

Abstract
MicroRNAs (miRNAs) are a class of non-coding RNAs 18–25 nucleotides in length, which play important roles in the regulation of cancer progression through gene silencing. miRNA (miR)-101 has been suggested to be associated with hepatocellular carcinoma (HCC). However, the detailed role of miR-101 in HCC metastasis and the underlying mechanism remain largely unclear. The present study demonstrated that the expression of miR-101 was significantly reduced in HCC tissues compared with that in matched normal adjacent tissues. miR-101 was also found to be downregulated in four HCC cell lines compared with its expression in a normal liver cell line. Vascular endothelial growth factor (VEGF)-C was further identified as a direct target of miR-101, and the protein expression of VEGF-C was downregulated by miR-101 in HepG2 HCC cells. Furthermore, the overexpression of miR-101 and the knockdown of VEGF-C significantly inhibited HepG2 cell migration and invasion, while restoration of VEGF-C reversed the inhibitory effect of miR-101 overexpression on HepG2 cell migration and invasion. Finally, the expression of VEGF-C was notably increased in HCC tissues and cell lines. These findings suggest that miR-101 exerts a suppressive effect on HCC cell migration and invasion, at least in part through the direct inhibition of VEGF-C protein expression. Therefore, the miR-101/VEGF-C axis may serve as a potential therapeutic target for HCC metastasis.
Keywords: hepatocellular carcinoma, microRNA-101, vascular endothelial growth factor-C, migration, invasion
Introduction
Hepatocellular carcinoma (HCC) is a major health concern and the third leading cause of cancer-related mortality, with an increasing incidence worldwide (1). As the deregulation of oncogenes or tumor suppressor genes is closely associated with HCC progression, the identification of molecular targets and the development of effective agents is urgently required for the treatment of HCC.
MicroRNAs (miRNAs) are a class of non-coding RNAs 18–25 nucleotides in length, which may lead to mRNA degradation or inhibit protein translation through directly binding to the 3′-untranslated region (UTR) of their target mRNAs. By mediating protein expression of their target genes, miRNAs play key roles in the regulation of cell survival, proliferation, differentiation and motility (2,3). Recently, the deregulation of miRNAs that serve as tumor suppressor genes or oncogenes has been found to be involved in the development and progression of various human malignancies (4). Among these, miRNA (miR)-101 generally plays a suppressive role in several types of cancer, including retinoblastoma and breast, endometrial, cervical, ovarian, gastric and non-small-cell lung cancer (5–9). Furthermore, several studies have investigated the role of miR-101 in HCC. Xu et al (10) previously demonstrated that miR-101 inhibited autophagy and enhanced cisplatin-induced apoptosis in HCC cells. In addition, Zhang et al (11) demonstrated that miR-101 was associated with a favorable prognosis in HCC via inhibition of SOX9-dependent tumorigenesis. However, the molecular mechanisms through which miR-101 regulates HCC cell migration and invasion remain largely unclear.
Vascular endothelial growth factor (VEGF)-C is a member of the VEGF family and has been suggested to possess important lymphangiogenic properties (12). The role of VEGF-C in human cancer, including HCC, has been previously investigated (13). However, no study has yet fully elucidated the association between miR-101 and VEGF-C in HCC cells. The present study mainly aimed to investigate the role of miR-101 in the regulation of cell migration and invasion, particularly focusing on the association between miR-101 and VEGF-C in HCC cells.
Materials and methods
Tissue specimen collection
A total of 20 samples from HCC tissues and their matched adjacent normal tissues were obtained from the Department of Gastroenterology and Hepatology (Jinshan Hospital, Fudan University). All the tissues were immediately snap-frozen in liquid nitrogen following surgical removal and stored at −70°C until use.
The study was approved by the Ethics Committee of Fudan University (Shanghai, China) and all the participants provided written informed consent for the use of their tissue specimens.
Cell culture
The human HCC cell lines HepG2, LH86, LMH and PLHC-1, and the normal liver cell line THLE-3, were obtained from the American Type Culture Collection (Manassas, VA, USA). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS) at 37°C in a humidified incubator containing 5% CO2.
