

Abstract
Small molecule inhibitors targeting the mitogen-activated protein kinase pathway (Braf/mitogen-activated protein kinase kinase/extracellular signal-regulated kinase) have had success in extending survival for patients with metastatic melanoma. Unfortunately, resistance may occur via cross-activation of alternate signaling pathways. One approach to overcome resistance is to simultaneously target the phosphoinositide 3-kinase/mammalian target of rapamycin signaling pathway. Recent reports have shown that GSK2126458 [2,4-difluoro-N-(2-methoxy-5-(4-(pyridazin-4-yl)quinolin-6-yl)pyridin-3-yl) benzenesulfonamide], a dual phosphoinositide 3-kinase/mammalian target of rapamycin inhibitor, can overcome acquired resistance to Braf and mitogen-activated protein kinase kinase inhibitors in vitro. These resistance mechanisms may be especially important in melanoma brain metastases because of limited drug delivery across the blood–brain barrier. The purpose of this study was to investigate factors that influence the brain distribution of GSK2126458 and to examine the efficacy of GSK2126458 in a novel patient-derived melanoma xenograft (PDX) model. Both in vitro and in vivo studies indicate that GSK2126458 is a substrate for P-glycoprotein (P-gp) and breast cancer resistance protein (Bcrp), two dominant active efflux transporters in the blood–brain barrier. The steady-state brain distribution of GSK2126458 was 8-fold higher in the P-gp/Bcrp knockout mice compared with the wild type. We also observed that when simultaneously infused to steady state, GSK212658, dabrafenib, and trametinib, a rational combination to overcome mitogen-activated protein kinase inhibitor resistance, all had limited brain distribution. Coadministration of elacridar, a P-gp/Bcrp inhibitor, increased the brain distribution of GSK2126458 by approximately 7-fold in wild-type mice. In the PDX model, GSK2126458 showed efficacy in flank tumors but was ineffective in intracranial melanoma. These results show that P-gp and Bcrp are involved in limiting the brain distribution of GSK2126458 and provide a rationale for the lack of efficacy of GSK2126458 in the orthotopic PDX model.
Introduction
Metastatic melanoma is an aggressive skin cancer with a propensity for brain metastasis. Over 74,000 new cases and approximately 10,000 deaths due to melanoma are expected in 2015 in the United States (Siegel et al., 2015). Localized melanoma is curable with a 5-year survival of greater than 90%, whereas metastatic melanoma has an extremely poor prognosis with a 5-year survival of about 15% (Balch et al., 2009). After lung and breast cancer, melanoma is the third most common cancer to metastasize to the brain (Gállego Pérez-Larraya and Hildebrand, 2014).
Patients with one to three brain metastases are often treated with surgical resection or stereotactic radiosurgery, whereas those with several brain metastases typically receive whole-brain irradiation (Gibney et al., 2012). Unfortunately, melanomas are highly resistant to radiation and chemotherapy, and patients with brain metastases have a dismal survival rate of approximately 4 months (Sampson et al., 1998; Fife et al., 2004). Moreover, 50%–70% of patients with melanoma have brain metastases at autopsy (Fife et al., 2004), confirming an important unmet medical need for prevention and treatment of metastatic melanoma in the brain.
Oncogenic driver mutations in the v-raf murine sarcoma viral oncogene homolog (BRAF) and the neuroblastoma ras viral oncogene homolog (RAS) in the mitogen-activated protein kinase (MAPK) signaling pathway, p53 mutations, and phosphatase and tensin homolog mutations have been identified as important to the progression of the disease (Hodis et al., 2012). The high prevalence of the valine to glutamic acid substitution at amino acid 600 (V600E) in the BRAF protein in melanoma led to the development and U.S. Food and Drug Administration (FDA) approval of two BRAF inhibitors: vemurafenib and dabrafenib. This mutation increases BRAF protein catalytic activity by approximately 50- to 200-fold compared with the wild type (WT), resulting in constitutive activation of the mitogen-activated protein kinase kinase (MEK) and extracellular signal-regulated kinase (ERK) downstream proteins (Davies et al., 2002; Karasarides et al., 2004; Wan et al., 2004). These two drugs were approved after they showed a remarkable initial efficacy against peripheral tumors (Chapman et al., 2011; Bollag et al., 2012; Falchook et al., 2012). Similarly, trametinib, a MEK inhibitor, was approved after it showed a 4.8-month progression-free survival (PFS) compared with 1.5-month PFS in the chemotherapy group in phase 3 clinical trials in patients with BRAF V600E mutation (Flaherty et al., 2012b). Much of the current clinical data suggest that patients taking BRAF inhibitors eventually stop responding to therapy due to the development of resistance and relapse of disease (Puzanov et al., 2011; Sullivan and Flaherty, 2013). Mutations in upstream signaling proteins such as RAS or compensatory signaling from other growth factor receptors such as phosphoinositide 3-kinase (PI3K)/mammalian target of rapamycin (mTOR), loss of phosphatase and tensin homolog, upregulation of cyclin D1, and downregulation of p27Kip1 may be driving the reactivation of the MAPK signaling pathway and strengthening the resistance to BRAF inhibitor therapy (Johannessen et al., 2010; Aplin et al., 2011; Gowrishankar et al., 2012).Therefore, combination therapy with multiple molecularly targeted agents is a promising approach to overcome resistance. In this context, the combination of dabrafenib and trametinib was approved by the FDA after the two-drug combination showed a 9.8-month PFS compared with 5.8 months in the dabrafenib monotherapy arm (Flaherty et al., 2012a). Unfortunately, resistance to BRAF and MEK inhibition also occurs eventually via acquired MEK mutations (Wagle et al., 2014). A combination of BRAF or MEK inhibitors with PI3K/mTOR inhibitors is known to overcome acquired resistance in vitro (Greger et al., 2012). GSK2126458 [2,4-difluoro-N-(2-methoxy-5-(4-(pyridazin-4-yl)quinolin-6-yl)pyridin-3-yl) benzenesulfonamide] (Fig. 1), a potent ATP competitive inhibitor of the PI3K family and the mTOR kinase (mTORC1 and mTORC2) is a promising candidate for such combinations (Knight et al., 2010). GSK2126458 was found to be a highly specific, orally available inhibitor with low picomolar inhibitory activity against PI3Ks and mTOR (Knight et al., 2010).
Fig. 1.

Chemical structure of GSK2126458.
The efficacy of molecularly targeted agents and combinations for the successful treatment of melanoma brain metastases requires the delivery of all agents in the combination across the blood–brain barrier (BBB) to all target sites in the brain, including the clinically undetectable tumor cells behind an intact BBB. We have previously demonstrated that targeted melanoma therapies vemurafenib, dabrafenib, and trametinib have limited brain distribution due to active efflux by P-glycoprotein (P-gp) and breast cancer resistance protein (Bcrp) expressed at the BBB (Mittapalli et al., 2012, 2013; Vaidhyanathan et al., 2014). We have explored the role of efflux by P-gp and Bcrp in the brain penetrance of GSK2126458 to better understand a significant limitation of an otherwise promising melanoma combination therapy.
