

Abstract
Human pluripotent stem cells (hPSCs) are rapidly emerging as a powerful tool for biomedical discovery. The advent of human induced pluripotent stem (hiPS) cells with human embryonic stem (hES) cell-like properties has led to hPSCs with disease-specific genetic backgrounds for in-vitro disease modeling, drug discovery, mechanistic and developmental studies. To fully realize this potential it will be necessary to modify the genome of hPSCs with precision and flexibility. Pioneering experiments utilizing site-specific double strand break (DSB)-mediated genome engineering tools, including Zinc Finger Nucleases (ZFNs) and Transcription Activator-Like Effector Nucleases (TALENs), have paved the way to genome engineering in previously recalcitrant systems such as hPSCs. However, these methods are technically cumbersome and require significant expertise, which limited adoption. A major recent advance involving the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) endonuclease has dramatically simplified the effort required for genome engineering and will likely be adopted widely as the most rapid and flexible system for genome editing in hPSCs. Herein, we describe commonly practiced methods for CRISPR endonuclease genomic editing of hPSCs to cell lines containing genomes altered by Insertion/Deletion (INDEL) mutagenesis or insertion of recombinant genomic DNA.
Keywords: Human Pluripotent Stem Cells, Genomic Engineering, CRISPR, Cas9
Introduction
Since the 1980s, basic techniques for homologous recombination of exogenous DNA sequence have existed, but were only adopted widely in specific species and cell types that happened to have high recombination frequencies (Capecchi, 1989). Homology directed repair (HDR) utilizes endogenous DNA repair machinery to site-specifically integrate exogenous DNA provided by a “targeting vector” in place of a sister chromatid template. The site-specificity is provided by Watson-Crick base-pairing between the genomic DNA and the targeting DNA 5′- and 3′-homology arms, while the intervening region of targeting vector sequence can be varied to include DNA to be inserted, deleted, or mutated. A second less flexible method utilizes another endogenous DNA repair system called non-homologous end joining (NHEJ). Upon the generation of DNA strand breaks, repair mechanisms are activated to re-connect the broken ends without the aid of a homologous template. This system is highly error prone and tends to insert or delete a small number of nucleotides from one or both ends, creating INDEL-mutants wherein the reading frame is altered. Interestingly, Rouet et al noted that dsDNA breaks can also be utilized to enhance the frequency of HDR (Rouet et al., 1994). This provided a motivation for identifying or creating nuclease systems that were accurate and precise enough to target single sites within large genomes.
The earliest modifiable site-specific nuclease system utilized for human genome engineering was the ZFN (Porteus and Baltimore, 2003; Alwin et al., 2005; Lombardo et al., 2007). For each site targeted, a novel protein had to be created by combining together amino acid sequences of several zinc fingers with the propensity to interact with the nucleotides in the target DNA sequence. A conceptually similar system involving TALENs was subsequently described; combining DNA-binding domain modules to give sequence specificity. The main advantage of the TALEN system was that module combination was easily systematized and that the resulting novel combinations of DNA-binding domain modules were more reliably functional (Boch et al., 2009; Hockemeyer et al., 2011). Still, these systems were commercially expensive, complicated, and laborious, limiting their widespread use.
Concurrent to the development of gene editing approaches, the emergence of hiPS cell technology provided access to hPSCs with a variety of previously inaccessible genomic backgrounds (Takahashi and Yamanaka, 2006; Takahashi et al., 2007; Yu et al., 2007; Yu et al. 2009; Okita et al., 2011). The hPSC field was also improved by the development of feeder free culture systems and efficient cell passage survival with ROCK inhibitor, but the limitations of genomic engineering initially precluded the generation of isogenic controls necessary to accurately determine consequences of individual genetic variation (Levenstein et al., 2006; Ludwig et al., 2006; Chen et al., 2011; Ohgushi and Sasai, 2011). By 2010, with the broad range of hPSCs in culture, researchers were readily able to alter human genomes with ZFNs and TALENs, providing an approach to correct or introduce specific genetic mutations, albeit with low efficiency.
In 2010, the CRISPR/Cas9 system was described. It is a multiplex bacterial adaptive defense system that uses guide RNAs (gRNAs) and a protospacer adjacent motif (PAM) to target and site-specifically cleave genomic DNA (Horvath and Barrangou, 2010). CRISPRs were found in several microbes with varying protein structures and PAM recognition sequences, each designed to degrade or inactivate invading nucleic acids. The CRISPR associated protein 9 of S. pyogenes (spCas9) proved exceptionally useful with its practical size and low-specificity ‘NGG’ PAM recognition sequence. It was quickly recognized that this system could be adopted as an alternative strategy for inducing DNA-breaks for mammalian genome engineering (Jinek et al., 2012; Jinek et al., 2013; Cong et al., 2013; Mali et al., 2013). Free online tools were soon developed for the design of guide RNAs with limited nonspecific activity (Internet Resources 4). Various tools have been developed leveraging CRISPRs specific genome localization activity, ranging from imaging for chromosomal localization to regulation of gene expression (Qi et al., 2013; Gilbert et al., 2013; Konermann et al., 2015). As such, CRISPR technology has revolutionized experiments involving genomic DNA and continues to evolve rapidly. With human codon-optimized spCas9 protein from the CRISPR system, we now have a technology for genomic DNA engineering that is simple, efficient, and easily accessible for biomedical research (Mali et al., 2013). In this unit, we provide current methods for hPSC genome engineering with spCas9 and subsequent high-throughput screening for clonal populations. These methods can be adapted to other cell lines with thoughtful modification.
Strategic Planning
Directing spCas9 with gRNA to desired genomic loci is an effective way to induce specific DSBs. Since each cell line will have unique genomes, researchers should consider sequencing the region of interest because single nucleotide polymorphisms (SNPs) have major consequences on target sequence efficiency. For gene Knock-Out experiments, researchers can induce the NHEJ mechanism for INDEL mutagenesis by directing DSB(s) to exons, preferentially the first common exon. They can alternatively use HDR mechanisms to insert stop codons or excise significant regions of DNA. For Knock-In experiments, researchers can introduce homologous-arm donor plasmids for HDR into loci flanked by DSBs (Internet Resources 1). Each system will require gRNAs but only those used for insertion of recombinant DNA will require large specialized donor plasmids present during repair. This unit will focus on full DSB nucleolytic spCas9 and will not discuss single-strand nickase or null variant applications. We find that full DSBs are efficient for in-vitro work with hPSCs and we encourage the use of this system.
If targeted genes are not expressed in hPSCs or have SNPs, screening for pure populations becomes impossible with respect to traditional selection methods such as immunocytochemistry, protein tags, fluorescent proteins or antibiotic resistance. In some cases, even a fraction of cells with genetic disruption can provide early clues in discovery. Furthermore, since hPSCs cannot be reliably plated as single cells, high throughput techniques for clonal enrichment using interim cryopreservation and genomic DNA analysis of serially picked and subcultured small clusters have been developed (Miyaoka et al., 2014). Descriptions of high throughput cryopreservation and genomic DNA purification have been included in this unit. In all cases, researchers must carefully consider the approach and tools that will be necessary for the editing event and the clonal purity required in downstream applications. This unit will broadly address Knock-Out and Knock-In approaches for hPSCs for the applications described below (see Table 1).
Table 1. Various Approaches for hPSC Genome Engineering.
| hPSC Genome Engineering | ||||
|---|---|---|---|---|
|
| ||||
| Knock-Out | Knock-In | |||
|
| ||||
| Approach | INDEL | Early Stop Codon/ Exon Excision | Transgene Insertion | Point Mutation/SNP |
| Mechanism | NHEJ | HDR | HDR | HDR |
| Min# gRNAs | 1 -3 | 2 | 2 | 2 |
| Donor Plasmid | No | Yes | Yes | Yes |
We do not elaborate the details for directed HDR Knock-Out or Point Mutation/SNP in this unit but donor plasmids with intervening in-frame stop codons or lacking critical exons may be designed as needed and applied with these protocols. For more detail on introducing Point Mutation/SNP, some researchers have found single DSBs with high concentration mutant DNA oligonucleotide donors to be effective. Please review Miyaoka et al., 2014 for more detail on that approach.
