

Abstract
Cranial neural crest cells (CNCCs) delaminate from embryonic neural folds and migrate to pharyngeal arches, which give rise to most mid-facial structures. CNCC dysfunction plays a prominent role in the etiology of orofacial clefts, a frequent birth malformation. Heterozygous mutations in SPECC1L have been identified in patients with atypical and syndromic clefts. Here, we report that in SPECC1L-knockdown cultured cells, staining of canonical adherens junction (AJ) components, β-catenin and E-cadherin, was increased, and electron micrographs revealed an apico-basal diffusion of AJs. To understand the role of SPECC1L in craniofacial morphogenesis, we generated a mouse model of Specc1l deficiency. Homozygous mutants were embryonic lethal and showed impaired neural tube closure and CNCC delamination. Staining of AJ proteins was increased in the mutant neural folds. This AJ defect is consistent with impaired CNCC delamination, which requires AJ dissolution. Further, PI3K-AKT signaling was reduced and apoptosis was increased in Specc1l mutants. In vitro, moderate inhibition of PI3K-AKT signaling in wildtype cells was sufficient to cause AJ alterations. Importantly, AJ changes induced by SPECC1L-knockdown were rescued by activating the PI3K-AKT pathway. Together, these data indicate SPECC1L as a novel modulator of PI3K-AKT signaling and AJ biology, required for neural tube closure and CNCC delamination.
Cranial neural crest cells (CNCCs) are specified in the dorsal neuroectoderm and delaminate from the neuroepithelium of the developing neural folds through a process that involves epithelial-mesenchymal transition (EMT)1,2,3. Premigratory epithelial CNCCs break down their cell-cell junctions and become migratory mesenchymal CNCCs, which populate the first and second pharyngeal arches, and give rise to the majority of craniofacial cartilage. Thus, genes that modulate CNCC function are frequently perturbed in the etiology of craniofacial congenital malformations such as orofacial clefts3,4,5,6,7, which are among the most frequent birth malformations affecting 1/800 births in the U.S. alone8.
CNCC delamination occurs simultaneously with anterior neural tube closure between mouse embryonic days 8.5 and 9.5. Many mouse mutants of genes associated with orofacial clefts also manifest some form of neural tube defects, including Irf69,10, Ghrl310, Cfl111 and Pdgfrα12. However, the neural tube closure and CNCC delamination processes can be considered independent since the Splotch (Pax3) mouse mutant shows neural tube closure defects without any effect on CNCC delamination or migration13,14. Additional mouse models with defects in CNCC delamination and neural tube closure will help delineate the shared molecular underpinnings of these two processes.
Delamination of CNCCs from the neural epithelium requires dissolution of adherens junctions (AJs), composed of a protein complex, containing E-cadherin, β-catenin, α-E-catenin and α-actinin among others, tethered to actin filaments2. Studies where E-cadherin is overexpressed in the neural folds, show reduced or delayed delamination of CNCCs. Conversely, down-regulation of E-cadherin results in early delamination15,16. Many factors that mediate EMT during CNCC delamination are transcription factors (AP2α, Id2, FOXD3, SNAIL, TWIST, SOX10) and extracellular matrix (ECM) remodeling proteins such as matrix metalloproteases (MMPs), however, direct cytoskeletal regulators of CNCC AJs are not yet known. The PI3K-AKT pathway is known to antagonize E-cadherin levels, primarily from cancer studies17. Recent studies show that loss of murine PDGFα based PI3K-AKT signaling leads to craniofacial malformations including cleft palate and neural tube defects12. However, a link between the PI3K-AKT pathway and AJ stability in CNCC delamination is not clear.
Previously, we identified SPECC1L as the first gene mutated in two individuals with a severe cleft that extends from the oral cavity to the eye, termed Oblique Facial Cleft (ObFC) or Tessier IV clefts18. SPECC1L mutations have since been identified in two multi-generation families with autosomal dominant Opitz G/BBB syndrome (OMIM #145410), where affected individuals manifest hypertelorism and cleft lip/palate19, and in a family with Teebi hypertelorism syndrome (OMIM #145420)20. More than half of Opitz G/BBB syndrome cases are X-linked (OMIM #300000), caused by mutations in MID1 gene21, which encodes a microtubule-associated cytoskeletal protein22. We proposed that SPECC1L, also a microtubule- and actin cytoskeleton-associated protein, may mediate transduction of signals required to remodel the actin cytoskeleton during cell adhesion and migration18. Using in vitro and in vivo studies, we now describe SPECC1L as a novel regulator of AJ stability through PI3K-AKT signaling. At the cellular level, SPECC1L deficiency resulted in reduced levels of pan-AKT protein and increased apico-basal AJ dispersion, which was rescued by chemical activation of the AKT pathway. In vivo, Specc1l-deficient embryos showed neural tube closure failure and reduced CNCC delamination. Thus SPECC1L functions in the highly regulated cell adhesion based signaling required for proper CNCC function during facial morphogenesis.
Results
SPECC1L is localized at cell boundaries in cultured cells upon confluency
To characterize the role of SPECC1L at the cellular level, we used the SPECC1L-deficient U2OS osteosarcoma stable cell line described previously18. These stable SPECC1L-knockdown (kd) U2OS cells have a moderate (60–70%) reduction in SPECC1L transcript and protein levels with defects in migration and actin cytoskeleton reorganization18. In contrast, a severe transient reduction in SPECC1L has been shown to cause mitotic defects23. Upon further characterization, we find that our stable SPECC1L-kd cells changed morphology upon very high confluency (Fig. 1). Individual control and kd cells at low confluency appeared similar (Fig. 1A,D). Twenty-four hours after confluency, control cells maintained their cuboidal shape (Fig. 1B,E), while SPECC1L-kd cells elongated (Fig. 1C,F). The extent of this cell shape change was captured by in vivo live-imaging of control and kd cells (Movie 1). To determine the role of SPECC1L in confluent cells, we first examined its expression. We found that SPECC1L protein level was increased upon confluency (Fig. 1G) without an increase in SPECC1L transcript levels (Fig. 1H). Furthermore, SPECC1L protein accumulated at cell-cell boundaries with increasing cell density (Fig. 2A–E), in a pattern overlapping with that of membrane-associated β-catenin (Fig. 2A’–E’). Given the association of SPECC1L with actin cytoskeleton18,23, we hypothesized that SPECC1L interacts with actin-based adherens junctions (AJs).
Figure 1. SPECC1L-knockdown cells elongate upon high confluency.

(A–F) Compared to control U2OS cells (A–C), SPECC1L-knockdown cells (D–F) elongate upon high confluency (F). Three (T1, T3, T6) of the six time-points we chose for various cell densities are shown here. (G) Western blot analysis shows that SPECC1L protein is stabilized at high, compared to low confluency, in control cells. SPECC1L immunoblot shows an expected 120 kD band and a higher molecular weight band (*) that is likely modified post-translationally. The western blot analyses were performed under the same conditions for low and high confluence. The images shown for SPECC1L at low and high confluence are taken from a single blot. The same blot was stripped and re-probed with β-actin antibody. (H) Quantitative RT-PCR analysis shows no significant change in SPECC1L transcript levels. Error bars represent SEM from four independent experiments.
Figure 2. SPECC1L is stabilized at cell-cell boundaries similarly to β-catenin.

