

Abstract
Hepatocyte growth factor (HGF) has been implicated in epithelial-mesenchymal transition (EMT) in numerous types of cancer. However, to the best of our knowledge, there has been no previous evidence that HGF has a role in prostate cancer. The present study aimed to investigate the effect of HGF on EMT and invasive potential, as well as the underlying molecular mechanisms, in a human prostate cancer cell line. Therefore, PC-3 cells were treated with various concentrations of HGF for varying durations. EMT-associated proteins, including E-cadherin and vimentin, were examined by western blot analysis. The effects of HGF on cell proliferation, migration, invasion and tumorigenicity were assessed using MTT, wound-healing, Transwell and soft-agar assays. Subsequently, the role of c-Met in the mediation of EMT-like changes was investigated using reverse transcription-polymerase chain reaction, western blot analysis and gene knockdown by small interfering RNA. Finally, western blot analysis was used to quantify the expression of a downstream transcription factor and extracellular signal-related kinase/mitogen activated protein kinase (ERK/MAPK) signaling pathway proteins. The results indicated that treatment with HGF induced EMT-like changes and enhanced the invasive potential of PC-3 cells. There was an increase in the expression of ERK, phosphorylated-ERK and zinc finger E-box binding homeobox-1 (Zeb-1), suggesting that EMT-like changes may be mediated through the ERK/MAPK and Zeb-1 signaling pathway. Furthermore, HGF-mediated EMT-like changes were associated with c-Met activation, and these changes were able to be blocked by c-Met knockdown. The present study demonstrated that HGF-induced EMT increased the invasive potential of PC-3 human prostate cancer cells through activating the ERK/MAPK and Zeb-1 signaling pathway.
Keywords: hepatocyte growth factor, epithelial-mesenchymal transition, prostate cancer, extracellular signal-related kinase/mitogen activated protein kinase signaling pathway, zinc finger E-box binding homeobox
Introduction
Worldwide, prostate cancer (PCa) is the second most commonly diagnosed type of cancer and sixth leadinwg cause of cancer-associated mortality among males (1). Mortality associated with PCa results from distant metastasis, particularly to bone. Specifically, ~80% of patients with PCa succumb to bone metastasis, and up to 80% of patients with PCa exhibit bone metastasis at autopsy (2,3). However, the mechanisms underlying the metastasis of PCa remain to be elucidated.
In recent years, the epithelial-mesenchymal transition (EMT) has been established as a regulator of tumor aggressiveness (4). EMT was originally identified during embryogenesis, where it was described as a crucial process involved in differentiation and morphogenesis (5). EMT has additionally been attributed to tumor progression and metastasis (6). During EMT, cancer cells lose epithelial characteristics and acquire mesenchymal properties, including fibroblastoid morphology, characteristic changes in gene expression and increased motility. Simultaneously, the cells develop characteristics of cancer stem cells (7). These changes promote cancer cell invasiveness, metastasis and resistance to chemotherapy (8–10).
Numerous factors induce EMT, including transforming growth factor-β (TGF-β), epidermal growth factor (EGF), fibroblast growth factor (FGF), hepatocyte growth factor (HGF), platelet-derived growth factor, insulin-like growth factor (IGF) (11), hypoxia (11,12) and micro RNA (13). These factors induce EMT via various signaling pathways, including Wnt, Hedgehog and Notch (14,15).
In the present study, the association between HGF and EMT in prostate cancer was investigated. Previous studies have reported that higher plasma levels of HGF are associated with advanced stage and poor prognosis in patients with prostate cancer (16,17). This may be mediated by the promotion of EMT by HGF in cancer cells. However, the mechanisms by which HGF induces EMT remain unclear. The present study utilized the PC-3 human prostate cancer cell line as an experimental model. PC-3 cells are negative for EMT (18–21) and positive for c-Met expression (22). The present study investigated the effects of HGF on the EMT and invasive potential of PC-3 cells. Furthermore, the potential signaling pathways mediating this effect were investigated.
Materials and methods
Cell culture and treatment
PC-3 cells (American Type Culture Collection, Manassas, VA, USA) were maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco Life Technologies, Carlsbad, CA, USA) supplemented with 10% (v/v) fetal bovine serum (FBS; Gibco Life Technologies) and incubated at 37°C in an atmosphere containing 5% CO2. Cells were treated with recombinant human HGF (Sigma-Aldrich, St. Louis, MO, USA) at various concentrations (20, 40 and 60 ng/ml) over varying time-periods (12, 24 and 36 h) following overnight starvation.
Cell transfection
c-Met small interfering RNA (siRNA) or control siRNA plasmids (Santa Cruz Biotechnology Inc., Dallas, TX, USA) were transfected into PC-3 cells using Lipofectamine® 2000 transfection reagent (Invitrogen Life Technologies, Carlsbad, CA, USA). Stable transfectants were selected in 10 mg/ml puromycin (Life Technologies, Grand Island, NY USA) 24 h following transfection. Subsequently, the selection medium was replaced every 3 days. Following 2 weeks of selection, resistant clones were isolated. Cells were treated with recombinant human HGF as described above.