INDEL Mutagenesis
Inducing deleterious and codon frame-shift breaks to disrupt functional protein expression is not trivial, but purifying the affected cells can be a challenge when the gene is not expressed in the cell being targeted. Targeting 1-3 optimally designed gRNAs to disrupt the first common exon(s) of the gene of interest is usually sufficient. While three gRNAs may be effective, it will save time to produce more than three and test several combinations of them. If the protein is normally expressed in the hPSC state, immunohistochemistry or flow cytometry of a replica sample can reveal effective knock-out early in experiments; but one must also consider hPSC homeostasis and the consequence of homozygous knock-out of important hPSC genes. Alternative gene disruption involves excising an exon by way of HDR of donor plasmids with early stop codons or plasmids lacking the exon between the two DSBs.
Recombination of Exogenous DNA
Insertion of transgenes or other recombinant DNA elements allows for powerful experimental control by way of Knock-In. Alternative Knock-Out strategies using HDR can be achieved with a donor plasmid containing 5′- and 3′-homology arms, but lacking an intervening exon, inserting an early stop codon, or adding exon-flanking CRE recombinase sites (LoxP) for conditional disruption of the gene. Working knowledge of the mechanistic events in DSB-mediated HDR can facilitate effective design strategies. From our collected experiences, we currently use the following working model:
First, the DSB ends are sensed by the cell and the 5′-ends are chewed back to expose 3′-overhangs. A 3′-overhanging end will anneal to an available repair template which we anticipate as the donor plasmid rather than the sister chromatid. The 3′-end of the 3′-overhang may be chewed back by an exonuclease if the tip does not anneal. The 3′-end is then extended by DNA polymerization along the template. After some random length of extension the strand will hop back onto the original chromosome given a region of homology. From this model, it should be clear that all DNA that is to be inserted or deleted should be present on one side of the CRIPSR cut site or between two CRISPR cut sites. Therefore, given [X] as the CRISPR cut site and [A] and [B] as exogenous sequence to be added, [X][A][B] and [A][B][X] are viable strategies, while [A][X][B] is not. Furthermore, if [X][A] [intervening region of homology][B] is attempted, the longer the intervening region of homology, the less likely the event will incorporate [B]. This is due to a tendency for the extending strand to hop back onto the original chromosome before reaching [B]. And finally, if one cuts at a site considerably distant from [A], as in [X][intervening region of homology][A], the efficiency of [A] being inserted will decrease as the length of the region of homology increases.
It is also wise to mutate the CRISPR site in the donor plasmid to protect it from CRISPR cutting; choose a cut site where mutagenesis is inconsequential or silent. HDR-mediated repair is less efficient than NHEJ and can vary by an order of magnitude between cell types or between the same cell type with different genetic backgrounds. It is also important to understand that HDR and NHEJ are competing processes. It is not uncommon to have HDR at one allele while the other allele is mutated by INDEL mutagenesis. In our experience the order of likely outcomes from CRIPSR targeting with a donor plasmid (attempted HDR) is NHEJ/NHEJ > NHEJ/HDR > HDR/HDR > +/HDR = +/NHEJ. One should design screening strategies that not only detect the presence of integrated DNA (successful HDR), but also determine the state of the second allele. While less frequent than on single alleles, homozygous recombination from one nucleofection event is still effective and can be gradually enriched with high throughput screening methods. When developing homozygous targeting strategies, using the same gRNAs for both alleles is effective and may be enhanced when the target site mutated in the donor sequence. For faster enrichment using traditional selection markers, consider applying two donor plasmids with distinct selection markers (e.g PuromycinR and NeomycinR). We find the frequency of homozygous recombination to be between 0.05-1.5%. Employing dual antibiotic selection or fluorescence proteins in screening can drastically reduce labor and financial costs during clone purification.
The workflow through this unit is shown in Figure 1 and will usually require the repeat of BP 4 and BP 5 for clonal populations:
Figure 1.

Workflow for this unit.
Basic Protocol 1
Design and Preparation of gRNA Plasmids
To use the spCas9 endonuclease system one first designs targeting vectors that express gRNAs for each DSB desired. We use the online tool from Feng Zhang's Lab (MIT/Broad institute) to find candidate targeting sequences that have low non-specific background activity. Genomic regions of interest are screened by the software and targeting sites are ranked. Desirable target sequences are ordered as oligos with directional overhangs, annealed, and then ligated into the Zhang lab's widely available pX330 plasmid prepared by digestion with BbsI.
Materials
pX330-U6-Chimeric_BB-CBh-hSpCas9 (pX330) (Addgene cat# 42230)
BbsI nuclease (New England Biolabs cat# R0539S)
Calf Intestine Alkaline Phosphatase (CIP) (New England Biolabs cat# M0290S)
1M Dithiothreitol (DTT)
T4 DNA Ligase Kit with Buffer (New England Biolabs cat# M0202S)
T4 Polynucleotide Kinase (PNK) (New England Biolabs cat# M0201S)
Ampicillin LB Agar Plates
Competent Cells
QIAGEN EndoFree Maxiprep Kit (QIAGEN cat# 12362)
Step 1.1: For each genomic region where gRNA will be needed, start a new analysis at http://crispr.mit.edu. Enter your name and e-mail address as the results will be stored on their server and the link to access these results later will be e-mailed to you.
Step 1.2: Whether targeting for INDEL mutagenesis or 5′ and 3′ homology regions for recombination, a significant region of genomic DNA sequence is optimal to find desirable options. Copy the sequence of your targeting region of interest (100-250 bases) to the sequence box. Set the sequence type to “Unique Genomic Region” and select “Human” for target genome. Click “Submit” to initiate the analysis.
The site allows for the input of 23-250 bases of DNA. Try to include at least 100-200 bases if your strategy will allow it.
Step 1.3: When the analysis is complete, target sequences will be prioritized from optimal to suboptimal listing those with the least number of off-target sites at the top. Scores represent an internal evaluation of off-target hits and if those targets are within genes. An explanation of this scoring system and other useful guidance can be found on the help page at http://crispr.mit.edu/about. Green target sequences are optimal and the highest scores should be prioritized. For INDEL experiments, select 3-4 guides within the exon you wish to target. For HDR recombination, select at least one target 5′ and one target 3′ to the region of interest. It is important to test more than one gRNA for each region of interest since DSB efficiency is variable.
The PAM recognition sequence for spCas9 is shown in green ‘NGG’ letters to the right of the target sequence and should *not* be included in the target sequence to be cloned into the gRNA-expression vector; this 3nt sequence in the genome is necessarily 3′ to the target sequence but is recognized by spCas9 protein and not gRNA.
Step 1.4: For each target selected you must order two oligos with directionally specific overhangs that will be annealed and ligated into the gRNA expression vector immediately upstream of the gRNA scaffold. The Forward Oligo is the 20nt target sequence preceded by CACC while the Reverse Oligo is the reverse complement of the 20nt target sequence preceded by AAAC. Once annealed, the unique sticky ends will preferentially ligate immediately after the U6 promoter and in-frame with the gRNA scaffold (Figure 2 A, B, C).
Figure 2. gRNA Plasmid Ligation Conditions.
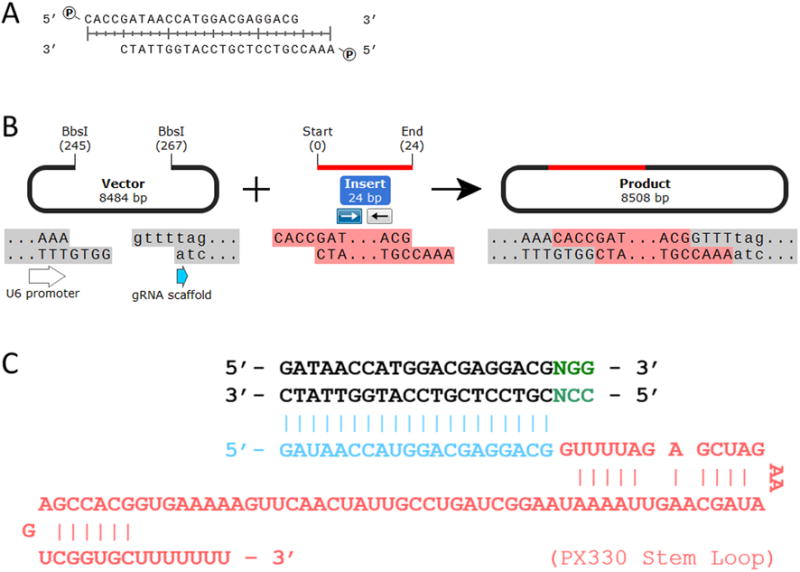
(A) Example of annealed and phosphorylated gRNA target sequence oligo with directional sticky 5′ overhangs. (B) Example of directional ligation into BbsI digested dephosphorylated pX330 vector. (C) Example of the completed sgRNA (blue/red) and how it will recognize the genomic target sequence (black) with Cas9 PAM binding motif (green).
