-value of student t-test lower than 0.05 and fold change higher than 1.5 or lower than 0.67 (or −1.5) were considered as differentially expressed proteins. As shown in Fig. 3b, 200 proteins (120 up-regulated and 80 down-regulated) were found significantly changed in the SIPI-8294/Oxa treatment group. The number is much higher than the sum of the numbers of differentially expressed proteins in the Oxa and the SIPI-8294 treatment groups (72 and 57 differentially expressed proteins respectively). On the other hand, 124 proteins (66 up-regulated and 58 down-regulated) were differentially expressed in the Ery/Oxa treatment group, whereas 55 and 72 proteins were changed in the Ery and the Oxa treatment groups, respectively. The Venn diagram in Fig. 3c shows the number of differentially expressed proteins that are unique to the combination treatment and the individual drug treatments. As shown, 154 differentially expressed proteins were unique to the combination treatment of the SIPI-8294/Oxa treatment group, compared to 66 for the Ery/Oxa treatment group. There are 89 proteins both differentially expressed in SIPI-8294/Oxa and Ery/Oxa group. Figure 3d shows the correlation of these 89 proteins, and still a number of them couldn’t correlate well. These results indicate that the cellular response of MRSA to SIPI-8294/Oxa is different and much more complicated than Ery/Oxa, hinting at some specific mechanisms for the synergistic effect. All the differentially expressed proteins are listed in Supplementary Table S1.
Figure 3.

(a) Volcano plot of differentially expressed proteins in the SIPI-8294/Oxa and Ery/Oxa groups; (b) Numbers of differentially expressed proteins in different treatment groups, SIPI-8294/Oxa, Ery/Oxa, SIPI8294, Ery and Oxa, compared with control (no drug treatment); (c) Venn diagram for differentially expressed proteins in the SIPI-8294/Oxa and the Ery/Oxa treatment groups compared with their individual drug treatment groups; (d) The correlation between the differentially expressed proteins in the SIPI-8294/Oxa and Ery/Oxa groups.
Hierarchical cluster analysis was performed for all the differentially expressed proteins for each treatment group and a simple addition of the effects of SIPI-8294 and oxacillin (Additive) (Fig. 4). The “Additive” data-set was generated by multiplying the corresponding fold changes of each differentially expressed protein in the SIPI-8294 treatment group and the Oxa treatment group, to mimic the situation that SIPI-8294 only has simple additive effect with oxacillin. As shown in Fig. 4, the differentially expressed proteins in the SIPI-8294, Oxa and Additive treatment groups clustered together and the differentially expressed proteins in the Ery treatment group clustered with that in the Ery/Oxa treatment group, while the differentially expressed proteins in the SIPI-8294/Oxa treatment group was obviously different from all the other groups. These results suggest that SIPI-8294/Oxa elicits a cellular response that is much different from what one would expect if SIPI-8294 and Oxa act independently. Again, this provides some confidence that the observed synergistic effect does have a molecular underpinning, of which some essential elements should be captured in our data. Moreover, it is important to note that the differentially expressed proteins in the SIPI-8294 treatment group did not cluster together with that in the Ery treatment group. This suggests that the cellular responses between SIPI-8294 and erythromycin are different even though they share similar structures (Fig. 1 in red color).
Figure 4. Hierarchical cluster analysis was conducted for all the differentially expressed proteins in the different treatment groups.

SIPI-8294/Oxa, SIPI-8294, Oxa, Ery and Ery/Oxa, as well as the outcome that would be expected if SIPI-8294 and Oxa acted independently (Additive). Each row indicates one treatment group and each column represents one differentially expressed protein (shown in the zoom-in image). The color indicates relative fold changes (up-regulation relative to mean fold change in red, and down-regulation relative to mean fold change in green). The “Additive” group is generated by multiplying the corresponding fold changes of each differentially expressed protein in the SIPI-8294 treatment group and the Oxa treatment group.
To investigate the relationships between the differentially expressed proteins and the synergistic effect, all the differentially expressed proteins in the SIPI-8294/Oxa treatment group were processed by bioinformatic tools such as Megan26 for KEGG pathway analysis and SEED27 protein classification (Supplementary Fig. S2 (a) and (b)). The result shows that most assigned proteins are involved in carbohydrate metabolism (30 proteins), followed by translation (22 proteins), amino acid metabolism (17 proteins), energy metabolism (16 proteins), and nucleotide metabolism (15 proteins) etc. Moreover, all the identified proteins in the SIPI-8294/Oxa treatment group (about 1000 proteins) were also processed by the same bioinformatics tool, Megan, for KEGG pathway analysis (Supplementary Fig. S2 (c)) and SEED protein classification (Supplementary Figure S2 (d)). The KEGG pathway analyses and SEED protein classifications for all the identified proteins and the differentially expressed proteins in the SIPI-8294/Oxa treatment group exhibit different profiles. This implies that the response of the bacteria to SIPI-8294/Oxa is global and cannot be readily captured by traditional analytical techniques. To figure out what proteins are related to the synergistic effect, the subsequent analysis focuses on the proteins involved in the known oxacillin resistance mechanism, and the proteins which are changed only under SIPI-8294/Oxa treatment.