MTT assay
The PC-3 cells (5×103/0.2 ml) were plated in 96-well plates and stimulated with HGF (60 ng/ml) for 0, 24, 48 or 72 h. The cultures were incubated with 5 mg/ml MTT (Sigma-Aldrich)for 4 h. The metabolic product was then dissolved in 200 µl buffered dimethyl sulfoxide (Sigma-Aldrich), and the absorbance at 570 nm was measured with a Bio-Rad microplate reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Western blot analysis
Cells were lysed using Extraction and Quantification ProteoJET Mammalian Cell Lysis Reagent (MBI Fermentas, Ontario, Canada) with a protease inhibitors (Roche Diagnostics, Basel, Switzerland). Total protein concentration was estimated using the BCA method (Pierce Biotechnology Inc., Rockford, IL, USA). A total of 30 µg clarified protein lysate was electrophoretically resolved by denaturing 12% SDS-PAGE and electrotransferred onto nitrocellulose membranes (Bio-Rad Laboratories, Inc.). The immunoblots were incubated in 3% bovine serum albumin (Sijiqing Biotech Co. Ltd., Hanzhou, China), 10 mM Tris-hydrochloride (pH 7.5), 1 mM EDTA and 0.1% Tween-20 (Sigma-Aldrich) at room temperature and probed for 1.5 h with appropriate primary antibodies, polyclonal rabbit anti-human c-Met (1:200), phosphorylated-c-Met (p-c-Met; 1:200), anti-human E-cadherin, zinc finger E-box binding homeobox-1 (Zeb-1; 1:150) and extracellular signal-related kinase (ERK; 1:200); monoclonal mouse anti-human vimentin (1:300) and phosphorylated ERK (p-ERK; 1:200; Santa Cruz Biotechnology, Inc.) at various dilutions. The membranes were then incubated for 1 h with secondary antibodies, horseradish peroxidase-conjugated goat anti-rabbit (1:500) and goat anti-mouse (1:200) immunoglobulin G (Boshide Biotech Co. Ltd., Kaohsiung City, Taiwan). Monoclonal mouse anti-human GAPDH antibody (1:10,000 dilution; Santa Cruz Biotechnology, Inc.) was used as the internal control. Blots were imaged using enhanced chemiluminescence detection system (Pierce Biotechnology, Inc.).
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated from cells using TRIzol reagent (Invitrogen Life Technologies). The isolated RNA was reverse-transcribed into complementary DNA (cDNA) using oligo (dT) primers and Avian Myeloblastosis Virus Reverse Transcriptase (Takara Bio, Inc., Shiga, Japan). cDNA (10 µl) was used as a template for PCR in a final reaction volume of 50 µl. Invitrogen primers were obtained from Thermofisher Scientific, Inc. (Carlsbad, CA, USA): The human C-Met primers (sense, 5′-GTTTCCCAATTTCTGACC-3′ and antisense, 5′-TATATCAAAGGTGTTTAC-3′) generated a 516 bp product. The β-actin primers (sense, 5′-TGGGCATGGGTCAGAAGGAT-3′ and antisense, 5′-AAGCATTTGCGGTGGACGAT-3′) generated a product of 991 bp. The DNA amplification conditions were as follows: An initial denaturation step at 95°C for 5 min, 30 cycles at 95°C for 30 sec, 60°C for 30 sec, and 72°C for 40 sec, and a final elongation step at 72°C for 7 min. The RT-PCR samples were electrophoresed on 1.5% agarose gel and stained with ethidium bromide (0.5 µg/ml); Sigma-Aldrich). Images of the gels were then captured using an ultraviolet transillumination system (Liuyi Biotech Co. Ltd., Beijing, China).
In vitro wound-healing assay
Cells were seeded in 6-well plates and grown to 60–70% confluence. Cells were then incubated in Gibco serum-free medium (Thermofisher Scientifi, Inc.) overnight and treated with HGF (60 ng/ml). Prior to the addition of HGF, 2-mm scratches were made in the confluent cell monolayer with a 200-µl pipette tip. Cell migration into the denuded area was assessed 12 and 24 h following treatment using a Type CK2 optical microscope (Olympus Corporation, Tokyo, Japan).
In vitro Transwell invasion assay
Polycarbonate filters (8 µm; EMD Millipore, Billerica, MA, USA) were coated with 50 µg/cm2 reconstituted Matrigel (Sigma-Aldrich). Cells (5×103) were seeded into the upper chamber in 300 µl serum-free growth medium. Cells were incubated under normoxic conditions and allowed to migrate toward the complete growth medium for 24 and 48 h. Non-invading cells were removed mechanically using cotton swabs and cells on the lower surface were subsequently counted microscopically.
Soft agar assay
Cells were resuspended in 2 ml top agar medium (DMEM containing 0.4% low-melting agarose and 10% FBS; Sigma-Aldrich) and then rapidly overlaid on 2 ml bottom agar medium (DMEM containing 0.8% low-melting agarose and 10% FBS) in 6-well culture plates. Following 2–3 weeks of incubation, colonies >0.1 mm in diameter were scored as positive. Colony-formation efficiency was evaluated using a Type CK2 optical microscope (Olympus Corporation).