The clinically relevant patient-derived xenograft (PDX) model provides an excellent platform for testing the efficacy of these agents to improve the treatment of melanoma brain metastases. PDX models involve the transplantation of patient tumor tissue directly into mice followed by serial passage in the mouse, hence preserving genetic and phenotypic features (Carlson et al., 2011). We have evaluated the critical factors influencing the brain distribution of GSK2126458 as a single agent and in combination with dabrafenib and trametinib. We have also evaluated the efficacy of GSK2126458 in a novel PDX melanoma mouse model with the goal that this information will guide the development of effective combinations for melanoma brain metastases.
Materials and Methods
Chemicals.
GSK2126458, trametinib (N-[3-[3-cyclopropyl-5-(2-fluoro-4-iodoanilino)-6,8-dimethyl-2,4,7-trioxopyrido[4,3-d]pyrimidin-1-yl]phenyl]acetamide), and dabrafenib (GSK2118436A, N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide) were purchased from Chemietek (Indianapolis, IN). Elacridar (N-[4-[2-(3,4-dihydro-6,7-dimethoxy-2(1H)-isoquinolinyl)ethyl]phenyl]-9,10-dihydro-5-methoxy-9-oxo-4-acridinecarboxamide) was purchased from Toronto Research Chemicals (Toronto, ON, Canada).[3H]-Prazosin was purchased from Perkin Elmer Life and Analytical Sciences (Waltham, MA). [3H]-Vinblastine was purchased from Moravek Biochemicals (La Brea, CA). Ko143 [(3S,6S,12aS)-1,2,3,4,6,7,12,12a-octahydro-9-methoxy-6-(2-methylpropyl)-1,4-dioxopyrazino(1′,2′:1,6)pyrido(3,4-b)indole-3-propanoic acid 1,1-dimethylethyl ester] was purchased from Tocris Bioscience (Ellisville, MO) and zosuquidar [LY335979, (R)-4-((1aR, 6R,10bS)-1,2-difluoro-1,1a,6,10b-tetrahydrodibenzo-(a,e)cyclopropa(c)cycloheptan-6-yl)-((5-quinoloyloxy)methyl)-1-piperazine ethanol, trihydrochloride] was kindly provided Eli Lilly and Co. (Indianapolis, IN). Cell culture reagents were purchased from Invitrogen (Carlsbad, CA). All other chemicals used were of high-performance liquid chromatography or reagent grade and were obtained from Sigma-Aldrich (St. Louis, MO).
In Vitro Studies.
In vitro studies were performed using polarized Madin-Darby canine kidney II (MDCK-II) cells. MDCKII-WT and Bcrp1-transfected (MDCKII-Bcrp1) cell lines were gifts from Dr. Alfred Schinkel (The Netherlands Cancer Institute, Amsterdam, The Netherlands). MDCKII-WT and MDR1-transfected (MDCKII-MDR1) cell lines were kindly provided by Dr. Piet Borst (The Netherlands Cancer Institute). Cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% (v/v) fetal bovine serum and antibiotics (100 U/ml penicillin, 100 μg/ml streptomycin, and 250 ng/ml amphotericin B). Cells were grown in 25-ml tissue culture–treated flasks before seeding for the experiments and were maintained at 37°C in a humidified incubator with 5% CO2. The flasks were cultured for a maximum of 10 passages to be used for in vitro studies. The growth media for MDCKII-MDR1 additionally contained 80 ng/ml colchicine to maintain positive selection pressure of P-gp expression.
In Vitro Accumulation Studies.
Intracellular accumulation was conducted in WT cells and in MDR1- and Bcrp1-overexpressing cells. Cells were seeded at a density of 2 × 105 cells per well in 12-well plates and the medium was changed every other day until confluent monolayers were achieved. On the day of the experiment, the cells were washed twice with warm cell assay buffer (122 mM NaCl, 25 mM NaHCO3, 10 mM glucose, 10 mM HEPES, 3 mM KCl, 2.5 mM MgSO4, 1.8 mM CaCl2, and 0.4 mM K2HPO4). Cells were then preincubated with cell assay buffer for 30 minutes at 37°C on an orbital shaker at 60 rpm. Cell assay buffer was replaced with either 1 ml of 2 μM GSK2126458 or 1 ml of 2 μM GSK2126458 and 1 µM LY335979 (P-gp inhibitor) (n = 6). Cells were incubated for 60 minutes. The experiment was terminated by aspirating the cell assay buffer or GSK2126458 treatment and washing two times with ice-cold phosphate-buffered saline. Cells were lysed by adding 500 μl lysis buffer (1% Triton-X100). Cell lysate was sampled from each well and the concentration of GSK2126458 was determined by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS). Concentrations for each sample were normalized to the cell count for each well using a Pierce BCA protein assay (ThermoScientific, Rockford, IL). This experiment was similarly conducted in the Bcrp1-transfected cells with the Bcrp inhibitor, Ko143 (0.2 µM), substituted for the P-gp inhibitor.
Bcrp and P-gp Inhibition Studies.
Inhibition assays were performed using radiolabeled prototypical probe substrates [3H]-prazosin (27 nCi/µl) for Bcrp and [3H]-vinblastine (113 nCi/µl) for P-gp. The intracellular accumulation of these probe substrates was evaluated in the presence of varying concentrations of GSK2126458 ranging from 0.1 to 50 μM. Briefly, the cells were preincubated with increasing concentrations of GSK2126458 for 30 minutes. After preincubation, the cells were incubated with substrates along with increasing concentrations of GSK2126458 for 60 minutes. At the end of the incubation period, the buffer was aspirated and cells were lysed using 1% Triton-X100. The radioactivity in solubilized cell fractions was determined by liquid scintillation counting (LS-6500; Beckman Coulter, Fullerton, CA). The increase in cellular accumulation of substrate compared with control (no treatment) was measured and reported as a function of GSK2126458 concentration.
In Vivo Studies.
All of the in vivo brain distribution studies were performed in Friend leukemia virus strain B (FVB) (WT), Mdr1a/b−/− (P-gp knockout), Bcrp1−/− (Bcrp knockout), and Mdr1a/b−/−Bcrp1−/− (triple knockout) mice of either sex from an FVB genetic background (Taconic Farms, Germantown, NY). All animals were aged 8–12 weeks at the time of the experiment. Animals were maintained in a 12-hour light/dark cycle with unlimited access to food and water. All studies were carried out in accordance with the guidelines set by the Principles of Laboratory Animal Care (National Institutes of Health, Bethesda, MD) and approved by the University of Minnesota Institutional Animal Care and Use Committee.
Efficacy studies were performed in athymic Nu/Nu mice that were implanted with M12 melanoma cells (transduced with green fluorescent protein and luciferase), which were implanted as flank, intracranial, or intracardiac xenografts. Xenografts were established as described in Carlson et al. (2011). Mice with established tumors were randomized into treatment groups. Efficacy studies were approved by the Mayo Animal Care and Use Committee (Rochester, MN).