Synergistic effect related to oxacillin resistance mechanism
Two major resistance mechanisms have been developed by bacteria to resist β-lactam antibiotics: one is the production of β-lactamase which can inactivate or degrade the β-lactam antibiotics before the antibiotics reach their targets; the other is the production of penicillin binding protein 2a (PBP2a) which has low affinity with β-lactam antibiotics to bypass the activities of antibiotics28,29. However, the β-lactam used in this work, oxacillin, cannot be hydrolyzed by β-lactamase. The main resistance mechanism to oxacillin depends on PBP2a.
As expected, PBP2a was up-regulated in all the oxacillin presented groups (the SIPI-8294/Oxa, Ery/Oxa and Oxa treatment group) and not differentially expressed in the controls, SIPI-8294 only and Ery only treatment groups. It is noted that in the SIPI-8294/Oxa and Ery/Oxa treatment groups the same doses of oxacillin were applied to the cells; however, the fold changes for PBP2a were around four times lower in the SIPI-8294/Oxa group than those in the Ery/Oxa group (Fig. 5A). Specifically, the fold changes for PBP2a were up-regulated 42.0 folds in the Ery/Oxa treatment groups whereas only up-regulated 11.8 folds in the SIPI-8294/Oxa group. These results indicate that the synergistic effect of SIPI-8294/Oxa may be due to the interference of the oxacillin resistance mechanism. Interestingly, the same phenomenon was also observed for β-lactamase even though β-lactamase should not be responsible for the resistance of oxacillin. The fold changes for β-lactamase were up-regulated 16.8 folds in the Ery/Oxa treatment group but only up-regulated 4 folds in the SIPI-8294/Oxa treatment group. To investigate whether this regulation occurs at the transcription or translation level, quantitative real-time PCR (Q-RT-PCR) was performed for both genes of PBP2a (mecA) and β-lactamase (blaZ). The results show that mRNA transcript levels of these two enzymes (mecA and blaZ) in the SIPI-8294/Oxa group were significantly lower than that in the Ery/Oxa group (t-test p < 0.05) as shown in Fig. 5b, following the same trend as the corresponding proteins. Therefore, this regulation appears to happen upstream of translation, and could not be explained by an overall reduction in protein synthesis potentially triggered by the macrolides.
Figure 5. Quantification results for the expression of PBP2a (gene: mecA) and β-lactamase (gene: blaZ) at protein level by label-free quantitative proteomics method (a) and at transcription level by Q-RT-PCR method (b).
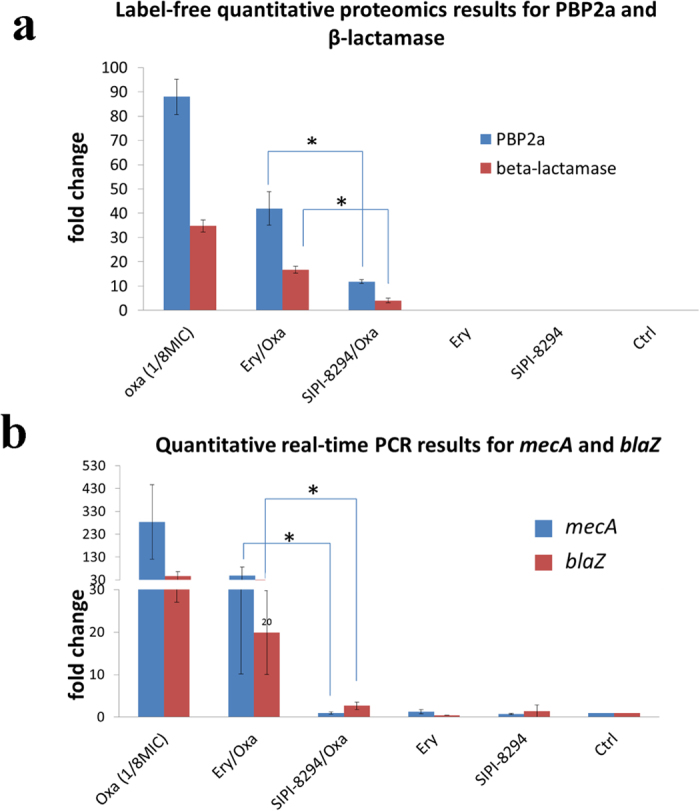
PBP2a and β-lactamase are the proteins involved in resistance mechanism of bacteria against β-lactam antibiotics. Label-free quantitative proteomics results show that the levels of PBP2a and β-lactamase are lower in the SIPI-8294/Oxa than Ery/Oxa and other treatment groups. The same trend was found at the mRNA level. These results suggest that the synergistic effect mechanism may be related to the interference with the oxacillin resistance mechanism. Asterisks (*) indicate differential expression at p < 0.05.
Synergistic effect related to other cellular pathways
We further focused our attention to the set of differentially expressed proteins that most strongly distinguish the SIPI-8294/Oxa and Ery/Oxa groups. Thirty-two proteins, among the 200 differentially expressed proteins in the SIPI-8294/Oxa group, exhibit a fold change over five and a ratio of (fold change of SIPI-8294/Oxa) to (fold change of Ery/Oxa) over 1.5 or lower than 0.67 as shown in Table 1. Several pathways caught our attention due to their potential roles in antibiotic responses, which we describe below.