(A–E) We picked six time-points (T1–T6) representing a range of cell densities to standardize analysis of cell shape and AJ change in SPECC1L-knockdown (kd) U2OS cells. First five of these time-points include individual cells (T1), 50–70% confluency with small cell clusters (T2), confluency without kd cell shape change (T3), shape change in kd cells (T4), and 24 hrs post shape change in kd cells (T5). SPECC1L protein is mostly dispersed within the cytoplasm at T1 (A), but is observed to accumulate at cell-cell boundaries at subsequent time-points (B–E), arrows). (F–J) β-catenin shows a similar accumulation at cell-cell boundaries in association with AJ complex. (A’–E’) SPECC1L and β-catenin show overlapping staining at cell boundaries at high cell densities (arrows). (F’–J’) In SPECC1L-kd cells, β-catenin staining appears normal at low cell densities (F’–H’), but is expanded upon cell shape change (I’,J’; arrows), indicating altered AJs. Bars = 10 μm.
SPECC1L deficient cells show abnormal staining and apico-basal dispersion of adherens junction proteins
Next we sought to determine the effects of SPECC1L deficiency on AJs. We used several markers associated with AJs, including canonical components F-Actin, Myosin IIb, β-catenin, and E-cadherin24,25,26,27. As previously reported, actin stress fibers were increased in SPECC1L-kd cells (Fig. 3A,B)18. Myosin IIb, associated with actin filaments, showed a similar increase in SPECC1L-kd cells in vitro (Fig. 3C,D). AJ-associated β-catenin, which binds to cadherins at the cell membrane, showed a normal “honey-comb” pattern of expression in control cuboidal cells (Fig. 3E,G). Interestingly, in planar images using confocal microscopy, β-catenin (Fig. 3E,F) and E-cadherin (Fig. 3G,H) staining at the cell membrane in confluent SPECC1L-deficient cells showed a drastically expanded staining pattern. This expansion in AJ-associated β-catenin staining in kd cells was most evident upon confluency, but appeared to precede the cell shape change (Fig. 2F–J,F’–J’). To determine the physical nature of this expanded AJ staining, we examined the cell boundaries in the apico-basal plane of SPECC1L-kd U2OS cells (Fig. 3I,J) using transmission electron microscopy (TEM). Compared to control cells (Fig. 3I), which had distinct electron-dense regions indicating AJs (arrowheads), the kd cells (Fig. 3J) showed a large contiguous region of high electron density, suggesting extensive dispersion of AJs along the apico-basal plane. Also, in lateral sections, we observed widespread ruffling of the cell membrane in the kd cells (Fig. S1A,B), accounting for the expanded striated pattern of β-catenin and E-cadherin staining (Fig. 3F,H). In support of a role for SPECC1L in AJs, β-catenin co-immunoprecipitated with SPECC1L in lysates from confluent U2OS cells (Fig. 3K). Together with expanded immunostaining of AJ markers, the TEM analysis is consistent with our hypothesis that SPECC1L deficiency increased apico-basal density and dispersion of AJs.
Figure 3. SPECC1L-kd U2OS cells show altered expression of adherens junction markers.
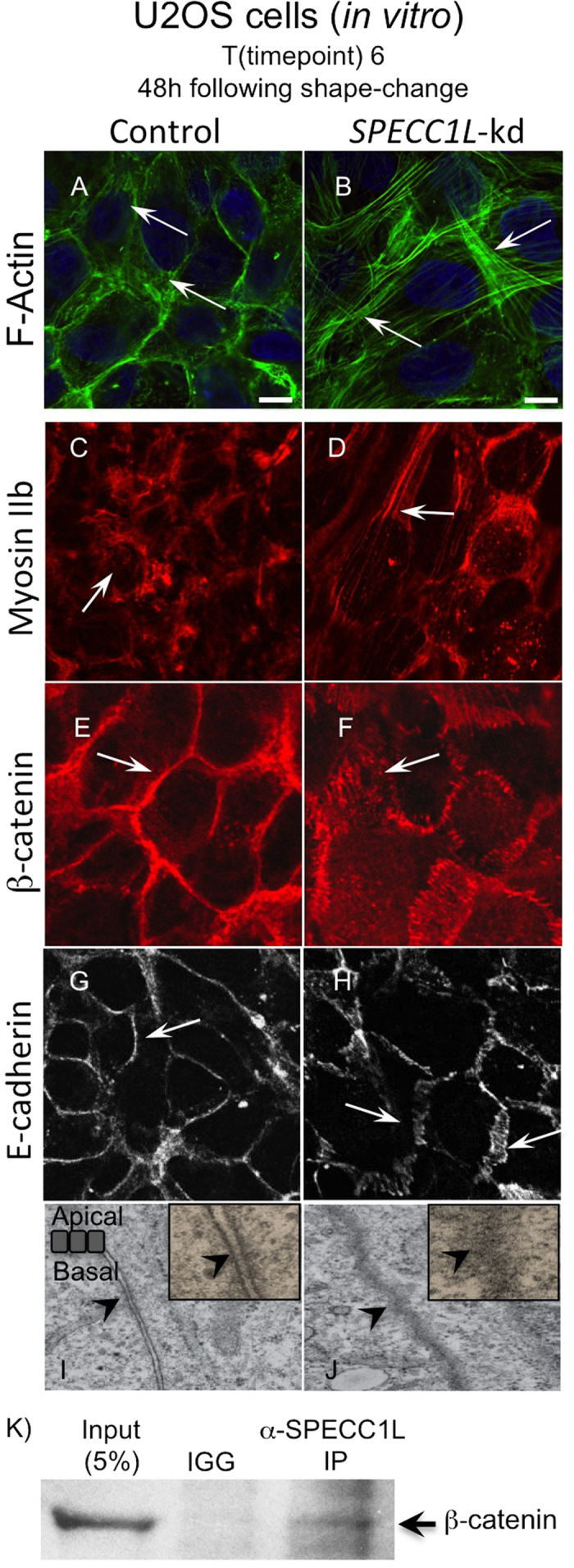
(A–H) Increased F-actin staining in kd cells at 48 hours post-confluency (T6; A,B). Altered F-actin associated Myosin IIb staining (C,D). Smooth pattern of β-catenin and E-cadherin membrane staining in control cells (E,G) is expanded in SPECC1L-kd cells (F,H). Bars = 10 μm. (I–J) Electron micrographs looking at apico-basal cell-cell boundary. Control cells show distinct electron-dense regions, indicating adherens junctions (I, arrow). In contrast, the entire apico-basal boundary appears electron-dense in SPECC1L-kd cells (J, arrows), suggesting increased density and dispersion of adherens junctions. (K) β-catenin co-immunoprecipitation with SPECC1L in lysates from confluent U2OS cells. The image is taken from a single blot, and represents one of four independent experiments.