Using an example target sequence of ‘GATAACCATGGACGAGGACGNGG’ our example gRNA insert Oligos should be:
Forward Oligo = 5′– CACCGATAACCATGGACGAGGACG – 3′
Reverse Oligo = 5′– AAACCGTCCTCGTCCATGGTTATC – 3′
Please review Figure 2 for a detailed explanation of the guide design and function against its targeted genomic sequence.
Step 1.5: Prepare the vector for ligation. Digest 2-4 ug of pX330 vector with BbsI in a 20uL reaction volume, incubate for 1 hour at 37°C. Spike in 1uL of CIP and incubate for 15 minutes at 37°C. Then heat-inactivate by incubating at 65°C for 15 minutes. Gel purify the digested dephosporylated vector and elute in 30uL of water or EB. When more gRNAs may be planned for the future it is useful to scale this reaction up and store the excess prepared vector at -20°C.
Step 1.6: Anneal and phosphorylate the target sequence oligos. Suspend oligos at [100uM] in nuclease free water or EB. Add 1uL 1M DTT to 9uL 10× T4 DNA Ligase Buffer to ensure its function(*). For each targeting sequence to be ligated, set up the following in a PCR strip tube:
| Forward Oligo (100 μM) | 1uL |
| Reverse Oligo (100 μM) | 1uL |
| 10× T4 DNA Ligase Buffer* | 1uL |
| dH2O | 6.5uL |
| T4 PNK | 0.5uL |
| Total Volume | 10uL |
Place the PCR strip(s) into a programmable thermocycler and run the following program to anneal the oligos to produce inserts: 37°C for 30 min, 95°C for 5 min, Ramp down to 25°C at ∼5°C/min.
Step 1.7: Dilute the annealed inserts 1:100 in a new PCR strip tube. Keep the original annealed 100× stock as backup.
Step 1.8: Ligate the diluted inserts for several hours or overnight at room temperature, as follows:
| Digested/Dephosphorylated pX330 (50-100ng) | 1uL |
| Phosphorylated and Annealed Insert (1:100 dilution) | 1uL |
| 10× T4 DNA Ligase Buffer* | 1uL |
| T4 DNA Ligase | 1uL |
| dH2O | 6uL |
| Total Volume | 10uL |
It is important to prepare a control ligation and transform it into competent cells in case undigested vector remains in your stock after gel purification.
Step 1.9: Transform 5uL of ligated product into competent cells of choice, per manufacturer's protocol. After transformation recovery, plate appropriately diluted transformed cells onto Ampicillin selection LB agar plates and incubate 37°C overnight.
Step 1.10: Screen clones for correct sequence by miniprep and DNA sequencing with U6 Forward or CMV Reverse primers as you desire based on the sequence data provided on Addgene. Look carefully for point mutations that may arise from ligation oligo production error. Most clones will have the correct insertion such that the 20nt guide sequence is preceded by CACC and immediately followed by the gRNA scaffold “GTTTTAG…” Using our example from step 1.4, the sequencing would contain:
5′ …aaaCACCGATAACCATGGACGAGGACGGTTTtag… 3′
Step 1.11: Correct gRNA clones should be prepared by midi or maxiprep with QIAGEN EndoFree kits or equivalent. High concentration DNA (above 2ug/uL) is required for efficient nucleofection, so be careful to elute/resuspend DNA in a low volume, typically ¼ of manufacturer recommendation. Lower concentration yields can be washed and resuspended in lower volumes using standard ethanol precipitation methods (Internet Resources 2).
Basic Protocol 2
Preparation of Recombinant DNA with Homology Arms
For homologous recombination experiments, we prepare the recombinant DNA sequences flanked with large regions of 5′ and 3′ homologous genomic DNA. DNA insertion experiments should be thoughtfully designed to extend past the 5′ and 3′ gRNA target sites. ∼500-1000 bp homology arms are amplified by PCR and recombinant DNA sequence is amplified and inserted using standard molecular cloning techniques.
Materials
Gateway® pENTR™ 1A Dual Selection Vector (Optional) (Life Technologies cat# A10462)
Cold Fusion Cloning Kit (suggested) (Systems Biosciences cat# MC100B-1)
Your preferred molecular cloning tools
High fidelity DNA Polymerase Kit of Choice
Kanamycin LB Agar Plates
QIAGEN EndoFree Maxiprep Kit (QIAGEN cat# 12362)
pENTR vectors are desirable for cloning since uncut vector contains a cell-death signal for standard bacteria and will not survive in standard selection. These vectors will not be used for Gateway cloning. The purpose is to readily generate donor plasmid DNA with the large homology arms flanking transgenes, and so researchers may opt for their own plasmids instead. TOPO vectors can also be used for insertion of homology arms.
Step 2.1: Decide which region of genomic DNA you wish to modify with recombinant DNA sequence. Include approximately 500-1000 bp of genomic DNA sequence on each of the 5′ and 3′ flank in your recombinant construct (Figure 3 A, B). gRNA target sites are preferable close to the internal ends of the flanks, but design of the total recombinant DNA can include genomic sequence internal to DSBs (Figure 3 A, B). If it will not affect gene expression, consider introducing silent base mutation of PAM or gRNA binding sequences in the donor construct to prevent spCas9 recognition of the recombinant construct in addition to genomic loci.
Figure 3. HDR Donor Plasmid Design and Assembly Workflow.
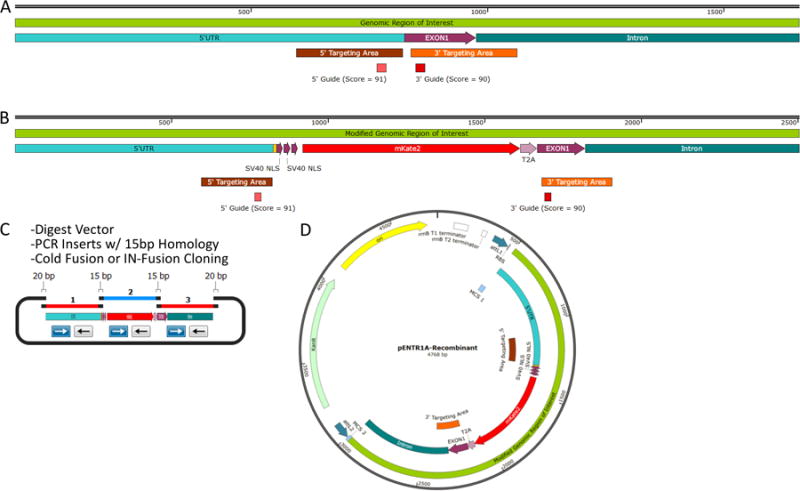
(A) Example genomic region of interest for targeted transgene recombination. (B) Theoretical recombinant genomic region of interest with 3× nuclear localized fluorescent reporter and 2A peptide sequences. (C) Diagram of 3 insert cloning with Cold Fusion/IN-Fusion with restriction enzyme digested pENTR1A vector. (D) Diagram of complete donor plasmid construct. See Li and Elledge, 2007, for the principles behind Cold Fusion/IN-Fusion technologies.
Avoid designing larger homology arms as this can cause complications during the genotyping PCR step.
Step 2.2: (Optional) When antibiotic selection will be utilized with homozygous recombination, design a second similar construct with a distinct antibiotic resistance gene (e.g. PuromycinR, NeomycinR, HygromycinR, BlasticidinR). While homozygous knock-in of one construct is usually effective, including a second selection marker may simplify purification of homozygous recombinant clones.
