

Abstract
The metalloid arsenic is a worldwide environmental toxicant, exposure to which is associated with many adverse outcomes. Arsenic is also an effective therapeutic agent in certain disease settings. Arsenic was recently shown to regulate the activity of the Hedgehog (HH) signal transduction pathway, and this regulation of HH signaling was proposed to be responsible for a subset of arsenic’s biologic effects. Surprisingly, these separate reports proposed contradictory activities for arsenic, as either an agonist or antagonist of HH signaling. Here we provide in vitro and in vivo evidence that arsenic acts as a modulator of the activity of the HH effector protein glioma-associated oncogene family zinc finger (GLI), activating or inhibiting GLI activity in a context-dependent manner. This arsenic-induced modulation of HH signaling is observed in cultured cells, patients with colorectal cancer who have received arsenic-based therapy, and a mouse colorectal cancer xenograft model. Our results show that arsenic activates GLI signaling when the intrinsic GLI activity is low but inhibits signaling in the presence of high-level GLI activity. Furthermore, we show that this modulation occurs downstream of primary cilia, evidenced by experiments in suppressor of fused homolog (SUFU) deficient cells. Combining our findings with previous reports, we present an inclusive model in which arsenic plays dual roles in GLI signaling modulation: when GLIs are primarily in their repressor form, arsenic antagonizes their repression capacity, leading to low-level GLI activation, but when GLIs are primarily in their activator form, arsenic attenuates their activity.
Introduction
Arsenic is a widespread environmental toxicant that poses a significant threat to human health (Rahman et al., 2009). Chronic exposure to arsenic has been linked to increased risk of a variety of adverse human health outcomes, including cancer and developmental defects (National Research Council, 1999; International Agency for Research on Cancer, 2004). In contrast, formulations of arsenic have previously exhibited efficacy against a variety of human diseases, such as syphilis, agues, malaria, and leukemia (Au, 2011). Arsenic trioxide (ATO) is currently approved by the U.S. Food and Drug Administration for the treatment of acute promyelocytic leukemia (Mi, 2011). Despite the long history of human exposure to and usage of arsenic, the exact mechanisms by which arsenic exerts these biologic effects remain obscure.
The Hedgehog (HH) signaling pathway is one of the primary signal transduction pathways that govern the rapid growth and patterning of vertebrate embryos and regulate adult tissue homeostasis (Ingham and McMahon, 2001). Consistent with the important role that HH signaling plays during development and tissue homeostasis, deregulation of HH signaling leads to an array of developmental disorders and many types of cancer (Ruiz i Altaba, 2006; Jiang and Hui, 2008; Barakat et al., 2010). The HH family of ligands bind to their primary cellular receptor Patched1 (PTCH1) to initiate signaling (Robbins and Hebrok, 2007). Upon HH binding, PTCH1 releases its inhibitory effect on Smoothened (SMO), which subsequently triggers a series of cellular events that regulate the stability, proteolytic processing, and activity of the glioma-associated oncogene family zinc finger (GLI) family of transcription factors (GLI1–GLI3) (Robbins and Hebrok, 2007; Ruiz i Altaba et al., 2007). In the absence of HH, the full-length forms of GLI2 and GLI3 (GLI-FL) are processed into truncated repressor forms (GLI-R). In the presence of HH, this partial proteolysis is attenuated and instead GLI-FL is converted into its activated form (GLI-A). GLI trafficking through primary cilia is required for the conversion of GLI-FL to either its GLI-R or GLI-A forms (Robbins et al., 2012).