Specc1l deficiency leads to incomplete neural tube closure and reduced CNCC delamination
To understand the role of SPECC1L in craniofacial morphogenesis, we created a mouse model of Specc1l deficiency using two independent gene-trap ES cell lines - DTM096 and RRH048 (BayGenomics, CA), which trap Specc1l transcripts in introns 1 and 15 respectively (Fig. 4A, Fig. S2). Genomic location of gene-trap vector insertion was identified by whole-genome sequencing and verified by PCR (Fig. S2). Both gene-trap constructs also afford in-frame Specc1l-lacZ reporter fusion upon trapping. Thus, lacZ expression, as determined by X-gal staining, was used as a proxy for Specc1l expression. Both alleles show a similar lacZ expression pattern with the DTM096 gene-trap in intron 1 showing stronger expression than RRH048 in intron 15 (not shown). Specc1l is expressed broadly, however, expression is particularly robust in the neural folds at E8.5 (Fig. 4B), the neural tube and facial prominences at E9.5 and E10.5 (Fig. 4C,D), and in the developing limbs and eyes at E10.5 (Fig. 4D). We previously reported that SPECC1L expression in the first pharyngeal arch at E10.5 is present in both the epithelium and the underlying mesenchyme18, consistent with CNCC lineage. To validate expression of SPECC1L in CNCCs, we co-stained for SPECC1L and NCC markers AP2-alpha (AP2A) and SOX10 in E8.5 neural folds (Fig. 4E–J) and in E9.5 cranial sections (Fig. 4K–P). At E8.5, SPECC1L stains the neural folds broadly (Fig. 4E,H), including cells stained with the NCC markers (Fig. 4G,J). At E9.5, SPECC1L (Fig. 4K,N) strongly stains migratory CNCCs co-stained with AP2A (Fig. 4L,M) or SOX10 (Fig. 4O,P).
Figure 4. Specc1l expression and overlap with the neural crest cell lineage.

(A) Schematic representation of murine Specc1l gene indicating insertion of genetrap vectors in ES cell clones DTM096 (intron 1) and RRH048 (intron 15). (B–D) Heterozygous Specc1lDTM096 embryos stained for lacZ, representing Specc1l expression, from E8.5 to E10.5. NE = neuroectoderm, NF = neural folds, PA1 = 1st pharyngeal arch. (E–P) Co-immunostaining of SPECC1L with NCC markers AP2A and SOX10 in E8.5 neural folds (NF; E–J) and in E9.5 cranial sections (K–P). SPECC1L staining is broadly observed in the E8.5 neural folds (E,H; arrows), including in cells marked by AP2A (F,G; arrows) and SOX10 (I,J; arrows). At E9.5, SPECC1L strongly stains migrating CNCCs (K,N; arrows) marked by AP2A (L,M; arrows) and SOX10 (O,P; arrows).
Intercross matings between Specc1lDTM096/+ and Specc1lRRH048/+ heterozygous mice showed that the two gene-trap alleles failed to complement each other, and that compound heterozygous and embryos homozygous for either gene trap allele are embryonic lethal (Table S1). The Mendelian ratios indicate reduced survival of heterozygotes at birth (1.34 vs. 2.0 expected). We noted a low incidence of perinatal lethality in heterozygotes; some with craniofacial malformations (Fig. S3). However, the low penetrance of these perinatal craniofacial phenotypes makes it difficult to study the underlying pathophysiological mechanism. Therefore, we focused on the embryonic lethal phenotype of the Specc1l homozygous mutant.
The majority of Specc1lDTM096/RRH048compound heterozygous or homozygous mutant embryos failed to thrive beyond E9.5–10.5 (Fig. 5A–D), with a failure of the neural tube to close anteriorly (Fig. 5B,D), and sometimes posteriorly (not shown). This cranial neural tube closure defect was accompanied by a large proportion of CNCCs, marked by DLX2, to remain in the neural folds at E10.5, suggesting a failure to delaminate (Fig. 5A’–D’). To determine if there was also a reduction in overall specification of CNCCs, we marked the CNCC lineage with GFP in our gene-trap lines with Wnt1-Cre and ROSAmTmG. We flow-sorted the GFP+ NCCs and GFP- (RFP+) non-NCCs from whole embryos. At E9.5, the proportion of flow-sorted GFP-labeled CNCCs did not change significantly between WT and mutant embryos (not shown), indicating normal CNCC specification. Thus, we hypothesized that the residual staining with Wnt1-Cre and DLX2 in the open neural folds (Fig. 5B’) was due to a defect in CNCC delamination, possibly due to increased density or dispersion of AJs, as observed in SPECC1L-kd cells. We used NCC markers SOX10, AP2A, and DLX2 to confirm the presence of CNCCs in the neural folds (Fig. 5E–R). At E8.5, neural fold staining is observed with all three NCC markers in both WT (Fig. 5E,G,I) and Specc1l mutant (Fig. 5F,H,J) sections. At E9.5, while NCC markers stain migratory NCCs in WT sections (Fig. 5M,O,Q), residual NCC staining is present in the open neural folds of Specc1l mutant embryos (Fig. 5N,P,R). Since SOX10 and DLX2 mark migratory CNCCs, this result indicates that SPECC1L deficient CNCCs attain post-migratory specification but fail to emigrate from the neural folds.
Figure 5. Specc1l deficiency leads to defects in neural tube closure, cranial neural crest cell delamination and AJs.

(A,B’) E9.5 WT (A) embryo with migratory cranial neural crest cells (CNCCs) marked with Wnt1-Cre (A’). In contrast, Specc1l mutant embryo shows open neural folds (B), arrow) with CNCCs that have still not migrated (B’, arrow). (C,D’) Brightfield images (C,D) and CNCC marker DLX2 immuno-stain (C’,D’) of E10.5 WT (C,C’) and Specc1l mutant (D,D’) embryos. In E10.5 WT embryo, DLX2 positive CNCCs populate the branchial arches (C’, arrowhead), while in the mutant significant staining remains in open neural folds (D’, arrow) with some staining in the 1st pharyngeal arch (D’, arrowhead), indicating poor CNCC delamination and migration. E-R) Sections from WT and Specc1l mutant embryos at E8.5 (E–L) and at E9.5 (M–R) were stained with NCC markers SOX10 (E,F,M,N), AP2A (G,H,O,P), and DLX2 (I,J,Q,R). At E8.5, NCC staining is observed in the neural folds (NF) of WT and mutant sections. Co-staining of SOX10 and β-catenin in E8.5 WT (K) and mutant (L) shows increased β-catenin staining at cell boundaries in the neural folds. At E9.5, migratory CNCC staining is observed in WT (M,O,Q), while in mutants undelaminated CNCCs stain the open neural folds (N,P,R). (S–Z) Analysis of AJ markers in vivo in WT and Specc1lDTM096/RRH048mutant E9.5 embryo coronal sections. The approximate plane of section is indicated in the top-right corner. Increased F-actin (S,T) and Myosin IIb (U,V) staining is observed in mutant tissue sections. Similarly to in vitro results in Fig. 3, expanded β-catenin (W,X) and E-cadherin (Y,Z) membrane staining is observed in vivo in mutant embryos. (AA-BB) Electron micrograph of WT embryo section looking at apico-basal cell boundary shows a distinct electron-dense region indicating adherens junction (AA, arrowhead). In contrast, the entire apico-basal boundary appears electron-dense in Specc1l mutant embryo section (BB, arrowheads), suggesting increased density and dispersion of adherens junctions.