Table 1. Differentially expressed proteins potentially related to synergistic mechanism.
| Uniprot_AC | Gene name | Protein name | Fold changes |
Cellular location | Pathway | |
|---|---|---|---|---|---|---|
| 8294+Oxa | Ery+Oxa | |||||
| A6QJN7 | narG | Nitrate reductase, alpha subunit | (−)73.4 (Repression) | (−)6.4 | Membrane | Nitrogen metabolism |
| A5IQF9 | adh | Alcohol dehydrogenase | (−)49.1 (Repression) | (−)13.1 | CytopPlasmic | Tyrosine metabolism; Fatty acid metabolism; Glycolysis/Gluconeogenesis; |
| E5QWJ9 | HMPREF0772_10696 | LPXTG-motif cell wall anchor domain protein | (−)44.1 | (−)9.1 | Cell wall | |
| A5IVI0 | nirB | Assimilatory nitrite reductase (NAD(P)H) large subunit (EC 1.7.1.4) | (−)35.8 | (−)6.0 | Cytoplasmic | |
| Q6GGX3 | ebh SAR1447 | Extracellular matrix-binding protein ebh (ECM-binding protein homolog) | (−)27.7 (Repression) | (−)4.8 | Cell wall | |
| A8Z0I5 | pfl | Formate C-acetyltransferase (EC 2.3.1.54) | (−)27.5 | (−)5.3 | Cytoplasmic | Butanoate metabolism; Propanoate metabolism; Pyruvate metabolism |
| E5QTK7 | sdrD | Serine-aspartate repeat-containing protein D | (−)24.3 | — | Cell wall | |
| A5IUP1 | sceD SaurJH9_2132 | Transglycosylase domain protein, probable transglycosylase SceD | (−)22.9 (Repression) | (−)12.5 (Repression) | Extracellular | |
| A5IVH7 | narH | Respiratory nitrate reductase β subunit | (−)21 (Repression) | (−)7.5 | CytoplasmicMembrane | Nitrogen metabolism |
| A5ISU2 | SaurJH9_1471 | Uncharacterized protein | (−)15.2 | (−)5.2 | Unknown | |
| A5IPA9 | ldh1 | L-lactate dehydrogenase 1 (L-LDH 1) (EC 1.1.1.27) | (−)11.9 | (−)3.5 | Cytoplasmic | Cysteine and methionine metabolism; Glycolysis/Gluconeogenesis; Propanoate metabolism; Pyruvate metabolism |
| E5QSI9 | sle | N-acetylmuramoyl-L-alanine amidase Sle1 (EC 3.5.1.28) | (−)7.3 | (−)3.5 | Cell wall | |
| A5IVG3 | SaurJH9_2406 | Pyridoxamine 5'-phosphate oxidase-related, FMN-binding | (−)6.2 | — | Unknown | |
| A5IQZ6 | SaurJH9_0817 | Cold-shock DNA-binding protein family | (−)5.1 | (−)2.9 | Cytoplasmic | |
| A5IWF1 | blaZ | β-Lactamase (EC 3.5.2.6) | (+)4 (Induction) | (+)16.8 (Induction) | Cell wall | β-Lactam resistance |
| E5QS79 | uvrB | UvrABC system protein B (Protein UvrB) (Excinuclease ABC subunit B) | (+)5.3 (Induction) | ND | Cytoplasmic | Nucleotide excision repair |
| A5IUI6 | SaurJH9_2077 | PfkB domain protein | (+)5.4 | ND | Cytoplasmic | Amino sugar and nucleotide sugar metabolism; Fructose and mannose metabolism; Starch and sucrose metabolism |
| A5IW34 | SaurJH9_2631 | AMP-dependent synthetase and ligase | (+)5.5 | — | Cytoplasmic | Carbon fixation pathways in prokaryotes; Glycolysis/Gluconeogenesis; Propanoate metabolism; Pyruvate metabolism |
| A5IST2 | lysA | Diaminopimelate decarboxylase (DAP decarboxylase) (DAPDC) (EC 4.1.1.20) | (+)5.5 (Induction) | — | Cytoplasmic | Lysine biosynthesis |
| A5ISH1 | SaurJH9_1347 | Uncharacterized protein | (+)5.7 | — | Cytoplasmic | |
| A5ISS3 | SaurJH9_1452 | Uncharacterized protein-like protein | (+)6.1 (Induction) | (+)3.8 (Induction) | Cytoplasmic | |
| A6QFW9 | pycA | Pyruvate carboxylase (EC 6.4.1.1) | (+)6.7 | ND | Cytoplasmic | Citrate cycle (TCA cycle); Carbon fixation pathways in prokaryotes; Pyruvate metabolism |
| E5QS29 | nadE | NH(3)-dependent NAD(+) synthetase (EC 6.3.1.5) | (+)8.4 | (+)5.5 (Induction) | Cytoplasmic | Nicotinate and nicotinamide metabolism |
| A5ISV5 | msrA | Peptide methionine sulfoxide reductase MsrA (EC 1.8.4.11) | (+)8.5 | — | Extracellular | |
| A6QI23 | prsA | Foldase protein PrsA (EC 5.2.1.8) | (+)9.6 | (+)4.3 | CytoplasmicMembrane | |
| A8Z1L4 | purH | Bifunctional purine biosynthesis protein PurH | (+)9.8 | ND | Cytoplasmic | One carbon pool by folate; Purine metabolism |
| A5INS0 | mecA | Penicillin binding protein 2a (EC 2.4.1.129) | (+)11.8 | (+)42.0 | CytoplasmicMembrane | β-Lactam resistance |
| A5IU99 | SaurJH9_1988 | ABC transporter related | (+)12.2 (Induction) | ND | CytoplasmicMembrane | ABC transporters |
| A5ISH6 | miaB | (Dimethylallyl)adenosine tRNA methylthiotransferase MiaB (EC 2.-.-.-) | (+)12.8 | ND | Cytoplasmic | |
| Q6GEW4 | mnaA | UDP-N-acetylglucosamine 2-epimerase (EC 5.1.3.14) | (+)13.8 | ND | Cytoplasmic | Amino sugar and nucleotide sugar metabolism |
| A5IV74 | ureG | Urease accessory protein UreG | (+)13.9 (Induction) | ND | Cytoplasmic | |
| A5INQ7 | SaurJH9_0016 | Primary replicative DNA helicase (EC 3.6.1.-) | (+)14.7 | ND | Cytoplasmic | DNA replication; Alanine, aspartate and glutamate metabolism; Purine metabolism |