We previously suggested that HH pathway activation underlies the etiology of some arsenic-induced diseases (Fei et al., 2010). We showed that both short-term and chronic arsenic exposure activates HH signaling in primary cells, established cell lines, and certain tissues of exposed mice. We further showed that arsenic exposure levels are positively associated with HH pathway activity in human bladder cancer, a malignancy previously linked to chronic arsenic exposure (Fei et al., 2010). More recently, other reports have shown that arsenic inhibits HH signaling initiated by either HH treatment or high GLI levels, and researchers concluded that arsenic is a functional inhibitor of HH/GLI signaling (Kim et al., 2010, 2013; Beauchamp et al., 2011; Han et al., 2013; Nakamura et al., 2013; Yang et al., 2013; You et al., 2014). Here, we provide in vitro and in vivo evidence suggesting that arsenic can both activate and inhibit GLI signaling, with the direction of GLI modulation dictated primarily by the starting levels of intrinsic GLI activity. Furthermore, we provide a unifying model that explains how arsenic acts mechanistically to modulate GLI activity.
Materials and Methods
Cell Culture, Reagents, and Assays.
NIH-3T3 cells (American Type Culture Collection, Manassas, VA) and the colorectal cancer (CRC) cell lines SW480, DLD-1, and HCT116 (gifts from Dr. Ethan Lee, Vanderbilt University, Nashville, TN) were cultured as previously described (Li et al., 2014b). ATO (Trisenox; Cephalon, Frazer, PA) was used at the indicated concentrations or combined with recombinant HH (rHH C25II; R&D Systems, Minneapolis, MN) on cultured cells. RNA extraction, quantitative reverse-transcription polymerase chain reaction (qRT-PCR), luciferase reporter assays, and plasmids expressing MYC-tagged human GLI1 or GLI2 were previously described (Fei et al., 2010; Li et al., 2014a). MYC-tagged GLI3-R (expressing truncated GLI3, which lacks the GLI transcriptional activation domain) was a gift from Dr. Ariel Ruiz i Altaba (University of Geneva Medical School, Geneva, Switzerland).
Mouse Experiments.
Animal experiments were conducted in accordance with protocols approved by the University of Miami Institutional Animal Care and Use Committee. HCT116 cells (2 × 106) were injected subcutaneously onto the left flank area of 4- to 5-week-old female nude mice (Crl:NU-Foxn1nu). When the xenograft tumor reached a size of approximately 200 mm3, the mice were randomized into two groups and treated by intraperitoneal injection with vehicle (phosphate-buffered saline) or 10 mg/kg of ATO. After 2 days of treatment, the tumors and kidneys were harvested and RNA was extracted for qRT-PCR analysis.
Patient Samples.
Details of the patient selection and treatment plan were previously described (ClinicalTrials.gov identifier NCT00449137) (Ardalan et al., 2010). Briefly, the enrolled patients harbored CRC at advanced metastatic stages and were refractory to at least two standard chemotherapy regimens including 5-fluorouracil. These patients were treated with ATO (0.15–0.20 mg/kg) for 2–10 treatment cycles, along with intermittent doses of 5-fluorouracil (Ardalan et al., 2010). Biopsies of CRC liver metastases and white blood cells (WBCs) were taken within 1 week before the start of ATO treatment, on day 23 of the first cycle, and after completion of the last treatment cycle in one patient (ID number 1). All fine-needle biopsies of CRC liver metastases were confirmed as malignant cells before further processing. Biopsies were stored in RNAlater (Ambion, Austin, TX) before total RNA isolation. WBCs were isolated as previously described (Subbarayan et al., 2002). Total RNA was isolated using TRIzol (Invitrogen, Carlsbad, CA) and purified using RNAeasy kits (Invitrogen), followed by gene expression analysis using qRT-PCR.
Statistical Analysis.
Experiments were independently performed multiple times. Statistical significance was determined by the t test unless otherwise indicated. In all analyses, a P value < 0.05 was considered statistically significant.
