

Abstract
Background
Human head and neck squamous carcinoma is the 6th most prevalent carcinoma worldwide. Although many novel therapies have been developed, the clinical treatment for patients remains non-ideal. Chloride intracellular channel 4 (CLIC4), one of the seven members of the CLIC family, is a newly found Cl− channel that participates in various biological processes, including cellular apoptosis and differentiation. Accumulating evidence has revealed the significant role of CLIC4 in regulating the apoptosis of different cancer cells. Here, we investigated the functional role of CLIC4 in the apoptosis of HN4 cells, a human head and neck squamous carcinoma cell line.
Results
In the present study, we used immunohistochemical staining to demonstrate that the expression level of CLIC4 is elevated in the tissue of human oral squamous carcinoma compared with healthy human gingival tissue. Specific CLIC4 small interfering RNA was used to knockdown the expression of CLIC4. The results showed that knockdown of CLIC4 with or without 100 μM adenosine triphosphate (ATP) treatment significantly increased the expression of Bax, active caspase 3, active caspase 4 and CHOP but suppressed Bcl-2 expression in HN4 cells. Moreover, the results from the TdT-mediated dUTP nick end labeling assay indicated that CLIC4 knockdown induced a higher apoptotic rate in HN4 cells under the induction of ATP. In addition, knockdown of CLIC4 dramatically enhanced ATP-induced mitochondrial membrane depolarization in HN4 cells. Moreover, intracellular Ca2+ measurement revealed that Ca2+ release induced by ATP and thapsigargin, a Ca2+-ATPase inhibitor of the endoplasmic reticulum, was significantly enhanced by the suppression of CLIC4 in HN4 cells.
Conclusions
Knockdown of CLIC4 enhanced ATP-induced apoptosis in HN4 cells. Both the pathways of mitochondria and endoplasmic reticulum stress were involved in CLIC4-mediated cell apoptosis. Based on our finding, CLIC4 may be a potential and valuable target for the clinical treatment of head and neck squamous carcinoma.
Electronic supplementary material
The online version of this article (doi:10.1186/s13578-016-0070-1) contains supplementary material, which is available to authorized users.
Keywords: Chloride intracellular channel 4, Head and neck squamous carcinoma, Apoptosis, Mitochondrial membrane potential, Endoplasmic reticulum stress, Adenosine triphosphate
Background
Head and neck cancer, the 6th most common carcinoma worldwide, severely deteriorates the health of elderly people [1]. Despite recent advances in therapy, clinical treatment for patients with head and neck squamous cell carcinoma (HNSCC) remains to be improved further [2]. Thus, exploring new ways to improve the treatment for HNSCC is required and of great significance [3]. In recent years, accumulating evidence has indicated the role of ion channels in the development of different cancers [4, 5]. Among various ion channels, the Cl− intracellular channel (CLIC) has been listed as one of the most active ones [5, 6]. CLIC is the most recently discovered family of Cl− channels. CLIC4, also known as p64H1, RS43, or mtCLIC, is one of the seven members of the CLIC family and is ubiquitously expressed in the brain, liver and skin at a particularly high level [5, 7, 8]. The literature has shown that the subcellular localization of CLIC4 varies among different cell types, including the inner mitochondrial membrane, cytoplasm, Golgi apparatus and endoplasmic reticulum (ER) [8].
Recent studies have shown that CLIC4 participates in the cell apoptotic process. However, the results are quite complex with a controversial effect [8]. Both suppression and overexpression of CLIC4 can induce apoptosis in keratinocytes [8]. Additionally, the apoptotic response mediated by CLIC4 occurs not only through the mitochondrial pathway accompanied with the increase of Bax/Bcl-2, activation of caspase 3 and release of cytochrome C, but through the ER stress pathway along with the increase in CHOP and cleaved caspase 4 expression [9]. Multiple studies have investigated the expression profile and regulatory effect on the apoptosis of CLIC4 in cancer cells [8, 10–13]. It has been suggested that the suppression of CLIC4 via CLIC4-antisense inhibits the growth of human osteosarcoma cells in vitro and in vivo, weakens cell proliferation and increases cell apoptosis [14]. However, other studies have reported that the loss of CLIC4 is a common phenotype in many human cancers and marks malignant progression; additionally, if CLIC4 was introduced into human breast cells, tumor growth is inhibited [15]. Therefore, the expression profile and regulatory effect on apoptosis of CLIC4 in each specific cancer cell may be different. Moreover, the effect of CLIC4 on HNSCC apoptosis has not yet been examined. Adenosine triphosphate (ATP) released by stressed and dying cells [16] is a potent extracellular apoptosis inducer that disrupts normal cellular metabolism by binding extracellular specific receptor and regulating intracellular metabolic products [17]. Because CLIC4 is indicated as an integral part of the cellular stress-response pathway and is a significant regulator of cell apoptosis and growth in live cells, we hypothesize that the regulation of CLIC4 protein expression may affect ATP-induced apoptosis.
The present study was undertaken to investigate the expression profile of CLIC4 in the surgical specimens of HNSCC patients and the effect of CLIC4 knockdown on ATP-induced apoptosis in the HNSCC cell line HN4.