Cutoff: Fold change (SIPI-8294/Oxa) >5; Fold change (SIPI-8294/Oxa) /Fold change (Ery/Oxa) >1.5 or <0.67. Note: The fold changes with (+) stand for up-regulated proteins and those with (−) stand for down-regulated proteins. The symbol of “−” stands for non-differentially expressed protein and “ND” stands for non-detected protein. The proteins which cannot be detected in the blank but detected in the drug treatment group are considered as “induction”; the proteins which can be detected in the blank but cannot be detected in the drug treatment group are considered as “repression”. For the induction or repression proteins, the spectral counts are set to 1 for non-detected proteins to calculate fold changes.
Oxidative stress
Two enzymes involved in nitrogen metabolism (Fig. 6), nitrate reductase (encoded by narG) and respiratory nitrate reductase (narH), which are responsible for reducing nitrate to nitrite were dramatically down-regulated 73.4 and 21.0 folds, respectively. This may be significant since nitrite is a key source of nitric oxide, an important gasotransmitter in bacteria which has close relationship with oxidative stress and antibiotic resistance30,31. Although the level of enzyme nitric oxide reductase (encoded by nirKS) which reduces nitrite to nitric oxide was below the detection limit in our experiments, it is possible that the nitric oxide level will be regulated because of the down-regulation of the up-stream enzymes. It was previously reported that elimination of endogenous nitric oxide in MRSA can sensitize the cells to oxidative stress32. It was also conjectured that NO-mediated antibiotic resistance is achieved through both the chemical modification of toxic compounds and the alleviation of the oxidative stress imposed by many antibiotics33. Besides, several other important oxidoreductases were also down-regulated dramatically. Alcohol dehydrogenase (encoded by adh), which catalyzes the reversible reduction of acetaldehyde to ethanol, was down-regulated 49.1 folds; and L-lactate dehydrogenase 1 (encoded by ldh1), responsible for catalyzing lactate formation from pyruvate, was down-regulated 11.9 folds in the SIPI-8294/Oxa group. Since both alcohol dehydrogenase and L-lactate dehydrogenase 1 convert NAD+ to NADH in their reactions, they play important role in maintaining the redox homeostasis in bacteria34. Therefore, these results suggest that the oxidation-reduction homeostasis has been disturbed by the SIPI-8294/Oxa combination, which might be related to its observed synergistic effect.
Figure 6. The differentially expressed proteins in the SIPI-8294/Oxa treatment group involved in nitrogen metabolism pathway classified by KEGG.

narG: nitrate reductase alpha subunit; narH: respiratory nitrate reductase β subunit; nirB: assimilatory nitrite reductase (NAD(P)H) large subunit; NirKS: nitric oxide reductase; glnA: glutamine synthase; gltBD: glutamate synthase. The symbol of (↓) stands for down-regulated protein, and the number ahead is the fold change. nirKS and gltBD were not detected in our experiment.
Cell wall biosynthesis
In nitrogen metabolism (Fig. 6), the enzyme assimilatory nitrite reductase (encoded by nirB), which reduces nitrite to ammonia, was also down-regulated by 35.8 folds. This may reduce the amount of ammonia available for synthesizing amino acids, in particular glutamine and glutamate35. Downstream of this pathway, glutamine synthase (encoded by glnA) was detected slightly down-regulated by 1.6 folds, and glutamate synthase (encoded by gltBD) from glutamine was below the detection limit. Glutamate and glutamine serve as the amino group donors for many nitrogen-containing compounds in at least 37 reactions35. Besides, glutamate is important in cell wall biosynthesis since glutamate serves as one of the amino acids in the penta-peptide component of the cell wall (Supplementary Fig. S3) and is also the monomer of poly-γ-glutamate, which is expressed on the surface of the S. aureus cell wall36. It has been reported that the inhibition of the expression and activity of glutamine synthase in Mycobacteria can lower the amount of poly-γ-glutamate and hamper bacteria replication37. Therefore, we surmise that the down-regulation of glutamate biosynthetic pathway may be detrimental to cell wall synthesis. Under the stress of the SIPI-8294/Oxa combination, it may become more difficult for the cell to maintain cell wall integrity or to produce new cell wall for replication. Since the known bactericidal effect of oxacillin is the interference with cell wall synthesis through inactivating penicillin-binding proteins (PBPs), the combination of SIPI-8294/Oxa may enhance that effect through depriving the cells of the building blocks of the cell wall. To test this hypothesis, we employed scanning electron microscope (SEM) to visualize outer surface of the cells. The SEM image for the cells in the SIPI-8294/Oxa treatment group clearly exhibit damages on the cell surface, while in other treatment conditions the cells are not visibly affected (Fig. 7).