Results
Because previous reports of arsenic’s effect on GLI/HH signaling have been contradictory (Fei et al., 2010; Kim et al., 2010, 2013; Beauchamp et al., 2011), we re-examined this question using experimental systems capable of showing both increases and decreases in GLI activity. Because ultimately our interest is in the effects of arsenic on human pathologies, we first examined the effects of ATO on GLI target genes in patient specimens obtained as part of an ATO clinical trial for CRC (Ardalan et al., 2010). Needle biopsies of CRC liver metastases, along with cognate nontumorigenic WBCs, were taken from these patients before, during, and after termination (in one patient) of ATO treatment. The expression of GLI target genes was subsequently examined in these primary tissue samples. For the patient from whom we had all three tissue samples (patient ID number 1), ATO attenuated GLI target gene expression and did so in a manner that was reversible upon ATO withdrawal (Fig. 1A). In contrast, ATO treatment activated GLI target genes in WBCs from this same patient (Fig. 1B). Notably, these effects on GLI signaling by ATO were also reversible upon ATO withdrawal (Fig. 1, A and B). When we extended this analysis to the five patients in our cohort for which we had such tissue samples (Fig. 1C), the expression of GLI/HH biomarkers was downregulated in the majority of ATO-treated patient tumor samples (four of five patients for GLI1 and two of five patients for PTCH1), whereas PTCH1 was upregulated in WBCs in three of five patients. Although the small numbers of patient samples examined precludes a generalization of these results, we were able to observe distinct modulation of GLI/HH signaling in tumors and WBCs within the same patient. This differential modulation of HH signaling in vivo occurred in response to therapeutically relevant doses of arsenic (Shen et al., 1997). Interestingly, GLI1 expression levels were higher in the tumor samples than in the matching WBC samples (Supplemental Fig. 1), consistent with previous reports showing higher levels of GLI activity in CRC metastases (Varnat et al., 2009).
Fig. 1.

ATO modulates GLI activity in patients with CRC. (A and B) RNA was extracted from the tumor (A) or associated WBCs (B) of a patient with CRC who received ATO in the clinic. The expression of GLI target genes relative to that of GAPDH was determined by qRT-PCR. Error bars indicate the S.E. of triplicate experiments using one sample. Similar non-normalized data are shown in Supplemental Fig. 1. (C) Summary of GLI target gene expression in the tumors and associated WBCs of a group of patients with CRC who received ATO treatment. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Because we were unable to make statistically relevant conclusions regarding GLI modulation using our small cohort of arsenic-treated patients with CRC, we developed a CRC xenograft mouse model and examined the effect of ATO on GLI activity in the CRC and mouse tissue. HCT116 cells were used in this assay because this cell line, along with two other CRC cell lines, requires high levels of GLI activity (Varnat et al., 2009; Mazumdar et al., 2011a,b) for viability and is responsive to ATO in vitro (Fig. 2, A and B). HCT116 cells were implanted subcutaneously onto the flanks of immunocompromised nude mice. Once the tumors reached a size of approximately 200 mm3, mice were randomized to two treatment groups and were given daily doses of ATO (10 mg/kg) or vehicle for 2 days. Tumor tissues or mouse kidneys, which we previously showed to respond to arsenic by increasing GLI signaling (Fei et al., 2010), were harvested from these mice, and the expression of GLI1 and PTCH1 was determined by qRT-PCR. ATO treatment dramatically decreased the expression of GLI target genes in the CRC xenograft tumors (Fig. 2C). However, ATO also increased basal GLI activity in the kidneys of these same mice (Fig. 2D). Combined, these data suggest that ATO treatment can both decrease and increase GLI activity in the same experimental system, albeit in a tissue-dependent manner.
Fig. 2.