Results
Enhanced CLIC4 expression in HNSCC tissue
Because growing evidence has indicated that CLIC4 is involved in the development of different cancers, including cutaneous cancer, breast cancer, ovarian cancer and others [11, 12, 15], we also investigated CLIC4 expression in surgical specimens from HNSCC patients. Immunohistochemical staining showed that CLIC4 expression in HNSCC tissue was obviously higher than that in normal gingival tissue (Fig. 1). Based on this finding, we hypothesized that CLIC4 might also have a relationship with the development of HNSCC.
Fig. 1.
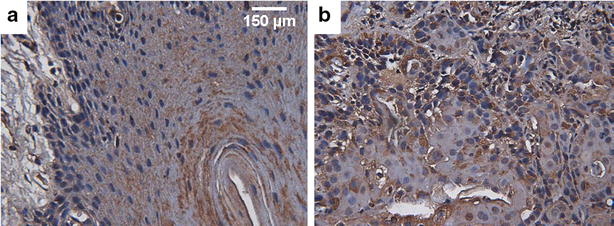
Expression levels of CLIC4 in human normal gingival and oral squamous carcinoma. Immunohistochemical staining images showing the expression level of CLIC4 in human normal gingival tissue (a) and human oral squamous carcinoma (b). CLIC4 protein was recognized by anti-CLIC4 mouse monoclonal primary antibody
Knockdown of CLIC4 enhances ATP-induced HN4 cell apoptosis via the mitochondrial pathway
Recent studies have reported that CLIC4 was a regulator for cancer cell apoptosis [8]. Therefore, we employed specific small interfering RNA (siRNA) to suppress CLIC4 expression to identify the role of CLIC4 in HN4 cell apoptosis. Our data indicated that CLIC4 siRNA significantly suppressed CLIC4 expression (Figs. 3a, 6a and Additional file 1: Figure S1). TdT-mediated dUTP nick end labeling (TUNEL) assay was utilized to detect apoptotic cells, and 100 μmol/L ATP was used to evoke cell apoptosis. Our data showed that the percentage of apoptotic cells was significantly increased in CLIC4-specific siRNA-transfected cells compared with scrambled siRNA-transfected ones (Fig. 2a, b).
Fig. 3.

Effect of CLIC4 on the mitochondrial apoptotic pathway. Representative images (a) and summarized data (b–g) showing the expression levels of CLIC4 (a), Bcl-2 (a, b–c), Bax (a, d–e) and active caspase 3 (a, f–g). HN4 cells were transfected with CLIC4 (+, CLIC4 siRNA) or scrambled (−, Con) siRNA, and treated with (+, +ATP) or without (−, −ATP) 100 μmol/L ATP for 3 h. β-Tubulin was used as a loading control. Values are shown as the mean ± SE. n = 3. *P < 0.05. vs. the control (Con) group
Fig. 6.
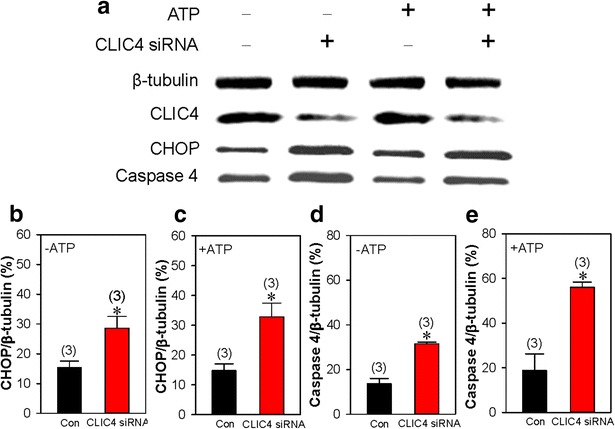
Effect of CLIC4 on the endoplasmic reticulum-stress apoptotic pathway. Representative images (a) and summarized data (b–e) showing the expression levels of CLIC4 (a), CHOP (a, b–c) and active caspase 4 (a, d–e). The HN4 cells were transfected with CLIC4 (+,CLIC4 siRNA) or scrambled (−, Con) siRNA, and treated with (+, +ATP) or without (−, −ATP) 100 μmol/L ATP for 3 h. β-Tubulin was used as a loading control. Values are shown as the mean ± SE. n = 3. *P < 0.05. vs. the control (Con) group
Fig. 2.
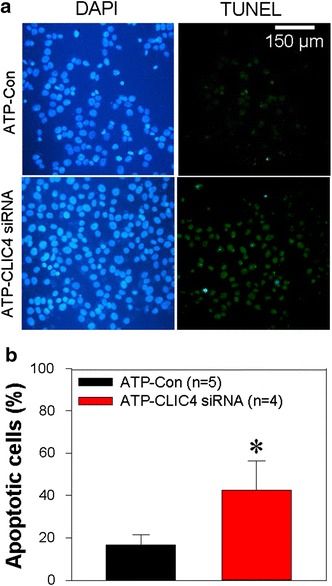
Effect of CLIC4 on the apoptosis of HN4 cells. a Representative images showing the cellular nucleus (DAPI) and apoptotic cells (TUNEL). The HN4 cells were transfected with CLIC4 (ATP-CLIC4 siRNA) or scrambled (ATP-Con) siRNA and treated with 100 μmol/L ATP for 3 h to facilitate apoptosis. b Summarized data showing the percentage rate of the apoptotic cells. The percentage of the apoptotic cells = apoptotic cell number/total cell number. Values are shown as the mean ± SE. n = 4–5. *P < 0.05 vs. the control (ATP-Con) group
It is well documented that mitochondrial signaling pathway crucially participates in cell apoptosis [18]. To elucidate the intracellular signaling pathway, we first examined the expression change in apoptosis-related proteins within the mitochondrial pathway. Immunoblotting data suggested that with or without 100 μmol/L ATP treatment, knockdown of CLIC4 via specific siRNA notably increased the expression of Bax and active caspase 3, which are the proverbial pro-apoptotic proteins, but decreased the expression of Bcl-2, which demonstrates as an anti-apoptotic effect, compared with scrambled siRNA (Fig. 3).
