

Abstract
The ω-3 fatty acid desaturase (fat-1) gene encodes the enzyme that converts ω-6 polyunsaturated fatty acids (PUFAs) to ω-3 PUFAs. Numerous studies have suggested that the ratio of ω-6/ω-3 PUFAs has an impact on tumorigenesis. To investigate the biological function of the fat-1 gene in human oral squamous cell carcinoma (OSCC), the fat-1 gene was introduced into OSCC cells by transfection. The uptake of the gene was confirmed by reverse transcription-polymerase chain reaction and analyzed using gas chromatography. The antitumor effects and mechanisms of the fat-1 gene were evaluated by studying cell survival and tumor growth in vitro and in vivo. Gas chromatography results revealed that the cells transfected with the fat-1 gene had a higher ω-3/ω-6 PUFA ratio than cells transfected with the control vector. An MTT and DNA fragmentation assay indicated that the presence of the fat-1 gene in vitro significantly decreased OSCC cell proliferation and significantly increased the rate of apoptosis. Similar antitumor effects of the fat-1 gene were also observed in vivo. Immunohistochemistry analysis confirmed that Tca8113 cell tumors displayed a significant reduction in cell growth and cell survival following the introduction of the fat-1 gene. The current study suggests that the inhibitory effect of the fat-1 gene on tumor growth may be a result of a reduction in the expression of the tumor survival protein β-catenin. The results also support the theory that the ratio of ω-3/ω-6 PUFAs has an impact on OSCC tumor growth. The findings of the study provide notable molecular insight into the theory suggesting that ω-3 PUFAs are an intermediate for the chemoprevention and treatment of human OSCC.
Keywords: oral squamous cell carcinoma, ω-3 polyunsaturated fatty acids, β-catenin
Introduction
Oral squamous cell carcinoma (OSCC) has been established as one of the most common types of cancer worldwide. It is estimated that in the United States, 3% of cancer cases in men and 2% in women are OSCC (1). Recognized risk factors for OSCC include heavy smoking and alcohol consumption, and current treatment options include surgery and radiation therapy (2). Patients with OSCC have a 5-year survival rate of <50% and the treatment of OSCC has not greatly improved during recent decades (3). Although it has been suggested that OSCC is a result of accumulated genetic mutations, a clear pathogenetic pathway for OSCC is yet to be defined (4–6).
Long-chain ω-3 polyunsaturated fatty acids (PUFAs) have been demonstrated to possess significant chemopreventive properties and therapeutic potential in the treatment of cancer (7,8). Dietary intake of ω-3 PUFAs has been reported to reduce the risk of several malignancies, including OSCC, by inhibiting the growth of tumors and metastatic lesions (9). It is suggested that the ratio of ω-6/ω-3 fatty acids, rather than the absolute levels of the two PUFAs, is the principal factor that results in the observed antitumor effects (10–12). In particular, it was reported that the introduction of the fat-1 gene, which converts ω-6 PUFAs to ω-3 PUFAs, inhibited lung and breast cancer cell growth in vitro (13,14). In addition to this, it was demonstrated that melanoma cell growth is reduced in fat-1 transgenic mice (15). However, the function of the fat-1 gene with regards to OSCC remains unknown. The present study was designed to evaluate the function and mechanisms of ω-3 PUFAs and the fat-1 gene (from the C. elegans worm) in OSCC cells.
Materials and methods
Cell culture
The Tca8113 OSCC cell line was obtained from the American Type Culture Collection (Rockville, MD, USA). All cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 100 µg/ml streptomycin and 100 U/ml penicillin (Invitrogen; Thermo Fisher Scientific, Inc., Beijing, China). Cells were cultured in 10-cm tissue culture dishes and maintained in a 37°C incubator equilibrated with 5% CO2 in humidified air.
Construction of the fat-1 gene expression vector
The mfat-1 gene was created from the coding region of the C. elegans fat-1 gene (GenBank: NM_001028389) by optimizing for mammalian cell expression. The mfat-1 cDNA was synthesized (Tiangen Biotech Co., Ltd., Beijing, China) and constructed into the control vector, named pCB0, which is from pcDNA3.1(+), using restriction enzyme digestion and ligation (NEB, Beijing, China. The cytomegalovirus (CMV) promoter was replaced with the CMV immediate-early enhancer/chicken β-actin hybrid promoter, and the constructed mfat-1 expression vector was named pCB1.