For all mouse pharmacokinetic studies, we used a mixture (50% male, 50% female, n = 4 to 5 mice) in each group; for survival studies, we used 100% female mice (n = 10). In addition, brain concentrations for all in vivo studies were corrected for residual drug in the brain vasculature, assumed to be 1.4% of the volume of the mouse brain (Dai et al., 2003).
Brain Distribution of GSK2126458 after an Oral Dose in FVB Mice.
WT and Mdr1a/b−/−Bcrp1−/− mice received an oral dose of 10 mg/kg GSK2126458. Blood and brain samples were collected after 0.5, 1, 2, 4, 6, and 8 hours postdose. The GSK2126458 oral dosing formulation was prepared in a vehicle containing 1% Methocel (Dow Chemical, Midland, MI) and 5% dimethylsulfoxide (DMSO). All GSK2126458 dosing suspensions were freshly prepared on the day of the experiment. At the end of the desired time point, the animals were euthanized using a CO2 chamber. Blood was collected via cardiac puncture in heparinized tubes. Plasma was separated by centrifuging whole blood at 3500 rpm for 15 minutes at 4°C. The whole brain was removed from the skull and washed with ice-cold water and superficial meninges were then removed by blotting with tissue paper. Both brain and plasma samples were stored at −80°C until further analysis.
Steady-State Brain Distribution of GSK2126458 and a Combination of Dabrafenib, Trametinib, and GSK2126458.
To determine the steady-state brain and plasma concentrations of GSK2126458, Alzet osmotic minipumps (Durect Corporation, Cupertino, CA) were loaded with GSK2126458 (2 mg/ml dissolved in DMSO) to be released for 48 hours at a rate of 1 µl/h. After initial GSK2126458 loading, minipumps were primed overnight in sterile saline at 37°C. Pumps were implanted in the peritoneal cavity of WT, Mdr1a/b−/−, Bcrp1−/−, and Mdr1a/b−/−Bcrp1−/− mice. Mice were briefly anesthetized using isoflurane and the abdominal cavity was shaved. A small midline incision was made in the lower abdominal wall under the rib cage. Then a small incision was made directly in the peritoneal membrane and the primed pump was inserted in the cavity. The incision was sutured and the skin was closed using surgical clips. The animals were allowed to recover on a heating pad; once they recovered, they were moved to their original cages. The animals were euthanized 48 hours after the implantation of the pumps, and brain and plasma samples were processed as described above.
Similarly, in another study, Alzet minipumps were loaded with GSK2126458, trametinib, and dabrafenib (1 mg/ml GSK2126458, 0.5 mg/ml trametinib, and 2.5 mg/ml dabrafenib dissolved in DMSO) to be simultaneously released for 48 hours at the rate of 1 μl/h. Pumps were primed overnight and implanted in the peritoneal cavity of WT, Mdr1a/b−/−, Bcrp1−/−, and Mdr1a/b−/−Bcrp1−/− mice. These animals were also euthanized 48 hours after the implantation of the pumps, and brain and plasma samples were processed as described previously.
Efficacy of GSK2126458 in a Melanoma Mouse Model.
We examined the efficacy of GSK212658 in athymic Nu/Nu mice implanted with patient-derived melanoma cells positive for the V600E mutation (M12 cells). Efficacy studies were conducted in mice after cells were implanted for flank tumors (5 × 106 cells subcutaneously), intracranial tumors (1 × 105 cells), or intracardiac tumors (1 × 105 cells). Mice receiving M12 cells via intracardiac injection had serial in vivo bioluminescent imaging performed to track the development of peripheral and intracranial metastases. Intracardiac mice developed both peripheral and brain tumors, which closely recapitulated the natural progression of metastatic melanoma. After tumors were established, mice were then randomized into treatment groups and received either vehicle or GSK2126458 (1.25 mg/kg p.o. once daily). Flank tumors were measured thrice weekly, and mice were euthanized when tumor volume exceeded 2000 mm3 or when tumors began to ulcerate. Mice with an intracranial or intracardiac xenograft were observed daily and were euthanized upon reaching a moribund state.
Influence of Elacridar on the Brain Distribution of GSK2126458.
Elacridar microemulsion was made by preparing a 3-mg/ml solution of elacridar in Cremaphor EL, Carbitol, and Captex 355 in a 6:3:1 ratio. This solution was diluted to 1 mg/ml with water prior to injection. WT mice received microemulsion vehicle or 10 mg/kg elacridar microemulsion via an intraperitoneal injection. All mice received a 10 mg/kg oral dose of GSK2126458 1 hour after elacridar or elacridar vehicle. Mice were euthanized 1 hour after administration of GSK2126458. Plasma and brains were harvested and samples were processed as described earlier.
Analysis of GSK2126458, Dabrafenib, and Trametinib Using LC-MS/MS.
The concentrations of GSK2126458, dabrafenib, and trametinib in plasma and brain homogenate were determined using a sensitive and specific LC-MS/MS assay. For brain analysis, three volumes of 5% bovine serum albumin were added and homogenized to obtain a uniform homogenate. For analysis of samples, an aliquot of cell lysate, brain homogenate, or plasma was spiked with 10 ng PLX4720 [N-(3-(5-chloro-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)propane-1-sulfonamide; N-[3-[(5-Chloro-1H-pyrrolo[2,3-b]pyridin-3-yl)carbonyl]-2,4-difluorophenyl]-1-propanesulfonamide] and 10 ng AG1478 [4-(3-chloroanilino)-6,7-dimethoxyquinazoline] as internal standards. The samples were then extracted by addition of 10 volumes of ethyl acetate followed by vigorous shaking for 5 minutes and centrifugation at 7500 rpm for 5 minutes at 4°C to separate the organic layer. The organic layer was transferred to microcentrifuge tubes and dried under a gentle stream of nitrogen. Samples were reconstituted in 100 µl mobile phase and transferred into high-performance liquid chromatography glass vials. Chromatographic analysis was performed using an Aquity UPLC system (Waters, Milford, MA). The chromatographic separation was achieved using an Agilent Technologies Eclipse XDB-C18 column (4.6 × 50 mm) with 1.8 µm Zorbax Rx-SIL as the stationary phase (Agilent, Santa Clara, CA). The mobile phase was delivered at 0.25 ml/min. The aqueous component (A) of the mobile phase consisted of 20 mM ammonium formate with 0.1% formic acid and the organic mobile phase (B) was acetonitrile. The mobile phase gradient was composed as follows: 45% B for the first 3.0 minutes, increased to 70% B during the next 0.5 minutes and maintained at 70% B for the next 2.5 minutes, decreased to 45% B within the next 0.5 minutes and maintained at 45% B until 11.0 minutes. The total run time was 11 minutes. The cassette method involved detection of dabrafenib in the positive ionization mode, and the mass-to-charge transitions were 520.1→307.1 and 316.1→299.9 for dabrafenib and AG1478, respectively. The detection of GSK2126458 and trametinib was in the negative ionization mode with mass-to-charge transitions of 412.3→304.8, 504.0→176.9, and 613.9→530.8 for PLX4720, GSK2126458, and trametinib, respectively. Retention times were as follows: AG1478 (4.82 minutes), GSK2126458 (5.25 minutes), PLX4720 (6.11 minutes), trametinib (6.96 minutes), and dabrafenib (7.26 minutes). The standard curve range was 1–500 ng/ml for GSK212645, 1.5–750 ng/ml for trametinib, and 1–500 ng/ml for dabrafenib. Our assay was sensitive and linear, with the coefficient of variation being less than 20% over the entire range.