The reduction of the mitochondrial membrane potential or enhanced mitochondrial membrane depolarization is an early sign of cell apoptosis ahead of DNA breakage and activation of caspase 3 [19]. ATP can increase [Ca2+]i and induce mitochondrial membrane depolarization [20]. Therefore, we assessed whether knockdown of CLIC4 affected ATP-induced mitochondrial membrane depolarization. The mitochondrial membrane potential was detected by rhodamine-123, which is a membrane potential-sensitive fluorescent indicator. Our results showed that the application of 100 μmol/L ATP induced mitochondrial membrane depolarization. Compared with scrambled siRNA, CLIC4 siRNA transfection dramatically enhanced ATP-induced mitochondrial membrane depolarization (Fig. 4a, b). These findings strongly indicate that knockdown of CLIC4 enhances ATP-induced HN4 cell apoptosis through the mitochondrial apoptotic pathway.
Fig. 4.

Effect of CLIC4 on ATP-induced mitochondrial membrane depolarization. Traces (a) and summarized data (b) showing 100 μmol/L ATP-induced changes in the HN4 cell mitochondrial membrane potential, which was detected by the cell-permeable membrane potential sensitive-fluorescent indicator rhodamine-123. The change in the membrane potential was indicated by the ratio of the fluorescence density before and after the application of ATP. The ratio increase represents the membrane potential depolarization. The HN4 cells were transfected with CLIC4 (CLIC4 siRNA) or scrambled (Con) siRNA. Values are shown as the mean ± SE. n = 5. *P < 0.05. vs. control (Con) group
Knockdown of CLIC4 enhanced ATP- and TG-induced intracellular Ca2+ release and ER stress-mediated apoptosis
Besides being distributed in the mitochondrial membrane, CLIC4 is also located on the membrane of ER, which is an alternative mediator of apoptosis. Previous studies have suggested that the depletion of ER Ca2+ stores leads to cell apoptosis and growth arrest [21, 22]. The Ca2+ stores can be depleted by both ATP and thapsigargin (TG), an ER Ca2+-ATPase inhibitor [20, 23]. Therefore, we examined the effect of CLIC4 on the ATP- and TG-induced Ca2+ release from ER Ca2+ stores. Our results showed that knockdown of CLIC4 significantly enhanced both ATP- and TG-induced Ca2+ release in HN4 cells, indicating that Ca2+ stores might be overloaded by the loss of CLIC4 (Fig. 5a–d). Because accumulating evidence has revealed that heavy and continued ER stress may activate the process of cell apoptosis [24], we hypothesize that ER stress may also participate in the apoptosis induced by CLIC4 knockdown. To address this issue, in HN4 cells transfected with CLIC4 or scrambled siRNAs and with or without ATP treatment, we examined the expression level of CHOP and active caspase 4, both of which play key roles in ER stress-induced apoptosis [9]. Data showed that knockdown of CLIC4 markedly elevated the expression level of CHOP and active caspase 4 in HN4 cells compared with the scrambled siRNA group regardless of the presence of ATP (Fig. 6). Together, these results indicate that ER stress along with CHOP and caspase 4 activation also participates in ATP-induced apoptosis of HN4 cells following CLIC4 knockdown.
Fig. 5.

Effect of CLIC4 on the Ca2+ store release. Representative traces (a and c) and summarized data (b and d) showing 100 μmol/L ATP− (a and b) and 2 μmol/L thapsigargin (TG, c and d) -induced HN4 cell intracellular Ca2+ ([Ca2+]i) rise in Ca2+-free saline solution. [Ca2+]i was detected by the cell-permeable Ca2+-sensitive fluorescent indicator Fluo-8 AM. The change in [Ca2+]i was indicated by the ratio of the fluorescence density before and after the application of agonists. The HN4 cells were transfected with CLIC4 (CLIC4 siRNA) or scrambled (Con) siRNA. Values are shown as the mean ± SE. n = 5. *P < 0.05. vs. the control (Con) group
Discussion
In the present study, we investigated the functional role of CLIC4, a member of the CLIC family, in the apoptosis of HN4 cells. The major findings in our study are as follows: (1) the CLIC4 expression level was elevated in HNSCC tissue compared with normal gingival tissue; (2) knockdown of CLIC4 via specific siRNA significantly enhanced ATP-induced apoptosis of HN4 cells; (3) knockdown of CLIC4 dramatically increased the expression level of Bax, CHOP, active caspase 3 and active caspase 4, but decreased Bcl-2 expression with or without ATP treatment; (4) suppression of CLIC4 notably strengthened ATP-induced mitochondrial membrane depolarization; (5) Ca2+ measurement data indicated that knockdown of CLIC4 significantly enhanced ATP- and TG-induced Ca2+ release. Taken together, our findings demonstrated that CLIC4 negatively regulated HN4 cell apoptosis via the mitochondrial and ER stress pathways.