Transfection of Tca8113 cells
Tca8113 cells were transfected with linearized pCB1 and pCB0 with Lipofectamine Plus reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. The cells were then selected with G418 (Invitrogen; Thermo Fisher Scientific, Inc.) at 350 µg/ml. After 2 weeks of selection, the G418-resistant colonies were pooled to reduce potential variations amongst different clones. The mfat-1 transgene expression and function were confirmed by reverse transcription-polymerase chain reaction (RT-PCR) and fatty acid profile analysis.
Analysis by RT-PCR
Total RNA was extracted using a SimplyP Total RNA Extraction kit (Bioer, Beijing, China). Reverse transcription was performed using random hexamer oligonucleotide primers using the BioRT Two-Step RT-PCR kit (Bioer) according to the manufacturer's protocol. Cells were then subjected to RT-PCR for the detection of mfat-1 mRNA expression. The sequences of the primers of mfat-1 were as follows: Sense 5′-CCGCCACCGCTCCCGTCAC-3′ and antisense 5′-TGCTTCCCAATCCTTAT-3′. The resulting amplification product was 527 bp in length. PCR primers for GAPDH were as follows: Sense 5′-CCATGGAGAAGGCTGGGG-3′ and antisense 5′-CAAAGTTGTCATGGATGACC-3′. GAPDH was used as the control, and the resulting amplification product was 194 bp. The PCR amplification was performed at 95°C for 30 sec, 60°C for 30 sec and 72°C for 30 sec for 30 cycles, with a final extension at 72°C for 6 min. The PCR products were separated on a 1.0% agarose gel (Tiangen Biotech Co., Ltd.) at 120 V and stained with ethidium bromide (Tiangen Biotech Co., Ltd.).
Gas chromatography
Lipids were extracted from the cells according to the technique of Bligh and Dyer (16). Briefly, the samples were homogenized in a mixture of methanol, chloroform, and water. After 15 min, chloroform was added and the samples were vortexed and centrifuged at 1,500 × g for 30 min. The lower phase was dried under nitrogen and resuspended in boron trifluoride methanol (Sigma-Aldrich, St. Louis, MO, USA). The samples were heated at 90°C for 30 min and extracted with 4.0 ml pentane (Beijing Chemical Works, Beijing, China) and 1.5 ml water. The mixtures were then vortexed and the upper phase was recovered. The extracts were dried, resuspended in heptane (Sigma-Aldrich), and injected into a capillary column (SP-2380, 105 m × 53 mm ID, 0.20-µm film thickness; Sigma-Aldrich). The gas chromatograph was performed on a Clarus 500 gas chromatograph (PerkinElmer, Inc., Waltham, MA, USA). The comparison of retention time to authentic standards (Sigma-Aldrich) were used to identify components.
MTT cell proliferation assay
Briefly, Tca8113 cells and the cells transfected with either pCB1 and pCB0 were plated in 96-well plates overnight. A density of 3,000 cells/well in 200 µl appropriate growth media was used, and these cells were incubated for 24, 48 and 72 h. Next, 20 µl/well MTT solution (Invitrogen; Thermo Fisher Scientific, Inc.) was added and the cells were incubated for another 3.5 h. The absorbance of each well at 570 nm, with the background subtraction at 650 nm, was read by a Multiskan GO Microplate Spectrophotometer (Thermo Fisher Scientific, Inc., Waltham, MA, USA). The average absorbance of 6 wells/experiment was used.
DNA fragmentation analysis
Apoptosis was detected and quantified by the Cell Death Detection ELISAPLUS kit (Roche Diagnostics, Shanghai, China). Briefly, cells were seeded in plates with a regular medium and allowed to grow until 70–80% confluent. Next, cells were washed twice with PBS and the regular media was changed to DMEM without serum. DNA fragmentation was calculated by the fold change and normalized with corresponding MTT results from identical culture conditions.
Xenograft model
To establish xenografts in nude mice, Tca8113 cells that were transfected with either pCB1 or pCB0 in log-phase growth were washed twice with PBS and then counted. Subcutaneous tumors were established by injecting 1.2×106 Tca8113 cells, which were resuspended in 200 µl PBS, onto each side of the dorsal surface on the BALB/c nude male mice. Six-week-old male BALB/c nude mice were obtained from Model Animal Research Center of Nanjing University (Nanjing, China). Mice were assigned to two groups (n=6 per group): Tca8113-pCB0 or Tca8113-pCB1, and were handled in accordance to institutional guidelines for animal care. The tumor size was measured once weekly for 5 weeks and the tumor volume (mm3) was calculated as follows: Tumor volume = LxW2×0.5. The tumor volume data were subjected to statistical analysis using either the Student's two-tailed t-test or the Mann-Whitney U test. The animal experiments were performed according to the protocol approved by the Jilin University Institutional Animal Care and Use Committee (#2012079; Changchun, China).