Reverse transcription quantitative polymerase chain reaction (RT-qPCR) assay
Total RNA was extracted using TRIzol® reagent (Ambion; Thermo Fisher Scientific, Carlsbad, CA, USA). For miR detection, a TaqMan miRNA Reverse Transcription kit (Thermo Fisher Scientific) was used to convert RNA into cDNA, according to the manufacturer's protocol. qPCR was then performed using an miRNA Q-PCR Detection kit (GeneCopoeia, Rockville, MD, USA) on an ABI 7500 thermocycler (Applied Biosystems; Thermo Fisher Scientific, Foster City, CA, USA). The U6 gene was used as an internal reference. For mRNA detection, a RevertAid First-Strand cDNA Synthesis kit (Fermentas, Carlsbad, CA, USA) was used to convert RNA into cDNA, according to the manufacturer's protocol. qPCR was then performed using iQTM SYBR® Green Supermix (Bio-Rad, Hercules, CA, USA) on the ABI 7500 thermocycler. The specific primers were as follows: VEGF-C: forward, 5′-GAG GAG CAG TTA CGG TCT GTG-3′ and reverse, 5′-TCC TTT CCT TAG CTG ACA CTT GT-3′; and GAPDH: forward, 5′-CTG GGC TAC ACT GAG CACC-3′ and reverse, 5′-AAG TGG TCG TTG AGG GCA ATG-3′. The relative mRNA expression of VEGF-C was normalized to GAPDH and analyzed using the 2−ΔΔCq method.
Western blot analysis
Tissues and cells were solubilized in cold radioimmunoprecipitation assay lysis buffer. The proteins were separated with 10% SDS-PAGE and transferred onto a polyvinylidene difluoride (PVDF) membrane. The PVDF membrane was incubated with phosphate-buffered saline containing 5% milk overnight at 4°C. Subsequently, the PVDF membrane was incubated with mouse monoclonal anti-VEGF-C primary antibody (dilution 1:200; ab191274; Abcam, Cambridge, MA, USA) and mouse monoclonal anti-GAPDH primary antibody (dilution 1:100; ab8245; Abcam) and at room temperature for 3 h, and then with rabbit anti-mouse-IgG (dilution 1:5,000; ab175743; Abcam) at room temperature for 1 h. An enhanced chemiluminescence kit (Pierce Chemical, Rockford, IL, USA) was then used for detection. The relative protein expression was analyzed by Image-Pro plus software 6.0 (Media Cybernetics, Inc., Bethesda, MD, USA) and presented as the density ratio versus GAPDH.
Transfection
Lipofectamine® 2000 (Invitrogen; Thermo Fisher Scientific) was used to perform cell transfection according to the manufacturer's protocol. For VEGF-C functional analysis, HepG2 cells were transfected with VEGF-C-specific small interfering (si)RNA or VEGF-C plasmid (Nlunbio, Changsha, China). For miR-101 functional analysis, HepG2 cells were transfected with scrambled miRNA as a negative control (NC), miR-101 mimics or a miR-101 inhibitor (Thermo Fisher Scientific).
Dual luciferase reporter assay
A Directed Mutagenesis kit (Stratagene, La Jolla, CA, USA) was used to generate a mutant (MUT) 3′-UTR of VEGF-C, according to the manufacturer's protocol. The wild-type (WT) or MUT 3′-UTR of VEGF-C were inserted into the psiCHECK™2 vector (Promega, Madison, WI, USA). For the luciferase reporter assay, HepG2 cells were cultured to ~60% confluence in a 24-well plate, and then transfected with psiCHECK™2-VEGF-C-3′-UTR or psiCHECK™2-mutant VEGF-C-3′-UTR vectors, with or without 100 nM miR-101 mimics. Following incubation for 48 h, a dual-luciferase reporter assay system (Promega) was used to determine the luciferase activity on a LD400 luminometer (Beckman Coulter, Fullerton, CA, USA). Renilla luciferase activity was normalized to firefly luciferase activity.
Cell migration and invasion assay
Cell migration and invasion assays were performed using Transwell® chambers (BD Biosciences, Frankin Lakes, NJ, USA). A cell suspension containing 5×105 cells/ml was prepared in serum-free media. For the cell migration assay, 300 µl of cell suspension was added into the upper Transwell® chamber. For the cell invasion assay, 300 µl of cell suspension was added into the upper Transwell® chamber pre-coated with Matrigel™ (BD Biosciences). Subsequently, 500 µl of DMEM with 10% FBS as the chemoattractant was added into the lower Transwell® chamber and the cells were incubated for 24 h. Subsequently, cells that did not migrate or invade through the pores were carefully removed using a cotton-tipped swab. The filters were fixed in 90% alcohol and stained with crystal violet. The cell number was determined in five randomly selected fields under an CX41 inverted microscope (Olympus, Tokyo, Japan) at 400X magnification.