Specc1l mutant tissue recapitulates in vitro adherens junction changes
To test our hypothesis that reduced delamination is due to altered AJs, we investigated the AJ markers in the open neural folds of Specc1l mutant embryos (Fig. 5S–Z). We observed increased actin stress fibers (Fig. 5S,T) and concomitant increased localization of Myosin IIB staining to the actin fibers (Fig. 5U,V). Importantly, we saw increased staining of β-catenin (Fig. 5W,X) and E-cadherin (Fig. 5Y,Z) at cell-cell boundaries. We also examined β-catenin staining of NCCs in the neural folds of E8.5 embryos (Fig. 5K,L). The β-catenin staining appears more robust in Specc1l mutant neural folds (Fig. 5L vs. K), suggesting that changes in AJs have commenced. In electron micrographs of cranial sections from E9.5 embryos, we again observed an increase in dispersed electron-dense staining in Specc1l mutant embryos compared to WT (Figs 5AA,BB and S1E–H). Taken together, these results validate our in vitro findings in SPECC1L-kd U2OS cells, and indicate that abnormal staining of AJs precedes CNCC delamination in our mutant embryos.
Reduced PI3K-AKT signaling and increased apoptosis in SPECC1L deficient cells
Given the known antagonistic relationship of AKT activity to E-cadherin stability17,28, we hypothesized involvement of PI3K-AKT signaling. In addition, we observed sub-epidermal blebbing in some of our mutant embryos that escape E9.5–10.5 lethality (<5%), and instead arrest at approximately E13.5 (Fig. S3). Sub-epidermal blebbing is a hallmark of reduced PDGFRα-based PI3K-AKT signaling12. Fantauzzo et al. (2014) reported that disruption of PDGFRα-based PI3K activation in PdgfraPI3K/PI3K mutant embryos results in sub-epidermal blebbing, neural tube defects, and cleft palate phenotypes. Indeed, pan-AKT and active phospho-Ser473-AKT levels were reduced in vivo in Specc1l mutant tissue (Fig. 6A–D), prior to embryonic arrest at E9.5. Reduced phospho-Ser473-AKT level was likely entirely due to reduced pan-AKT levels in vivo (Fig. 6E) and in vitro (Fig. 6F). The in vitro reduction was only present at high confluency of U2OS cells upon cell shape and AJ density changes (Fig. 6D). Thus, our data suggest SPECC1L is a novel positive regulator of PI3K-AKT signaling in craniofacial morphogenesis.
Figure 6. Specc1l mutant embryos show reduced PI3K-AKT signaling and increased cell death.

(A–E) E8.5 (A,B) and E9.5 (C,D) cranial sections or E9.5 lysates (E) from Specc1l mutant embryos show decreased levels of active phospho-S473-AKT and pan-AKT protein, compared to WT control. The western blot analyses were performed under the same conditions for WT and mutant lysates. The images shown for SPECC1L are taken from a single blot. The same blot was stripped and re-probed with pan-AKT and then β-actin antibodies. Pan-AKT level in E8.5 neural folds (A,B) and phospho-S473-AKT level in E9.5 cranial section are markedly reduced. (F) A similar reduction is seen in pan-AKT levels in SPECC1L-kd U2OS cell lysates collected at high confluency. The error bars represent SEM from quantitation of three independent western blots. (G–J) Sections of WT embryos at E9.5 show cell proliferation (G,G’) with negligible apoptotic activity (H,H’) through KI67 and cleaved Caspase 3 staining, respectively. In Specc1l mutant embryos, there is comparable cell proliferation (I), however, there is a striking increase in cells undergoing apoptosis (J).
We next examined markers of proliferation and apoptosis. We did not observe any differences in proliferation in E9.5 embryos (Fig. 6E,G vs. I) as measured by KI67 staining, with a proliferative index of 82.5% for WT and 86.5% for Specc1l mutant (p < 0.56, Fisher exact test). Similarly, we did not observe any difference in apoptosis as measured by cleaved Caspase 3 staining in neural folds at E8.5, prior to embryonic arrest (not shown). In contrast, apoptosis was markedly increased throughout the E9.5 mutant embryo (Fig. 6F,H vs. J). This global increase in apoptosis is consistent with reduced PI3K-AKT signaling and early embryonic lethality29,30,31.
Upregulation of PI3K-AKT pathway rescues SPECC1L-kd phenotype
Next, to confirm a causal role for PI3K-AKT signaling in AJ change in our kd cells, we chemically altered the pathway in control and kd cells (Fig. 7A–F). We used the cell shape change phenotype observed in confluent SPECC1L-kd cells as a marker, which we quantified using the ratio of the longest dimension (length) and the corresponding perpendicular dimension (width). A relatively round or cuboidal cell would be expected to give a ratio of 1 (Fig. 7G). In addition to cell shape, we confirmed the effect on AJs through β-catenin staining (Fig. 7A’–F’). Inhibition of the PI3K-AKT pathway using Wortmannin was sufficient to alter cell shape (Fig. 7A,C) and AJs (Fig. 7A’) in control cells. The PI3K-AKT activator, SC-79, did not affect cell shape (Fig. 7A,E) or AJ expansion (Fig. 7A’) in control cells. In SPECC1L-kd cells, further down-regulation of PI3K-AKT pathway resulted in increased apoptosis (Fig. 7B,D) with drastically expanded staining for β-catenin (Fig. 7B’), consistent with our severe mutants in vivo. Importantly, upregulation of the PI3K-AKT pathway drastically ameliorated the cell shape (Fig. 7B,F) and AJ (Fig. 7B’’) phenotypes. As stated above, changes in cell shape were quantitated as a cell circularity ratio (CCR), and compared for significance (Fig. 7G). Indeed, in control cells (Fig. 7G, CCR = 1.56), Wortmannin treatment was sufficient to significantly change cell shape (Fig. 7G, CCR = 3.61, p < 2.4 × 10−9), which was similar in extent to that seen in SPECC1L-kd cells (Fig. 7G, CCR = 3.46). Wortmannin treatment of SPECC1L-kd cells (Fig. 7G, CCR = 3.60, not significant) did not affect cell elongation any further than untreated kd cells (Fig. 7G, CCR = 3.46, not significant) or Wortmannin-treated control cells (Fig. 7G, CCR = 3.61, not significant). Most important, SC-79 AKT activator rescued the elongated cell shape phenotype of SPECC1L-kd cells (Fig. 7G, CCR = 1.74, p < 6.2 × 10−12). These results substantiate that SPECC1L regulates PI3K-AKT signaling, and indicate that moderate reduction in SPECC1L affects cell adhesion, while severe reduction can lead to apoptosis (Fig. 8).
Figure 7. Upregulation of PI3K-AKT pathway can rescue SPECC1L-kd phenotype in vitro.