Immunostaining
Xenograft tumors were harvested and placed in 10% formalin, embedded in paraffin, and sectioned at 5 µm thickness using an RM2255 microtome (Leica Microsystems, Inc., Buffalo Grove, IL, USA). Paraffin-embedded tumor blocks were cut at serial sections and the obtained slides were used for hematoxylin and eosin (H&E) staining and immunohistochemical analysis, as described by Lu et al (17). Tumor cell proliferation was studied by immunostaining with a Ki-67 antibody (#PA121520; Invitrogen; Thermo Fisher Scientific, Inc.). Similarly, apoptosis was detected using an Apoptag terminal deoxynucleotidyl-transferase-mediated dUTP nick end labeling (TUNEL) assay kit (EMD Millipore, Billerica, MA, USA), counterstained with methyl green (Sigma-Aldrich). The rates of proliferation and apoptosis were quantitatively determined by counting the percentage of positively stained cell bodies from three histologically similar fields viewed at ×400 magnification using a Labophot-2 microscope (Nikon Corporation, Tokyo, Japan), which were randomly selected from each slide. Slides of 3 fields per animal and 5 animals per group were used for comparison.
Immunoblotting
Cell samples were measured for total protein concentration using the bicinchoninic acid (BCA) assay (Pierce Biotechnology, Inc., Rockford, IL, USA). A total of 40 µg protein/samples were prepared and separated on 12% SDS-PAGE gels at 100 V prior to transfer to polyvinylidine difluoride membranes (Bio-Rad Laboratories, Inc., Beijing, China). The membranes were blocked and shaken at room temperature for 1 h and then incubated overnight with rabbit anti-mouse primary antibodies against caspase-3 (#9662), caspase-9 (#9504), GSK-3β (#9315), p-GSK-3β (#9322), β-catenin (#9562), Akt (#9272) and p-Akt (#9275; Cell Signaling Technology, Inc., Danvers, MA, USA), all at a dilution of 1:1,000. The next day, the blots were washed 3 times with PBS-Tween-20 (PBS-T), then incubated with the corresponding goat anti-rabbit polyclonal horseradish peroxidase-conjugated secondary antibodies (1:5,000: #12348; Sigma-Aldrich) for 1 h at room temperature. Following another 3 washes with PBS-T, the blots were subjected to the ECL Western blotting detection reagents and analysis system (GE Healthcare Life Sciences, Piscataway, NJ, USA) to visualize the protein bands.
Statistical analysis
Statview software, version 4.5 (Abacus Concepts, Berkley, CA, USA) was used for the statistical analysis. Analysis of variance was used for initial analysis, followed by Fisher's protected least significant difference for post hoc analyses. All experiments were performed at least in triplicate and the results are presented as the mean ± standard deviation. P<0.05 was considered to indicate a statistically significant difference.
Results
Expression of the mfat-1 gene decreased the ratio of ω-6/ω-3 PUFAs
Following transfection of the Tca8113 cells with the mfat-1 gene (pCB1), RT-PCR with mfat-1-specific primers indicated that the cells transfected with pCB1 expressed high levels of the mfat-1 gene (Fig. 1A). As expected, the mfat-1 gene was not expressed in the parental cells or the cells that were transfected with the control vector (pCB0). To analyze the function of the mfat-1 gene, gas chromatography was performed to assess the composition of PUFAs in the cells. The level of ω-3 PUFAs was significantly increased in cells transfected with pCB1, compared with cells transfected with pCB0 (Table I). The level of PUFAs in cells transfected with pCB1 and pCB0 are presented in Table I as a percentage of the total cellular lipids. The expression of the mfat-1 gene led to a significant increase in ω-3 PUFAs, accompanied by a decrease in ω-6 PUFAs (Table I). The ratio of ω-6/ω-3 PUFAs with or without exogenous mfat-1 were 0.381 and 1.275, respectively (P<0.001; Fig. 1B). This indicates that the expression of mfat-1 can convert ω-6 PUFAs into the corresponding ω-3 forms. It can be predicted that the change in the ω-6/ω-3 PUFA ratio affects the endogenous ω-3 PUFA production mechanism, while reducing the level of ω-6 PUFA production.
Figure 1.