Statistical analysis
All data are expressed as mean ± standard deviation. SPSS 17.0 software (SPSS, Inc., Chicago, IL, USA) was used to perform the statistical analysis. The differences were analyzed using one-way analysis of variance and P<0.05 was considered to indicate statistically significant differences.
Results
miR-101 is downregulated in HCC tissues and cell lines
The expression level of miR-101 was determined using RT-qPCR in HCC tissues and matched normal adjacent tissues. As shown in Fig. 1A, the expression level of miR-101 was significantly reduced in HCC tissues compared with that in matched adjacent normal tissues. As shown in Fig. 1B, miR-101 was also downregulated in HCC cell lines compared with the normal liver cell line THLE-3. Furthermore, HepG2 cells exhibited the most significant decrease in miR-101 expression. Accordingly, HepG2 cells were used for subsequent investigations.
Figure 1.

Reverse transcription quantitative polymerase chain reaction analysis was performed to examine the determine expression of miR-101 in (A) HCC tissues and matched normal adjacent tissues (**P<0.01 vs. normal); and in (B) human HCC cell lines HepG2, LH86, LMH and PLHC-1, and the normal liver cell line THLE-3 (**P<0.01 vs. THLE-3). HCC, hepatocellular carcinoma; miR, microRNA.
VEGF-C is a target gene of miR-101
As described above, bioinformatics analysis suggested that VEGF-C is a potential target of miR-101. To verify this hypothesis, WT and MUT VEGF-C 3′-UTR were generated as shown in Fig. 2A. The luciferase reporter assay was subsequently performed to confirm whether miR-101 was able to directly bind to their seed sequences in the VEGF-C 3′-UTR in HepG2 cells. As shown in Fig. 2B, luciferase activity was significantly reduced in cells co-transfected with the WT VEGF-C 3′-UTR and miR-101 mimics; however, the luciferase activity was not different in cells co-transfected with the MUT VEGF-C 3′UTR and miR-101 mimics compared with the control group. These findings indicate that VEGF-C is a direct target of miR-101 in HepG2 cells.
Figure 2.

Seed sequences and luciferase report assay data. (A) Seed sequences of miR-101 in the WT or MUT 3′-UTR of VEGF-C are indicated. (B) Luciferase report assay data demonstrated that co-transfection of HepG2 cells with miR-101 and WT VEGF-C 3′-UTR caused a significant decrease in luciferase activity, whereas co-transfection with MUT VEGF-C 3′-UTR and miR-101 mimics exhibited no difference compared with the control group (cells co-transfected with blank vector and WT VEGF-C 3′-UTR or MUT VEGF-C 3′-UTR). **P<0.01 vs. the control group. WT, wild-type; MUT, mutant; 3′-UTR, 3′-untranslated region; miR, microRNA; VEGF-C, vascular endothelial growth factor-C; NC, negative control.
VEGF-C expression is downregulated by miR-101 at the post-transcriptional level
We further investigated the role of miR-101 in the regulation of VEGF-C expression in HepG2 HCC cells. Following transfection of HepG2 cells with scrambled miR, miR-101 mimics or a miR-101 inhibitor, the expression level of miR-101 was first determined in each group, and the transfection efficiency was confirmed to be satisfactory (Fig. 3A). Subsequently, the protein level of VEGF-C in each group was determined using western blot analysis. As shown in Fig. 3B, the upregulation of miR-101 induced a decrease in the protein expression of VEGF-C, while knockdown of miR-101 promoted VEGF-C protein expression in HepG2 cells. Therefore, the protein expression of VEGF-C was downregulated by miR-101 in HepG2 HCC cells.
Figure 3.