ATO modulates GLI signaling in a CRC xenograft mouse model. (A and B) CRC cells (DLD-1, SW480, and HCT116) were treated with the indicated concentrations of ATO for 48 hours, and GLI1 (A) or PTCH1 (B) expression relative to that of GAPDH was determined by qRT-PCR. (C and D) HCT116 cells (2 × 106) were subcutaneously implanted into nude mice. When the tumor size reached approximately 200 mm3, 10 mg/kg ATO or vehicle control was injected for 2 consecutive days. Tumor (C) and kidney samples (D) were then harvested for RNA extraction and the expression of GLI1 and PTCH1 relative to that of GAPDH was determined by qRT-PCR. Error bars represent the S.D. (n = 10). *P < 0.05 calculated using a Wilcoxon signed-rank test. Normalized data are shown here because of the inherent difficulty in comparing mouse expression levels with human expression levels. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
We hypothesized that ATO might modulate GLI activity in a manner dependent on the basal levels of GLI activity, which would be dictated by the GLI-R/GLI-A ratio (Ruiz i Altaba et al., 2007). To test this hypothesis, we examined the effects of ATO on GLI activity in NIH-3T3 fibroblasts, whose GLI signaling level can be modulated by HH ligands or SMO agonists (Fei et al., 2010; Kim et al., 2010). We treated NIH-3T3 cells with increasing doses of ATO in the absence of HH ligand and noted that ATO elevated the expression of GLI biomarkers (Fig. 3A, left), consistent with our previous report (Fei et al., 2010). However, when NIH-3T3 cells were treated with ATO in the presence of HH, which activates GLI-A but attenuates GLI-R activity, GLI1 and PTCH1 levels were attenuated in a dose-dependent manner (Fig. 3A, right). These results support the hypothesis that ATO can both activate and attenuate GLI signaling depending on its basal state; in this case, it does so in a single cell type. An alternative possibility is that cells with different GLI signaling levels may have differential rates of arsenic transport or methylation and thus production of arsenic metabolites, which may have distinct effects on GLI signaling. However, we tested the expression of the major arsenite transporters, AQP7 and AQP9, and arsenite methyltransferase, AS3MT, in wild-type (WT) mouse embryonic fibroblasts (MEFs) and did not find it to be significantly different (Supplemental Fig. 2) under conditions of low-level versus high-level GLI signaling.
Fig. 3.

ATO exerts context-dependent modulation of GLI signaling and does so independently of primary cilia. (A) NIH-3T3 cells were treated with the indicated dose of ATO with or without 1 µg/ml recombinant HH for 48 hours, and the expression of the indicated target genes relative to that of GAPDH was determined. (B) WT or SUFU−/− MEFs were treated with the indicated dose of ATO for 48 hours. The expression of GLI1 relative to that of GAPDH was then determined by qRT-PCR. (C) NIH-3T3 cells were treated with vehicle, 3 µM ATO, or 100 nM SAG for 6 hours, and PTCH1 expression relative to that of GAPDH was determined by qRT-PCR. (D and E) NIH-3T3 cells were treated with either 100 nM SAG or 3 µM ATO for 6 hours, and full-length GLI3 and GLI3-R levels were determined by immunoblotting. A representative immunoblot (D) and a quantification of multiple independent immunoblots (E) are shown. The top left panel (vehicle-treated NIH-3T3 cells or WT MEFs) in each figure was set as 1 for direct comparison in all figures. Error bars represent S.E.M (n = 3). *P < 0.05. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
We speculated that a unified mechanism to explain the seemingly conflicting effects of ATO on GLI activity might exist. One plausible hypothesis is that ATO targets the primary cilium, which is suggested to play dual roles in HH signaling (Han et al., 2009). Furthermore, one previous report indicated that ATO decreased the trafficking of GLI2 through primary cilia (Kim et al., 2010). We therefore tested the ability of ATO to attenuate HH signaling in MEFs lacking suppressor of fused homolog (SUFU), a pivotal negative regulator of HH signaling, whose increased levels of GLI activity are activated independently of primary cilia (Zhang et al., 2009). ATO attenuated the constitutive GLI activity of SUFU−/− MEFS, but activated GLI activity in WT MEFs (Fig. 3B). Therefore, ATO can attenuate GLI activity downstream of SMO, independently of primary cilia, inconsistent with the hypothesis that ATO attenuates GLI-A by affecting its trafficking through the primary cilium.