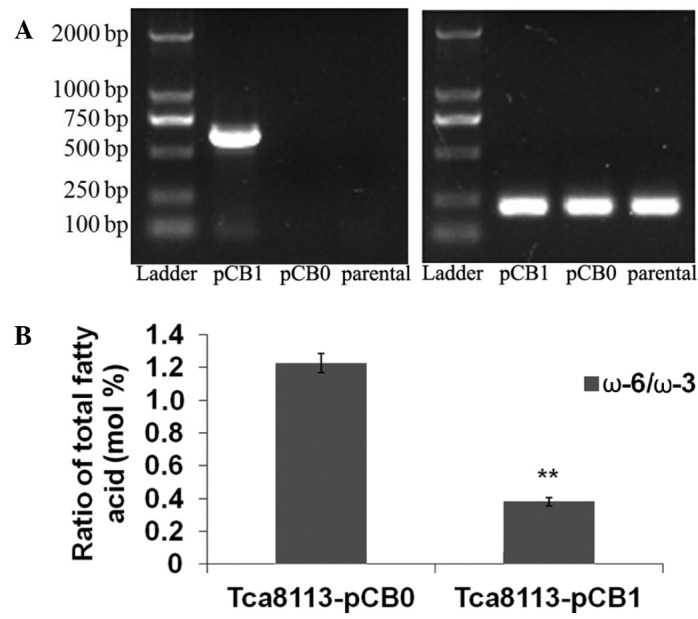
Expression of the mfat-1 gene reduces the ω-6/ω-3 ratio in Tca8113 cells. (A) Reverse transcription-polymerase chain reaction of mfat-1 (left) and GAPDH (right) gene expression in cells transfected with pCB1 (the mfat-1 expression vector) or pCB0 (the control vector), compared with the parental Tca8113 cells. (B) The ratio of ω-6 to ω-3 polyunsaturated fatty acids in Tca8113 transfected with pCB0 and pCB1 vectors. Error bars indicate standard deviation. **P<0.001 vs. pCB0.
Table I.
PUFA composition of total cellular lipids from the cells that were transfected with the mfat-1 cDNA or the control vector.
| % of total fatty acid | Tca8113-pCB1 | Tca8113-pCB0 |
|---|---|---|
| ω-6 polyunsaturates | ||
| 18:2 | 7.68 | 4.31 |
| 20:4 | 10.88 | 16.95 |
| Total | 18.56 | 20.96 |
| ω-3 polyunsaturates | ||
| 18:3 | 1.62 | 0.32 |
| 20:5 | 22.58 | 2.29 |
| 22:5 | 13.86 | 4.68 |
| 22:6 | 10.61 | 9.15 |
| Total | 48.67 | 16.44 |
| ω-3/ω-6 ratio | 0.38a | 1.27 |
Compositions of ω-6 or ω-3 PUFAs are expressed using the relative distribution area of the peak in gas chromatography assays. All data are presented as the mean ± standard error; n=3.
P<0.001, pCB1 vs. pCB0. PUFA, polyunsaturated fatty acid; pCB1, fat-1 gene expression vector; pCB0, control expression vector.
Expression of the mfat-1 gene decreased cell growth and increased apoptosis in vitro
To determine whether the change in the ratio of ω-6/ω-3 PUFAs in Tca8113 cells inhibits tumor cell proliferation in vitro, cells were cultured for different time periods and MTT cell proliferation assays were performed. Expression of the mfat-1 gene significantly inhibited the Tca8113 cell proliferation in cells that were transfected with pCB1, compared with the cells that transfected with pCB0 at 48 and 72 h (P<0.001; Fig. 2A). To evaluate whether mfat-1 gene expression in Tca8113 cells promotes programmed cell death, a DNA fragmentation ELISA assay was performed. The results demonstrated that mfat-1 gene expression enhances apoptosis in Tca8113 cells at 24 and 48 h, compared with cells transfected with the control vector (P<0.001; Fig. 2B).
Figure 2.