HNSCC is one of the six most prevalent cancers that threatens the health of the global population with approximately 600,000 new cases annually [1–3]. Conventional treatment no longer satisfies a better cure rate; therefore, seeking new therapeutic approaches is of great need and significance [2, 25]. It is well documented that Cl− channels undertake essential tasks to maintain the hemostasis of the internal environment and play important roles in biological processes, including the regulation of the cell membrane potential, acidification of organelles, cell proliferation and apoptosis [6]. Among various Cl− channels, CLIC, a newly found class of Cl− channels, was suggested to participate in cancer development and apoptosis [5]. More importantly, CLIC4 gained more attention for its participation in malignant transformation, cellular stress and apoptosis of cancer cells [5]. Several studies have suggested that CLIC4 regulates cancer cell apoptosis and proliferation even it displays an opposite effect in different cancer cells [7–10, 12, 14, 15]. Our immunohistochemical staining and TUNEL assay showed that CLIC4 was elevated in HNSCC tissue, and knockdown of CLIC4 via the transfection of specific siRNA enhanced ATP-induced apoptosis in HN4 cells. ATP is able to be released from the stressed and dying cells [16], for example the cells in the central part of solid tumor. In cancer cells, chemotherapy also can elicit ATP release [26]. Therefore, the results indicated that the loss of CLIC4 may inhibit the development of HNSCC and strengthened curative effect of chemotherapy.
CLIC4 is widely distributed inside cells, particularly on the membrane of two essential organelles, mitochondria and ER, both of which are involved in two major apoptotic pathways [5, 8, 9]. In the mitochondrial pathway, Bcl-2 family proteins undertake an essential task during the regulation of apoptosis. The fate of cells is determined by the ratio of pro-apoptotic and anti-apoptotic Bcl-2 family members [27]. Activated Bax forms oligomers and inserts into the mitochondrial membrane causing dissipation of the mitochondrial membrane potential and cytochrome C release [28]. Subsequently, the apoptotic cascade is activated, whereas Bcl-2 prevents Bax activity via exerting its pro-apoptotic effect [28]. A previous study revealed that the suppression of CLIC4 enhanced the apoptosis induced by H2O2 in glioma C6 cells along with the dissipation of the mitochondrial membrane potential and nuclear translocation of CLIC4 but without the change of Bax/Bcl-2 ratio [10]. In our study, we found that CLIC4 knockdown not only dramatically elevated the expression level of Bax and active caspase 3 but notably down-regulated the Bcl-2 expression with or without ATP treatment in HN4 cells, revealing that Bax/Bcl-2 ratio participated in CLIC4-mediated apoptosis. The mitochondrial membrane potential, which is formed by the normal function of proton pumps plays key roles in maintaining oxidative phosphorylation and ATP production [29]. Enhanced mitochondrial membrane depolarization is a well-recognized sign of apoptosis in the early stage [20]. Our data show that the knockdown of CLIC4 significantly enhanced ATP-induced mitochondrial membrane depolarization in HN4 cells, further proving that the knockdown of CLIC4 exerts a pro-apoptotic effect through the mitochondrial apoptosis pathway and strengthened ATP-induced apoptosis in HN4 cells.
A recent study reported that the inhibition of CLIC4 triggers mitochondria- and ER stress-mediated apoptosis in human glioma U251 cells under starvation [9]. ER stress, which refers to a subcellular pathological state of the ER manifesting as Ca2+ dyshomeostasis and the accumulation of unfolded proteins, is essential in the process of cell apoptosis [30–32]. Our [Ca2+]i measurement results suggested that the knockdown of CLIC4 enhanced ATP- and TG-induced intracellular Ca2+ release, indicating that Ca2+ overload possibly caused by CLIC4 knockdown may contribute to the apoptotic progress of HN4 cells. Heavy and continued ER stress may activate downstream apoptotic signaling molecules, including CHOP and active caspase 4, further leading to cell apoptosis [33–36]. In the present study, we found that the expression levels of ER stress-related proteins, including CHOP and active caspase 4, were strongly increased following the knockdown of CLIC4 with or without ATP treatment. Based on our total findings, the apoptotic response mediated by CLIC4 occurs through both the mitochondrial pathway accompanied with the enhancement of mitochondrial membrane depolarization and activation of caspases 3, and the ER stress pathway with the elevated expression of CHOP and active caspase 4. Accumulating evidence has revealed the synergistic roles played by both the mitochondria and ER in the process of CLIC4-mediated cancer cell apoptosis [9], findings that were consistent with ours.
Conclusions
In conclusion, we demonstrate that the expression level of CLIC4 was elevated in oral squamous carcinoma tissues, and the knockdown of CLIC4 enhanced ATP-induced apoptosis via both the mitochondrial and ER stress pathways in HN4 cells. Our finding indicates that CLIC4 may be considered as a potential target for treating HNSCC and sheds light on future clinical therapy. But the effect of CLIC4 overexpression in the apoptosis of HN4 cells still remains. The future study will benefit this issue.