(A–F’) Control (A,C,E) and SPECC1L-kd (B,D,F) cells were treated with PI3K-AKT pathway inhibitor Wortmannin (C,D) or activator SC-79 (E,F). Untreated control cells are cuboidal (A) with normal honey-comb pattern of β-cat staining (A’), while kd cells are elongated (B) with expanded β-cat staining (B’). Upon down-regulation of PI3K-AKT pathway, control cells are elongated (C) with β-cat expansion (C’), while kd cells begin to undergo apoptosis (D), similarly to our severe mutant embryos, and show extremely expanded β-cat staining (D’). Upon up regulation of PI3K-AKT pathway, control cells remain cuboidal (E) with normal β-cat staining (E’), while kd cells show much improved cell shape (F) and β-cat staining (F’), indicating rescue. (G) The extent of cell shape change in (A–F) were quantitated using a cell circularity ratio (CCR) of longest dimension (length) and the corresponding perpendicular dimension (width) using MetaMorph software. The no treatment (NT) SPECC1L-kd cells (CCR = 3.46) were significantly elongated than control cells (CCR = 1.56, p < 6.1 × 10−13). Wortmannin (Wort) inhibition of PI3K-AKT pathway in control cells was sufficient to cause a similar elongation of cell shape (CCR = 3.61, p < 2.4 × 10−9). Similarly, AKT activation with SC-79 in SPECC1L-kd cells was able to rescue the elongated cell shape back to control level (CCR = 1.74, p < 6.2 × 10−12). Wortmannin treatment of SPECC1L-kd cells resulted in increased apoptosis, but did not cause further increase in cell shape change (CCR = 3.60) beyond that observed in untreated kd (CCR = 3.46, ns) or Wortmannin-treated control cells (3.61). ns = not significant. Measurement of 50 cells +/− S.E.M. is shown. Statistical differences were calculated using a Student’s t-test.
Figure 8. Model of SPECC1L modulation of PI3K-AKT pathway and AJs.
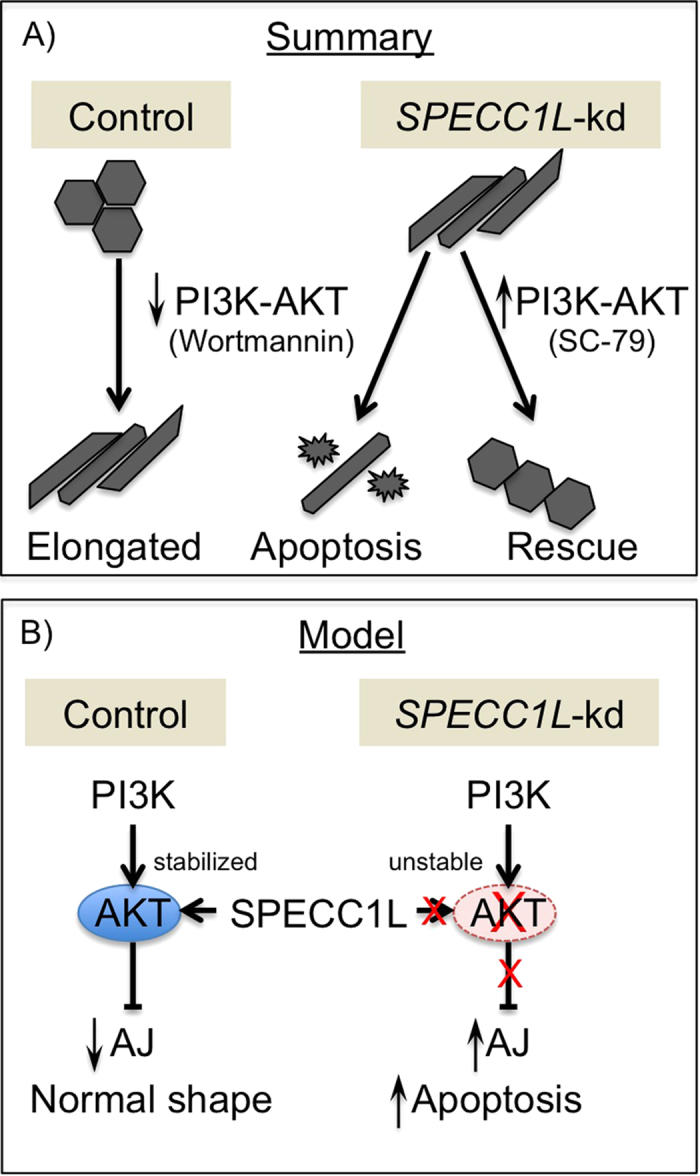
(A) Schematic summary of PI3K-AKT pathway inhibition and activation leading to AJ change and rescue, respectively. (B) A model proposing that SPECC1L stabilizes AKT protein.
Discussion
Dissolution of AJs is required for pre-migratory CNCCs to delaminate from the neural epithelium of the anterior neural folds1,15,32. The increased staining of AJ components and loss of apico-basal asymmetrical distribution of AJs in SPECC1L-deficient cells in vitro and in vivo, combined with physical proximity of SPECC1L with β-catenin indicates a role for SPECC1L in maintaining localized stability of AJs by properly organizing the actin cytoskeleton. The association of SPECC1L with the actin cytoskeleton and β-catenin combined with an increase in condensed actin filaments upon SPECC1L deficiency are consistent with the observed increase in AJ density. Another possibility is that increased actin fibers in SPECC1L deficient cells lead to altered tension between cells. As cell tension can impact AJ dynamics33, a change in tension may lead to more dispersed AJs34. Either alteration can consequently affect CNCC delamination.
Wnt1 is expressed in the early neural folds that give rise to neural crest cells. Thus, Wnt1-cre lineage tracing marks both pre-migratory and migratory NCCs35. However, Wnt1 also marks lineage of dorsal brain tissues that also arise from the early neural folds35,36, leaving a possibility that the Wnt1-marked staining in our E9.5 mutant open neural folds were not CNCCs. Our positive staining with NCC markers AP2A and SOX10 confirms that the open neural folds of the Specc1l mutant embryos indeed contain CNCCs. Furthermore, since AP2A and SOX10 are markers for early migrating NCCs, the positive staining indicates that these cells are post-migratory CNCCs that fail to delaminate by E9.5.
Our data indicate that the molecular regulation of AJs by SPECC1L is mediated by PI3K-AKT signaling. AKT signaling is reduced in SPECC1L-deficient cells and tissue. A direct role for PI3K-AKT signaling in craniofacial morphogenesis is supported by the findings of Fantauzzo et al. (2014) showing that lack of PDGFRα-based activation of PI3K-AKT signaling leads to cleft palate phenotype. We also showed that inhibition of the PI3K-AKT pathway is sufficient to alter AJs and cell shape in U2OS cells. Consistent with our findings, Cain et al.37 have shown that in endothelial cells down-regulation of PI3K α110 subunit led to a similarly expanded pericellular β-catenin staining, termed as increased “junctional index”. However, in their endothelial cells with already highly organized actin filaments, the down-regulation of PI3K-AKT pathway resulted in a relaxation of cell shape. In contrast, the SPECC1L-kd U2OS cells show elongated cell shape. This difference is likely cell-type specific. While down-regulation of PI3K-AKT signaling consistently affects the actin cytoskeleton, the effect on cell shape is dictated by altered tension resulting from changes in central actin fiber density and organization. In U2OS cells, we used the cell shape change only as a marker of the SPECC1L-deficient AJ change and rescue. Taken together, we propose that suppression of AKT pathway upon SPECC1L deficiency increases AJ stability and reduces delamination of the CNCCs.