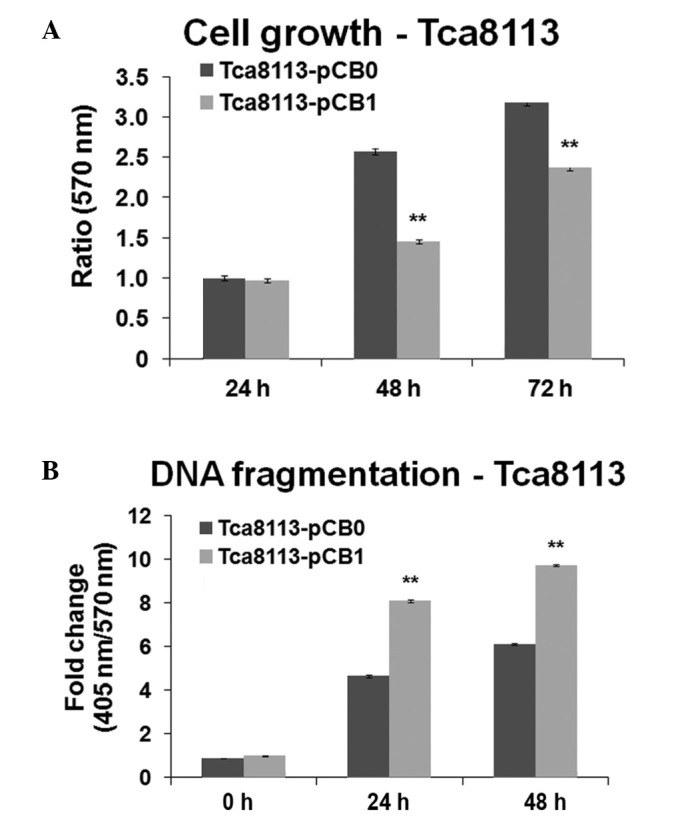
Expression of mfat-1 gene inhibited cell growth and increased apoptosis in Tca8113 cells. (A) Cell proliferation measured by MTT. (B) Apoptosis measured by DNA fragmentation assay. **P<0.001 vs. pBC0. pCB0, control expression vector; pCB1, fat-1 gene expression vector.
Expression of the mfat-1 gene inhibited tumor growth in vivo
To determine the antitumor effect of the mfat-1 gene in vivo, Tca8113 cells transfected with either pCB1 or pCB0 were implanted into nude mice. The cells were allowed 5 weeks for tumor growth. The results in Fig. 3 demonstrate that mfat-1 gene expression significantly reduces Tca8113 cell tumor volume at 4 and 5 weeks subsequent to implantation, compared with cells that were transfected with the control vector (P<0.05; Fig. 3A).
Figure 3.

Expression of the mfat-1 gene reduces subcutaneous tumor growth of Tca8113 cells. (A) Tumor volume in the mice implanted with mfat-1-transfected Tca8113 cells and the control. *P<0.05. vs. pCB0. (B) Hematoxylin and eosin staining. (C) mfat-1 gene expression inhibited proliferation of the tumor. Quantified data were determined by the number of Ki-67-positive cells divided by the total number of cells in five randomly selected fields under light microscopy (×400). **P<0.001 vs. pCB0. (D) mfat-1 gene expression led to increased apoptosis in tumor cells. Similar sections were stained by TUNEL. Black arrows indicate Ki-67-positive nuclei. Quantified data were determined by the number of apoptotic nuclei divided by the total number of cells in five randomly selected fields under light microscopy (×400). **P<0.001 vs. pCB0. pCB0, control expression vector; pCB1, fat-1 gene expression vector.
Expression of the mfat-1 gene reduced the number of viable Tca8113 cells and increased necrosis
To determine the mechanism of tumor growth inhibition by the mfat-1 gene, histological analyses were performed. H&E staining demonstrated that the overexpression of the mfat-1 gene was associated with a reduced number of viable cells and an increased area of necrosis (Fig. 3B). The number of proliferating cells, identified by Ki-67-positive nuclei, was significantly reduced in the tumor cells transfected with the mfat-1 gene compared with the control-transfected cells (P<0.001; Fig. 3C). It was also observed, using a TUNEL assay, that the introduction of the mfat-1 gene to Tca8113 cells was associated with an increased number of positively stained apoptotic cells. Cell counts indicated that 50–60% of cells transfected with the mfat-1 gene were apoptotic, whereas 10–20% of cells were apoptotic in the control group, and this difference was demonstrated to be statistically significant (P<0.001; Fig. 3D).
Expression of the mfat-1 gene induced caspase-3-mediated cell apoptosis and inhibited Tca8113 cell proliferation
To further investigate the mechanism of mfat-1 gene expression on the inhibition of tumor cell growth, the protein expression of caspase-3, caspase-9, p-GSK-3β, GSK-3β, β-catenin, p-Akt and Akt was examined (Fig. 4). It was demonstrated that the cleavage of caspase-9 and caspase-3 was enhanced in cells transfected with the mfat-1 gene, and mfat-1-transfected cells presented a reduced level of GSK-3β phosphorylation and β-catenin expression. No alterations were observed in Akt expression or phosphorylation.