Methods
Materials
Specific siRNA for human CLIC4 (accession numbers, NM_013943; corresponding to the cDNA sequence 5-GCTGAAGGAGGAGGACAAAGA-3) and scrambled siRNA (5-ACGCGUAACGCGGGAAUUU-3) were designed and obtained from Biomics Company [9]. For cell transfection, Lipofectamine 2000 reagent and opti-MEM I reduced serum medium were purchased from Invitrogen Company. Cell counting kit CCK8 was obtained from Beyotime Company. Anti-β-tubulin, anti-CHOP (GADD-153), anti-Bax, anti-Bcl-2, anti-caspase 3 and anti-caspase 4 rabbit polyclonal primary antibodies and anti-CLIC4 mouse monoclonal primary antibody were purchased from Santa Cruz Biotechnology. The TUNEL bright green apoptosis detection kit was purchased from Vazyme Biotechnology. Rhodamine-123 was obtained from invitrogen.
Immunohistochemistry
Each specimen was obtained with written informed consent from each patient involved. The procedures were conducted in line with the Declaration of Helsinki and the Good Clinical Practice [37, 38]. The surgical specimens of human normal gingival and oral squamous carcinoma were fixed in 4 % poly-formaldehyde for 1 to 2 d. Next, the specimens were dehydrated and embedded in paraffin following routine methods. Paraffin-embedded tissues were sliced into sections with 5 μm thickness. The specimens were treated by deparaffinization and rehydration routinely. Heat-mediated antigen retrieval was performed with citrate buffer (0.01 M, pH 6.0) in a microwave oven. Specimens were treated for 10 min with 3 % peroxide-methanol for quenching endogenous peroxidase ablation and blocked with 10 % normal goat serum for 20 min at room temperature. Next, the specimens were incubated with mouse anti-CLIC4 primary antibody (1:50, sc-135739, Santa Cruz) overnight at 4 °C. After the specimens were washed out and incubated with goat anti-mouse IgG secondary antibody conjugated with horseradish peroxidase for 30 min, avidin–biotin-peroxidase reagents were added. Peroxidase activity was visualized by incubating the specimens with 3, 3′-diaminobenzidine tetrahydrochloride (DAB, Japan) for 10 min, and hematoxylin was used to visualize the cell nucleus. The negative control was performed by incubating with normal mouse serum to replace the primary antibody. Cells exhibiting brown particle staining were considered positive.
Cell culture and transfection
HN4, an HNSCC cell line, was derived from patients with HNSCC [39]. HN4 cells were cultured in Dulbecco’s modified Eagle medium (DMEM, 4.5 g/L glucose) (Life Technologies, USA) supplemented with 10 % fetal bovine serum and antibiotics (100 KU/L penicillin and 100 mg/L streptomycin) in an incubator at 37 °C with 5 % CO2. HN4 cells were transiently transfected with CLIC4 siRNA using Lipofectamine 2000 following the manufacturer’s instruction and then were cultured for another 48 h before the following experiment.
CCK8 assay
The viability of the HN4 cells was determined by the CCK8 assay. After transfection with CLIC4 and scrambled siRNA for 1 d, respectively, HN4 cells were trypsinized and seeded in 96-well plates at an equal density of 6 × 103 cells/well. On the following day, after the treatment was based on experimental design, 10 μl of CCK8 was added to each well with 4 h of incubation in an incubator at 37 °C with 5 % CO2. The absorbance was recorded at a wavelength of 450 nm. The cell viability is proportional to the value of the absorbance value.
Immunoblotting
Western blotting was performed as previously described [40]. Briefly, HN4 cells were lysed by lysis buffer containing 20 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1 % NP-40, 1 % sodium deoxycholate, and 2.5 mM sodium pyrophosphate, plus protease inhibitor cocktail tablets, and total proteins were extracted. Next, 30 μg of proteins was loaded in each well and separated by 12 % SDS-PAGE with a voltage of 100 V for 1 h and then transferred to PVDF membranes. After blocking with 5 % non-fat milk diluted by PBST for 1 h at room temperature, the membrane was incubated at 4 °C overnight with the appropriate primary antibodies. On the following day, the primary antibodies were washed out, and the membrane was incubated with horseradish peroxidase-conjugated secondary antibodies. Each band for specific protein was visualized by ECL. The optical density of each blot was normalized to that of β-actin, analyzed within the same lane and represented as the relative optical density.
TUNEL assay
Cell apoptosis was detected by the TUNEL assay. Experiments were performed according to the manufacturer’s protocol. Briefly, after appropriate experimental treatment, HN4 cells on the cover slip were fixed with 4 % paraformaldehyde solution and then incubated with proteinase K solution (2 mg/ml) for 5 min at 37 °C to inactivate endogenous peroxidase. Next, the cells were treated with equilibration buffer for 20 min at room temperature. After washing with PBS for three times, the cells were incubated in TdT buffer for another 1 h at 37 °C. Following washing with PBS, HN4 cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) for 10 min. Finally, the cells were dried and analyzed with a fluorescence microscope. Each cell was visualized by blue fluorescence (DAPI) at the wavelength of 460 nm, and the apoptotic cells were identified by green fluorescence at the wavelength of 520 nm. Apoptotic percentage = apoptotic cell number/total cell number × 100 %.
Mitochondrial membrane potential measurement
Rhodamine-123 was utilized to measure the mitochondrial membrane potential in HN4 cells [41]. HN4 cells transfected with scrambled or CLIC4 siRNAs were trypsinized and seeded onto circular cover glass placed in 6-well plates. On the following day, the cells were incubated with 2 μmol/L rhodamine-123 for 40 min at 37 °C. After washing, the fluorescence intensity of rhodamine-123 was recorded by confocal laser microscopy. The fluorescent dye was exited at 507 nm, and the signal was collected at an emission wavelength of 529 nm. Each data of the fluorescence intensity were the average of 20–30 cells.