RT-qPCR and western blot analysis. (A) RT-qPCR was performed to determine the relative expression of miR-101 and (B) western blot analysis was performed to determine the protein level of VEGF-C in HepG2 cells transfected with NC scrambled miR, miR-101 mimics and a miR-101 inhibitor. GAPDH was used as an internal reference. Control, HepG2 cells without any transfection. **P<0.01 vs. control. RT-qPCR, reverse transcription quantitative polymerase chain reaction; miR, microRNA; VEGF-C, vascular endothelial growth factor-C; NC, negative control.
miR-101 suppresses HCC cell migration through targeting VEGF-C
The roles of miR-101 and VEGF-C in the regulation of HCC cell migration were further investigated. The findings demonstrated that transfection with miR-101 mimics or VEGF-C siRNA notably suppressed the migration of HepG2 cells. However, the suppressive effect of miR-101 overexpression on HepG2 cell migration was significantly reversed by the upregulation of VEGF-C (Fig. 4), suggesting that miR-101 inhibits HepG2 cell migration via directly targeting VEGF-C.
Figure 4.

The Transwell® assay was performed to determine the migration capacity of HepG2 cells transfected with miR-101 mimics, VEGF-C siRNA, or co-transfected with miR-101 mimics and VEGF-C plasmid. Control, HepG2 cells without any transfection. **P<0.01 vs. control. miR, microRNA; VEGF-C, vascular endothelial growth factor-C; siRNA, small interfering RNA.
miR-101 suppresses HCC cell invasion through targeting VEGF-C
The roles of miR-101 and VEGF-C in the regulation of HCC cell invasion were further investigated. It was demonstrated that transfection with miR-101 mimics or VEGF-C siRNA notably suppressed the invasion of HepG2 cells. However, the suppressive effect of miR-101 overexpression on HepG2 cell invasion was significantly reversed by the upregulation of VEGF-C (Fig. 5), suggesting that miR-101 inhibits HepG2 cell invasion via directly targeting VEGF-C.
Figure 5.

The Transwell® assay was performed to determine the invasion capacity of HepG2 cells transfected with miR-101 mimics, VEGF-C siRNA, or co-transfected with miR-101 mimics and VEGF-C plasmid. Control, HepG2 cells without any transfection. **P<0.01 vs. control. miR, microRNA; VEGF-C, vascular endothelial growth factor-C; siRNA, small interfering RNA.
VEGF-C is upregulated in HCC tissues
The expression levels of VEGF-C mRNA and protein were determined using RT-qPCR and western blot analysis, respectively, in HCC tissues and matched normal adjacent tissues. As shown in Fig. 6A and B, the mRNA and protein expression of VEGF-C was significantly increased in HCC tissues compared with that in matched adjacent normal tissues.
Figure 6.
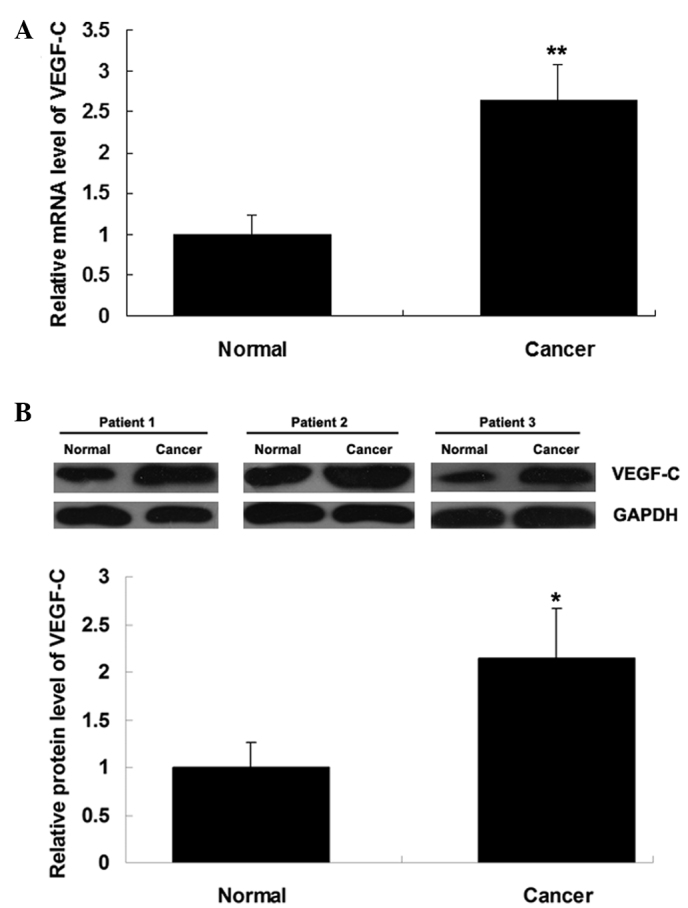
VEGF-C mRNA and protein expression. (A) Reverse transcription quantitative polymerase chain reaction analysis was performed to determine the relative mRNA expression and (B) western blot assay was performed to determine the relative protein expression of VEGF-C in hepatocellular carcinoma tissues and matched normal adjacent tissues. GAPDH was used as the internal reference for western blotting; three representative results are shown. *P<0.05 and **P<0.01 vs. normal. VEGF-C, vascular endothelial growth factor-C.