Figure 4.
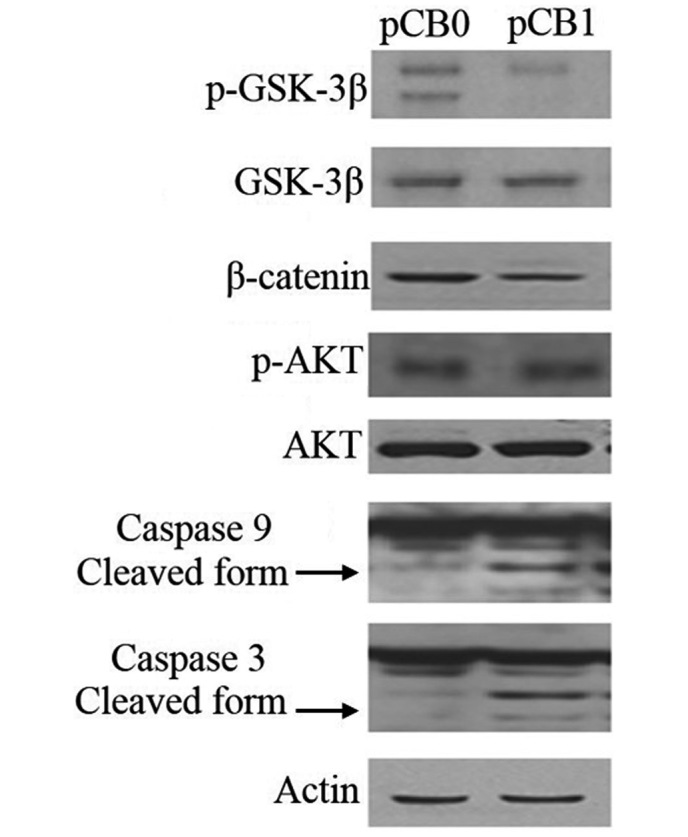
Expression of the mfat-1 gene induced caspase-3-mediated cell apoptosis and inhibited GSK-3β phosphorylation in the Tca8113 cells. Expression of mfat-1 gene induced caspase-3 and caspase-9 cleavage, and reduced Tca8113 cell proliferation through reduction of GSK3β phosphorylation and subsequent downregulation of β-catenin expression. Caspase-3, caspase-9, GSK-3β, p-GSK-3β, β-catenin, p-Akt and Akt protein levels were examined by western blot. β-actin served as an equal loading control.
Discussion
Previous experimental data have demonstrated that the ω-3 fatty acids antagonize the stimulative roles of ω-6 fatty acids in carcinogenesis, tumor growth and metastasis (10,13,17). In the current study, in vitro and in vivo models were used to evaluate the effect of ω-3 PUFAs on Tca8113 cell growth. A codon-optimized C. elegans gene, mfat-1, encoding the enzyme ω-3 fatty acid desaturase, was transfected into Tca8113 cells. Since this enzyme converts endogenous ω-6 PUFAs into the corresponding ω-3 forms, there is no requirement for a traditional, lengthy feeding regimen to achieve stable increases in cellular ω-3 PUFAs. This model serves as a good platform for the analysis of the effect of ω-3 PUFAs on the proliferation and viability of OSCC cells. The present study confirms that mfat-1 expression increases the level of ω-3 PUFAs and decreases the level of ω-6 PUFAs in OSCC cells. Additionally, the study demonstrated that mfat-1 expression inhibits cell proliferation and enhances cell apoptosis in vitro. To investigate the feasibility of using an mfat-1 transgene as a potential therapeutic approach, mfat-1-transfected OSCC cells were implanted into athymic nude mice and the tumor volume was monitored for 5 weeks. It was demonstrated that mfat-1 gene expression reduced tumor growth in vivo via the induction of apoptosis and the inhibition of cell proliferation. It can therefore be hypothesized that ω-3 PUFAs may have chemopreventive and antitumorigenic effects.