[Ca2+] measurement
Intracellular Ca2+ concentration ([Ca2+]i) was measured using methods described elsewhere [42]. Briefly, HN4 cells were seeded on circular coverslips and incubated with 10 μmol/L Fluo-8 with 0.02 % pluronic acid F-127 at 37 °C for 30 min. Ca2+ release was evoked by treating HN4 cells with 4 μmol/L TG or 100 μmol/L ATP for 10 min in Ca2+-free PBS that contained (in mmol/L) 140 mmol/L NaCl, 1 mmol/L MgCl2, 5 mmol/L KCl, 10 mmol/L glucose, 0.2 mmol/L EGTA, and 5 mmol/L HEPES, pH 7.4. The real-time change of fluorescence indicating [Ca2+]i was recorded by Nikon fluorescence microscopy with a 488 nm excitation and 515 nm long pass emission wavelength. Changes in [Ca2+]i were displayed as the ratio of fluorescence relative to the intensity before the application of ATP or TG (F1/F0). Each data of the fluorescence intensity were the average of 20–30 cells.
Statistical analysis
Collected data are presented as mean ± SE. The two-tailed independent t test was used to compare the results in different groups. A P value <0.05 was considered a significant difference. All of the statistical analyses were performed by the software SigmaPlot 11.0.
Authors’ contributions
HX, JL, YL, JD and BS designed experiments and analyzed data; HX, JL, JL, RY, KW and JW performed experiments; HX, JL and BS wrote the manuscript; HX, JD and BS supervised the project. All of the authors read and approved the final manuscript.
Acknowledgements
We kindly thank Ms. Lele Wu and Mr. Chao Fang for cell culture. This work was supported by grants from the Natural Science Foundation of China (Grant No. 81570403, 81371284); Scientific Research Grant of Anhui Medical University (Grant No. 2015xkj090); Outstanding Young Investigator of Anhui Medical University; Anhui Provincial Natural Science Foundation (Grant No. 1408085MH157); Supporting Program for Excellent Young Talents in Universities of Anhui Province.
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- HNSCC
head and neck squamous cell carcinoma
- CLIC
Cl− intracellular channel
- ER
endoplasmic reticulum
- [Ca2+]i
intracellular Ca2+ concentration
- siRNA
small interfering RNA
- TG
thapsigargin
- ATP
adenosine triphosphate
- DAPI
4′,6-diamidino-2-phenylindole
- TUNEL
TdT-mediated dUTP nick end labeling
Additional file
10.1186/s13578-016-0070-1 Effect of CLIC4 or scrambled siRNA on CLIC4 expression. Effect of CLIC4 or scrambled siRNA on CLIC4 expression. Summarized data showing the expression level of CLIC4. HN4 cells were transfected with CLIC4 or scrambled siRNA, and then were treated with (ATP-Con, ATP-CLIC4) or without (Con, Con-CLIC4) 100 μmol/L ATP for 3 h. β-Tubulin was used as a loading control. Values are shown as the mean ± SE. n = 3. *P < 0.05. vs. the control (Con) group,# P < 0.05 vs. the ATP control (ATP-Con) group.
Footnotes
Haowei Xue and Jinsen Lu contributed equally to this work
Contributor Information
Haowei Xue, Email: xuehaowei@ahmu.edu.cn.
Jinsen Lu, Email: 759567097@qq.com.
Renxiang Yuan, Email: 1255632088@qq.com.
Jinli Liu, Email: liujinli77@126.com.
Yehai Liu, Email: 13856906986@126.com.
Kaile Wu, Email: wukaile@126.com.
Jing Wu, Email: 276288863@qq.com.
Juan Du, Email: dujuan@ahmu.edu.cn.
Bing Shen, Phone: +86-0551-65161132, Email: shenbing@ahmu.edu.cn.