Figure 7. SEM images for MRSA under different drug treatments.

Ctrl: Normal methicillin-resistance S aureus (MRSA) with no treatment; Oxa: MRSA treated with 1/8 MIC oxacillin (8 μg/ml); SIPI-8294: MRSA treated with 8 μg/ml SIPI-8294; SIPI-8294/Oxa: MRSA treated with the combination of SIPI-8294 and oxacillin (8 μg/ml for SIPI-8294 and 0.03125 μg/ml for oxacillin).
Other differentially expressed proteins were found to involve in pathways such as glycolysis/gluconeogenesis, TCA cycle, pyruvate metabolism, pyrimidine/purine metabolism, and DNA mismatch repair (Supplementary Fig. S4). The vast number and diversity of cellular pathways affected by the SIPI-8294/Oxa treatment again confirmed that antibiotic response in bacteria is global rather than isolated, consistent with previous studies14,24.
Conclusions
In this study, a new drug candidate SIPI-8294, derived from erythromycin and found to have synergistic effect with oxacillin against MRSA, was investigated. In order to understand the synergistic mechanism of SIPI-8294 and oxacillin, spectral counting based label-free quantitative proteomics was applied. Based on well-developed label-free quantitative proteomics workflow, sub-MIC doses of the drug combination of SIPI-8294/Oxa and Ery/Oxa as well as SIPI-8294 only, oxacillin only and erythromycin only were applied to MRSA. The differentially expressed proteins were obtained by comparing the drug treatment groups with the untreated control group. Hierarchical clustering analysis shows that SIPI-8294/Oxa elicits very different responses in the cell than those by the individual drugs or the Ery/Oxa combination, which shows no synergistic effect.
Moreover, the differentially expression levels of PBP2a was four times lower in the SIPI-8294/Oxa group than those in the Ery/Oxa group in label-free quantitative proteomics results. In addition, mRNA transcription levels also have the same trend. These results indicate that the synergistic mechanism may be related to the interference with oxacillin resistance mechanism. We also observed large fold changes in proteins spanning many different pathways, indicating a global response of the bacteria to the antibiotics. Although we cannot distinguish between cause (the action of the antibiotic) and effect (the response of the bacteria to combat such action) using our data, we identified oxidation-reduction homeostasis and cell wall biosynthesis to be possible players in mechanism of the synergistic effect of SIPI-8294 and oxacillin. Since the cells were still growing under our conditions, it would be expected that the cells can counteract the stress imposed on them sufficiently to restore the homeostatic balance required for continued growth. However, our data would reveal which aspects of the cellular machinery have been perturbed, from which potential hypotheses about cause and effect can be formulated.
Methods
Minimum inhibitory concentration (MIC) tests
The S. aureus strains ATCC 43300 (MRSA) was obtained from the American Type Culture Collection. The bacteria were cultured in cation-adjusted Mueller-Hinton broth at 35 ± 2 °C. Minimum inhibitory concentration (MIC) test was performed according to the Clinical and Laboratory Standards Institute (CLSI) standard to verify the antibiotic effect. The MIC was determined to be the lowest concentration at which no visible growth of bacteria can be observed after incubation for 24 hours.
The synergistic effect was evaluated by the fractional inhibitory concentration (FIC) index based on the Loewe additivity zero-interaction theory38. For the combination A and B, it was calculated as FIC = MIC(A in combination)/MIC(A alone) + MIC(B in combination)/MIC(B alone). When FIC index is lower than or equal to 0.5, the combination is considered to have a synergistic effect. The measured MIC and FIC values were given in Supplementary Table S2.
Cell culture and drug treatment
The bacteria were grown in cation-adjusted Mueller-Hinton broth (Sigma-Aldrich, St. Louis, MO, USA). The bacteria without dosing any antibiotics were considered as untreated control. For antibiotic treatment, 1/8 MIC of antibiotics were added to 100 mL of medium and cultured together with 106/mL seeded bacteria. For the combination of SIPI-8294 and oxicillin (SIPI-8294/Oxa) treatment group, 8 μg/mL of SIPI-8294 was used in this work because the lowest FIC index was obtained at this concentration (as shown in Supplementary Table S2). The concentration for oxacillin in the SIPI-8294/Oxa group was taken at 1/8 MIC of oxacillin in the presence of 8 μg/mL SIPI-8294, which is 0.03125 μg/mL. For the combination of erythromycin and oxacillin (Ery/Oxa) treatment group, the concentrations used in this work were 32 μg/mL and 0.03125 μg/mL for erythromycin and oxacillin, respectively. For the single drug treatment, SIPI-8294 was added at the concentration of 8 μg/mL; oxacllin was added at the concentration of 8 μg/mL; erythromycin was added at the concentration of 32 μg/mL.
Three biological replicates were performed for each condition and two technical replicates were performed for each biological replicate. Bacteria growth was measured by OD600 (optical density at 600 nm). The cells were harvested when OD600 reached 0.1.