Interestingly, pan-AKT levels are reduced in addition to phospho-473-AKT levels in vitro and in vivo upon SPECC1L deficiency, thus indicating regulation of PI3K-AKT signaling at the level of AKT protein stability or turnover. Both SPECC1L and MID1 genes, implicated in Opitz/GBBB Syndrome, encode proteins that stabilize microtubules18,22. The mechanism through which SPECC1L and MID1 mediate microtubule stabilization is not entirely clear. In the case of SPECC1L, this stabilization involves enhanced acetylation of a subset of microtubules18. Perhaps SPECC1L utilizes a similar mechanism to stabilize other proteins such as AKT. Acetylation of lysine residues in AKT protein has been shown to result in reduced membrane localization and phosphorylation38. In addition, K63 chain ubiquitination of the same lysine residues on AKT is required for its membrane localization and activation39,40. Of the few SPECC1L protein interactors identified in different high throughput yeast two-hybrid screens, four-CCDC841, ECM2942, APC and UBE2I43 - participate in protein turnover or stability through ubiquitination or sumoylation. It is possible that SPECC1L participates in the post-translational modification of AKT lysine residues, affecting AKT stability. However, a definitive role for SPECC1L in AKT protein localization and stability is yet to be elucidated.
A severe deficiency in SPECC1L expression in vivo results in increased AJ marker staining and defective CNCC delamination, as well as in increased apoptosis and early embryonic lethality. Previous reports have shown that mouse mutants with increased levels of apoptosis have associated neural tube44,45,46,47 and craniofacial defects48. It is proposed that excessive cell death in the neural folds or pharyngeal arches may result in insufficient number of cells required for proper morphogenetic movement48,49,50. In contrast, our SPECC1L-deficient cell lines with moderate reduction in SPECC1L expression show only AJ changes without evidence of increased cell death. However, chemical inhibition of PI3K-AKT pathway in these kd cells does indeed lead to increased apoptosis. Thus, a moderate reduction in SPECC1L expression or function allows cell viability. This is consistent with the observation that rare Specc1l mutant embryos that escape E9.5 arrest - likely due to reduced gene-trapping efficiency - are able to close their neural tubes and arrest later in development, frequently with craniofacial defects (Fig. S3). Also consistent with this is the rare occurrence of Specc1l heterozygous embryos with craniofacial malformation - likely due to increased gene-trapping efficiency - and findings in zebrafish where morphants for one of two SPECC1L orthologs (specc1lb) lead to late embryonic phenotypes including a loss of mandible and bilateral clefts51. Thus, heterozygous SPECC1L loss-of-function mutations identified in human patients may lead to mild perturbation in SPECC1L function during craniofacial morphogenesis, sufficient to account for their orofacial clefts. SPECC1L-based modulation of cell-cell contacts may also play a role in palatogenesis and pharyngeal arch fusion. Further studies of SPECC1L function will help elucidate the role of transient cell-cell contacts in neuroepithelial cell movement during neural tube closure and in CNCCs during craniofacial morphogenesis.
Materials and Methods
Cell lines and antibodies
U2OS osteosarcoma control and SPECC1L-kd cells were previously described (Saadi et al. 2011). Anti-SPECC1L antibody was also previously characterized (Saadi et al. 2011). Antibodies against β-catenin (rabbit; 1:1000; Santa Cruz, Dallas TX) (mouse; 1:1000; Cell Signaling Technology, Danvers, MA), Myosin IIb (1:1000; Sigma-Aldrich, St. Louis, MO), E-cadherin (1:1000; Abcam, Cambridge, MA), AP2A (1:1000; Novus Biologicals, Littleton, CO), SOX10 (1:1000; Aviva Systems Biology, San Diego, CA), DLX2 (1:1000; Abcam, Cambridge, MA), phospho-Ser473-AKT (1:1000; Cell Signaling Technology, Danvers, MA), pan-AKT (1:1000; ThermoFisher Scientific, Waltham, MA), KI67 (1:1000; Cell Signaling Technology, Danvers MA), cleaved Caspase 3 (1:1000; Cell Signaling Technology, Danvers, MA), and β-actin (1:2500; Sigma-Aldrich, St. Louis, MO) were used as described. Actin filaments were stained using Acti-stain rhodamine phalloidin (Cytoskeleton, Denver, CO).
Cell shape change in U2OS cells
U2OS control and SPECC1L-kd cells were cultured in standard DMEM high glucose media supplemented with 10% fetal bovine serum (Life technologies, Carlsbad, CA). For AJ change, 2 × 105 cells were plated on 0.1% porcine gelatin treated glass (Sigma-Aldrich, St. Louis, MO), and observed for cell shape change. Cells were collected at various indicated timepoints: 4 hours post plating (t = 1), 24 hours post plating (t = 2), confluency without cell shape change (t = 3), cell shape change (t = 4), 24 hours following cell shape change (t = 5), and 48 hours following cell shape change (t = 6) (Figs. 1, 2, 3). To modulate the PI3K-AKT pathway, cells were cultured in described concentrations of PI3K-AKT inhibitor, Wortmannin (TOCRIS Biosciences, Minneapolis, MN), or activator, SC-79 (TOCRIS Biosciences, Minneapolis, MN). Media with chemicals was replaced daily.
Live-imaging analysis
Time-lapse recordings were performed of live control and kd cells in regular culture conditions with a phase contrast image collected every 10 minutes over a period of seven days. Images were taken with a computer-controlled Leica DM IRB inverted microscope equipped with a powered stage and a 10 × N-PLAN objective, coupled to a QImaging Retiga-SRV camera. During imaging, cell cultures were kept at 37C in a humidified 5% CO2 atmosphere.
Mouse strains
Two gene trap ES cells lines, DTM096 and RRH048, from Mutant Mouse Regional Resource Center (UC Davis, CA) were used to generate Specc1l deficient mouse strains, designated Specc1lgtDTM096 and Specc1lgtRRH046. Briefly, 129/REJ ES cells were injected into C57BL6 blastocysts. The resulting chimeric male mice were crossed to C57BL6 females to identify progeny with agouti coat color. Presence of gene trap vector insertion was used to identify heterozygotes. The mice are maintained on a mixed 129/REJ; C57BL6 background. The location of the genetrap vector insertion site was verified by RT-PCR, genomic sequencing, and genetic complementation (Suppl. Fig. 1). For lineage-tracing of CNCCs, Specc1lGT double heterozygous mice with ROSAmTmG (#007576) and Wnt1-Cre (#003829) mice (Jackson Laboratories, Bar Harbor, ME) were intercrossed to generate Specc1l mutant embryos also carrying both ROSAmTmG and Wnt1-Cre alleles. All mouse experiments were carried out in accordance with protocols approved by the Institutional Animal Care and Use Committee at the University of Kansas Medical Center.
X-gal staining
Embryos were fixed in (1% formaldehyde, 0.2% gluteraldehyde, 2 mM MgCl2, 0.02% NP-40, 5 mM EGTA) for 60 minutes at room temperature. Following fixation, color development in X-gal staining solution (5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 2 mM MgCl2, 0.01% sodium deoxycholate, 0.02% NP-40, 1 mg/ml X-gal) was performed at 37 °C for 1–6 hours. Embryos were post-fixed in 4% PFA and imaged.