With regards to the mechanism underlying the antitumor effects of the mfat-1 gene in OSCC cells, it was observed that mfat-1 expression reduces GSK3β phosphorylation and the expression of β-catenin. β-catenin is a key molecule in the Wnt signaling pathway, and serves a significant role in the regulation of multiple cellular functions in embryogenesis and tumorigenesis (18). Wnt proteins are a large family of secreted glycoproteins that have been reported to be responsible for the activation of multiple pathways, including the canonical Wnt pathway through β-catenin, the non-canonical Wnt pathway through planar cell polarity, and the Wnt-Ca2+ pathway (19). In particular, the canonical Wnt pathway requires the stabilization of β-catenin (18). However, in the absence of a Wnt signal, β-catenin resides within the cytosol in the form of a β-catenin destruction complex, where it is phosphorylated and destined for proteasome-dependent degradation (20). Upon the activation of Wnt signaling, this β-catenin destruction complex separates, resulting in the cytoplasmic accumulation of β-catenin followed by its translocation into the nucleus (21,22). β-catenin then associates with T-cell factor/lymphoid enhancer factor transcription factors in the nucleus and stimulates the transcription of target genes involved in cell proliferation, differentiation and apoptosis (23,24). The dysregulation of the Wnt-β-catenin signaling pathway has been reported in several types of cancer in humans, including multiple myeloma (25) and gastrointestinal cancer (26). However, its implications in oral cancer remain uncertain. The present study suggests that the overexpression of the mfat-1 gene in OSCC cells inhibits cell proliferation through the inhibition of the Wnt/β-catenin signaling pathway.
In summary, the current study describes preclinical evidence for the use of mfat-1 and ω-3 PUFAs in the chemoprevention and treatment of OSCC.
Acknowledgements
The present work was supported by the Youth Foundation of the Jilin Science and Technology Commission (grant no. 3D512J526604).
References
- 1.Lambert R, Sauvaget C, de Camargo Cancela M, Sankaranarayanan R. Epidemiology of cancer from the oral cavity and oropharynx. Eur J Gastroenterol Hepatol. 2011;23:633–641. doi: 10.1097/MEG.0b013e3283484795. [DOI] [PubMed] [Google Scholar]
- 2.Bhide SA, Ahmed M, Newbold K, Harrington KJ, Nutting CM. The role of intensity modulated radiotherapy in advanced oral cavity carcinoma. J Cancer Res Ther. 2012;8(Suppl 1):S67–S71. doi: 10.4103/0973-1482.92218. [DOI] [PubMed] [Google Scholar]
- 3.Khan Z, Bisen PS. Oncoapoptotic signaling and deregulated target genes in cancers: Special reference to oral cancer. Biochim Biophys Acta. 2013;1836:123–145. doi: 10.1016/j.bbcan.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 4.Braakhuis BJ, Leemans CR, Brakenhoff RH. A genetic progression model of oral cancer: Current evidence and clinical implications. J Oral Pathol Med. 2004;33:317–322. doi: 10.1111/j.1600-0714.2004.00225.x. [DOI] [PubMed] [Google Scholar]
- 5.Califano J, van der Riet P, Westra W, Nawroz H, Clayman G, Piantadosi S, Corio R, Lee D, Greenberg B, Koch W, Sidransky D. Genetic progression model for head and neck cancer: Implications for field cancerization. Cancer Res. 1996;56:2488–2492. [PubMed] [Google Scholar]
- 6.Choi S, Myers JN. Molecular pathogenesis of oral squamous cell carcinoma: Implications for therapy. J Dent Res. 2008;87:14–32. doi: 10.1177/154405910808700104. [DOI] [PubMed] [Google Scholar]
- 7.Hardman WE. (n-3) fatty acids and cancer therapy. J Nutr. 2004;134(Suppl):3427S–3430S. doi: 10.1093/jn/134.12.3427S. [DOI] [PubMed] [Google Scholar]
- 8.Larsson SC, Kumlin M, Ingelman-Sundberg M, Wolk A. Dietary long-chain n-3 fatty acids for the prevention of cancer: A review of potential mechanisms. Am J Clin Nutr. 2004;79:935–945. doi: 10.1093/ajcn/79.6.935. [DOI] [PubMed] [Google Scholar]