Statistical analysis
All values are expressed as the mean ± standard deviation of at least three independent experiments. Statistical analysis was performed using Student's t-test. P<0.05 was considered to indicate a statistically significant difference.
Results
HGF induces EMT-like changes in PC-3 cells. Characteristic changes associated with EMT include downregulation of epithelial markers and upregulation of mesenchymal markers. These changes are associated with the scattered growth of cancer cells, enabling cell-cell dissociation, migration and motility (23). In the present study, western blot analysis revealed that HGF treatment downregulated E-cadherin expression and upregulated vimentin expression in PC-3 cells in a time- and dose-dependent manner. PC-3 cells acquired stable, EMT-like changes following incubation with HGF (60 ng/ml) for 36 h (Fig. 1A). These EMT-like changes were not observed at other time-points or HGF concentrations. The changes lasted for 7 days following withdrawal of HGF (Fig. 1B). These results indicate that HGF promotes reversible changes in the expression of EMT markers in PC-3 cells. Based on these results, PC-3 cells were treated with 60 ng/ml HGF for 36 h in all subsequent experiments.
Figure 1.
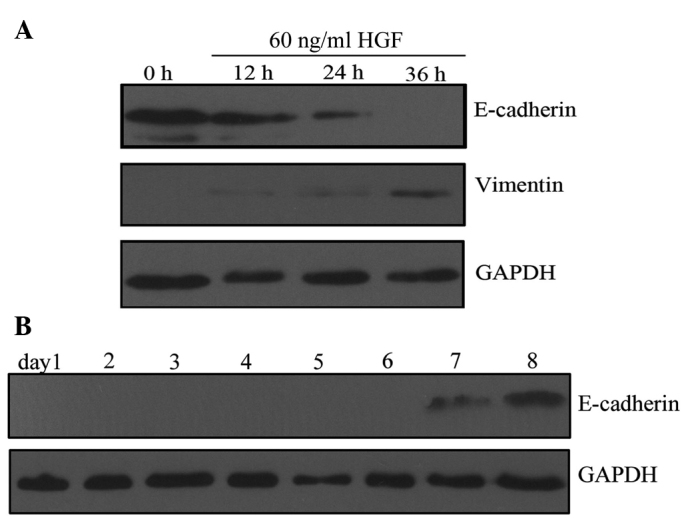
HGF induces epithelial-mesenchymal transition-like changes in PC-3 cells. (A) HGF (60 ng/ml) treatment downregulates E-cadherin and upregulates vimentin in a time-dependent manner, compared with untreated PC-3 cells (control). Changes were not observed with treatemnet with 20 or 40 ng/ml HGF. (B) E-cadherin expression was restored 7 days subsequent to the withdrawal of HGF. HGF, hepatocyte growth factor.
c-Met expression is enhanced following HGF treatment
To investigate the role of HGF in inducing EMT-like changes in PC-3 cells, messenger RNA (mRNA) and protein expression levels of c-Met, the receptor for HGF, were measured. RT-PCR analysis demonstrated an upregulation of c-Met transcription following HGF treatment for 36 h (Fig. 2A). Furthermore, c-Met was activated by HGF-mediated phosphorylation (p-c-Met) and activated c-Met is able to regulate various downstream target genes. Western blot analysis indicated that HGF treatment increased the expression of c-Met and p-c-Met (Fig. 2B). Together, these results suggest that HGF upregulates c-Met at the mRNA and protein levels.
Figure 2.
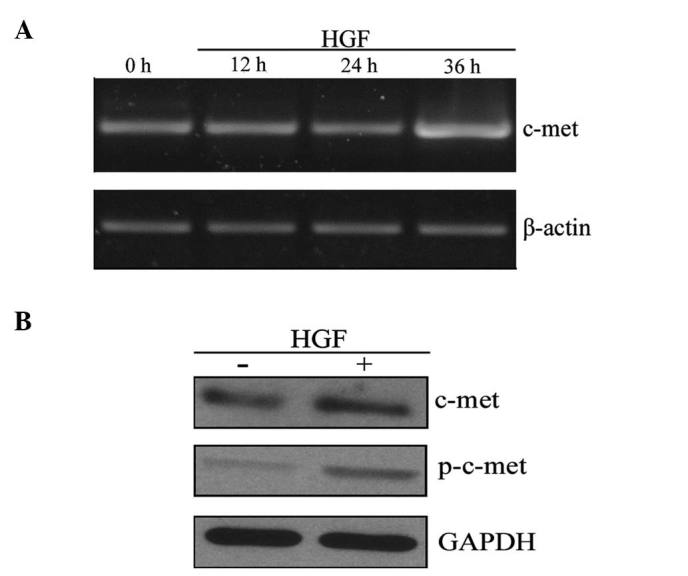
HGF treatment increases the expression of c-Met. PC-3 cells were treated with 60 ng/ml HGF, and c-Met expression was measured at the (A) messenger RNA level (treatment for 0, 12, 24 and 36 h) and (B) protein level (treatment for 36 h). HGF, hepatocyte growth factor; p, phosphorylated.