Pharmacokinetic Calculations.
Pharmacokinetic parameters and metrics from the concentration-time data in plasma and brain were obtained by noncompartmental analysis performed using Phoenix WinNonlin 6.2 (Pharsight, Mountain View, CA). The area under the curve (AUC) concentration-time profiles for plasma (AUCplasma) and brain (AUCbrain) were calculated using the linear trapezoidal method. The sparse sampling module in WinNonlin was used to estimate the standard error around the mean of the AUCs. The drug targeting index (DTI) was calculated using the following equation:
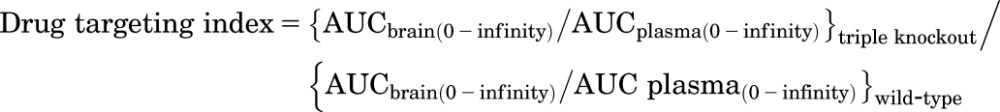 |
Statistical Analysis.
Data in all experiments represent means ± S.D. unless otherwise indicated. Comparisons between two groups were made using an unpaired t test. One-way analysis of variance followed by Boneferronis multiple comparison test for comparisons between two different groups or Dunnets test for comparing versus control was performed. The log-rank (Mantel–Cox) test was used to determine significance in survival studies. A significance level of P < 0.05 was used for all experiments (GraphPad Prism 5.01 software; GraphPad, San Diego, CA).
Results
Intracellular Accumulation of GSK2126458.
The intracellular accumulation of GSK2126458 was studied in MDCKII-WT and P-gp– or Bcrp–overexpressing cell lines. The cellular accumulation of [3H]-prazosin and [3H]-vinblastine was used as the positive control for Bcrp– and P-gp–mediated efflux transport, respectively. The accumulation of [3H]-prazosin (Fig. 2A) was approximately 70% lower in Bcrp-overexpressing cells (WT: 100% ± 3.96%; Bcrp: 29.87% ± 7.72%; P < 0.0001) compared with respective WT cells. Similarly, the accumulation of [3H]-vinblastine (Fig. 2B) in P-gp–overexpressing cells was approximately 82% lower compared with WT cells (WT: 99.98% ± 9.03%; MDR1: 18.25% ± 2.12%; P < 0.0001). GSK2126458 accumulation was approximately 84% lower in Bcrp-overexpressing cells compared with WT cells (WT: 100% ± 13.41%; Bcrp: 15.65% ± 7.97%; P < 0.0001). Addition of specific Bcrp inhibitor Ko143 significantly increased the accumulation of GSK2126458 in the Bcrp-transfected cells (Bcrp: 15.65% ± 7.97%; Bcrp with Ko143: 59.43% ± 16.91%). Similarly, the accumulation of GSK2126458 was approximately 61% lower in the P-gp–overexpressing line compared with its WT control (WT: 100.0% ± 11.21%; MDR1: 38.63% ± 5.90%; P < 0.0001). When a specific P-gp inhibitor (LY335979) was added (Fig. 2B), there was a significant increase in the accumulation of GSK2126458 in the MDR1-transfected cells (MDR1: 38.63% ± 5.90%; MDR1 with LY: 86.07% ± 12.11%). These cellular accumulation data indicate that GSK2126458 is a substrate for both P-gp and Bcrp in vitro.
Fig. 2.

In vitro cellular accumulation of GSK2126458. (A) The accumulation of prazosin (prototypical Bcrp probe substrate; positive control) and GSK2126458 (2 μM) in MDCKII-WT and Bcrp1-transfected cells with and without specific Bcrp inhibitor Ko143 (0.2 μM). n = 3–6. (B) The accumulation of GSK2126458 (2 μM) and vinblastine (probe substrate for P-gp) in MDR1 cells with and without specific P-gp inhibitor LY335979 (1 μM). n = 3–6. (C and D) Intracellular accumulation of [3H]-prazosin (Bcrp probe substrate) in Bcrp1-transfected cells (C) and [3H]-vinblastine in MDR1-transfected cells (D) with increasing concentrations of GSK2126458 from 0.1 µM to 50 µM. n = 3. Data represent means ± S.D. *P < 0.05, compared with respective WT controls; ***P < 0.0001, compared with respective WT controls; †P < 0.05, compared with transfected line without inhibitor; †††P < 0.0001, compared with transfected line without inhibitor.
Competition Assays Using Prototypical Probe Substrates.
The effect of increasing concentrations of GSK2126458 on probe substrate accumulation was assessed in Bcrp1-transfected and MDR1-transfected MDCKII cells. Increasing concentrations of GSK2126458 significantly increased the accumulation of prazosin in the Bcrp1-transfected cells at GSK2126458 concentrations of 25 μM and above (Fig. 2C). We observed an approximately 3-fold and 4-fold increase in prazosin accumulation at 25 μM and 50 μM GSK2126458. Similarly, increasing concentrations of GSK2126458 significantly increased (approximately 3-fold at 15 μM GSK2126458) the accumulation of vinblastine in the MDR1 cells (Fig. 2D). The results of these competitive inhibition studies suggest that GSK2126458 may share a binding site on Bcrp with prazosin and a binding site on P-gp with vinblastine.
Plasma and Brain Pharmacokinetics of GSK2126458.
The plasma and brain pharmacokinetics of GSK2126458 were studied in WT and Mdr1a/b−/−Bcrp1−/− mice after oral administration of 10 mg/kg. Figure 3 shows the plasma and brain concentrations of GSK2126458 in the two genotypes at 0.5, 1, 2, 4, 6, and 8 hours after a single oral dose. The plasma concentrations (Fig. 3A) were not significantly different between WT and Mdr1a/b−/−Bcrp1−/− mice. In the WT mice, the plasma concentrations were approximately 2 log units higher than the corresponding brain concentrations, indicating the severely restricted brain distribution of GSK2126458. The brain concentrations of GSK2126458 were 2-to 11-fold higher in the Mdr1a/b−/−Bcrp1−/− mice compared with WT, with a DTI of approximately 10 (Fig. 3B). In addition, the AUC in the brain in the triple knockout mice was approximately 6-fold higher than the AUC in the brain of WT mice. These data show the significant role played by P-gp and Bcrp in restricting the brain distribution of GSK2126458.
Fig. 3.