We previously reported that the levels of GLI3-R were decreased in cells treated chronically with ATO (Fei et al., 2010), suggesting that the agonist effect of ATO may come from the degradation of GLI3-R protein. However, we found here that the agonist effect of ATO could be observed as early as 6 hours (Fig. 3C), before any detectable GLI3-R degradation (Fig. 3, D and E). To further explore the relationship between ATO exposure and GLI activity, we took advantage of immortalized GLI2−/−;GLI3−/− MEFs that lack the major repressor and activator GLI forms and consequently do not respond to HH (Lipinski et al., 2008). We transfected plasmids expressing GLI1 or GLI2, the major GLI-A forms, into these cells to activate GLI signaling. This exogenous GLI activity was subsequently attenuated by ATO (Fig. 4A). We next investigated the mechanism of GLI activation by ATO using WT and GLI2−/−;GLI3−/− MEFs, validating that ATO’s activation of GLI signaling requires GLI2 and/or GLI3 (Fig. 4B) (Fei et al., 2010). To further assess the effect of ATO on the dominant GLI-R (GLI3), we used GLI3−/− MEFs. In these cells, the loss of GLI3-R activity results in a net increase in basal GLI activity (Lipinski et al., 2008), driven by unrepressed, basal GLI2 activity. Transfection of a plasmid expressing GLI3-R into these cells repressed PTCH1 expression to levels comparable with those of WT MEFs. ATO was capable of reversing this GLI3-R activity (Fig. 4C) and did so in a manner that did not decrease GLI3-R protein levels (Fig. 4D). This result is consistent with ATO acting directly to repress GLI3-R activity, with degradation of GLI3-R being a secondary effect of ATO subsequent to attenuation of its activity. Taken together, these results suggest that ATO is capable of attenuating both GLI-A and GLI-R activities, depending on which one is dominant in the cells.
Fig. 4.

ATO attenuates the activity of the activator and repressor forms of GLI proteins. (A) GLI2−/−;GLI3−/− MEFs were transfected with a plasmid expressing a GLI luciferase reporter gene with or without GLI1 or GLI2 expression plasmids and then treated with the indicated doses of ATO. Luciferase activity relative to total protein level was measured after 48 hours of treatment. The activity of a vehicle-treated and mock-transfected sample (left) was set as 1 for comparison. Error bars represent the S.E.M. (n = 3). *P < 0.05. (B) GLI2−/−;GLI3−/− or WT MEFs were transfected with a plasmid expressing a GLI luciferase reporter gene and then treated with the indicated doses of ATO. Luciferase activity relative to total protein level was then determined. The activity of a vehicle-treated sample (left, WT MEFs) was set as 1 for comparison. Error bars represent the S.E.M. (n = 3). *P < 0.05. (C) GLI3−/− MEFs were transfected with increasing amounts of a GLI3-R expressing plasmid and then treated with vehicle or 3 µM ATO. PTCH1 expression relative to that of GAPDH was measured as a readout of GLI activity. PTCH1 expression relative to that of GAPDH in WT MEFs was also determined as a control. Expression in a vehicle-treated, mock-transfected sample (left) was set as 1 for comparison. Error bars represent the S.E.M. (n = 3). *P < 0.05. (D) GLI3−/− MEFs were transfected with the indicated amounts of GLI3-R expression plasmids and then treated with vehicle or 3 µM ATO. GLI3-R (a long and short exposure) or GAPDH levels were revealed by immunoblotting. A representative result is shown. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Discussion
The mechanism and direction of modulation of GLI/HH signaling by arsenic has been controversial. We have previously shown, and also demonstrate here, that short and long-term exposure to arsenic activates GLI signaling. However, a number of other groups have argued that arsenic attenuates GLI signaling rather than activates it (Fei et al., 2010; Kim et al., 2010, 2013; Beauchamp et al., 2011; Han et al., 2013; Nakamura et al., 2013; Yang et al., 2013; You et al., 2014). We now show, in agreement with these previous groups, that arsenic can also antagonize GLI/HH signaling. Furthermore, we advance this work by showing that GLI activity can be activated or attenuated by arsenic in the same experimental systems, in vitro and in vivo. The disparate modulation of GLI activity we observe in these identical experimental systems is consistent with the modulation of GLI activity being context dependent, rather than resulting from experimental vagaries that can sometimes occur between different laboratories. Our NIH-3T3 data show that altering arsenic concentrations can both increase and decrease Gli signaling in a dose-dependent manner, depending on the presence or absence of HH. It is therefore unlikely that differential expression levels of arsenic transporters or arsenite methyltransferase is the primary mechanism responsible for the dual regulatory capacity of GLI/HH signaling by arsenic, at least in a single cell line. However, we cannot rule out that organic arsenic metabolites may be involved in the regulation of GLI activity by arsenic in more complicated systems such as human cancer patient samples, although inorganic arsenic has been reported to directly bind to GLI proteins (Beauchamp et al., 2011; Han et al., 2013), suggesting that inorganic arsenic is the main active arsenic species that modulates GLI activity.