Antibiotic selection should be carried out semi-sequentially because dual-selection at full selection concentration may be too stressful on cells. When using antibiotic selection it is important to find good selection and maintenance concentrations in your current hPSC culture conditions with kill curves.
Step 2.3: Using standard high-fidelity PCR and DNA cloning techniques, generate the full construct with recombinant DNA and homology arms and ligate it into digested pENTR1A vector (Figure 3). In cases where constructs are in excess of 5kb, design intermediate plasmids with unique restriction sites and clone additional DNA into the intermediate.
For fast, multi-fragment vector insertion, we recommend the ‘cut-and-paste’ Cold Fusion (Systems Biosciences) or IN-FUSION (Clontech) cloning systems that utilize 15bp homology between adjacent inserts and vector (Li and Elledge, 2007). With these systems high quality DNA is required for workable efficiency; UV exposure should be minimized and inserts/vectors should be purified with significant washing.
Step 2.4: Sequence verify miniprep clones of recombinant DNA vectors. Correctly targeting clones should be prepared by maxiprep with QIAGEN EndoFree maxiprep or equivalent.
High concentration DNA (above 2ug/uL) is required for efficient nucleofection, so be careful to elute/resuspend DNA in a low volume, typically ¼ of manufacturer recommendation. Lower concentration yields can be washed and resuspended in lower volumes using standard ethanol precipitation methods (Internet Resources 2).
Basic Protocol 3
Human Pluripotent Stem Cell Culture and Nucleofection
hPSCs are cultured in preparation for the nucleofection of spCas9/gRNA and donor plasmids. Nucleofection reagents, plating conditions, and plasmids are prepared in advance. Exponentially growing hPSCs in culture are suspended in buffer with plasmids expressing spCas9, gRNA, and optional homologous recombination templates and then nucleofected. Cells are immediately suspended in fresh hES cell media with [10uM] ROCK inhibitor and plated onto a Matrigel substrate coated plate for subsequent culture, picking, and clonal enrichment of desired populations.
Materials
(Optional) Donor vector(s) from Basic Protocol 2
pX330 gRNA/spCas9 plasmids from Basic Protocol 1
Amaxa Nucleofector 2b
Human Stem Cell Nucleofector® Kit 1 (Lonza/Amaxa cat# VPH-5012)
Program A-023 (Mouse ES cell program)
10ug total Plasmid DNA per sample, not to exceed 10uL volume.
Matrigel Substrate Coated 6W or 100mm plate for each sample
hES cell media (We prefer mTeSR1)
ROCK inhibitor (Y-27632, stocked at [10mM]) (SelleckChem cat# S1049)
Accutase (Millipore cat# SCR005)
Plating Media (hES cell media with [10uM] ROCK Inhibitor, prepared on day of use)
DPBS (Ca/Mg free)
100mm, 150mm, and 6W plates as needed
Step 3.1: (Feeder Free Culture Step 1/2) hPSC culture should be stably established on feeder free systems. Consider the passaged dilutions described in Step 3.2 and coat target plate(s) with 1.6mL Matrigel substrate for every 10cm2 for 1 hour, room temperature, and set aside. Prepare plating media by adding ROCK inhibitor to a final concentration of [10uM] in hES cell media.
Step 3.2: (Feeder Free Culture Step 2/2) Aspirate media from hPSC stock culture and wash briefly with DPBS. Aspirate DPBS and apply 1mL Accutase for every 10cm2 for approximately 1 minute. Aspirate the Accutase and wash cells briefly with DPBS. Aspirate the DPBS and apply 1.6-2 mL plating media for every 10cm2. Scrape the cells and triturate gently 4-5 times to break the colonies down to smaller aggregates. Dilute between 1:4 and 1:10 passage in an appropriate volume of plating media for the Matrigel substrate coated plate(s). Aspirate the Matrigel and apply diluted cells in plating media gently. Disperse the cells evenly, incubate overnight at 37°C. Feed fresh hES cell media every day thereafter until cells become 70-80% confluent. Replate and culture sequentially until feeder cells are absent and hPSCs are abundant for cryopreservation and downstream experimentation.
Passaging as in steps 3.1 and 3.2 every few days is also a standard feeder-free hPSC culture technique. Maintain healthy cultures with no differentiating cells.
Step 3.3: Continue to passage cells on Matrigel substrate coated plates to ensure a 100mm plate backup stock is always ready and a 100mm or 150mm working stock is amplified for nucleofection.
Always use [10uM] ROCK inhibitor in plating media while replating hPSCs.
Step 3.3: As the working stock of cells approaches 80% confluent, cells will be ready and abundant for nucleofection. Feed cells fresh hES media approximately two hours in advance to nucleofection.
A 100mm stock plate will generally provide enough cells for 4-6 nucleofections, with 1-2 million cells needed per nucleofection.
Step 3.4: Prepare the nucleofection materials. Set the Nucleofector 2b in the hood and set it to program A-023. Ensure the nucleofection buffer is prepared by adding the supplement completely and evenly. For each sample, prewarm 10mL of plating media in the incubator and a coat a 100mm plate with 10mL Matrigel substrate for at least 1 hour at room temperature. For each nucleofection add 10ug of plasmid DNA (max 10uL) to a 1.5mL Eppendorf tube and then add 100uL of complete nucleofection buffer:
-INDEL Mutagenesis samples: 1-3 pX330 sgRNA plasmids with 2.5-5ug each.
-Homologous Recombination samples: 2ug of each pX330 gRNA targeting plasmid and 6ug of homologous recombination plasmid (or 3ug of each when using two unique selection for homozygous targeting).
Step 3.5: Prepare hPSCs for nucleofection. Aspirate hES media from hPSC stock culture and wash briefly with DPBS. Aspirate DPBS and apply 1mL Accutase for every 10cm2, incubate 3-5 minutes at 37°C. Cells should be near single-cell suspension in small clusters of ∼2-5 cells since large clumps will have low nucleofection efficiency. Add an equal volume of plating media to Accutase, triturate gently to release the cells and then centrifuge for 3 minutes at 200 × g, room temperature.
Freshly thawed Accutase is recommended for timely digestion of hPSCs for nucleofection. Replating particularly adherent hPSCs the day before nucleofection can help loosen the cells for Accutase treatment on the day of nucleofection.
Step 3.6: Resuspend the cell pellet in 1.2 mL DPBS. Count the cells and aliquot 1-2 × 106 hPSCs into sterile Eppendorf tubes for each nucleofection reaction. Centrifuge the Eppendorf tubes for 3 minutes at 300 × g, room temperature. Move quickly to aspirate the DPBS from the cell pellets.
The cell pellet is loose, so be careful not to approach it aggressively with the aspirator tip.
Step 3.7: For each nucleofection sample, transfer the plasmid DNA/nucleofection buffer from Step 3.4 to the cell pellet and resuspend the pellet gently, minimizing trituration. Transfer the entire ∼110uL volume of DNA/buffer/hPSCs to a nucleofection cuvette and place the cap on the cuvette.
Step 3.8: For each sample, place the cuvette in the Nucleofector 2b and push start. The cuvette rotates into position, the voltage is briefly applied, and then the cuvette rotates back out. When processing multiple samples, continue to process the others quickly. Add 500uL of plating media to each cuvette.
Step 3.9: For each sample, remove all contents from each cuvette and mix gently with 10mL of prewarmed plating media. Aspirate the Matrigel substrate from a 100mm plate and disperse the ∼10.5mL sample evenly and gently. Alternatively, for 6W plates, seed cells evenly or in serial dilution to reduce material costs as you see fit. Incubate overnight at 37°C.
Do not disturb the plate after placing it in the incubator.
Step 3.10: The next day most of the cells should be attached and the plate will be 10-15% confluent. Feed fresh hES cell media every day.
When using antibiotic selection, wait 2-3 days for CRISPR targeting to occur and antibiotic resistance proteins to be expressed. Antibiotic selection will kill most of the cells due to low recombination and so culturing for colony picking can take up to 10 days after plating. Do not split the cells during the selection period to avoid generation of pseudoclones.