Brain distribution of GSK2126458 in FVB WT and Mdr1a/b−/−Bcrp1−/− mice. (A–C) Plasma (A), brain (B), and brain-to-plasma concentration ratios (C) of GSK2126458 in WT and Mdr1a/b−/−Bcrp1−/− mice after an oral dose of 10 mg/kg. Plasma and brain concentrations of GSK2126458 at 0.5,1,2, 4, 6, and 8 hours postdose were determined using LC-MS/MS. Data represent means ± S.D. n = 3 to 4. *P < 0.05, compared with WT; **P < 0.01; ***P ≤ 0.0001, compared with WT.
Steady-State Brain Distribution of GSK2126458.
The steady-state brain distribution of GSK2126458 was examined after a continuous intraperitoneal infusion using Alzet osmotic pumps for 48 hours at 2 µg/h (Fig. 4). The brain concentrations were approximately 5-fold higher in the Mdr1a/b−/−Bcrp1−/− mice compared with WT (Fig. 4A). The brain concentrations were not significantly different in the Mdr1a/b−/− and Bcrp1−/− mice compared with WT and were significantly lower than their corresponding plasma concentrations (Fig. 4A). As shown in Fig. 4B, the steady-state brain-to-plasma ratios were 0.06 ± 0.02, 0.05 ± 0.03, and 0.46 ± 0.23 in the WT, Mdr1a/b−/−, and Mdr1a/b−/−Bcrp1−/− mice, respectively (brain concentration not detectable in Bcrp1−/− mice). The brain-to-plasma ratios at steady state were approximately 8-fold higher in the Mdr1a/b−/−Bcrp1−/− mice compared with WT mice. These data further confirm that the brain distribution of GSK2126458 is significantly impaired due to active efflux at the BBB.
Fig. 4.

Steady-state distribution of GSK2126458 at 2 μg/h for 48 hours. Steady-state brain-to-plasma ratio of GSK2126458 in WT, Mdr1a/b−/−, Bcrp1−/−, and Mdr1a/b−/−Bcrp1−/− mice. GSK2126458 was delivered at a constant infusion rate of 2 µg/h for 48 hours using Alzet osmotic pumps implanted in the peritoneal cavity. Data represent means ± S.D.,= n = 3–5. *P < 0.05, compared with WT; ***P < 0.001, compared with WT. N.D., not detectable (not detected after correction for amount in brain microvessel, GSK2126458 LC- MS/MS assay lower limit of quantification = 1 ng/ml).
Steady-State Brain Distribution of GSK2126458, Dabrafenib, and Trametinib in Combination.
We then examined the steady-state brain distribution of GSK2126458 (1 µg/h), dabrafenib (2.5 µg/h), and trametinib (0.5 µg/h) after a 48-hour simultaneous intraperitoneal infusion of the three-drug combination. This combination was infused in WT, Mdr1a/b−/−, Bcrp1−/−, and Mdr1a/b−/−Bcrp1−/− mice. The steady-state brain-to-plasma concentration ratios of GSK2126458, dabrafenib, and trametinib were significantly higher in the Mdr1a/b−/−Bcrp1−/− mice compared with WT (Fig. 5). Plasma and brain concentrations of dabrafenib, trametinib, and GSK2126458 are reported in Supplemental Fig. 1. In addition, the steady-state brain-to-plasma concentration ratios of dabrafenib and trametinib were significantly higher in the Mdr1a/b−/− mice compared with WT (Fig. 5). These data suggest that all three drugs in the combination suffer from limited brain distribution due to active efflux at the BBB.
Fig. 5.

Steady-state distribution of GSK2126458, dabrafenib, and trametinib after 48-hour simultaneous infusion. Steady-state brain-to-plasma ratios of GSK2126458, dabrafenib, and trametinib in WT, Mdr1a/b−/−, Bcrp1−/−, and Mdr1a/b−/−Bcrp1−/− mice. GSK2126458, dabrafenib, and trametinib were simultaneously delivered at a constant rate of 1 µg/h, 2.5 µg/h, and 0.5 µg/h, respectively, for 48 hours using Alzet osmotic pumps implanted in the peritoneal cavity. Data represent means ± S.D. n = 3–5. *P < 0.05, compared with corresponding brain-to-plasma ratio in WT.
Efficacy of GSK2126458 in the Novel M12 Melanoma PDX Model.
We examined the efficacy of GSK2126458 in a flank (Fig. 6), intracranial, and intracardiac PDX mouse model. There was a significant reduction in flank tumor growth in GSK2126458-treated mice compared with the vehicle-treated group (Fig. 6A) (P < 0.0001). We also observed that GSK2126458 treatment was ineffective in the M12 intracranial xenograft compared with vehicle treatment (Fig. 6C) (no statistically significant difference in median survival, 23 versus 26 days). However, we observed a significantly improved median survival with GSK2126458 treated mice compared with vehicle treatment (53 versus 37 days) when the mice were implanted with M12 cells via intracardiac injections (Fig. 6B). These data suggest that GSK2126458 is more effective in peripheral tumors and is ineffective in the brain. The improved survival with the intracardiac PDX model may be attributed to the better efficacy observed in peripheral tumors.
Fig. 6.

Efficacy of GSK2126458 in M12 xenograft model. (A) Tumor volume after flank implantation (means ± S.E.M.) with and without GSK2126458 (1.25 mg/kg daily). (B and C) Efficacy of GSK2126458 (1.25 mg/kg daily) in intracardiac (B) and intracranial (C) tumors. n = 6–10.
Influence of Elacridar Microemulsion on the Brain Distribution of GSK2126458.
Next we examined the influence of elacridar on the brain distribution of GSK2126458. WT mice were pretreated with vehicle or elacridar (10 mg/kg) via an intraperitoneal injection 1 hour before an oral dose of GSK2126458 (10 mg/kg). The brain-to-plasma concentration ratio of GSK2126458 was approximately 7-fold higher in the elacridar-pretreated group compared with the vehicle-treated group (Fig. 7). These results demonstrate that the administration of elacridar as a microemulsion formulation improves the brain distribution of GSK2126458.
Fig. 7.