Label-free quantitative proteomics
The cells were collected and washed with phosphate buffered saline (PBS) buffer twice. Cell disruption was completed by ultra-sonication in the lysis buffer (8 M urea) for 5 min under ice-water bath. Then the cell lysate was centrifuged at 4000 rpm for 10 min at 4 °C and the supernatant was subjected to cold acetone precipitation. The precipitated proteins were dissolved in 4 M urea and 30 mM Tris-HCl (pH 6.5) and the protein concentration was measured by Bradford Protein assay (Bio-Rad, Hercules, CA, U. S. A.).
Thirty micrograms of proteins were used for the proteomics sample preparation, which was reduced by dithiothreitol (DTT) and alkylated by iodoacetamide. The resulting proteins were then digested by trypsin (1: 50 w/w, Promega, Madsion, WI) overnight at 37 °C. After digestion, the peptide sample was desalted by C18 reverse-phase ZipTip (Millipore, Darmstadt, Germany) and dried by SpeedVac (Eppendorf, Hamburg, Germany). Unless otherwise noted, all reagents were purchased from Sigma-Aldrich (St. Louis, MO, U.S.A).
The peptide samples were analyzed on a Thermo Scientific LTQ VelosTM platform (Thermo Fisher Scientific, Bremen, Germany) coupled with a Thermo Accela LC. One microgram of peptides were enriched on a trap column (Zorbax X300 SB-C18, 5 × 0.3 mm, 5 μm particle size) and separated on a C18 column (Thermo Bio-Basic-18, 150 × 0.1 mm, 300 Å pore size, 5 μm particle size) at a flow rate of 150 μL/min and 150 min LC run. For the MS parameters, the top ten most intense ions observed in the MS1 scan were set to acquire MS2 spectra. The dynamics exclusion was set as 60 seconds and the normalized collision energy was set at 30%.
MM file conversion (v3.9)39 was utilized to convert all the raw data into mgf file, and OMSSA40 was applied to search all the files against MRSA database. The database was constructed by combining complete proteome of 23 strains MRSA in UniProt, 1:1 ratio shuffled decoy protein sequences and common contaminants. Carbamidomethylation on cysteine and oxidation on methionine were set as fix modification and variable modification, respectively. The search results were further processed by the Trans-Proteomic Pipeline (TPP)41. The spectral counts of the two technical replicates from the same biological replicate with protein FDR lower than 0.01 were combined.
For the label-free comparative statistical analysis, the proteins identified confidently in at least two out of three biological replicates and the average spectral counts equal or over five were included in the statistical analysis. The spectral count of each protein was normalized by the total spectral counts of the biological replicate42,43. The statistical analysis (Student’s t-test and G-test) was conducted by PepC44 on every two conditions: antibiotic treated versus untreated control group. The differentially expressed proteins were filtered by the following cutoff: p-value (t-test) was lower than 0.05; and the fold changes were higher than 1.5 fold.
Bioinformatic analysis
Protein hierarchical analysis was done by Matlab. Protein localization prediction was done by the automatic bioinformatic pipeline named SLEP (surface localization of extracellular proteins)45 and PSORTb v3.046. Differentially expressed proteins were blasted against the NCBI-NR database of non-redundant protein sequences, and input into MEGAN47 to perform functional analysis using the SEED classification48 of subsystems and functional roles or the KEGG classification of pathways and enzymes.
Q-RT-PCR analysis of mecA and blaZ gene expression
The expression of two oxacillin resistance-related genes (mecA, coding for PBP2a, and blaZ, coding for β-lactamase) was quantified by real-time PCR. MRSA cells were cultured in the same condition as the proteomics experiments with five drug treatment groups and one untreated control group. In order to stabilize RNA, RNALater® (Ambion, Austin, TX) was added to the cells immediately after the cells being collected. Total RNA was extracted using AllPrep DNA/RNA Mini Kit (Qiagen, California, U.S.A) according to the manufacturer’s instruction. Q-RT-PCR was performed by a two-step process. RNA was first reverse transcribed to cDNA (Invitrogen, Carlsbad, CA); and RT-PCR was conducted on 7500 Fast RT-PCR (Applied Biosystems, California, U.S.A) using KAPA SYBR® FAST qPCR Kit with 40 cycles of denaturation for 5 seconds at 95 °C, annealing for 30 seconds at 50 °C, and extension for 20 seconds at 72 °C PCR primers for the mecA gene were (F: GTTAGATTGGGATCATAGCGTCATT) and (R: GCCTAATCTCATATGTGTTC CTGTAT); for blaZ gene were (F: CGTCTAAAAGAACTAGGAG) and (R: GCTTAA TTTTCCATTTGCGATAAG) and for 16S rRNA were (F: TCCGGAATTATTGGGCGTAA) and (R: CCACTTTCCTCTTCTGCACTCA). The melting curve analysis was performed immediately after amplification to verify the specificity of the PCR amplification products.
Fluorescence was measured at the end of the annealing-extension phase of each cycle. A threshold value for the fluorescence of all samples was set manually. The reaction cycle at which the PCR product exceeds this fluorescence threshold was identified as the threshold cycle (CT). The relative quantitation was calculated by the 2−ΔΔCT method49.