Basic Protocol 4
Colony Picking and Subclone Selection
At this point, genome-edited cells should be growing among unaffected hPSCs. 7-14 days post nucleofection, colonies will be of sufficient size for manual picking and enrichment. When employing antibiotic selection strategies, the initial culture will take longer, but desired clones are more frequently enriched and readily purified.
Materials
hES cell media
ROCK inhibitor (Y-27632, stocked at [10mM]) (SelleckChem cat# S1049)
Plating Media (hES cell media with [10uM] ROCK Inhibitor, prepared on day of use)
Accutase (Millipore cat# SCR005)
DPBS (Ca/Mg free)
Matrigel Substrate
24W plates
96W plate (for manual colony dispersion)
Picking Microscope
Step 4.1: After nucleofection feed the cells in the 6W or 100mm plate the appropriate volume of hES cell media every day at a similar time. With antibiotic selection, hPSCs will grow initially and then die-off drastically before resistant cells expand further. To accomplish full dual-selection, one or two passages may be required in the interim. If so, ensure the last passage before colony picking will be toward a lower concentration of small clusters of 5-15 cells if possible. Monitor the culture for picking closely. When colonies approach sufficient size, coat all of the wells of 1-2 24W plates with 0.5mL Matrigel substrate for at least 1 hour, room temperature.
Depending on desired outcomes (homozygous, heterozygous, selection markers, etc.), 10-48 colonies should be picked at first for each spCas9 engineered sample. This work is laborious and manual picking can be done in 2-4 rounds with media change in between to reduce the total time outside of the incubator. An experienced researcher can pick about one colony per minute.
Step 4.2: Select a sample plate and identify/mark colonies of sufficient size (100-400 cells) that are not in contact with other colonies and have a high density of homogenous refractive hES-like morphology with a clear defined edge (Figure 4). Incubate the sample at 37°C until picking begins.
Figure 4. An example of ideal feeder-free hPSC colony for picking.

Step 4.3: Before picking, aspirate the Matrigel substrate from the 24W plate(s) and then replace with 0.5mL plating media per well. Incubate at 37°C until picking begins.
Step 4.4: Prepare a 96W plate with 20uL plating media in as many wells as will be picked (10-48 per sample plate). Picked colonies will be manually pipetted into smaller clumps here before plating in the 24W wells.
Step 4.5: Remove the sample plate that was observed in Step 4.2 and aspirate the media. Wash gently with 10mL DPBS and then aspirate the DPBS. Apply 5mL plating media.
The low volume is used to aid with picking and reduce lateral dispersion of cell debris into the other areas to be picked. If debris becomes substantial gently agitate the plate, aspirate the media, and replace with fresh plating media.
Step 4.6: Colony picking technique. Move your 100mm plate of colonies, a 24W Matrigel coated plate of plating media, and the 96W colony dispersion plate into a laminar flow hood with a picking microscope. Use a new 200uL barrier tip on a P200 pipette for each clone. Set the pipette to 30uL. Without pipetting, gently start at one side (right or left depending on handedness) at the edge of the colony and imagine gently lifting a sticky tear-able pancake from one side to the other. First, gently modulate laterally along the edge of the colony with the tip against the plate. Drift your lateral modulus longitudinally to span the colony, working slowly from one side to the other. Do not completely detach the colony from the plate. Once the colony is about 80-90% detached from the plate it will sway gently in the media but remain localized. Operate the pipette set at 30uL with the tip directly touching the colony. The colony should enter the tip while detaching from the plate. Move this to a 20uL media well of the 96W plate and pipette 7-10 times against the base of the well at 30uL to prevent bubbles. Adjust the pipette and then draw up the 50uL of cells and disperse evenly into one well of the 24W SNL feeder layer plate. Mark off the 96W and 24W plates so as not to reuse those wells. Reset the pipette to 30uL.
It is very important to fully stabilize the plate in its position during picking. If the plate can slide around it will do so under the friction of your tip against the plate.
Step 4.7: Repeat Step 4.6 for 10-48 total colonies from the sample. If picking is in excess of 30 minutes, aspirate the plating media from the sample and feed fresh hES media with incubation for 1-2 hours at 37°C in between. When enough colonies are picked, incubate the 24W plates overnight at 37°C.
Unpicked colonies can be pooled for passage or cryopreservation as a polyclonal population.
Repeat Steps 4.2 to 4.8 for each sample.
Step 4.8: Starting the next day, feed hES cell media every day for 3-5 days until clones approach 70-90% confluent. At this point researchers should move to Basic Protocol 5, or wells can be optionally split into two wells for downstream immunohistochemistry in parallel to Basic Protocol 5.
Basic Protocol 5
Cryopreservation and Genomic DNA Preparation
Large numbers of picked colonies require high throughput cryopreservation and genomic DNA preparation for genotyping. With multiple lines, mixed colonies, and numerous candidates, it is efficient to use 96W plates. When approaching confluent, colonies are suspended from the 24W plate and aliquots are cryopreserved and prepared for genomic DNA extraction for genotyping.
Materials
24W Plates of 70-90% confluent hPSCs
96W plates
Cryopreservation Solution
Lysis Buffer
Accutase (Millipore cat# SCR005)
Sterile Filtered Mineral Oil (Sigma cat# M5310-100ML)
70% Ethanol in Nuclease Free Water
[75mM] NaCL in Ethanol, Ice Cold
Step 5.1: Aspirate the media from each 24W of hPSCs to be stocked and replace with 250uL of Accutase. Incubate 2-3 minutes at 37°C until individual cells are apparent.
Step 5.2: Colonies should be visible with naked eye. Use a pipet to scrape and disperse cells from the bottom of the well.
Step 5.3: Transfer 100uL from each well to a unique well on a 96W plate and note its location for potential thawing and culture downstream.
Use of an adjustable multichannel pipette is effective for rapid processing of many clones in an appropriate time frame for cell health.
Step 5.4: Add 150uL of Cryopreservation Solution to each well and pipette gently to ensure even suspension. Avoid creating bubbles.
Step 5.5: Gently pipette 75uL of Sterile Filtered Mineral Oil to cover each well. Place the plate in a Styrofoam box and transfer to -80°C freezer.
The oil seals the well and prevents sample evaporation during cryopreservation; the Styrofoam box is necessary to slow the rate of freezing to prevent ice-crystal formation in cells. An optional PCR adhesive film can be applied over the wells to prevent lateral spilling/contamination.
Step 5.6: To extract genomic DNA, for each sample suspended in Step 5.1, add 150uL of Lysis Buffer per well in a 96W plate. Transfer 100uL of each sample into a Lysis Buffer well.
Step 5.7: Incubate the plate(s) overnight at 55-60°C in a plastic container with a small amount of water to prevent evaporation.
Step 5.8: The next day, add 100uL ice-cold [75mM] NaCl/Ethanol solution to each sample. Incubate the plate for 2 hours at room temperature.
It is best to keep the salt/ethanol buffer at -20°C for storage and immediately before use.
Step 5.9: Invert the plate over a waste container to discard the solution.
The DNA will remain adherent to the plastic bottom of the well.
Step 5.10: Using the multichannel pipette, wash by adding 100uL 70% Ethanol to each well, briefly. Invert the plate over a waste container to discard the wash. Repeat this wash 2 times.
Step 5.11: After the last wash, set out a clean sheet of paper. Invert the plate and slam it down onto the paper to remove the residual ethanol.
Step 5.12: Turn the plate over and cover it with another clean sheet of paper to prevent dust particle contamination. Allow the plate to air-dry for 30-45 minutes.
Step 5.13: Resuspend with your preferred DNA buffer that is compatible with downstream applications and store the DNA at -20°C as you see fit in Eppendorf tubes, PCR strips, etc.
Colonies are now stocked with an accompanying genomic DNA prep for each population. Genomic DNA can then be sourced for PCR evaluation to detect recombinant inserts, DNA sequencing, or digital droplet PCR evaluation of enrichment as discussed previously in Miyaoka et al. 2014. When pure or near-pure populations are identified, thaw enriched wells in hES cell media with [10uM] ROCK inhibitor and expand/subclone akin to Basic Protocols 4 & 5, repeating these high throughput steps as necessary until pure populations are isolated and genotyped. Once several candidate clonal populations are isolated, researchers should karyotype, repeat genotype, and validate pluripotency with ICC or teratoma formation.