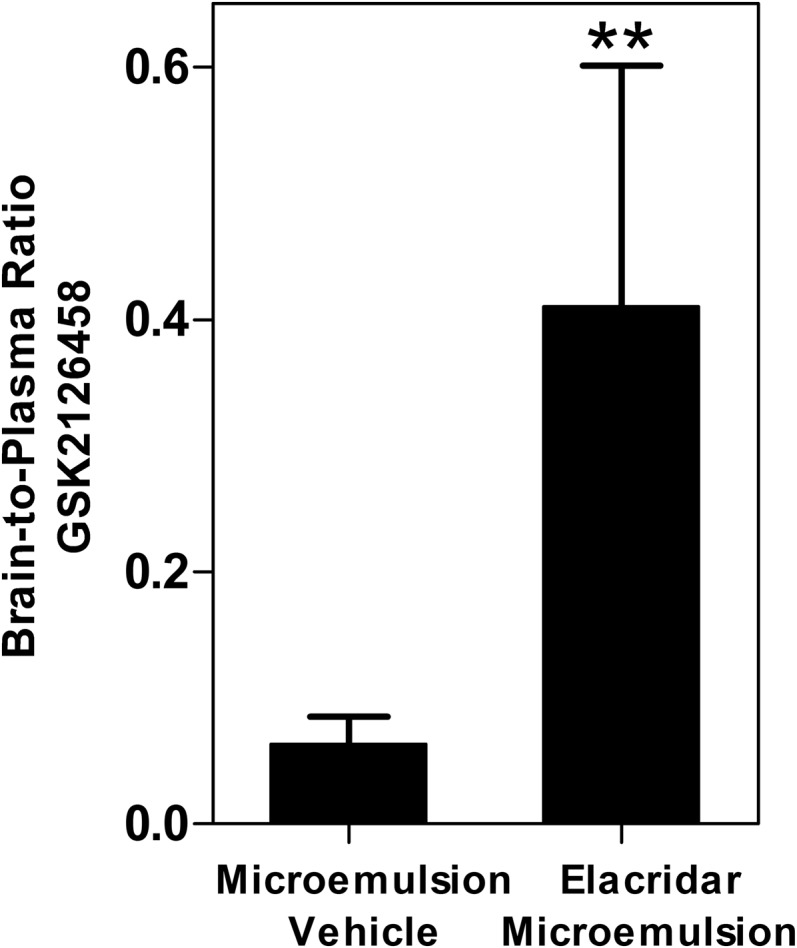
Influence of elacridar in a microemulsion formulation on the brain distribution of GSK2126458. Brain-to-plasma ratio of GSK212658 2 hours after pretreatment with either vehicle or elacridar microemulsion (10 mg/kg i.p.) and 1 hour after administration of GSK2126458 (10 mg/kg p.o.) in WT mice. Data represent means ± S.D. n = 3 to 4. **P < 0.01, compared with corresponding brain-to-plasma ratio in vehicle-treated mice.
Discussion
Brain metastases are a significant cause of mortality in patients with metastatic melanoma (Fife et al., 2004; Davies et al., 2011). The FDA approval of vemurafenib and dabrafenib (BRAF inhibitors), trametinib (MEK inhibitor), and the dabrafenib-trametinib combination has dramatically changed the treatment options for advanced melanoma. However, the remarkable initial efficacy of both mono- and combination therapy is eventually followed by relapse due to the development of resistance (Nazarian et al., 2010; Gowrishankar et al., 2012). Given this, it is well recognized that a combination of agents targeting multiple signaling pathways has the potential to delay or overcome acquired resistance. It has been previously suggested that targeting MEK, which is downstream of BRAF in the MAPK signaling pathway, or targeting PI3K is a valid therapeutic approach to overcome resistance to chronic BRAF inhibition (Villanueva et al., 2010). In this context, the FDA-approved combination of dabrafenib and trametinib has been shown to provide a more durable response than vemurafenib (BRAF inhibitor) monotherapy (Robert et al., 2015). However, concomitant MEK mutations observed with the dabrafenib-trametinib combination suggest that tumor proliferation can continue through alternate signaling pathways such as the PI3K/AKT/mTOR pathway. Chronic BRAF inhibition has been known to enhance PI3K/AKT activity via insulin-like growth factor 1 receptor signaling (Villanueva et al., 2010). Importantly, GSK2126458 in combination has been shown to overcome acquired resistance to dabrafenib in vitro (Greger et al., 2012) .
These data defining resistance mechanisms strongly indicate that the successful treatment of melanoma brain metastases will require that all agents in a rational combination must be delivered to all metastatic sites, especially those that reside behind an intact BBB. These metastatic cells are protected by the BBB that has both tight junctions and efflux transporters that will limit the brain distribution of most molecularly targeted agents tested thus far. We have previously shown that vemurafenib, dabrafenib, and trametinib are significantly limited in brain distribution due to their interaction with P-gp and Bcrp (Mittapalli et al., 2012, 2013; Vaidhyanathan et al., 2014). We have also shown that the brain distribution of the dabrafenib-trametinib combination is significantly higher in Mdr1a/b−/−Bcrp−/− mice compared with WT, suggesting that this combination also suffers from limited brain distribution due to active efflux at the BBB (Vaidhyanathan et al., 2014).
In this study, using in vitro and in vivo models, we demonstrate that GSK2126458 is a substrate for P-gp and Bcrp. This is the first report investigating the interaction of GSK2126458 with P-gp and Bcrp and examining its efficacy in melanoma PDX models. We also show that the combination of GSK2126458 with dabrafenib and trametinib suffers from active efflux at the BBB.
The experiments performed in vitro in MDCKII cells that overexpress either human MDR1 or murine Bcrp confirm that GSK2126458 is a substrate for both P-gp and Bcrp (Fig. 2). We observed a significantly lower accumulation of GSK2126458 in P-gp– and Bcrp–overexpressing cells compared with WT cells (Fig. 2). In the presence of specific inhibitors of P-gp and Bcrp (LY335979 and Ko143, respectively), this difference in intracellular accumulation was significantly decreased (Fig. 2). In P-gp–transfected cells, using a prototypical P-gp probe substrate vinblastine, we observed a significant increase in intracellular accumulation with increasing concentrations of GSK2126458 starting at 15 μM (Fig. 2D). In Bcrp-transfected cells, there was a significant increase in accumulation of Bcrp probe substrate prazosin at GSK2126458 concentrations of 25 μM and above (Fig. 2C). These data suggest that GSK2126458 may share binding sites on P-gp and Bcrp with vinblastine and prazosin, respectively. These in vitro results conclusively show that GSK2126458 is a substrate for both P-gp and Bcrp.
We conducted in vivo experiments to investigate the translation of our in vitro findings on the P-gp and BCRP substrate status of GSK2126458 to the brain distribution of GSK2126458 in a mouse model. The concentration-time profile of GSK2126458 given orally at 10 mg/kg confirms that there are significantly higher brain concentrations in Mdr1a/b−/−Bcrp−/− mice compared with WT mice. Plasma concentrations were not significantly different between the two genotypes (Fig. 3). The ratio of AUCbrain/AUCplasma given by the concentration-time profile is 0.086 for Mdr1a/b−/−Bcrp−/− mice and 0.009 for WT mice, resulting in a DTI of approximately 10. It is important to note that even though there was a 10-fold increase in brain distribution in the Mdr1a/b−/−Bcrp−/− mouse, there is still only a 10% distribution. This may be due to other heretofore unknown efflux transporters for GSK2126458.