Our results suggest that ATO acts as an unbiased GLI inhibitor, attenuating both its transcriptional activation and repressor activity. On the basis of results from a number of laboratories, including ours, we suggest that ATO attenuates GLI-A and GLI-R activity by binding directly to their zinc finger domains to disrupt their transcriptional regulatory functions. These effects occur downstream of primary cilia, on GLI proteins bound to the regulatory elements of their target genes. Alternatively, because arsenic has been implicated in global epigenetic modulation (Ren et al., 2011), ATO might regulate the activity of GLI target genes via epigenetic mechanisms. We do not favor this latter mechanism, because ATO is also capable of modulating the effects of exogenous GLI-A and GLI-R on a GLI-driven luciferase reporter gene, which should not be regulated by epigenetic mechanisms. We present here a unifying model to explain these cell context–specific responses to arsenic (Fig. 5). In cellular contexts with low GLI activity, GLI3-R plays a dominant role in repressing target gene transcription (Litingtung and Chiang, 2000; Pan et al., 2006) and ATO consequently binds to GLI3-R to attenuate this repression, which leads to increased GLI signaling. In cellular contexts harboring high GLI-A activity, ATO binds to GLI-A to attenuate GLI activity.
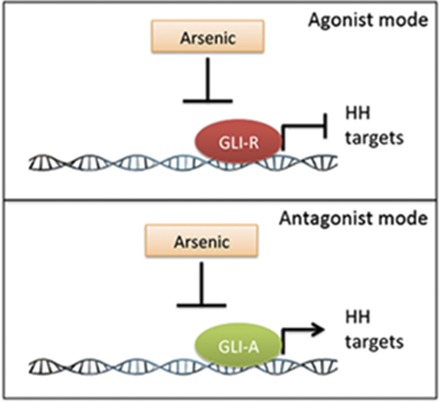
Fig. 5.

A schematic model of how ATO modulates GLI/HH signaling in different contexts. ATO suppresses the activity of GLI proteins in both their activator and repressor forms. In cells with a low level of HH signaling, arsenic inhibits the repressor forms of GLI and thus results in a net increase in basal HH signaling (agonist mode). In cells with high levels of HH signaling, arsenic serves as an inhibitor of the activator forms of GLI (antagonist mode) and therefore decreases HH signaling.
Supplementary Material
Acknowledgments
The authors thank the members of the Robbins’ laboratory and Drs. M. Karagas and E. Dmitrovsky for helpful discussions during the course of this study.
Abbreviations
- ATO
arsenic trioxide
- GLI
glioma-associated oncogene family zinc finger
- HH
Hedgehog signaling pathway
- MEF
mouse embryonic fibroblast
- PTCH1
primary cellular receptor Patched1
- qRT-PCR
quantitative reverse-transcription polymerase chain reaction
- SUFU
suppressor of fused homolog
- WBC
white blood cell
- WT
wild type
Authorship Contributions
Participated in research design: Li, Giambelli, Tang, Winterbottom, Z. Wang, Fei, Nguyen, Athar, Subbarayan, Rai, Ardalan, Capobianco, Robbins.