SEM
The cells were grown in SIPI-8294/Oxa (8/0.03125 μg/mL), SIPI-8294 (8 μg/mL), Oxacillin (8 μg/mL) and untreated control medium to OD 0.1. The cells were fixed in 2.5% glutaraldehyde (PBS buffer) for 2 hours, and washed with PBS buffer twice after fixation. Then the cells were dehydration in an ethanol series with increasing concentration (30%, 40%, 50%, 60%, 70%, 80%, 90%, pure ethanol) for 15 min, and finally suspend in pure tert-butanol for 15 min. The cells were air-dried and coated with gold followed by scanning electron microscope analysis (JSM-6390 Scanning Electron Microscope).
Additional Information
How to cite this article: Liu, X. et al. Proteomic response of methicillin-resistant S. aureus to a synergistic antibacterial drug combination: a novel erythromycin derivative and oxacillin. Sci. Rep. 6, 19841; doi: 10.1038/srep19841 (2016).
Supplementary Material
Acknowledgments
We thank Joyce Wong for technical assistance with the LC-MS/MS, HKUST Biosciences Central Research Facility (BioCRF) for providing mass spectrometry services and Materials Characterization and Preparation Facility (MCPF) for providing scanning electron microscope. We also thank Prof. Shunyi Shen in Shanghai Institute of Pharmaceutical Industry for providing SIPI-8294 for the mechanism study. This research is supported by the Research Grant Council of the Hong Kong Special Administrative Region Government, China (Grant No. 16302614 and 16100415), and the National Natural Science Foundation of China (Grant No. 81273413 and 81573329).
Footnotes
Author Contributions X.L. and P.P. contributed equally to the paper. X.L. and P.P. designed and conducted the proteomics experiments and the scanning electron microscopy. X.L., P.P. and Y.H. performed the data analysis. X.L. and W.Z. conducted the qPCR experiments. X.D. conducted the MIC tests. D.C., P.Q. and H.L. conceived of the study and devised the overall study strategy. X.L. and P.P. wrote the manuscript with input from all authors.
References
- Kluytmans J., Van Belkum A. & Verbrugh H. Nasal carriage of Staphylococcus aureus: epidemiology, underlying mechanisms, and associated risks. Clin Microbiol Rev 10, 505–520 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubes G. The bacteria fight back. Science (New York, NY) 321, 356 (2008). [DOI] [PubMed] [Google Scholar]
- Klevens R. M. et al. Invasive methicillin-resistant staphylococcus aureus infections in the united states. JAMA 298, 1763–1771 (2007). [DOI] [PubMed] [Google Scholar]
- Morens D. M. & Fauci A. S. Emerging Infectious Diseases: Threats to Human Health and Global Stability. PLoS Pathogens 9, e1003467 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLeo F. R. & Chambers H. F. Reemergence of antibiotic-resistant Staphylococcus aureus in the genomics era. J. Clin Invest 119, 2464–2474 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers H. F. The changing epidemiology of Staphylococcus aureus? Emerg Infect Dis 7, 178 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A. J. et al. Open PHACTS: semantic interoperability for drug discovery. Drug Discov Today 17, 1188–1198 (2012). [DOI] [PubMed] [Google Scholar]
- Mangili A., Bica I., Snydman D. & Hamer D. Daptomycin-resistant, methicillin-resistant Staphylococcus aureus bacteremia. Clin Infect Dis 40, 1058–1060 (2005). [DOI] [PubMed] [Google Scholar]
- Tsiodras S. et al. Linezolid resistance in a clinical isolate of Staphylococcus aureus. Lancet 358, 207–208 (2001). [DOI] [PubMed] [Google Scholar]
- Chait R., Craney A. & Kishony R. Antibiotic interactions that select against resistance. Nature 446, 668–671 (2007). [DOI] [PubMed] [Google Scholar]
- Alekshun M. N. & Levy S. B. Molecular mechanisms of antibacterial multidrug resistance. Cell 128, 1037–1050 (2007). [DOI] [PubMed] [Google Scholar]
- Berger-Bachi B. Resistance mechanisms of gram-positive bacteria. Int J Med Microbiol 292, 27–35 (2002). [DOI] [PubMed] [Google Scholar]
- Jia J. et al. Mechanisms of drug combinations: interaction and network perspectives. Nature reviews. Drug Discov 8, 111–128 (2009). [DOI] [PubMed] [Google Scholar]
- Lima T. B. et al. Bacterial resistance mechanism: what proteomics can elucidate. FASEB J 27, 1291–1303 (2013). [DOI] [PubMed] [Google Scholar]
- Wenzel M. & Bandow J. E. Proteomic signatures in antibiotic research. Proteomics 11, 3256–3268 (2011). [DOI] [PubMed] [Google Scholar]
- Burchmore R. Mapping pathways to drug resistance with proteomics. Expert Rev Proteomics 11, 1–3 (2014). [DOI] [PubMed] [Google Scholar]
- Shatalin K., Shatalina E., Mironov A. & Nudler E. H2S: a universal defense against antibiotics in bacteria. Science 334, 986–990 (2011). [DOI] [PubMed] [Google Scholar]
- Fajardo A. & Martinez J. L. Antibiotics as signals that trigger specific bacterial responses. Curr Opin Microbiol 11, 161–167 (2008). [DOI] [PubMed] [Google Scholar]
- Belenky P. & Collins J. J. Antioxidant strategies to tolerate antibiotics. Sci Signal 334, 915 (2011). [DOI] [PubMed] [Google Scholar]
- Cho H., Uehara T. & Bernhardt T. G. Beta-Lactam Antibiotics Induce a Lethal Malfunctioning of the Bacterial Cell Wall Synthesis Machinery. Cell 159, 1300–1311 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohanski M. A., Dwyer D. J., Hayete B., Lawrence C. A. & Collins J. J. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130, 797–810 (2007). [DOI] [PubMed] [Google Scholar]