At steady state, achieved using the osmotic pump delivery, the brain concentrations in WT, Mdr1a/b−/−, and Bcrp−/− mice were significantly lower than in Mdr1a/b−/−Bcrp−/− mice. The steady-state brain-to-plasma ratio increased from approximately 0.056 in the WT mice to 0.46 in the Mdr1a/b−/−Bcrp−/− mice (Fig. 4). This nearly 8-fold increase in the targeted brain distribution of GSK2126458 further confirms the effect of P-gp and Bcrp on the central nervous system penetration of GSK2126458. Importantly, we did not observe a significant change in the brain distribution of GSK2126458 in the Mdr1a/b−/− or Bcrp−/− (single knockout) mice compared with WT mice. This suggests the cooperative role of these two transporters in excluding GSK2126458 from the central nervous system. In the absence of either P-gp or Bcrp, modeled by the single knockout animals, the other transporter can compensate and limit substrate brain distribution; therefore, there can be a disproportionate enhancement in brain distribution of dual substrates in Mdr1a/b−/−Bcrp−/− mice (de Vries et al., 2007; Polli et al., 2009; Kodaira et al., 2010; Agarwal et al., 2011; Agarwal and Elmquist, 2012). Development of resistance to targeted melanoma monotherapy via pathway cross-activation suggests that combination therapy may be used to obtain a durable response. An understanding of the molecular mechanisms of resistance provides the rationale for combining GSK2126458 with either dabrafenib or trametinib (Greger et al., 2012). Given this, success in the treatment of brain metastases will require all agents in the combination to be effectively delivered across the BBB. Simultaneous infusion of GSK2126458, dabrafenib, and trametinib yielded significantly higher brain-to-plasma ratios in the Mdr1a/b−/−Bcrp−/− mice compared with WT mice for all three drugs (Fig. 5). These data confirm that the brain distribution of all three drugs in combination is limited by active efflux. In addition, the brain distributions of dabrafenib and trametinib were significantly higher in the single knockout Mdr1a/b−/− mice. This supports our previous observation that the brain distribution of trametinib was limited primarily by P-gp (Vaidhyanathan et al., 2014).
Based on our observation from efficacy studies in the M12 PDX models, we conclude that GSK2126458 may be a candidate for the treatment of peripheral melanoma metastases (Fig. 6). The improved survival with GSK2126458 treatment in the intracardiac xenograft model can be correlated with the improved efficacy observed in the flank tumor model based on tumor growth data. These data suggest that GSK2126458 is efficacious in treating peripheral melanoma metastases and is unlikely to be effective in the treatment of brain metastases, as observed from the lack of efficacy in the intracranial xenograft. This observation can be attributed to our findings with regard to the limited brain distribution of GSK2126458 due to its interaction with P-glycoprotein and Bcrp at the BBB.
Given the limited brain distribution of GSK2126458 and its potential use in combination therapy for the treatment of melanoma brain metastases, we investigated the influence of elacridar on its brain distribution. An intraperitoneal injection of an elacridar microemulsion 1 hour prior to oral administration of GSK2126458 led to a nearly 7-fold increase in the brain-to-plasma ratio of GSK2126458 compared with mice receiving blank microemulsion vehicle (Fig. 7). The results from this study suggest that the inhibition of P-gp and Bcrp with elacridar could be a potential strategy for improving the brain distribution of GSK2126458. This finding is of clinical relevance, and any modality that can lead to the enhancement of brain distribution of all drugs in the combination therapy is a possible strategy for enhancing efficacy of combination therapies that suffer from active efflux at the BBB.
The potential for active efflux at the BBB must be taken into consideration when developing combinations of agents for success in treating brain metastases. If one or more drugs in the combination do not reach the brain, there is an increased potential for resistance by creating a sanctuary site in the brain for unchecked growth and the development of further metastases. It is imperative that in addition to understanding molecular resistance mechanisms, the potential for active efflux at the BBB also must be considered prior to the development of combination therapies. Our findings are clinically relevant for the choice of rational combinations to ensure effective treatment of melanoma brain metastases.
Supplementary Material
Acknowledgments
The authors thank Jim Fisher (Clinical Pharmacology Analytical Services Laboratory, University of Minnesota) for help and support in the development of the LC-MS/MS assay.
Abbreviations
- AG1478
4-(3-chloroanilino)-6,7-dimethoxyquinazoline
- AUC
area under the curve
- BBB
blood–brain barrier
- DMSO
dimethylsulfoxide
- DTI
drug targeting index
- ERK
extracellular signal-regulated kinase
- FDA
U.S. Food and Drug Administration
- FVB
Friend leukemia virus strain B
- GSK2118436A
N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide
- GSK2126458
2,4-difluoro-N-(2-methoxy-5-(4-(pyridazin-4-yl)quinolin-6-yl)pyridin-3-yl) benzenesulfonamide
- Ko143
(3S,6S,12aS)-1,2,3,4,6,7,12,12a-octahydro-9-methoxy-6-(2-methylpropyl)-1,4-dioxopyrazino(1′,2′:1,6)pyrido(3,4-b)indole-3-propanoic acid 1,1-dimethylethyl ester
- LC-MS/MS
liquid chromatography coupled to tandem mass spectrometry
- LY335979
(R)-4-((1aR,6R,10bS)-1,2-difluoro-1,1a,6,10b-tetrahydrodibenzo-(a,e)cyclopropa(c)cycloheptan-6-yl)-α-((5-quinoloyloxy) methyl)-1-piperazine ethanol, trihydrochloride
- MAPK
mitogen-activated protein kinase
- MDCKII
Madin-Darby canine kidney II
- MEK
mitogen-activated protein kinase kinase
- mTOR
mammalian target of rapamycin
- PDX
patient-derived xenograft
- PFS
progression-free survival
- P-gp
P-glycoprotein
- PI3K
phosphoinositide 3-kinase
- PLX4720
N-(3-(5-chloro-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)propane-1-sulfonamide, N-[3-[(5-Chloro-1H-pyrrolo[2,3-b]pyridin-3-yl)carbonyl]-2,4-difluorophenyl]-1-propanesulfonamide
- V600E
amino acid 600
- WT
wild type
Authorship Contributions
Participated in research design: Vaidhyanathan, Wilken-Resman, Ma, Parrish, Mittapalli, Sarkaria, Elmquist.
Conducted experiments: Vaidhyanathan, Wilken-Resman, Ma, Parrish, Mittapalli, Carlson.
Performed data analysis: Vaidhyanathan, Wilken-Resman, Ma, Sarkaria, Elmquist.
Wrote or contributed to the writing of the manuscript: Vaidhyanathan, Wilken-Resman, Ma, Parrish, Mittapalli, Sarkaria, Elmquist.
Footnotes
This research was supported by the National Institutes of Health National Cancer Institute [Grants R01CA138437 and P50CA108961] and the National Institutes of Health National Institute of Neurological Disorders and Stroke [Grant R01NS077921]. S.V. was supported by a Ronald J. Sawchuk Fellowship.