Discussion
The present study demonstrated that the expression of miR-101 was markedly reduced in HCC tissues and cell lines. VEGF-C was identified as a novel target gene of miR-101, and the protein expression of VEGF-C was downregulated by miR-101 in HCC cells. The investigation of the underlying molecular mechanism revealed that miR-101 inhibited HCC cell migration and invasion, at least in part via direct inhibition of VEGF-C protein expression. Finally, VEGF-C expression was found to be significantly upregulated in HCC tissues.
miRNAs are frequently deregulated in malignant tumors. The expression of miRNAs such as miR-124 and miR-203 was previously found to be reduced in HCC, and restoration of their expression significantly inhibited HCC cell growth (14). In the present study, we investigated the expression of miR-101, which has been reported to be downregulated in several types of cancer (8,15), and found that it was reduced in HCC tissues and cell lines. Su et al (16) investigated the expression of 308 miRNAs in human HCC and normal hepatic tissues and identified 29 differentially expressed miRNAs, including miR-101; they found that miR-101 was downregulated in HCC tissues, which was consistent with our findings, and suggested that miR-101 induces HCC cell apoptosis via directly targeting myeloid cell leukemia-1 (16). As the differential expression of miR-101 suggests that it may be involved in HCC development, we further investigated the regulation of miR-101 in HCC cells and observed that restoration of miR-101 expression significantly inhibited HCC cell migration and invasion. Sheng et al (17) also suggested that miR-101 is involved in the regulation of HCC cell migration. However, the molecular mechanism underlying the effect of miR-101 on HCC cell migration and invasion remains to be fully elucidated.
In this study, VEGF-C was identified as a novel target of miR-101, and it was indicated that VEGF-C may be involved in the effect of miR-101 on HCC cell migration and invasion. VEGF-C is a member of the VEGF family, which plays an important role in angiogenesis via affecting endothelial cell proliferation and motility and vascular permeability (18). VEGFs are commonly expressed in aggressive cancers and significantly affect the prognosis of cancer patients (12). In addition to its angiogenic role in endothelial cells (19), VEGF-C also promotes lymphangiogenesis through VEGFR-2 and −3 (20). With respect to the role of VEGF-C in malignant tumors, VEGF-C is associated with lymph node metastasis in several human malignancies, including prostate cancer, melanoma, gastric adenocarcinoma and esophageal squamous cell carcinoma (21–26). Knockdown of VEGF-C expression was found to significantly inhibit cancer metastasis (27,28), higher VEGFR-2 and −3 expression levels were found to be associated with a higher risk of lymph node metastasis and poor prognosis of HCC, whereas inhibition of VEGFR-2 and −3 were associated with reduced HCC growth and metastasis (13,29,30). In this study, the overexpression of miR-101 notably reduced the protein level of VEGF-C and suppressed HCC cell migration and invasion, suggesting that VEGF-C also acts through a non-lymphangiogenic mechanism to promote the progression of HCC. Further investigation should focus on the downstream VEGF-C/VEGFR-2 and VEGF-C/VEGFR-3 axes that promote tumor cell mobility.