Reagents and Solutions
Commentary
Background Information
Advances in Genome Engineering and hPSC Accessibility
Through the 1980s, waves of advances in mammalian genome engineering set the tone for hopes of genomic malleability in human cells. Exogenous DNA was shown to recombine into genomes and the subsequent work to induce DNA breaks helped scientists enhance and characterize the endogenous repair mechanisms responsible for the event. Mario Capecchi, Martin Evans, and Oliver Smithies received the 2007 Nobel Prize in Physiology or Medicine for these revelations after 20+ years of anticipation. Still, adoption of these genome engineering applications in hPSCs was limited for technical reasons. James Thomson and colleagues established hES cell culture techniques and concomitant feeder-free cell culture improvements, but ethical concerns surrounding hES cell derivation for studying human diseases left researchers with few options to work with until the breakthroughs in iPS cell technology by Shinya Yamanaka. The ability to generate hiPS cells revolutionized hPSC research with applications in disease modeling, drug screening, and regenerative medicine.
In recent years, as hPSC research became easier and widely used, the need for site-specific targeted DNA breaks arrived with ZFNs as modular DNA binding nucleolytic tools. Although costly and complicated, several elite labs started to apply the technology to hPSCs. Targeted DSBs allowed researchers to approach genomic loci of interest and change it to meet research needs. Soon after, TALENs arrived with an analogous and somewhat preferable approach and hPSC genome engineering continued to grow. Most recently, CRISPR technology has been widely adopted due to its simplicity, precision, efficiency and ability to be multiplexed.
CRISPR Endonucleases – A Rising Star in Genomic DNA Engineering and Beyond
CRISPRs such as spCas9 have justly won the attention of researchers around the world. For the purposes of genomic DNA engineering, both DSB inducing and DNA strand nicking forms can be used. One step further, a nucleolytically dead form termed ‘dCas9′ has been engineered to seek and bind targeted genomic loci to localize numerous molecular tools for regulating gene expression, imaging, and more. Multiplexing is easily done with the application of several co-expressed sgRNAs or the parsing of a single multi-guide transcript with CSY4 expression (Nissim et al., 2014). Several reports have questioned the precision of CRISPRs in genomic engineering and have demonstrated unforeseen off-target events that have attenuated some of the excitement (Fu et al., 2013; Tsai et al., 2015). Still, the signal to noise ratio remains positive and the reports using CRISPRs continue to accelerate.
Critical Parameters and Troubleshooting
CRISPR/Cas9 engineering of hPSC genomes is effective and accessible. Still, these experiments require careful attention to detail and working experience with molecular cloning, hPSC culture, and molecular evaluations such as genotyping. Here, we provide insight into critical parameters and troubleshooting for several inflexible or deleterious facets of this unit.
Basic Protocol 1: Design and Preparation of gRNA Plasmids
gRNA selection and design is the crux upon which these experiments rely. It is important to select gRNA target sequences that are in the region of interest, within less than 100bp of the desired ideal cut site, and with low non-specific activity (Internet Resources 4). Using the Zhang Lab tool it is easy to screen for non-specific activity, but in some cases there may not be high quality gRNAs at the most convenient sites for initial plans. Still, one should avoid guides with significant nonspecific activity at all costs because such activity may confound downstream experiments. It is better to select less convenient high quality gRNAs and work with them. If this leads to larger transgenic constructs with genomic DNA internal to the DSBs, there likely remains headroom in the total size one can insert into the genome. Prioritizing accurate molecular tools can prevent the need to compensate for off-target effects.
Ligating target sequences into the pX330 vector is straightforward when they are carefully prepared and the correct sticky overhangs are included. Ensure that the sequence of the forward and reverse oligos are complementary and include the 4nt overhangs as in example described in Step 1.4. When screening minipreps for correct sequences, make sure you did not include the PAM in the gRNA sequence. In ligation negative controls look for significant colonies from undigested vector contamination and prepare a fresh BbsI digest of pX330 allowing longer incubation time for complete digestion. A diagnostic digest (such as EcoRI and EcoRV or EcoRI & XboI) is highly recommended for quality control of the plasmid DNA. When few clones arise from ligation, CIP activity on digested vector, oligo annealing, T4 DNA ligase buffer DTT freshness, or oligo phosphorylation may be the culprit. Make sure your reagents are stored properly within expiration dates and that you add DTT fresh to T4 DNA ligation buffer. Avoid repeated freeze thaw cycles of T4 DNA ligase buffer by preparing aliquots for ∼5-10 reactions and discarding the aliquot after each ligation experiment. While annealing, make sure the oligos are protected from hydrolysis at high temps by being in buffer; the temperature is gradually reduced for efficient base-pairing. Lastly, handle competent cells with care and allow for correct heat shock and recovery before plating at high and low densities.
Basic Protocol 2: Preparation of Recombinant DNA with Homology Arms
We have successfully cloned in large transgenic donor constructs containing many different recombinant markers and tools. With careful design, large 5′ and 3′ genome homology in transgenic donor plasmids flanking the DSBs, and efficient nucleofection, desirable clones can be generated and purified. Think of the construct as having homologous handles on the outside of the editing event that hold it together with desired HDR edits in between the handles at the gRNA target sites. These ∼500-1000bp handles are taken directly from the target cell genome and will localize the recombination event once the DSBs are directed (Figure 3 B).
Per genome, the repair mechanism during recombination is directional from each DSB: this is why two insertion-flanking gRNAs are highly recommended (Figure 3 A). Efforts to knock-in constructs with one DSB may yield incomplete and omnidirectional recombination (Figure 5). When the DNA sequence between the DSB sites contains significant genomic sequence, it is probable that some cells will have had one DSB and preferentially return to wild-type sequence prematurely; recombination of the donor can stop too soon because the construct and the genome match. Using selection markers in this situation is useful, but results can be confounded when a single DSB yields correctly inserted marker but lacking downstream elements. This can be prevented with careful design of 5′ and 3′ genotyping analyses during culture purification and finally DNA sequencing of the full region of interest. Look out for mixed population genotype results where bands appear as wildtype and mutant (larger) (Figure 5), with a third amplicon exactly 50% between them; this band may be a PCR artifact from wildtype and mutant amplifications annealing together.
Figure 5.
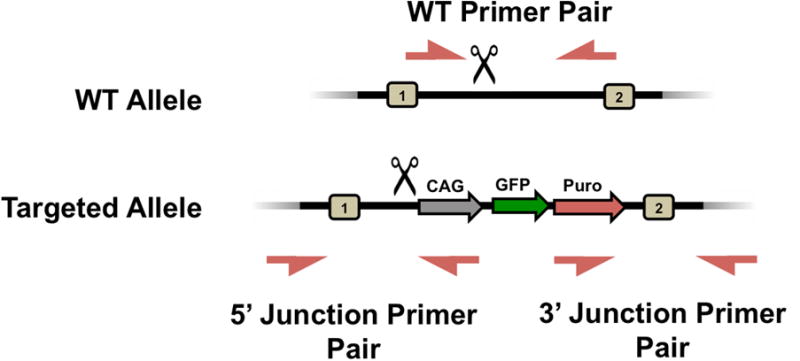
Genomic DNA PCR Screening Design for WT vs Mutant from HDR targeted with one DSB. With one sgRNA/DSB induced at the scissors, HDR to the left of the scissors will be unsuccessful while HDR to the right may be successful and may include donor DNA.
Basic Protocol 3: Human Pluripotent Stem Cell Culture and Nucleofection
Culture
Access to desirable hPSCs can be variable among labs but the generation of hiPS cells can open new avenues of research. We have previously provided a current protocol for integration-free hiPSC generation from human dermal fibroblast or CD34+ peripheral blood mononuclear cells. Transition from feeder layers to feeder free systems is helpful and is explained in product technical literature. Once established, hPSC culture is technically demanding and will require significant practice to gain confidence. Always make stocks of low passage cultures with no signs of differentiation to fall back on because cell culture pitfalls will occur. Regularly karyotype your cells to avoid the spread of chromosomal abnormities in the iPS cell line. hPSCs are more sensitive to environmental conditions than most cell lines and may die or differentiate seemingly spontaneously. To minimize user error, follow these best practices:
-Store media at 4°C for no longer than one week.