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Agarwal S, Elmquist WF. (2012) Insight into the cooperation of P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) at the blood-brain barrier: a case study examining sorafenib efflux clearance. Mol Pharm 9:678–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S, Hartz AM, Elmquist WF, Bauer B. (2011) Breast cancer resistance protein and P-glycoprotein in brain cancer: two gatekeepers team up. Curr Pharm Des 17:2793–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aplin AE, Kaplan FM, Shao Y. (2011) Mechanisms of resistance to RAF inhibitors in melanoma. J Invest Dermatol 131:1817–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, Buzaid AC, Cochran AJ, Coit DG, Ding S, et al. (2009) Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol 27:6199–6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollag G, Tsai J, Zhang J, Zhang C, Ibrahim P, Nolop K, Hirth P. (2012) Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat Rev Drug Discov 11:873–886. [DOI] [PubMed] [Google Scholar]
- Carlson BL, Pokorny JL, Schroeder MA, Sarkaria JN. (2011) Establishment, maintenance and in vitro and in vivo applications of primary human glioblastoma multiforme (GBM) xenograft models for translational biology studies and drug discovery. Curr Protoc Pharmacol Chapter 14:Unit 14 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et al. BRIM-3 Study Group (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364:2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai H, Marbach P, Lemaire M, Hayes M, Elmquist WF. (2003) Distribution of STI-571 to the brain is limited by P-glycoprotein-mediated efflux. J Pharmacol Exp Ther 304:1085–1092. [DOI] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. (2002) Mutations of the BRAF gene in human cancer. Nature 417:949–954. [DOI] [PubMed] [Google Scholar]
- Davies MA, Liu P, McIntyre S, Kim KB, Papadopoulos N, Hwu WJ, Hwu P, Bedikian A. (2011) Prognostic factors for survival in melanoma patients with brain metastases. Cancer 117:1687–1696. [DOI] [PubMed] [Google Scholar]
- de Vries NA, Zhao J, Kroon E, Buckle T, Beijnen JH, van Tellingen O. (2007) P-glycoprotein and breast cancer resistance protein: two dominant transporters working together in limiting the brain penetration of topotecan. Clin Cancer Res 13:6440–6449. [DOI] [PubMed] [Google Scholar]
- Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, Hamid O, Infante JR, Millward M, Pavlick AC, et al. (2012) Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet 379:1893–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fife KM, Colman MH, Stevens GN, Firth IC, Moon D, Shannon KF, Harman R, Petersen-Schaefer K, Zacest AC, Besser M, et al. (2004) Determinants of outcome in melanoma patients with cerebral metastases. J Clin Oncol 22:1293–1300. [DOI] [PubMed] [Google Scholar]
- Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, et al. (2012a) Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 367:1694–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, Demidov LV, Hassel JC, Rutkowski P, Mohr P, et al. METRIC Study Group (2012b) Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 367:107–114. [DOI] [PubMed] [Google Scholar]
- Gállego Pérez-Larraya J, Hildebrand J. (2014) Brain metastases. Handb Clin Neurol 121:1143–1157. [DOI] [PubMed] [Google Scholar]
- Gibney GT, Forsyth PA, Sondak VK. (2012) Melanoma in the brain: biology and therapeutic options. Melanoma Res 22:177–183. [DOI] [PubMed] [Google Scholar]
- Gowrishankar K, Snoyman S, Pupo GM, Becker TM, Kefford RF, Rizos H. (2012) Acquired resistance to BRAF inhibition can confer cross-resistance to combined BRAF/MEK inhibition. J Invest Dermatol 132:1850–1859. [DOI] [PubMed] [Google Scholar]
- Greger JG, Eastman SD, Zhang V, Bleam MR, Hughes AM, Smitheman KN, Dickerson SH, Laquerre SG, Liu L, Gilmer TM. (2012) Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol Cancer Ther 11:909–920. [DOI] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, et al. (2012) A landscape of driver mutations in melanoma. Cell 150:251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, et al. (2010) COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 468:968–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasarides M, Chiloeches A, Hayward R, Niculescu-Duvaz D, Scanlon I, Friedlos F, Ogilvie L, Hedley D, Martin J, Marshall CJ, et al. (2004) B-RAF is a therapeutic target in melanoma. Oncogene 23:6292–6298. [DOI] [PubMed] [Google Scholar]
- Knight SD, Adams ND, Burgess JL, Chaudhari AM, Darcy MG, Donatelli CA, Luengo JI, Newlander KA, Parrish CA, Ridgers LH, et al. (2010) Discovery of GSK2126458, a Highly Potent Inhibitor of PI3K and the Mammalian Target of Rapamycin. ACS Med Chem Lett 1:39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodaira H, Kusuhara H, Ushiki J, Fuse E, Sugiyama Y. (2010) Kinetic analysis of the cooperation of P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp/Abcg2) in limiting the brain and testis penetration of erlotinib, flavopiridol, and mitoxantrone. J Pharmacol Exp Ther 333:788–796. [DOI] [PubMed] [Google Scholar]
- Mittapalli RK, Vaidhyanathan S, Dudek AZ, Elmquist WF. (2013) Mechanisms limiting distribution of the threonine-protein kinase B-RaF(V600E) inhibitor dabrafenib to the brain: implications for the treatment of melanoma brain metastases. J Pharmacol Exp Ther 344:655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittapalli RK, Vaidhyanathan S, Sane R, Elmquist WF. (2012) Impact of P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) on the brain distribution of a novel BRAF inhibitor: vemurafenib (PLX4032). J Pharmacol Exp Ther 342:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee M-K, Attar N, Sazegar H, et al. (2010) Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 468:973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polli JW, Olson KL, Chism JP, John-Williams LS, Yeager RL, Woodard SM, Otto V, Castellino S, Demby VE. (2009) An unexpected synergist role of P-glycoprotein and breast cancer resistance protein on the central nervous system penetration of the tyrosine kinase inhibitor lapatinib (N-3-chloro-4-[(3-fluorobenzyl)oxy]phenyl-6-[5-([2-(methylsulfonyl)ethyl]aminomethyl)-2-furyl]-4-quinazolinamine; GW572016). Drug Metab Dispos 37:439–442. [DOI] [PubMed] [Google Scholar]
- Puzanov I, Burnett P, Flaherty KT. (2011) Biological challenges of BRAF inhibitor therapy. Mol Oncol 5:116–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L, et al. (2015) Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 372:30–39. [DOI] [PubMed] [Google Scholar]
- Sampson JH, Carter JH, Jr, Friedman AH, Seigler HF. (1998) Demographics, prognosis, and therapy in 702 patients with brain metastases from malignant melanoma. J Neurosurg 88:11–20. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. (2015) Cancer statistics, 2015. CA Cancer J Clin 65:5–29. [DOI] [PubMed] [Google Scholar]
- Sullivan RJ, Flaherty KT. (2013) Resistance to BRAF-targeted therapy in melanoma. Eur J Cancer 49:1297–1304. [DOI] [PubMed] [Google Scholar]
- Vaidhyanathan S, Mittapalli RK, Sarkaria JN, Elmquist WF. (2014) Factors influencing the CNS distribution of a novel MEK-1/2 inhibitor: implications for combination therapy for melanoma brain metastases. Drug Metab Dispos 42:1292–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu X, Gimotty PA, Kee D, et al. (2010) Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 18:683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagle N, Van Allen EM, Treacy DJ, Frederick DT, Cooper ZA, Taylor-Weiner A, Rosenberg M, Goetz EM, Sullivan RJ, Farlow DN, et al. (2014) MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov 4:61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, et al. Cancer Genome Project (2004) Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116:855–867. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.