Conducted experiments: Li, Giambelli, Tang, Winterbottom, Long, Jin, Fei, Subbarayan.
Contributed new reagents or analytical tools: Giambelli, B. Wang.
Performed data analysis: Li, Giambelli, L. Wang, Capobianco, Robbins.
Wrote or contributed to the writing of the manuscript: Li, Winterbottom, Subbarayan, Capobianco, Robbins.
Footnotes
This research was supported by the National Institutes of Health National Institute of Environmental Health Sciences [Grant P01ES01817502], the National Institutes of Health National Institute of General Medical Sciences [Grant GM64011], and the National Institutes of Health National Cancer Institute [Grant 1R21CA117116-01]
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Ardalan B, Subbarayan PR, Ramos Y, Gonzalez M, Fernandez A, Mezentsev D, Reis I, Duncan R, Podolsky L, Lee K, et al. (2010) A phase I study of 5-fluorouracil/leucovorin and arsenic trioxide for patients with refractory/relapsed colorectal carcinoma. Clin Cancer Res 16:3019–3027. [DOI] [PubMed] [Google Scholar]
- Au WY. (2011) A biography of arsenic and medicine in Hong Kong and China. Hong Kong Med J 17:507–513. [PubMed] [Google Scholar]
- Barakat MT, Humke EW, Scott MP. (2010) Learning from Jekyll to control Hyde: Hedgehog signaling in development and cancer. Trends Mol Med 16:337–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchamp EM, Ringer L, Bulut G, Sajwan KP, Hall MD, Lee YC, Peaceman D, Ozdemirli M, Rodriguez O, Macdonald TJ, et al. (2011) Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J Clin Invest 121:148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei DL, Li H, Kozul CD, Black KE, Singh S, Gosse JA, DiRenzo J, Martin KA, Wang B, Hamilton JW, et al. (2010) Activation of Hedgehog signaling by the environmental toxicant arsenic may contribute to the etiology of arsenic-induced tumors. Cancer Res 70:1981–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JB, Sang F, Chang JJ, Hua YQ, Shi WD, Tang LH, Liu LM. (2013) Arsenic trioxide inhibits viability of pancreatic cancer stem cells in culture and in a xenograft model via binding to SHH-Gli. Onco Targets Ther 6:1129–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YG, Kim HJ, Dlugosz AA, Ellison DW, Gilbertson RJ, Alvarez-Buylla A. (2009) Dual and opposing roles of primary cilia in medulloblastoma development. Nat Med 15:1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham PW, McMahon AP. (2001) Hedgehog signaling in animal development: paradigms and principles. Genes Dev 15:3059–3087. [DOI] [PubMed] [Google Scholar]
- International Agency for Research on Cancer (2004) Some drinking-water disinfectants and contaminants, including arsenic, in IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, pp 37–270, IARC Press, Lyon, France. [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Hui CC. (2008) Hedgehog signaling in development and cancer. Dev Cell 15:801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Aftab BT, Tang JY, Kim D, Lee AH, Rezaee M, Kim J, Chen B, King EM, Borodovsky A, et al. (2013) Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell 23:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lee JJ, Kim J, Gardner D, Beachy PA. (2010) Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc Natl Acad Sci USA 107:13432–13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Fei DL, Flaveny CA, Dahmane N, Baubet V, Wang Z, Bai F, Pei XH, Rodriguez-Blanco J, Hang B, et al. (2014a) Pyrvinium attenuates Hedgehog signaling downstream of smoothened. Cancer Res 74:4811–4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Flaveny CA, Giambelli C, Fei DL, Han L, Hang BI, Bai F, Pei XH, Nose V, Burlingame O, et al. (2014b) Repurposing the FDA-approved pinworm drug pyrvinium as a novel chemotherapeutic agent for intestinal polyposis. PLoS One 9:e101969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski RJ, Bijlsma MF, Gipp JJ, Podhaizer DJ, Bushman W. (2008) Establishment and characterization of immortalized Gli-null mouse embryonic fibroblast cell lines. BMC Cell Biol 9:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litingtung Y, Chiang C. (2000) Specification of ventral neuron types is mediated by an antagonistic interaction between Shh and Gli3. Nat Neurosci 3:979–985. [DOI] [PubMed] [Google Scholar]