References
- 1.Machiels JP, Lambrecht M, Hanin FX, Duprez T, Gregoire V, Schmitz S, et al. Advances in the management of squamous cell carcinoma of the head and neck. F1000Prime Rep. 2014;6:44. doi: 10.12703/P6-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Behera M, Owonikoko TK, Kim S, Chen Z, Higgins K, Ramalingam SS, et al. Concurrent therapy with taxane versus non-taxane containing regimens in locally advanced squamous cell carcinomas of the head and neck (SCCHN): a systematic review. Oral Oncol. 2014;50(9):888–894. doi: 10.1016/j.oraloncology.2014.06.014. [DOI] [PubMed] [Google Scholar]
- 3.Pancari P, Mehra R. Systemic therapy for squamous cell carcinoma of the head and neck. Surg Oncol Clin N Am. 2015;24(3):437–454. doi: 10.1016/j.soc.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 4.Lehen’kyi V, Shapovalov G, Skryma R, Prevarskaya N. Ion channnels and transporters in cancer. 5. Ion channels in control of cancer and cell apoptosis. Am J Physiol Cell Physiol. 2011;301(6):C1281–C1289. doi: 10.1152/ajpcell.00249.2011. [DOI] [PubMed] [Google Scholar]
- 5.Peretti M, Angelini M, Savalli N, Florio T, Yuspa SH, Mazzanti M. Chloride channels in cancer: Focus on chloride intracellular channel 1 and 4 (CLIC1 AND CLIC4) proteins in tumor development and as novel therapeutic targets. Biochim Biophys Acta. 2015;1848((10 Pt B)):2523–2531. doi: 10.1016/j.bbamem.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 6.Jentsch TJ, Stein V, Weinreich F, Zdebik AA. Molecular structure and physiological function of chloride channels. Physiol Rev. 2002;82(2):503–568. doi: 10.1152/physrev.00029.2001. [DOI] [PubMed] [Google Scholar]
- 7.Fernandez-Salas E, Suh KS, Speransky VV, Bowers WL, Levy JM, Adams T, et al. mtCLIC/CLIC4, an organellular chloride channel protein, is increased by DNA damage and participates in the apoptotic response to p53. Mol Cell Biol. 2002;22(11):3610–3620. doi: 10.1128/MCB.22.11.3610-3620.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mutoh M, Suh KS, Gerdes M, Yuspa SH. CLIC4, an intracellular chloride channel protein, is a novel molecular target for cancer therapy. J Investig Dermatol Symp Proc. 2005;10:105–109. doi: 10.1111/j.1087-0024.2005.200402.x. [DOI] [PubMed] [Google Scholar]
- 9.Zhong J, Kong X, Zhang H, Yu C, Xu Y, Kang J, et al. Inhibition of CLIC4 enhances autophagy and triggers mitochondrial and ER stress-induced apoptosis in human glioma U251 cells under starvation. PLoS One. 2012;7(6):e39378. doi: 10.1371/journal.pone.0039378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu Y, Kang J, Yuan Z, Li H, Su J, Li Y, et al. Suppression of CLIC4/mtCLIC enhances hydrogen peroxide-induced apoptosis in C6 glioma cells. Oncol Rep. 2013;29(4):1483–1491. doi: 10.3892/or.2013.2265. [DOI] [PubMed] [Google Scholar]
- 11.Tang HY, Beer LA, Tanyi JL, Zhang R, Liu Q, Speicher DW. Protein isoform-specific validation defines multiple chloride intracellular channel and tropomyosin isoforms as serological biomarkers of ovarian cancer. J Proteomics. 2013;89:165–178. doi: 10.1016/j.jprot.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suh KS, Malik M, Shukla A, Ryscavage A, Wright L, Jividen K, et al. CLIC4 is a tumor suppressor for cutaneous squamous cell cancer. Carcinogenesis. 2012;33(5):986–995. doi: 10.1093/carcin/bgs115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng DL, Huang QL, Zhou F, Huang QJ, Lin JY, Lin X. PA28beta regulates cell invasion of gastric cancer via modulating the expression of chloride intracellular channel 1. J Cell Biochem. 2012;113(5):1537–1546. doi: 10.1002/jcb.24022. [DOI] [PubMed] [Google Scholar]
- 14.Suh KS, Mutoh M, Gerdes M, Crutchley JM, Mutoh T, Edwards LE, et al. Antisense suppression of the chloride intracellular channel family induces apoptosis, enhances tumor necrosis factor {alpha}-induced apoptosis, and inhibits tumor growth. Cancer Res. 2005;65(2):562–571. [PubMed] [Google Scholar]
- 15.Suh KS, Crutchley JM, Koochek A, Ryscavage A, Bhat K, Tanaka T, et al. Reciprocal modifications of CLIC4 in tumor epithelium and stroma mark malignant progression of multiple human cancers. Clin Cancer Res. 2007;13(1):121–131. doi: 10.1158/1078-0432.CCR-06-1562. [DOI] [PubMed] [Google Scholar]
- 16.Selzner N, Selzner M, Graf R, Ungethuem U, Fitz JG, Clavien PA. Water induces autocrine stimulation of tumor cell killing through ATP release and P2 receptor binding. Cell Death Differ. 2004;11(Suppl 2):S172–S180. doi: 10.1038/sj.cdd.4401505. [DOI] [PubMed] [Google Scholar]
- 17.Humphreys BD. SAPK/JNK activation and apoptotic induction by the macrophage P2X7 nucleotide receptor. J Biol Chem. 2000;275:26792–26798. doi: 10.1074/jbc.M002770200. [DOI] [PubMed] [Google Scholar]
- 18.Walia V, Kakar S, Elble R. Micromanagement of the mitochondrial apoptotic pathway by p53. Front Biosci (Landmark Ed) 2011;16:749–758. doi: 10.2741/3717. [DOI] [PubMed] [Google Scholar]
- 19.Ly JD, Grubb DR, Lawen A. The mitochondrial membrane potential (deltapsi(m)) in apoptosis; an update. Apoptosis. 2003;8(2):115–128. doi: 10.1023/A:1022945107762. [DOI] [PubMed] [Google Scholar]