HGF treatment increases the invasive potential of PC-3 cells
The effect of HGF treatment on the invasive potential of PC-3 cells was examined. An MTT assay demonstrated that HGF treatment increased cancer cell proliferation and doubling time reduced (Fig. 3A). In addition, HGF treatment increased the number of tumor colonies that developed in the soft-agar assay (Fig. 3B). In the wound-healing assay, HGF-treated PC-3 cells demonstrated increased migratory capacity compared with that of the untreated cells (Fig. 3C). In the Transwell assay, HGF-treated cells displayed increased invasion beneath the insert surface and through the collagen matrix (Fig. 3D and E). Taken together, these results demonstrate that HGF increased the invasive potential of PC-3 cells.
Figure 3.

HGF enhances cell proliferation, tumorigenicity, migration and invasion. (A) HGF stimulated PC-3 cell proliferation in the MTT assay. (B) HGF-treated PC-3 cells exhibited increased tumorigenicity in the soft-agar assay (t=2.773; *P<0.05 vs. HGF). (C) HGF-treated cells demonstrate increased migration potential in the wound-healing assay. (D and E) In contrast to control PC-3 cells, HGF-treated cells displayed significantly increased invasiveness in the Transwell assay (t=2.481 and 2.532; P<0.05 vs. HGF at *24 and **48 h, respectively). Magnification, ×400. Data are expressed as the mean ± standard deviation. HGF, hepatocyte growth factor; OD, optical density.
ERK/mitogen activated protein kinase (MAPK) signaling is involved in HGF-induced EMT
To investigate the molecular mechanism underlying HGF-induced EMT, the changes in ERK/MAPK expression levels following HGF incubation were measured. Western blot analysis indicated that HGF treatment increased the expression levels of ERK and p-ERK. In addition, HGF treatment increased the expression of Zeb-1, a direct suppressor of E-cadherin. Thus, the ERK/MAPK signaling pathway was involved in HGF-induced EMT (Fig. 4).
Figure 4.
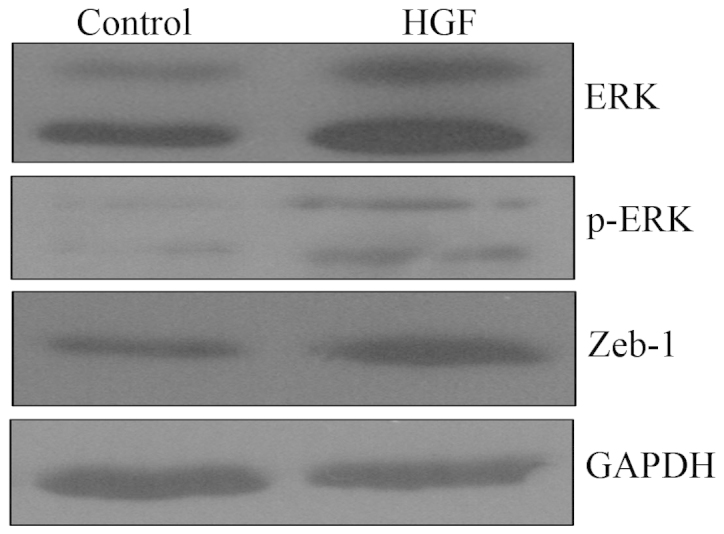
ERK/mitogen activated protein kinase signaling is involved in HGF-induced epithelial-mesenchymal transition. PC-3 cells were treated with HGF (60 ng/ml) for 36 h, and ERK, p-ERK and Zeb-1 expression levels were examined by western blotting. HGF, hepatocyte growth factor; ERK, extracellular signal-related kinase; Zeb-1, zinc finger E-box binding homeobox-1; p, phosphorylated.
c-Met siRNA inhibits HGF-induced EMT-like changes
The effect of c-Met knockdown by siRNA on HGF-induced EMT-like changes was assessed. Transfection with c-Met siRNA inhibited c-Met expression in PC-3 cells as compared with untreated PC-3 cells and cells treated with control siRNA (Fig. 5A). Following incubation with HGF, PC-3 cells and cells treated with control siRNA exhibited downregulation of E-cadherin and upregulation of vimentin as compared with cells treated with c-Met siRNA. This demonstrated the role of c-Met in mediating HGF-induced EMT-like changes (Fig. 5B). There were similar changes observed in the ERK/MAPK and Zeb-1 signaling pathways. HGF treatment upregulated ERK, p-ERK and Zeb-1 in PC-3 cells and cells treated with control siRNA, however, this was not observed in cells treated with c-Met siRNA (Fig. 5C). Together, these data suggest that HGF induces EMT-like changes in a c-Met-dependent manner.
Figure 5.