- Mazumdar T, DeVecchio J, Agyeman A, Shi T, Houghton JA. (2011a) The GLI genes as the molecular switch in disrupting Hedgehog signaling in colon cancer. Oncotarget 2:638–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumdar T, DeVecchio J, Shi T, Jones J, Agyeman A, Houghton JA. (2011b) Hedgehog signaling drives cellular survival in human colon carcinoma cells. Cancer Res 71:1092–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi J. (2011) Current treatment strategy of acute promyelocytic leukemia. Front Med 5:341–347. [DOI] [PubMed] [Google Scholar]
- Nakamura S, Nagano S, Nagao H, Ishidou Y, Yokouchi M, Abematsu M, Yamamoto T, Komiya S, Setoguchi T. (2013) Arsenic trioxide prevents osteosarcoma growth by inhibition of GLI transcription via DNA damage accumulation. PLoS One 8:e69466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Research Council (1999) Arsenic in Drinking Water, National Academy Press, Washington, DC. [Google Scholar]
- Pan Y, Bai CB, Joyner AL, Wang B. (2006) Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol Cell Biol 26:3365–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman MM, Ng JC, Naidu R. (2009) Chronic exposure of arsenic via drinking water and its adverse health impacts on humans. Environ Geochem Health 31 (Suppl 1):189–200. [DOI] [PubMed] [Google Scholar]
- Ren X, McHale CM, Skibola CF, Smith AH, Smith MT, Zhang L. (2011) An emerging role for epigenetic dysregulation in arsenic toxicity and carcinogenesis. Environ Health Perspect 119:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins DJ, Fei DL, Riobo NA. (2012) The Hedgehog signal transduction network. Sci Signal 5:re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins DJ, Hebrok M. (2007) Hedgehogs: la dolce vita. Workshop on Hedgehog-Gli Signaling in Cancer and Stem Cells. EMBO Rep 8:451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz i Altaba A. (2006) Hedgehog-Gli Signaling in Human Disease, 1st ed, Springer, New York. [Google Scholar]
- Ruiz i Altaba A, Mas C, Stecca B. (2007) The Gli code: an information nexus regulating cell fate, stemness and cancer. Trends Cell Biol 17:438–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, Zhu J, Tang W, Sun GL, Yang KQ, et al. (1997) Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood 89:3354–3360. [PubMed] [Google Scholar]
- Subbarayan PR, Sarkar M, Ardalan B. (2002) Isolation of genomic DNA from human whole blood. Biotechniques 33: 1231–1234. [DOI] [PubMed] [Google Scholar]
- Varnat F, Duquet A, Malerba M, Zbinden M, Mas C, Gervaz P, Ruiz i Altaba A. (2009) Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol Med 1:338–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Cao F, Ye X, Zhao H, Liu X, Li Y, Shi C, Wang H, Zhou J. (2013) Arsenic trioxide inhibits the Hedgehog pathway which is aberrantly activated in acute promyelocytic leukemia. Acta Haematol 130:260–267. [DOI] [PubMed] [Google Scholar]
- You M, Varona-Santos J, Singh S, Robbins DJ, Savaraj N, Nguyen DM. (2014) Targeting of the Hedgehog signal transduction pathway suppresses survival of malignant pleural mesothelioma cells in vitro. J Thorac Cardiovasc Surg 147:508–516. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Shi Q, Chen Y, Yue T, Li S, Wang B, Jiang J. (2009) Multiple Ser/Thr-rich degrons mediate the degradation of Ci/Gli by the Cul3-HIB/SPOP E3 ubiquitin ligase. Proc Natl Acad Sci USA 106:21191–21196. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.