In conclusion, miR-101 expression profiling was conducted in human HCC tissues and cells to identify the targets of abnormally expressed miR-101. The findings suggest that miR-101 exerts an inhibitory effect on HCC cell migration and invasion, at least in part by inhibiting the protein expression of its target, VEGF-C. These findings may contribute to the development of molecular-targeted therapies based on miRNAs.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 3.Bartel DP. MicroRNAs: Genomics, biogenesis, mechanism and function. Cell. 2004;116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 4.Baer C, Claus R, Plass C. Genome-wide epigenetic regulation of miRNAs in cancer. Cancer Res. 2013;73:473–477. doi: 10.1158/0008-5472.CAN-12-3731. [DOI] [PubMed] [Google Scholar]
- 5.Wang L, Li L, Guo R, Li X, Lu Y, Guan X, Gitau SC, Wang L, Xu C, Yang B, et al. miR-101 promotes breast cancer cell apoptosis by targeting Janus kinase 2. Cell Physiol Biochem. 2014;34:413–422. doi: 10.1159/000363010. [DOI] [PubMed] [Google Scholar]
- 6.Konno Y, Dong P, Xiong Y, Suzuki F, Lu J, Cai M, Watari H, Mitamura T, Hosaka M, Hanley SJ, et al. MicroRNA-101 targets EZH2, MCL-1 and FOS to suppress proliferation, invasion and stem cell-like phenotype of aggressive endometrial cancer cells. Oncotarget. 2014 doi: 10.18632/oncotarget.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liang X, Liu Y, Zeng L, Yu C, Hu Z, Zhou Q, Yang Z. miR-101 inhibits the G1-to-S phase transition of cervical cancer cells by targeting Fos. Int J Gynecol Cancer. 2014;24:1165–1172. doi: 10.1097/IGC.0000000000000187. [DOI] [PubMed] [Google Scholar]
- 8.Lei Q, Shen F, Wu J, Zhang W, Wang J, Zhang L. MiR-101, downregulated in retinoblastoma, functions as a tumor suppressor in human retinoblastoma cells by targeting EZH2. Oncol Rep. 2014;32:261–269. doi: 10.3892/or.2014.3167. [DOI] [PubMed] [Google Scholar]
- 9.Guo F, Cogdell D, Hu L, Yang D, Sood AK, Xue F, Zhang W. MiR-101 suppresses the epithelial-to-mesenchymal transition by targeting ZEB1 and ZEB2 in ovarian carcinoma. Oncol Rep. 2014;31:2021–2028. doi: 10.3892/or.2014.3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu Y, An Y, Wang Y, Zhang C, Zhang H, Huang C, Jiang H, Wang X, Li X. miR-101 inhibits autophagy and enhances cisplatin-induced apoptosis in hepatocellular carcinoma cells. Oncol Rep. 2013;29:2019–2024. doi: 10.3892/or.2013.2338. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, Guo X, Xiong L, Kong X, Xu Y, Liu C, Zou L, Li Z, Zhao J, Lin N. MicroRNA-101 suppresses SOX9-dependent tumorigenicity and promotes favorable prognosis of human hepatocellular carcinoma. FEBS Lett. 2012;586:4362–4370. doi: 10.1016/j.febslet.2012.10.053. [DOI] [PubMed] [Google Scholar]
- 12.Goel HL, Mercurio AM. VEGF targets the tumour cell. Nat Rev Cancer. 2013;13:871–882. doi: 10.1038/nrc3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou S, Tan C, Dai Z, Zhu H, Xu M, Zhou Z, Wang W, Zhao Y, Fu X, Zhou J, et al. Tacrolimus enhances the invasion potential of hepatocellular carcinoma cells and promotes lymphatic metastasis in a rat model of hepatocellular carcinoma: Involvement of vascular endothelial growth factor-C. Transplant Proc. 2011;43:2747–2754. doi: 10.1016/j.transproceed.2011.06.040. [DOI] [PubMed] [Google Scholar]
- 14.Furuta M, Kozaki KI, Tanaka S, Arii S, Imoto I, Inazawa J. miR-124 and miR-203 are epigenetically silenced tumor-suppressive microRNAs in hepatocellular carcinoma. Carcinogenesis. 2010;31:766–776. doi: 10.1093/carcin/bgp250. [DOI] [PubMed] [Google Scholar]
- 15.Lin X, Guan H, Li H, Liu L, Liu J, Wei G, Huang Z, Liao Z, Li Y. miR-101 inhibits cell proliferation by targeting Rac1 in papillary thyroid carcinoma. Biomed Rep. 2014;2:122–126. doi: 10.3892/br.2013.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Su H, Yang JR, Xu T, Huang J, Xu L, Yuan Y, Zhuang SM. MicroRNA-101, down-regulated in hepatocellular carcinoma, promotes apoptosis and suppresses tumorigenicity. Cancer Res. 2009;69:1135–1142. doi: 10.1158/0008-5472.CAN-08-2886. [DOI] [PubMed] [Google Scholar]