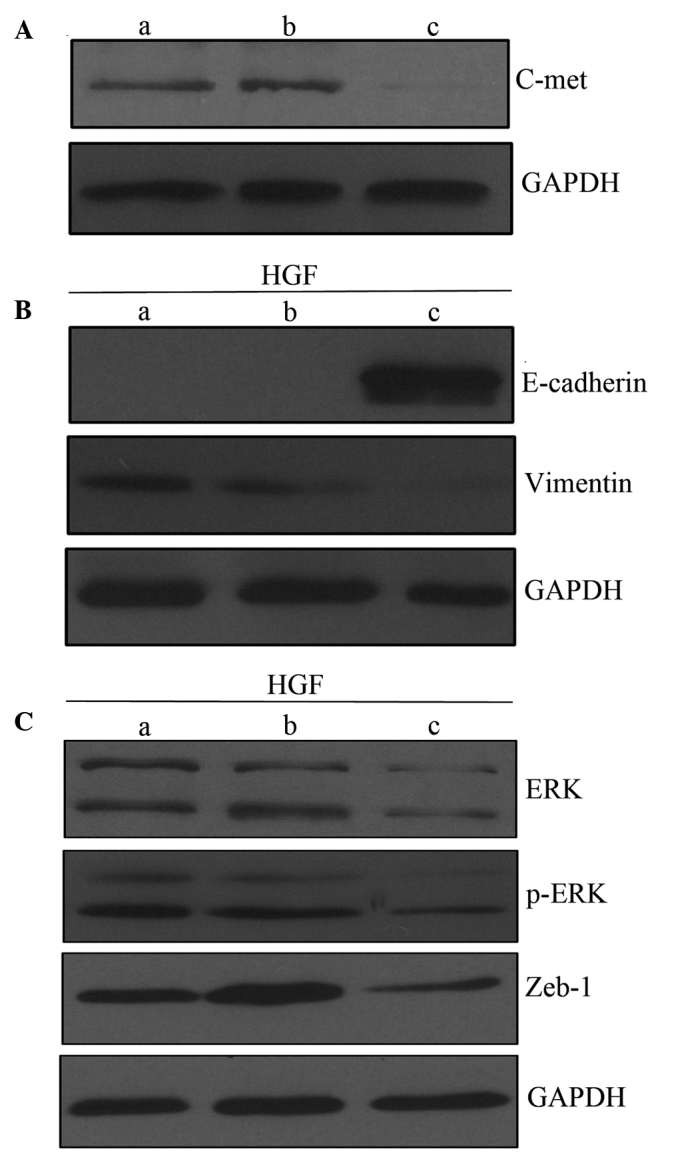
Role of c-Met in HGF-induced EMT. (A) Western blot analysis of c-Met expression. (B) c-Met knockdown inhibits EMT-like changes induced by HGF (60 ng/ml), compared with untreated PC-3 cells and cells treated with control siRNA. (C) HGF treatment upregulates ERK, p-ERK and Zeb-1 in PC-3 cells and cells treated with control siRNA cells, but not in cells treated with c-Met siRNA. Lanes: a, PC-3 cells; b, cells treated with control siRNA; and c, cells treated with c-Met siRNA. EMT, epithelial-mesenchymal transition; siRNA, small interferingRNA; HGF, hepatocyte growth factor; ERK, extracellular signal-related kinase; Zeb-1, zinc finger E-box binding homeobox-1; p, phosphorylated.
Discussion
HGF binds to its receptor, c-Met, and activates it through auto-phosphorylation, which induces the transcription of downstream target genes. Under normal physiological conditions, the HGF/c-Met signaling pathway regulates tissue and organ regeneration. Furthermore, HGF is significant in the modulation of cell morphology, and induction of angiogenesis and lymphangiogenesis (24).
Previous studies have indicated that HGF stimulates proliferation, migration and invasion in numerous types of cancer, including colon, stomach, lung, bladder and prostate cancer (17). For example, HGF levels are elevated in the serum of patients with prostate cancer. Furthermore, elevated HGF levels are associated with metastatic disease independent of prostate-specific antigen levels or age, and are associated with a decrease in overall survival rate (25,26). In addition, Duhon et al (27) reported that HGF treatment of DU145 prostate tumor cells stimulated the phosphoinositide 3-kinase (PI3K) and MAPK signaling pathways, leading to increased cell scattering, motility and invasion. These effects were prevented by treatment with epigallocatechin-3-gallate.
Although HGF accelerates the progression of prostate cancer, the underlying mechanisms remain to be elucidated. The association between HGF and EMT has been demonstrated in various cancer models (28,29). However to the best of our knowledge, no such association has previously been reported in prostate cancer. One study demonstrated that HGF induced EMT in DU145 cells (30); however, DU145 cells are EMT-positive (18,31,32). Therefore, the present study investigated the effect of HGF on EMT induction in PC-3 cells.
Typical characteristics of EMT include downregulation of epithelial markers, for example E-cadherin, and upregulation of mesenchymal markers, including vimentin, N-cadherin and α-smooth muscle actin (33,34). In particular, downregulation of E-cadherin is a key step in the induction of EMT (35). Intercellular adhesions are critical for maintaining the epithelial phenotype, and since E-cadherin is essential for adherent junctions, downregulation results in the loss of cell polarity and abnormal differentiation, thus facilitating EMT (9,36).
In the present study, treatment of PC-3 cells with HGF resulted in EMT-like changes, as indicated by the downregulation of E-cadherin and upregulation of vimentin. Thus, HGF induced an EMT-like phenotype in PC-3 cells in a time- and concentration-dependent manner. Further studies indicated that HGF stimulation increased the proliferation, migration, invasion and tumorigenicity of cancer cells. The EMT-like changes were reversible following withdrawal of HGF for 7 days, which was similar to the EMT phenotype induced by TGF-β1 (37). These results suggested that growth factors are required to maintain the EMT phenotype. Numerous growth factors, including FGF, IGF, TGF-β and HGF, are secreted from stromal cells (38). Under continued stimulation from these growth factors, cancer cells acquire a stable EMT phenotype. Therefore, the results of the present study demonstrate the bidirectional interaction and co-evolution of tumors and their stroma in cancer progression.