- 20.Perveen S, Yang JS, Ha TJ, Yoon SH. Cyanidin-3-glucoside inhibits ATP-induced intracellular free Ca(2+) concentration, ROS formation and mitochondrial depolarization in PC12 cells. Korean J Physiol Pharmacol. 2014;18(4):297–305. doi: 10.4196/kjpp.2014.18.4.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Contreras L, Drago I, Zampese E, Pozzan T. Mitochondria: the calcium connection. Biochim Biophys Acta. 2010;1797(6–7):607–618. doi: 10.1016/j.bbabio.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 22.Bernardi P, Rasola A. Calcium and cell death: the mitochondrial connection. Subcell Biochem. 2007;45:481–506. doi: 10.1007/978-1-4020-6191-2_18. [DOI] [PubMed] [Google Scholar]
- 23.Yang Y, Zhu J, Wang X, Xue N, Du J, Meng X, et al. Contrasting Patterns of Agonist-induced Store-operated Ca2+ Entry and Vasoconstriction in Mesenteric Arteries and Aorta With Aging. J Cardiovasc Pharmacol. 2015;65(6):571–578. doi: 10.1097/FJC.0000000000000225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Srinivasan V, Sundar Rajan S, Balasubramanyam M, Tatu U. Endoplasmic reticulum (ER) stress & diabetes. Indian J Med Res. 2007;125:411–424. [PubMed] [Google Scholar]
- 25.Zhu W, Wang X, Zhou Y, Wang H. C2-ceramide induces cell death and protective autophagy in head and neck squamous cell carcinoma cells. Int J Mol Sci. 2014;15(2):3336–3355. doi: 10.3390/ijms15023336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334(6062):1573–1577. doi: 10.1126/science.1208347. [DOI] [PubMed] [Google Scholar]
- 27.Wong WW, Puthalakath H. Bcl-2 family proteins: the sentinels of the mitochondrial apoptosis pathway. IUBMB Life. 2008;60(6):390–397. doi: 10.1002/iub.51. [DOI] [PubMed] [Google Scholar]
- 28.Ranganathan V, Murphy KM, Farnsworth ML, Kavallaris M, Lock RB. BCL-2 inhibits BAX translocation from cytosol to mitochondria during drug-induced apoptosis of human tumor cells. Cell Death Differ. 2002. [DOI] [PubMed]
- 29.Gogol P, Trzcińska M, Bryła M. Motility, mitochondrial membrane potential and ATP content of rabbit spermatozoa stored in extender supplemented with GnRH analogue [des-Gly10, D-Ala6]-LH-RH ethylamide. Pol J Vet Sci. 2014;17(4):571–575. doi: 10.2478/pjvs-2014-0085. [DOI] [PubMed] [Google Scholar]
- 30.Wang WA, Groenendyk J, Michalak M. Endoplasmic reticulum stress associated responses in cancer. Biochim Biophys Acta. 2014;1843(10):2143–2149. doi: 10.1016/j.bbamcr.2014.01.012. [DOI] [PubMed] [Google Scholar]
- 31.Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, et al. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300(5616):135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- 32.Oakes SA, Opferman JT, Pozzan T, Korsmeyer SJ, Scorrano L. Regulation of endoplasmic reticulum Ca2+ dynamics by proapoptotic BCL-2 family members. Biochem Pharmacol. 2003;66(8):1335–1340. doi: 10.1016/S0006-2952(03)00482-9. [DOI] [PubMed] [Google Scholar]
- 33.Fels DR, Koumenis C. The PERK/eIF2α/ATF4 module of the UPR in hypoxia resistance and tumor growth. Cancer Biol Ther. 2006;5:723–728. doi: 10.4161/cbt.5.7.2967. [DOI] [PubMed] [Google Scholar]
- 34.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/S0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 35.Jiang HY, Wek RC. Phosphorylation of the alpha-subunit of the eukaryotic initiation factor-2 (eIF2alpha) reduces protein synthesis and enhances apoptosis in response to proteasome inhibition. J Biol Chem. 2005;280(14):14189–14202. doi: 10.1074/jbc.M413660200. [DOI] [PubMed] [Google Scholar]
- 36.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11(4):381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 37.World Medical Association World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310:2191–2194. doi: 10.1001/jama.2013.281053. [DOI] [PubMed] [Google Scholar]
- 38.Grimes DA, Hubacher D, Nanda K, Schulz KF, Moher D, Altman DG. The good clinical practice guideline: a bronze standard for clinical research. Lancet. 2005;366(9480):172–174. doi: 10.1016/S0140-6736(05)66875-4. [DOI] [PubMed] [Google Scholar]
- 39.Kim SY, Chu KC, Lee HR, Lee KS, Carey TE. Establishment and characterization of nine new head and neck cancer cell lines. Acta Otolaryngol. 1997;117(5):775–784. doi: 10.3109/00016489709113477. [DOI] [PubMed] [Google Scholar]
- 40.Ma Y, Zhang P, Li J, Lu J, Ge J, Zhao Z, et al. Epoxyeicosatrienoic acids act through TRPV4-TRPC1-KCa1.1 complex to induce smooth muscle membrane hyperpolarization and relaxation in human internal mammary arteries. Biochim Biophys Acta. 2015;1852(3):552–559. doi: 10.1016/j.bbadis.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 41.Chen J, Liao W, Gao W, Huang J, Gao Y. Intermittent hypoxia protects cerebral mitochondrial function from calcium overload. Acta Neurol Belg. 2013;113(4):507–513. doi: 10.1007/s13760-013-0220-8. [DOI] [PubMed] [Google Scholar]
- 42.Shen B, Zhu J, Zhang J, Jiang F, Wang Z, Zhang Y, et al. Attenuated mesangial cell proliferation related to store-operated Ca2+ entry in aged rat: the role of STIM 1 and Orai 1. Age (Dordr). 2013;35(6):2193–2202. doi: 10.1007/s11357-013-9511-5. [DOI] [PMC free article] [PubMed] [Google Scholar]