- 9.Nikolakopoulou Z, Nteliopoulos G, Michael-Titus AT, Parkinson EK. Omega-3 polyunsaturated fatty acids selectively inhibit growth in neoplastic oral keratinocytes by differentially activating ERK1/2. Carcinogenesis. 2013;34:2716–2725. doi: 10.1093/carcin/bgt257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gago-Dominguez M, Yuan JM, Sun CL, Lee HP, Yu MC. Opposing effects of dietary n-3 and n-6 fatty acids on mammary carcinogenesis: The Singapore Chinese Health Study. Br J Cancer. 2003;89:1686–1692. doi: 10.1038/sj.bjc.6601340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maillard V, Bougnoux P, Ferrari P, Jourdan ML, Pinault M, Lavillonnière F, Body G, Le Floch O, Chajès V. N-3 and N-6 fatty acids in breast adipose tissue and relative risk of breast cancer in a case-control study in Tours, France. Int J Cancer. 2002;98:78–83. doi: 10.1002/ijc.10130. [DOI] [PubMed] [Google Scholar]
- 12.Simonsen N, van't Veer P, Strain JJ, Martin-Moreno JM, Huttunen JK, Navajas JF, Martin BC, Thamm M, Kardinaal AF, Kok FJ, Kohlmeier L. Adipose tissue omega-3 and omega-6 fatty acid content and breast cancer in the EURAMIC study. European Community Multicenter Study on Antioxidants, Myocardial Infarction, and Breast Cancer. Am J Epidemiol. 1998;147:342–352. doi: 10.1093/oxfordjournals.aje.a009456. [DOI] [PubMed] [Google Scholar]
- 13.Xia SH, Wang J, Kang JX. Decreased n-6/n-3 fatty acid ratio reduces the invasive potential of human lung cancer cells by downregulation of cell adhesion/invasion-related genes. Carcinogenesis. 2005;26:779–784. doi: 10.1093/carcin/bgi019. [DOI] [PubMed] [Google Scholar]
- 14.Ge Y, Chen Z, Kang ZB, Cluette-Brown J, Laposata M, Kang JX. Effects of adenoviral gene transfer of C. elegans n-3 fatty acid desaturase on the lipid profile and growth of human breast cancer cells. Anticancer Res. 2002;22(2A):537–543. [PubMed] [Google Scholar]
- 15.Xia S, Lu Y, Wang J, He C, Hong S, Serhan CN, Kang JX. Melanoma growth is reduced in fat-1 transgenic mice: Impact of omega-6/omega-3 essential fatty acids. Proc Natl Acad Sci USA. 2006;103:12499–12504. doi: 10.1073/pnas.0605394103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 17.Lu Y, Nie D, Witt WT, Chen Q, Shen M, Xie H, Lai L, Dai Y, Zhang J. Expression of the fat-1 gene diminishes prostate cancer growth in vivo through enhancing apoptosis and inhibiting GSK-3 beta phosphorylation. Mol Cancer Ther. 2008;7:3203–3211. doi: 10.1158/1535-7163.MCT-08-0494. [DOI] [PubMed] [Google Scholar]
- 18.Veeman MT, Axelrod JD, Moon RT. A second canon. Functions and mechanisms of beta-catenin-independent Wnt signaling. Dev Cell. 2003;5:367–377. doi: 10.1016/S1534-5807(03)00266-1. [DOI] [PubMed] [Google Scholar]
- 19.Bovolenta P, Rodriguez J, Esteve P. Frizzled/RYK mediated signalling in axon guidance. Development. 2006;133:4399–4408. doi: 10.1242/dev.02592. [DOI] [PubMed] [Google Scholar]
- 20.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 21.Gordon MD, Nusse R. Wnt signaling: Multiple pathways, multiple receptors, and multiple transcription factors. J Biol Chem. 2006;281:22429–22433. doi: 10.1074/jbc.R600015200. [DOI] [PubMed] [Google Scholar]
- 22.Hoppler S, Kavanagh CL. Wnt signalling: Variety at the core. J Cell Sci. 2007;120:385–393. doi: 10.1242/jcs.03363. [DOI] [PubMed] [Google Scholar]
- 23.Lim K, Han C, Dai Y, Shen M, Wu T. Omega-3 polyunsaturated fatty acids inhibit hepatocellular carcinoma cell growth through blocking beta-catenin and cyclooxygenase-2. Mol Cancer Ther. 2009;8:3046–3055. doi: 10.1158/1535-7163.MCT-09-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moon RT, Kohn AD, De Ferrari GV, Kaykas A. WNT and beta-catenin signalling: Diseases and therapies. Nat Rev Genet. 2004;5:691–701. doi: 10.1038/nrg1427. [DOI] [PubMed] [Google Scholar]
- 25.Chim CS, Pang R, Fung TK, Choi CL, Liang R. Epigenetic dysregulation of Wnt signaling pathway in multiple myeloma. Leukemia. 2007;21:2527–2536. doi: 10.1038/sj.leu.2404939. [DOI] [PubMed] [Google Scholar]
- 26.White BD, Chien AJ, Dawson DW. Dysregulation of Wnt/β-catenin signaling in gastrointestinal cancers. Gastroenterology. 2012;142:219–232. doi: 10.1053/j.gastro.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]