The effect of HGF on the expression of its receptor c-Met, at the mRNA and protein levels, was investigated. c-Met overexpression has been identified in the majority of human cancers (39,40). In the present study, c-Met expression was promoted by HGF-dependent transcriptional upregulation. This result is consistent with the findings of Boccaccio and Comoglio (41) regarding prostate cancer. Notably, in the present study, there was a marked elevation in p-c-Met following HGF treatment, demonstrating that HGF activates c-Met in prostate cancer cells. Knockdown of c-Met by siRNA prevented HGF-induced EMT-like changes. These results demonstrate that HGF induced EMT in a c-Met-dependent manner in PC-3 cells.
Various oncogenic effects of HGF and c-Met are mediated by a complex downstream signaling network, most prominently the MAPK and PI3K/Akt signaling pathways (42). In the present study, ERK was phosphorylated by HGF, and PC-3 cells expressed high basal levels of p-ERK and ERK. These changes were blocked by c-Met knockdown using siRNA. These data suggested that the functional expression of ERK is significant in HGF-induced EMT in PC-3 cells. A comparable effect was observed in HGF-induced EMT in hepatocellular cancer (43).
The present study demonstrated that HGF upregulated Zeb-1 in PC-3 cells. As with other zinc finger transcription factors, including SNAIL and SLUG, Zeb-1 has been linked to E-cadherin repression (44). Repression of E-cadherin enhances the ability of cancer cells to migrate to distant sites (45). HGF interacts with early growth response factor-1 through the MAPK signaling pathway, which binds to the Snail promoter, leading to rapid induction and execution of EMT (46). Another study revealed that the Zeb gene was activated upon activation of SNAIL (47). SW480 colorectal cancer cells possess a mesenchyme-like phenotype, which is characterized by loosely attached cells that lack membranous E-cadherin. Silencing of Zeb-1 by siRNA resulted in a cellular phenotype resembling the mesenchymal-epithelial transition (48). The results of the present study are consistent with the above-mentioned studies, and demonstrate the role of Zeb-1 in EMT in prostate cancer.
In conclusion, the results of the present study revealed that HGF directly promotes EMT and carcinogenic properties in prostate cancer via the ERK signaling pathway. Specific molecular targeting of this signaling pathway may provide therapeutic benefit in patients exhibiting prostate cancer.
Acknowledgements
The present study was supported by the National Natural Science Foundation of China (grant nos. 30700968 and 81341066).
References
- 1.Jin JK, Dayyani F, Gallick GE. Steps in prostate cancer progression that lead to bone metastasis. Int J Cancer. 2011;128:2545–2561. doi: 10.1002/ijc.26024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jacobs SC. Spread of prostatic cancer to bone. Urology. 1983;21:337–344. doi: 10.1016/0090-4295(83)90147-4. [DOI] [PubMed] [Google Scholar]
- 3.Shah RB, Mehra R, Chinnaiyan AM, Shen R, Ghosh D, Zhou M, Macvicar GR, Varambally S, Harwood J, Bismar TA, et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: Lessons from a rapid autopsy program. Cancer Res. 2004;64:9209–9216. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- 4.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 5.Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-mesenchymal transitions: The importance of changing cell state in development and disease. J Clin Invest. 2009;119:1438–1449. doi: 10.1172/JCI38019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 7.Scheel C, Weinberg RA. Cancer stem cells and epithelial-mesenchymal transition: Concepts and molecular links. Semin Cancer Biol. 2012;22:396–403. doi: 10.1016/j.semcancer.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One. 2008;3:e2888. doi: 10.1371/journal.pone.0002888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsuoka J, Yashiro M, Doi Y, Fuyuhiro Y, Kato Y, Shinto O, Noda S, Kashiwagi S, Aomatsu N, Hirakawa T, et al. Hypoxia stimulates the EMT of gastric cancer cells through autocrine TGFβ signaling. PLoS One. 2013;8:e62310. doi: 10.1371/journal.pone.0062310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luo Y, He DL, Ning L, Shen SL, Li L, Li X, Zhau HE, Chung LW. Over-expression of hypoxia-inducible factor-1alpha increases the invasive potency of LNCaP cells in vitro. BJU Int. 2006;98:1315–1319. doi: 10.1111/j.1464-410X.2006.06480.x. [DOI] [PubMed] [Google Scholar]