- 17.Sheng Y, Li J, Zou C, Wang S, Cao Y, Zhang J, Huang A, Tang H. Downregulation of miR-101-3p by hepatitis B virus promotes proliferation and migration of hepatocellular carcinoma cells by targeting Rab5a. Arch Virol. 2014;159:2397–2410. doi: 10.1007/s00705-014-2084-5. [DOI] [PubMed] [Google Scholar]
- 18.Lee HS, Jun JH, Jung EH, Koo BA, Kim YS. Epigalloccatechin-3-gallate inhibits ocular neovascularization and vascular permeability in human retinal pigment epithelial and human retinal microvascular endothelial cells via suppression of MMP-9 and VEGF activation. Molecules. 2014;19:12150–12172. doi: 10.3390/molecules190812150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao Y, Linden P, Farnebo J, Cao R, Eriksson A, Kumar V, Qi JH, Claesson-Welsh L, Alitalo K. Vascular endothelial growth factor C induces angiogenesis in vivo. Proc Natl Acad Sci USA. 1998;95:14389–14394. doi: 10.1073/pnas.95.24.14389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joukov V, Pajusola K, Kaipainen A, et al. A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J. 1996;15:1751. [PMC free article] [PubMed] [Google Scholar]
- 21.Kostis G, Ioannis L, Helen K, Helen P. The expression of vascular endothelial growth factor-C correlates with lymphatic microvessel density and lymph node metastasis in prostate carcinoma: An immunohistochemical study. Urol Ann. 2014;6:224–230. doi: 10.4103/0974-7796.134275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peppicelli S, Bianchini F, Calorini L. Inflammatory cytokines induce vascular endothelial growth factor-C expression in melanoma-associated macrophages and stimulate melanoma lymph node metastasis. Oncol Lett. 2014;8:1133–1138. doi: 10.3892/ol.2014.2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L, Li HG, Wen JM, Peng TS, Zeng H, Wang LY. Expression of CD44v3, erythropoietin and VEGF-C in gastric adenocarcinomas: correlations with clinicopathological features. Tumor. 2014;100:321–327. doi: 10.1700/1578.17216. [DOI] [PubMed] [Google Scholar]
- 24.Omoto I, Matsumoto M, Okumura H, Uchikado Y, Setoyama T, Kita Y, Owaki T, Kijima Y, Shinchi H, Ishigami S, et al. Expression of vascular endothelial growth factor-C and vascular endothelial growth factor receptor-3 in esophageal squamous cell carcinoma. Oncol Lett. 2014;7:1027–1032. doi: 10.3892/ol.2014.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang ZS, Xu YF, Huang FF, Ding GF. Associations of nm23H1, VEGF-C and VEGF-3 receptor in human prostate cancer. Molecules. 2014;19:6851–6862. doi: 10.3390/molecules19056851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karaman S, Detmar M. Mechanisms of lymphatic metastasis. J Clin Invest. 2014;124:922–928. doi: 10.1172/JCI71606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang H, Yin Y, Zhang L, et al. The effects of vascular endothelial growth factor C knockdown in esophageal squamous cell carcinoma. J Cancer Res Clin Oncol. 2012;138:133–139. doi: 10.1007/s00432-011-1079-9. [DOI] [PubMed] [Google Scholar]
- 28.Chaudary N, Milosevic M, Hill RP. Suppression of vascular endothelial growth factor receptor 3 (VEGFR3) and vascular endothelial growth factor C (VEGFC) inhibits hypoxia-induced lymph node metastases in cervix cancer. Gynecol Oncol. 2011;123:393–400. doi: 10.1016/j.ygyno.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 29.Zhao ZC, Zheng SS, Wan YL, Jia CK, Xie HY. The molecular mechanism underlying angiogenesis in hepatocellular carcinoma: The imbalance activation of signaling pathways. Hepatobiliary Pancreat Dis Int. 2003;2:529–536. [PubMed] [Google Scholar]
- 30.Zhuang PY, Shen J, Zhu XD, et al. Prognostic roles of cross-talk between peritumoral hepatocytes and stromal cells in hepatocellular carcinoma involving peritumoral VEGF-C, VEGFR-1 and VEGFR-3. PLoS One. 2013;8:e64598. doi: 10.1371/journal.pone.0064598. [DOI] [PMC free article] [PubMed] [Google Scholar]