-Feed hPSCs daily at routine intervals and *never* skip a day.
-Passage hPSCs before or at 80% confluent to prevent the accumulation of differentiation signals.
-Freshly add [10uM] ROCK inhibitor to hES media when passing or thawing cells.
-Thaw Matrigel on ice at 4°C and keep the prepared substrate solution on ice between uses.
-Use no less than the recommended volumes of Matrigel and do not use after one week.
-If cells begin differentiating and a backup stock cannot be sourced, scrape free the undesired cells and wash away just before passage. If differentiation persists, the culture is potentially primed to differentiate and may never be rescued.
Nucleofection
In this protocol we describe parameters for nucleofection using the Amaxa Nucleofector 2b and Lonza Kits as they are reliable for these applications. With this kit a common pitfall leading to poor nucleofection is to use an inadequate amount of nucleofection solution or its supplement. When preparing a smaller amount, make sure to mix the source vials evenly and then use 82uL of solution and 18uL of supplement for each 100uL of nucleofector solution required per sample. Prepare an excess so as to ensure exactly 100uL for each sample. When the supplement is added the complete solution will remain stable at 4°C for up to 3 months. A second point that must be upheld is the maximal 10uL DNA volume applied to each 100uL suspension of cells in nucleofection solution. Any increase above 10uL will drastically impact nucleofection efficiency and outcomes as it will alter the molecular concentrations of the solution beyond its intended range. We find that high concentration DNA in the lowest volume necessary is associated with improved results.
While preparing cells for nucleofection takes 10-15 minutes, the actual event requires a momentary mixing of cells in solution with a prepared tube of DNA, brief electrical disruption of cell membranes, and then plating. Since this last step can take less than two minutes per sample, please consider pelleting more cells initially and suspending them in an appropriate amount of complete nucleofection buffer. With good timing, 5-8 samples can be nucleofected from the same cell stock given there are enough cells to begin with. Splitting the task between two people with one mixing cells to nucleofect and the other carrying out subsequent plating is yet more efficient to keep cells healthy and increase desired outcomes. Be cautious not to keep cells in nucleofection buffer for prolonged periods as it will reduce cell viability. There exist other methods for nucleofection and these protocols could be adapted to alternate technologies, but be careful to scale accordingly and look for similar efficiency; different technologies may have varying efficiency or cellular toxicity. Our nucleofection conditions are adapted from optimized Lonza protocols that are available on the internet (3).
Basic Protocol 4: Colony Picking and Subclone Selection
Picking the first round of colonies will take a significant amount of time and may be daunting at first. Try to pick high quality colonies with ES-like properties that have no signs of differentiation and are not close to other colonies (Figure 4). Heterozygous knock-ins are frequent, and so screening 30-40 clones will yield one or more populations to enrich pure clones from. Homozygous knock-ins are less frequent by fourfold or more and selection markers for purification are strongly suggested throughout this protocol if possible. Obviating or purifying cells with selection markers saves time and money. Without markers, one can weigh the value of immediacy and start with more clones from small clusters that may enrich faster, or less clones from larger pools that require more rounds of screening. With many candidates at first we can assume they are likely mixed and so time can be saved by not changing aspirator tips between wells in the early culture steps. Make sure to use unique tips during entry into Basic Protocol 5 and for subsequent culture rounds thereafter.
Basic Protocol 5: Cryopreservation and Genomic DNA Preparation
In this protocol, cells are suspended in Accutase and combined with cryopreservation solution. This works because cells are very soon cooling to freezing temperatures and will not be harmed or differentiate. The sterile filtered mineral oil seals each well with low density hydrophobic chemical properties. When moving samples from 24W to 96W format, it is important to move quickly. Using an adjustable multichannel pipette can drastically help this process. The plate is placed in a Styrofoam container so that the temperature drops gradually to reduce ice-formation in cells akin to isopropanol cryopreservation containers. After the first round of genotyping, desirable wells and alternates should be carefully noted because the full plate will thaw at the same time and subculture must be initiated. Be sure to thaw into fresh prewarmed hES cell media with [10uM] ROCK inhibitor. When thawing cells using the mineral oil freezing technique, trace amounts of mineral oil will float on the surface of the media. The trace amount of oil can be washed away on the second day by gently washing with DPBS. Trace amounts of mineral oil do not affect hPSC and are eventually cleared from the media.
Preparation of genomic DNA for genotyping is straightforward. The DNA will precipitate on the plastic well as described in Steps 5.8 to 5.9, but the plate should be handled carefully while washing so as not to loosen it. The final slam of the plate to remove the wash solution should be done in one complete motion. Allow the samples to dry fully to prevent ethanol contamination issues in analysis experiments. When some genomic DNA is very dry it can be left to re-enter solution at room temperature for several hours or kept at 4°C overnight.
Enrichment and Analyses of Colonies and Subcultures
While the genomic DNA for candidate wells will be available for analysis, each genome engineering experiment will allow or exclude enrichment evaluation experiments based on design. For INDEL-derived clones, immunostaining or western blots from samples can be employed. For recombination of large constructs PCR genotyping is usually very helpful. Traditional selection markers can simplify subculture and enrichment. While designing the experiment, researchers should imagine what their output from mixed populations may look like during evaluation and determine which experiments will distinguish wild-type, undesired, and desired cells. As noted, genomic SNPs are one of the most difficult outcomes to purify. Recent technological advances in digital droplet PCR have simplified SNP analyses for mixed populations and a complete protocol for this analysis is referenced here in Miyaoka et al., 2014.
Anticipated Results
Plasmid Generation
Cloning annealed oligos into directional Type IIs cut sites from BbsI is simple and accurate. Researchers should expect dozens to hundreds of pX330-ligated colonies on Ampicillin LB Agar plates. When the vector is digested fully with low/no vector-only negative control colonies, we can rely on the directionality of the ligation and quality of oligos to preferentially ligate. While many clones may be available, screen (sequence verify and perform diagnostic digest) two from each desired gRNA plasmid for good measure.
Nucleofection Survival
Nucleofection survival can vary significantly between cell lines or experiments, but within a given sample preparation on the same day one should expect a similar nucleofection event to have similar survival. These protocols are optimized for cell survival well between 60-80%. A number of technical factors play a role in survival but in with these experiments researchers should expect at least 40-50% survival after nucleofection. We noted in Basic Protocol 3 that, for many applications, plating serial dilutions of nucleofected cells on 6W plates can save on reagents and reveal optimal densities for events to follow.
Genome Engineering Outcomes
The contingencies affecting the frequency of desired outcomes are different for each branch of genomic engineering. It is important to compensate for this by screening excess clones at first. For NHEJ based INDEL experiments with 1-3 sgRNAs, researchers may expect 10-30% of their population to have successful heterozygous or homozygous disruption. In some cases the proportion of INDEL genetic disruption in initial mixed populations are significant to reveal experimental phenotypes, but enrichment for pure populations can be initiated from picking 10-20 colonies at first. In contrast, the frequency of desirable cells from large donor construct HDR experiments is lower. We have seen wide variation from CRISPR targeting within hPSC lines; heterozygous HDR may be found in 2-7% of the population, and homozygous HDR targeting seems to vary between 0.05-1.5%. We typically nucleofect constructs with antibiotic resistance into 1-2 × 106 hPSCs and plate them on 100mm plates with antibiotics starting two days later. From this, we usually observe ∼100 distinct colonies if antibiotic selection is employed. Within the protocols, genotype and karyotype analysis are recommended for desirable clones but it is also important to consider the potential for off-target DSBs predicted in the Zhang Lab software. Furthermore, recent reports show that unpredicted targets may also be detected using a novel GUIDE-Seq method (Tsai et al., 2015). All experiments involving nucleofection are dependent on nucleofection efficiency, and so control nucleofection experiments are highly recommended. Researchers should aim for at least 30-40% or more nucleofection efficiency.