Western blotting and immunostaining
For western blotting, cells were lysed in passive lysis buffer (Promega, Fitchburg, WI) with HALT protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO) added. Lysates were electrophoresed on 12% polyacrylamide Mini-PROTEAN TGX precast gels (Bio-Rad, Hercules, CA) and transferred onto an Immobilon PVDF membrane (EMD Millipore, Billerica, MA). Membranes were blocked in 5% milk in PBS with 0.1% Tween. Antibodies were incubated either overnight at 4 °C or for one hour at room temperature. Femto SuperSignal West ECL reagent (Thermo Scientific, Waltham, MA) was used to develop the signal. For immunostaining, embryos were fixed in 4% PFA/PBS overnight and cryopreserved. Tissue cryosections were blocked in PBS containing 1% normal goat serum (Thermo Scientific, Waltham, MA) and 0.1% Triton X-100 (Sigma-Aldrich, St. Louis, MO), followed by incubation in primary antibody overnight at 4 °C, and fluorescent secondary antibody (1:1000) for one hour at 4 °C. Stained sections were mounted in ProLong gold mounting media (Thermo Scientific, Waltham MA), and planar images were obtained using a Leica TCS SPE confocal microscope. Each immunostaining was performed as three independent experiments on cyrosections from at least two mutant embryos. A representative experiment is shown.
Coimmunoprecipitation
Cells were lysed in a modified RIPA buffer (20 mM Tris-HCl pH8.0, 1% NP-40, 130 mM NaCl, 10% glycerol, 2 mM EDTA, and HALT protease inhibitors (Sigma-Aldrich, St. Louis, MO)). Briefly, lysates were pre-cleared with protein G magnetic beads (Life technologies, Carlsbad, CA) followed by incubation with anti-SPECC1L antibody or IGG overnight at 4 °C. Protein G magnetic beads were used to pull-down SPECC1L and western blotting was performed using β-catenin antibodies described above. The co-IP experiment shown is representative of four independent experiments.
TEM analysis
Fixed cultured cells or mouse embryonic tissues were provided to the University of Kansas Medical Center Electron Microscopy Core. Briefly, samples were embedded in EMbed 812 resin (Electron Microscopy Sciences, Ft. Washington, PA), polymerized at 60 °C overnight, and sectioned with a Leica UC7 ultramicrotome equipped with a diamond knife, at 80 nm. Sections were imaged using a JEOL JEM-1400 Transmission Electron Microscope equipped with a Lab6 gun, at 100 kV.
Additional Information
How to cite this article: Wilson, N. R. et al. SPECC1L deficiency results in increased adherens junction stability and reduced cranial neural crest cell delamination. Sci. Rep. 6, 17735; doi: 10.1038/srep17735 (2016).
Supplementary Material
Acknowledgments
Funding: This project was supported in part by the National Institutes of Health Center of Biomedical Research Excellence (COBRE) grant (National Institute of General Medical Sciences P20 GM104936, I.S.), Kansas IDeA Network for Biomedical Research Excellence grant (National Institute of General Medical Sciences P20 GM103418, I.S.), Kansas Intellectual and Developmental Disabilities Research Center grant (P30 Eunice Kennedy Shriver National Institute of Child Health and Human Development, HD 002528, I.S.). We wish to acknowledge the Electron Microscopy Research Lab (EMRL) facility for assistance with the electron microscopy. The EMRL is also supported in part by COBRE grant P20 GM104936. The JEOL JEM-1400 TEM used in the study was purchased with funds from NIH grant S10RR027564.
Footnotes
Author Contributions N.R.W. and I.S. conceived and designed the experiments. N.R.W., A.J.O., D.S.A., K.P., E.G.H., E.K., K.M.S., G.J.S., E.K., L.P., B.C.B., A.C. and I.S. performed the experiments. N.R.W., A.J.O., E.C.L., B.C.B., A.C. and I.S. analyzed the data. B.C.B., E.C.L. and A.C. edited the manuscript. N.R.W and I.S. wrote the paper. All authors reviewed the manuscript.
References
- Le Douarin N. The neural crest . (Cambridge University Press, 1982). [Google Scholar]
- Saint-Jeannet J.-P. Neural crest induction and differentiation . (Springer Science + Business Media; Landes Bioscience/Eurekah.com, 2006). [Google Scholar]
- Trainor P. A. Neural crest cells: evolution, development and disease . (Elsevier/AP, 2014). [Google Scholar]
- Cordero D. R. et al. Cranial neural crest cells on the move: their roles in craniofacial development. American journal of medical genetics. Part A 155A, 270–279, doi: 10.1002/ajmg.a.33702 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolande R. P. Neurocristopathy: its growth and development in 20 years. Pediatr. Pathol. Lab. Med. 17, 1–25 (1997). [PubMed] [Google Scholar]
- Mangold E., Ludwig K. U. & Nothen M. M. Breakthroughs in the genetics of orofacial clefting. Trends in molecular medicine 17, 725–733, doi: 10.1016/j.molmed.2011.07.007 (2011). [DOI] [PubMed] [Google Scholar]
- Minoux M. & Rijli F. M. Molecular mechanisms of cranial neural crest cell migration and patterning in craniofacial development. Development 137, 2605–2621, doi: 10.1242/dev.040048 (2010). [DOI] [PubMed] [Google Scholar]
- Dixon M. J., Marazita M. L., Beaty T. H. & Murray J. C. Cleft lip and palate: understanding genetic and environmental influences. Nature reviews. Genetics 12, 167–178, doi: 10.1038/nrg2933 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingraham C. R. et al. Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6). Nat. Genet. 38, 1335–1340, doi: 10.1038/ng1903 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyrard-Janvid M. et al. Dominant mutations in GRHL3 cause Van der Woude Syndrome and disrupt oral periderm development. Am J Hum Genet 94, 23–32, doi: 10.1016/j.ajhg.2013.11.009 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris M. J. & Juriloff D. M. An update to the list of mouse mutants with neural tube closure defects and advances toward a complete genetic perspective of neural tube closure. Birth defects research. Part A, Clinical and molecular teratology 88, 653–669, doi: 10.1002/bdra.20676 (2010). [DOI] [PubMed] [Google Scholar]
- Fantauzzo K. A. & Soriano P. PI3K-mediated PDGFRalpha signaling regulates survival and proliferation in skeletal development through p53-dependent intracellular pathways. Genes Dev. 28, 1005–1017, doi: 10.1101/gad.238709.114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copp A. J., Greene N. D. & Murdoch J. N. Dishevelled: linking convergent extension with neural tube closure. Trends Neurosci. 26, 453–455, doi: 10.1016/S0166-2236(03)00212-1 (2003). [DOI] [PubMed] [Google Scholar]
- Murdoch J. N. & Copp A. J. The relationship between sonic Hedgehog signaling, cilia, and neural tube defects. Birth defects research. Part A, Clinical and molecular teratology 88, 633–652, doi: 10.1002/bdra.20686 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneyhill L. A. To adhere or not to adhere: the role of Cadherins in neural crest development. Cell adhesion & migration 2, 223–230 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theveneau E. & Mayor R. Cadherins in collective cell migration of mesenchymal cells. Curr. Opin. Cell Biol. 24, 677–684, doi: 10.1016/j.ceb.2012.08.002 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larue L. & Bellacosa A. Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene 24, 7443–7454, doi: 10.1038/Sj.Onc.1209091 (2005). [DOI] [PubMed] [Google Scholar]