- 13.Lamouille S, Subramanyam D, Blelloch R, Derynck R. Regulation of epithelial-mesenchymal and mesenchymal-epithelial transitions by microRNAs. Curr Opin Cell Biol. 2013;25:200–207. doi: 10.1016/j.ceb.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 15.Chung LW, Huang WC, Sung SY, Wu D, Odero-Marah V, Nomura T, Shigemura K, Miyagi T, Seo S, Shi C, et al. Stromal-epithelial interaction in prostate cancer progression. Clin Genitourin Cancer. 2006;5:162–170. doi: 10.3816/CGC.2006.n.034. [DOI] [PubMed] [Google Scholar]
- 16.Hashem M, Essam T. Hepatocyte growth factor as a tumor marker in the serum of patients with prostate cancer. J Egypt Natl Canc Inst. 2005;17:114–120. [PubMed] [Google Scholar]
- 17.Yasuda K, Nagakawa O, Akashi T, Fujiuchi Y, Koizumi K, Komiya A, Saiki I, Fuse H. Serum active hepatocyte growth factor (AHGF) in benign prostatic disease and prostate cancer. Prostate. 2009;69:346–351. doi: 10.1002/pros.20890. [DOI] [PubMed] [Google Scholar]
- 18.Luo Y, He DL, Ning L. Expression of ‘epithelial-mesenchymal transition’ associated proteins in prostate cancer cell lines with different metastatic potentials and its significance. Zhonghua Nan Ke Xue. 2006;12:696–700. (In Chinese) [PubMed] [Google Scholar]
- 19.Gu X, Zerbini LF, Otu HH, Bhasin M, Yang Q, Joseph MG, Grall F, Onatunde T, Correa RG, Libermann TA. Reduced PDEF expression increases invasion and expression of mesenchymal genes in prostate cancer cells. Cancer Res. 2007;67:4219–4226. doi: 10.1158/0008-5472.CAN-06-3689. [DOI] [PubMed] [Google Scholar]
- 20.Veveris-Lowe TL, Lawrence MG, Collard RL, Bui L, Herington AC, Nicol DL, Clements JA. Kallikrein 4 (hK4) and prostate-specific antigen (PSA) are associated with the loss of E-cadherin and an epithelial-mesenchymal transition (EMT)-like effect in prostate cancer cells. Endocr Relat Cancer. 2005;12:631–643. doi: 10.1677/erc.1.00958. [DOI] [PubMed] [Google Scholar]
- 21.Whitbread AK, Veveris-Lowe TL, Lawrence MG, Nicol DL, Clements JA. The role of kallikrein-related peptidases in prostate cancer: Potential involvement in an epithelial to mesenchymal transition. Biol Chem. 2006;387:707–714. doi: 10.1515/BC.2006.089. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Yue D, Li K, Liu YL, Ren CS, Wang P. The role of TRPC6 in HGF-induced cell proliferation of human prostate cancer DU145 and PC3 cells. Asian J Androl. 2010;12:841–852. doi: 10.1038/aja.2010.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang J, Weinberg RA. Epithelial-Mesenchymal Transition: At the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 24.Martin TA, Mason MD, Jiang WG. Hepatocyte growth factor signaling in cancer metastasis. Curr Signal Transduct Ther. 2011;6:180–190. doi: 10.2174/157436211795659991. [DOI] [Google Scholar]
- 25.Naughton M, Picus J, Zhu X, Catalona WJ, Vollmer RT, Humphrey PA. Scatter factor-hepatocyte growth factor elevation in the serum of patients with prostate cancer. J Urol. 2001;165:1325–1328. doi: 10.1016/S0022-5347(01)69893-8. [DOI] [PubMed] [Google Scholar]
- 26.Humphrey PA, Halabi S, Picus J, Sanford B, Vogelzang NJ, Small EJ, Kantoff PW. Prognostic significance of plasma scatter factor/hepatocyte growth factor levels in patients with metastatic hormone-refractory prostate cancer: Results from cancer and leukemia group B 150005/9480. Clin Genitourin Cancer. 2006;4:269–274. doi: 10.3816/CGC.2006.n.006. [DOI] [PubMed] [Google Scholar]
- 27.Duhon D, Bigelow RL, Coleman DT, Steffan JJ, Yu C, Langston W, Kevil CG, Cardelli JA. The polyphenol epigallocatechin-3-gallate affects lipid rafts to block activation of the c-Met receptor in prostate cancer cells. Mol Carcinog. 2010;49:739–749. doi: 10.1002/mc.20649. [DOI] [PubMed] [Google Scholar]
- 28.Previdi S, Maroni P, Matteucci E, Broggini M, Bendinelli P, Desiderio MA. Interaction between human-breast cancer metastasis and bone microenvironment through activated hepatocyte growth factor/Met and b-catenin/Wnt pathways. Eur J Cancer. 2010;46:1679–1691. doi: 10.1016/j.ejca.2010.02.036. [DOI] [PubMed] [Google Scholar]
- 29.Ogunwobi OO, Liu C. Hepatocyte growth factor upregulation promotes carcinogenesis and epithelial-mesenchymal transition in hepatocellularcarcinoma via Akt and COX-2 pathways. Clin Exp Metastasis. 2011;28:721–731. doi: 10.1007/s10585-011-9404-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fram ST, Wells CM, Jones GE. HGF-induced DU145 cell scatter assay. Methods Mol Biol. 2011;769:31–40. doi: 10.1007/978-1-61779-207-6_3. [DOI] [PubMed] [Google Scholar]