- Van Oudenhove L. & Devreese B. A review on recent developments in mass spectrometry instrumentation and quantitative tools advancing bacterial proteomics. Appl Microbiol Biotechnol 97, 4749–4762 (2013). [DOI] [PubMed] [Google Scholar]
- Robinson J. L., Adolfsen K. J. & Brynildsen M. P. Deciphering nitric oxide stress in bacteria with quantitative modeling. Curr Opin Microbiol 19, 16–24 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranakis I. et al. Proteome studies of bacterial antibiotic resistance mechanisms. J Proteomics 97, 88–99 (2014). [DOI] [PubMed] [Google Scholar]
- Zhonglei L. et al. New erythromycin derivatives can enhance beta-lactam antibiotics against Methicillin-resistant Staphylococcus aureus. Lett Appl Microbiol 60 (4), 352–358 (2015). [DOI] [PubMed] [Google Scholar]
- Huson D. H., Mitra S., Ruscheweyh H. J., Weber N. & Schuster S. C. Integrative analysis of environmental sequences using MEGAN4. Genome Res 21, 1552–1560, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R. et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res 33, 5691–5702 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole K. Resistance to β-lactam antibiotics. Cell Mol Life Sciences CMLS 61, 2200–2223 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J. & Davies D. Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev: MMBR 74, 417–433 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumft W. G. Cell biology and molecular basis of denitrification. Microbiol Mol Biol Rev 61, 533–616 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane B. R., Sudhamsu J. & Patel B. A. Bacterial nitric oxide synthases. Annu Rev Biochem 79, 445–470 (2010). [DOI] [PubMed] [Google Scholar]
- Van Sorge N. M. et al. Methicillin-resistant Staphylococcus aureus Bacterial Nitric-oxide Synthase Affects Antibiotic Sensitivity and Skin Abscess Development. J Biolog Chem 288, 6417–6426 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusarov I., Shatalin K., Starodubtseva M. & Nudler E. Endogenous nitric oxide protects bacteria against a wide spectrum of antibiotics. Science 325, 1380–1384, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson A. R., Libby S. J. & Fang F. C. A Nitric Oxide–Inducible Lactate Dehydrogenase Enables Staphylococcus aureus to Resist Innate Immunity. Science 319, 1672–1676 (2008). [DOI] [PubMed] [Google Scholar]
- Gunka K. & Commichau F. M. Control of glutamate homeostasis in Bacillus subtilis: a complex interplay between ammonium assimilation, glutamate biosynthesis and degradation. Mol Microbiol 85, 213–224 (2012). [DOI] [PubMed] [Google Scholar]
- Candela T. & Fouet A. Poly-gamma-glutamate in bacteria. Mol Microbiol 60, 1091–1098 (2006). [DOI] [PubMed] [Google Scholar]
- Harth G., Zamecnik P. C., Tang J.-Y., Tabatadze D. & Horwitz M. A. Treatment of Mycobacterium tuberculosis with antisense oligonucleotides to glutamine synthetase mRNA inhibits glutamine synthetase activity, formation of the poly-L-glutamate/glutamine cell wall structure, and bacterial replication. Proc Nat Acad Sci 97, 418–423 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewe S. The problem of synergism and antagonism of combined drugs. Arzneimittelforschung 3, 285–290 (1953). [PubMed] [Google Scholar]
- Xu H. & Freitas M. A. Monte Carlo simulation-based algorithms for analysis of shotgun proteomic data. J Proteome Res 7, 2605–2615 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geer L. Y. et al. Open mass spectrometry search algorithm. J Proteome Res 3, 958–964 (2004). [DOI] [PubMed] [Google Scholar]
- Deutsch E. W. et al. A guided tour of the Trans-Proteomic Pipeline. Proteomics 10, 1150–1159 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho P. C., Fischer J. S., Chen E. I., Yates J. R. & Barbosa V. C. PatternLab for proteomics: a tool for differential shotgun proteomics. BMC Bioinformatics 9 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho P. C., Hewel J., Barbosa V. C. & Yates J. R. Identifying differences in protein expression levels by spectral counting and feature selection. Genet Mol Res 7, 342–356 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinecke N., Pratt B., Vaisar T. & Becker L. PepC: proteomics software for identifying differentially expressed proteins based on spectral counting. Bioinformatics 26, 1574–1575 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giombini E., Orsini M., Carrabino D. & Tramontano A. An automatic method for identifying surface proteins in bacteria: SLEP. BMC bioinformatics 11, 39 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu N. Y. et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26, 1608–1615 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson D. H., Mitra S., Ruscheweyh H.-J., Weber N. & Schuster S. C. Integrative analysis of environmental sequences using MEGAN4. Genome Res 21, 1552–1560 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R. et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res 33, 5691–5702 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen T. D. & Livak K. J. Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3, 1101–1108 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.