- Saadi I. et al. Deficiency of the cytoskeletal protein SPECC1L leads to oblique facial clefting. Am J Hum Genet 89, 44–55, doi: 10.1016/j.ajhg.2011.05.023 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruszka P. et al. Mutations in SPECC1L, encoding sperm antigen with calponin homology and coiled-coil domains 1-like, are found in some cases of autosomal dominant Opitz G/BBB syndrome. J. Med. Genet. , doi: 10.1136/jmedgenet-2014-102677 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhoj E. J. et al. Expanding the SPECC1L mutation phenotypic spectrum to include Teebi hypertelorism syndrome. American journal of medical genetics. Part A , doi: 10.1002/ajmg.a.37217 (2015). [DOI] [PubMed] [Google Scholar]
- So J. et al. Mild phenotypes in a series of patients with Opitz GBBB syndrome with MID1 mutations. American journal of medical genetics. Part A 132A, 1–7, doi: 10.1002/ajmg.a.30407 (2005). [DOI] [PubMed] [Google Scholar]
- Short K. M. & Cox T. C. Subclassification of the RBCC/TRIM superfamily reveals a novel motif necessary for microtubule binding. J. Biol. Chem. 281, 8970–8980, doi: 10.1074/jbc.M512755200 (2006). [DOI] [PubMed] [Google Scholar]
- Mattison C. P., Stumpff J., Wordeman L. & Winey M. Mip1 associates with both the Mps1 kinase and actin, and is required for cell cortex stability and anaphase spindle positioning. Cell cycle 10, 783–793 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza-Schorey C. Disassembling adherens junctions: breaking up is hard to do. Trends Cell Biol. 15, 19–26, doi: 10.1016/j.tcb.2004.11.002 (2005). [DOI] [PubMed] [Google Scholar]
- Stepniak E., Radice G. L. & Vasioukhin V. Adhesive and signaling functions of cadherins and catenins in vertebrate development. Cold Spring Harbor perspectives in biology 1, a002949, doi: 10.1101/cshperspect.a002949 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T. & Takeichi M. Remodeling of the adherens junctions during morphogenesis. Curr. Top. Dev. Biol. 89, 33–54, doi: 10.1016/S0070-2153(09)89002-9 (2009). [DOI] [PubMed] [Google Scholar]
- Strobl-Mazzulla P. H. & Bronner M. E. Epithelial to mesenchymal transition: new and old insights from the classical neural crest model. Semin. Cancer Biol. 22, 411–416, doi: 10.1016/j.semcancer.2012.04.008 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas-Tikhonenko A. Cancer genome and tumor microenvironment . (Springer, 2010). [Google Scholar]
- Zeitlin S., Liu J. P., Chapman D. L., Papaioannou V. E. & Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat. Genet. 11, 155–163, doi: 10.1038/ng1095-155 (1995). [DOI] [PubMed] [Google Scholar]
- Mueller A. G. et al. Embryonic lethality caused by apoptosis during gastrulation in mice lacking the gene of the ADP-ribosylation factor-related protein 1. Mol. Cell. Biol. 22, 1488–1494 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladdy R. A., Nutter L. M., Kunath T., Danska J. S. & Guidos C. J. p53-Independent apoptosis disrupts early organogenesis in embryos lacking both ataxia-telangiectasia mutated and Prkdc. Molecular cancer research: MCR 4, 311–318, doi: 10.1158/1541-7786.MCR-05-0258 (2006). [DOI] [PubMed] [Google Scholar]
- Theveneau E. & Mayor R. Can mesenchymal cells undergo collective cell migration? The case of the neural crest. Cell adhesion & migration 5, 490–498, doi: 10.4161/cam.5.6.18623 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris A. R., Daeden A. & Charras G. T. Formation of adherens junctions leads to the emergence of a tissue-level tension in epithelial monolayers. J. Cell Sci. 127, 2507–2517, doi: 10.1242/jcs.142349 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engl W., Arasi B., Yap L. L., Thiery J. P. & Viasnoff V. Actin dynamics modulate mechanosensitive immobilization of E-cadherin at adherens junctions. Nature cell biology 16, 587–594, doi: 10.1038/ncb2973 (2014). [DOI] [PubMed] [Google Scholar]
- Chai Y. et al. Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development 127, 1671–1679 (2000). [DOI] [PubMed] [Google Scholar]
- Brault V. et al. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development 128, 1253–1264 (2001). [DOI] [PubMed] [Google Scholar]
- Cain R. J., Vanhaesebroeck B. & Ridley A. J. The PI3K p110alpha isoform regulates endothelial adherens junctions via Pyk2 and Rac1. J. Cell Biol. 188, 863–876, doi: 10.1083/jcb.200907135 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaresan N. R. et al. The deacetylase SIRT1 promotes membrane localization and activation of Akt and PDK1 during tumorigenesis and cardiac hypertrophy. Science signaling 4, ra46, doi: 10.1126/scisignal.2001465 (2011). [DOI] [PubMed] [Google Scholar]
- Yang W. L. et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 325, 1134–1138, doi: 10.1126/science.1175065 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C. H. et al. Posttranslational regulation of Akt in human cancer. Cell Biosci 4, 59, doi: 10.1186/2045-3701-4-59 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson D., Stevens A., Murray P. G., Black G. C. & Clayton P. E. Identifying biological pathways that underlie primordial short stature using network analysis. J. Mol. Endocrinol. 52, 333–344, doi: 10.1530/JME-14-0029 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbea C. et al. A protein interaction network for Ecm29 links the 26 S proteasome to molecular motors and endosomal components. J. Biol. Chem. 285, 31616–31633, doi: 10.1074/jbc.M110.154120 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay S. et al. A human MAP kinase interactome. Nature methods 7, 801–805 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homanics G. E. et al. Exencephaly and hydrocephaly in mice with targeted modification of the apolipoprotein B (Apob) gene. Teratology 51, 1–10, doi: 10.1002/tera.1420510102 (1995). [DOI] [PubMed] [Google Scholar]
- Ikeda A., Ikeda S., Gridley T., Nishina P. M. & Naggert J. K. Neural tube defects and neuroepithelial cell death in Tulp3 knockout mice. Hum. Mol. Genet. 10, 1325–1334 (2001). [DOI] [PubMed] [Google Scholar]
- Ruland J. et al. Bcl10 is a positive regulator of antigen receptor-induced activation of NF-kappaB and neural tube closure. Cell 104, 33–42 (2001). [DOI] [PubMed] [Google Scholar]
- Migliorini D. et al. Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol. Cell. Biol. 22, 5527–5538 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J., Mao J., Tenzen T., Kottmann A. H. & McMahon A. P. Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev. 18, 937–951, doi: 10.1101/gad.1190304 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copp A. J. Neurulation in the cranial region–normal and abnormal. J. Anat. 207, 623–635, doi: 10.1111/j.1469-7580.2005.00476.x (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris M. J. & Juriloff D. M. Mouse mutants with neural tube closure defects and their role in understanding human neural tube defects. Birth defects research. Part A, Clinical and molecular teratology 79, 187–210, doi: 10.1002/bdra.20333 (2007). [DOI] [PubMed] [Google Scholar]
- Gfrerer L. et al. Functional Analysis of SPECC1L in Craniofacial Development and Oblique Facial Cleft Pathogenesis. Plast. Reconstr. Surg. 134, 748–759, doi: 10.1097/PRS.0000000000000517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.