Time Considerations
Despite its power, human genome engineering and hPSC culture is both expensive and laborious. Time and resources will vary based on the frequency of the desired outcome and the purification analyses available. In most cases the enrichment of desired clones will usually require several rounds of splitting to small clusters, picking, and subculture; all the while involving daily media changes followed by cryopreservation/genotyping. An estimation of a typical experiment from gRNA/Donor Plasmid cloning to successful clone purification is outlined in Table 3:
Table 3. Estimated Times for hPSC Genome Engineering and Purification Steps.
| STAGE | DAYS |
|---|---|
| VECTOR CLONING | 7-10 |
| NUCLEOFECTION (FROM WORKING STOCK) | 1 |
| SELECTION/COLONY GROWTH | 7-14 |
| CLONE PICKING -> FIRST ROUND | 6 |
| GENOTYPING FIRST ROUND | 2 |
| THAW AND SUBCLONING -> SECOND ROUND | 8 |
| GENOTYPING SECOND ROUND | 2 |
| THAW AND SUBCLONING -> THIRD ROUND | 8 |
| GENOTYPING THIRD ROUND | 2 |
| THAW AND EXPANSION OF PURIFIED CLONE | 14-21 |
| TOTAL | 57-74 |
Once several candidate clonal populations are isolated, researchers should karyotype, genotype, and validate pluripotency with ICC or teratoma formation.
Table 2. Materials Used in This Unit.
| MATERIAL | Provider | Catalog # | Amount |
|---|---|---|---|
| hES Cell Media (Option 1) Store for 1 week at 4°C | 500mL | ||
| MTesR1 | StemCell Technologies | 05850 | |
| hES Cell Media (Option 2) Store for 1 week at 4°C | 500mL | ||
| Essential 8 Medium | Life Technologies | A1517001 | |
| Cryopreservation Solution | 100mL | ||
| 0.22uM Filtered FBS | any | any | 90mL |
| Dimethyl Sulfoxide (DMSO) | any | any | 10mL |
| Matrigel Substrate Store for 1 week at 4°C | |||
| Corning® Matrigel® hESC-Qualified Matrix *See lot-by-lot dilution instructions, generally ∼100×;Thaw on ice at 4°C for 4 hours or overnight to reduce precipitation. | Corning (BD) | 354277 | Variable* |
| Knockout DMEM | Life Technologies | 10829-018 | Variable* |
| Lysis Buffer | As Needed | ||
| Tris pH7.5 | any | any | [10 mM] |
| EDTA pH 8.0 | any | any | [10 mM] |
| NaCl | any | any | [10 mM] |
| N-Lauroylsarcosine | any | any | [0.5% vol/vol] |
| Proteinase K - Add immediately before use. | any | any | [1 mg/ml] |
Acknowledgments
We gratefully acknowledge the Gladstone Institute Stem Cell Core and the many Gladstone scientists whom we have worked with for intellectual and material support. Furthermore, we are grateful to the world of scientists involved in hPS cell and genome engineering research. M.A.M. is supported by a Postdoctoral Fellowship from the Canadian Institutes of Health Research, 129844. B.R.C. received support from the U.S. National Heart, Lung, and Blood Institute, National Institutes of Health, U01-HL100406, U01-GM09614, R01-HL108677, U01-HL098179, and R01-HL060664. S.Y. is a scientific advisor of iPS Academia Japan without salary.
Literature Cited
- Alwin S, Gere MB, Guhl E, Effertz K, Barbas CF, Segal DJ, Weitzman MD, Cathomen T. Custom Zinc-Finger Nucleases for Use in Human Cells. Molecular Therapy. 2005;12:610–617. doi: 10.1016/j.ymthe.2005.06.094. [DOI] [PubMed] [Google Scholar]
- Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, Bonas U. Breaking the Code of DNA Binding Specificity of TAL-Type III Effectors. Science. 2009;326:1509–1512. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- Capecchi MR. Science. Vol. 244. New York, N.Y.: 1989. Altering the genome by homologous recombination; pp. 1288–1292. [DOI] [PubMed] [Google Scholar]
- Chen G, Gulbranson DR, Hou Z, Bolin JM, Ruotti V, Probasco MD, Smuga-Otto K, Howden SE, Diol NR, Propson NE, et al. Chemically defined conditions for human iPSC derivation and culture. Nature Methods. 2011;8:424–429. doi: 10.1038/nmeth.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature Biotechnology. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D, Wang H, Kiani S, Lai CS, Gao Q, Cassady JP, Cost GJ, Zhang L, Santiago Y, Miller JC, et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nature Biotechnology. 2011;29:731–734. doi: 10.1038/nbt.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath P, Barrangou R. CRISPR/Cas, the Immune System of Bacteria and Archaea. Science. 2010;327:167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA-programmed genome editing in human cells. eLife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenstein ME, Ludwig TE, Xu RH, Llanas RA, VanDenHeuvel-Kramer K, Manning D, Thomson JA. Basic Fibroblast Growth Factor Support of Human Embryonic Stem Cell Self-Renewal. STEM CELLS. 2006;24:568–574. doi: 10.1634/stemcells.2005-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MZ, Elledge SJ. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nature Methods. 2007;4:251–256. doi: 10.1038/nmeth1010. [DOI] [PubMed] [Google Scholar]
- Lombardo A, Genovese P, Beausejour CM, Colleoni S, Lee YL, Kim KA, Ando D, Urnov FD, Galli C, Gregory PD, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nature Biotechnology. 2007;25:1298–1306. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- Ludwig TE, Levenstein ME, Jones JM, Berggren WT, Mitchen ER, Frane JL, Crandall LJ, Daigh CA, Conard KR, Piekarczyk MS, et al. Derivation of human embryonic stem cells in defined conditions. Nature Biotechnology. 2006;24:185–187. doi: 10.1038/nbt1177. [DOI] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-Guided Human Genome Engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyaoka Y, Chan AH, Judge LM, Yoo J, Huang M, Nguyen TD, Lizarraga PP, So PL, Conklin BR. Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nature Methods. 2014;11:291–293. doi: 10.1038/nmeth.2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissim L, Perli SD, Fridkin A, Perez-Pinera P, Lu TK. Multiplexed and Programmable Regulation of Gene Networks with an Integrated RNA and CRISPR/Cas Toolkit in Human Cells. Molecular Cell. 2014;54:698–710. doi: 10.1016/j.molcel.2014.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohgushi M, Sasai Y. Lonely death dance of human pluripotent stem cells: ROCKing between metastable cell states. Trends in Cell Biology. 2011;21:274–282. doi: 10.1016/j.tcb.2011.02.004. [DOI] [PubMed] [Google Scholar]
- Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S, Hong H, Nakagawa M, Tanabe K, Tezuka K, et al. A more efficient method to generate integration-free human iPS cells. Nature Methods. 2011;8:409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nature Biotechnology. 2013;31:839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porteus MH, Baltimore D. Chimeric Nucleases Stimulate Gene Targeting in Human Cells. Science. 2003;300:763–763. doi: 10.1126/science.1078395. [DOI] [PubMed] [Google Scholar]
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, et al. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouet P, Smih F, Jasin M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Molecular and Cellular Biology. 1994;14:8096–8106. doi: 10.1128/mcb.14.12.8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, Wyvekens N, Khayter C, Iafrate AJ, Le LP, et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature Biotechnology. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic Stem Cell Lines Derived from Human Blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Yu J, Hu K, Smuga-Otto K, Tian S, Stewart R, Slukvin II, Thomson JA. Human Induced Pluripotent Stem Cells Free of Vector and Transgene Sequences. Science. 2009;324:797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
Internet Resources
- 1.Addgene: CRISPR in the Lab: A Practical Guide. [Accessed May 18, 2015]; Available at: http://www.addgene.org/crispr/guide/
- 2.Addgene: Handling Plasmids from Addgene - Purifying Plasmid DNA. [Accessed Accessed April 19, 2015]; Available at: https://www.addgene.org/plasmid-protocols/purify-plasmid-dna/#ethanol.
- 3.Amaxa Human Human Stem Cell Nucleofector Starter Kit | Lonza. [Accessed May 19, 2015]; Available at: http://bio.lonza.com/fileadmin/groups/marketing/Downloads/Protocols/Generated/Optimized_Protocol_203.pdf.
- 4.Optimized CRISPR Design. [Accessed May 18, 2015]; Available at: http://crispr.mit.edu/