- 31.Baritaki S, Huerta-Yepez S, Sahakyan A, Karagiannides I, Bakirtzi K, Jazirehi A, Bonavida B. Mechanisms of nitric oxide-mediated inhibition of EMT in cancer: Inhibition of the metastasis-inducer Snail and induction of the metastasis-suppressor RKIP. Cell Cycle. 2010;9:4931–4940. doi: 10.4161/cc.9.24.14229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yates CC, Shepard CR, Stolz DB, Wells A. Co-culturing human prostate carcinoma cells with hepatocytes leads to increased expression of E-cadherin. Br J Cancer. 2007;96:1246–1252. doi: 10.1038/sj.bjc.6603700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang J, Weinberg RA. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 34.Cano A, Perez-Moreno MA, Rodrigo I, Locasioa A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 35.Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 2004;303:1483–1487. doi: 10.1126/science.1094291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shimada S, Mimata A, Sekine M, Mogushi K, Akiyama Y, Fukamachi H, Jonkers J, Tanaka H, Eishi Y, Yuasa Y. Synergistic tumour suppressor activity of E-cadherin and p53 in a conditional mouse model for metastatic diffuse-type gastric cancer. Gut. 2012;61:344–353. doi: 10.1136/gutjnl-2011-300050. [DOI] [PubMed] [Google Scholar]
- 37.Odero-Marah VA, Wang R, Chu G, Zayzafoon M, Xu J, Shi C, Marshall FF, Zhau HE, Chung LW. Receptor activator of NF-kappaB Ligand (RANKL) expression is associated with epithelial to mesenchymal transition in human prostate cancer cells. Cell Rese. 2008;18:858–870. doi: 10.1038/cr.2008.84. [DOI] [PubMed] [Google Scholar]
- 38.Chung LW, Baseman A, Assikis V, Zhau HE. Molecular insights into prostate cancer progression: The missing link of tumor microenvironment. J Urol. 2005;173:10–20. doi: 10.1097/01.ju.0000141582.15218.10. [DOI] [PubMed] [Google Scholar]
- 39.Jiang WG, Davies G, Martin TA, Parr C, Watkins G, Mason MD, Mokbel K, Mansel RE. Targeting matrilysin and its impact on tumor growth in vivo: The potential implications in breast cancer therapy. Clin Cancer Res. 2005;11:6012–6019. doi: 10.1158/1078-0432.CCR-05-0275. [DOI] [PubMed] [Google Scholar]
- 40.You WK, McDonald DM. The hepatocyte growth factor/c-Met signaling pathway as a therapeutic target to inhibit angiogenesis. BMB Rep. 2008;41:833–839. doi: 10.5483/BMBRep.2008.41.12.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boccaccio C, Comoglio PM. Invasive growth: A MET-driven genetic programme for cancer and stem cells. Nat Rev Cancer. 2006;6:637–645. doi: 10.1038/nrc1912. [DOI] [PubMed] [Google Scholar]
- 42.Migliore C, Giordano S. Molecular cancer therapy: Can our expectation be MET? Eur J Cancer. 2008;44:641–651. doi: 10.1016/j.ejca.2008.01.022. [DOI] [PubMed] [Google Scholar]
- 43.Ogunwobi OO, Liu C. Hepatocyte growth factor upregulation promotes carcinogenesis and epithelial-mesenchymal transition in hepatocellular carcinoma via Akt and COX-2 pathways. Clin Exp Metastasis. 2011;28:721–731. doi: 10.1007/s10585-011-9404-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nieto MA, Cano A. The epithelial - mesenchymal transition under control: Global programs to regulate epithelial plasticity. Semin Cancer Biol. 2012;22:361–368. doi: 10.1016/j.semcancer.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 45.Kwok WK, Ling MT, Lee TW, Lau TC, Zhou C, Zhang X, Chua CW, Chan KW, Chan FL, Glackin C, et al. Upregulation of TWIST in prostate cancer and its implication as a therapeutic target. Cancer Res. 2005;65:5153–5162. doi: 10.1158/0008-5472.CAN-04-3785. [DOI] [PubMed] [Google Scholar]
- 46.Grotegut S, von Schweinitz D, Christofori G, Lehembre F. Hepatocyte growth factor induces cell scattering through MAPK/Egr-1-mediated upregulation of Snail. EMBO J. 2006;25:3534–3545. doi: 10.1038/sj.emboj.7601213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peña C, García JM, García V, Silva J, Domínguez G, Rodríguez R, Maximiano C, de Herreros García A, Muñoz A, Bonilla F. The expression levels of the transcriptional regulators p300 and CtBP modulate the correlations between SNAIL, ZEB1, E-cadherin and vitamin D receptor in human colon carcinomas. Int J Cancer. 2006;119:2098–2104. doi: 10.1002/ijc.22083. [DOI] [PubMed] [Google Scholar]
- 48.Spaderna S, Schmalhofer O, Hlubek F, Berx G, Eger A, Merkel S, Jung A, Kirchner T, Brabletz T. A transient, EMT-linked loss of basement membranes indicates metastasis and poor survival in colorectal cancer. Gastroenterology. 2006;131:830–840. doi: 10.1053/j.gastro.2006.06.016. [DOI] [PubMed] [Google Scholar]