

Abstract
Intracellular lipid binding proteins, including fatty acid binding proteins (FABPs) 1 and 2, are highly expressed in tissues involved in the active lipid metabolism. A zebrafish model was used to demonstrate differential expression levels of fabp1b.1, fabp1b.2, and fabp2 transcripts in liver, anterior intestine, and brain. Transcription levels of fabp1b.1 and fabp2 in the anterior intestine were upregulated after feeding and modulated according to diet formulation. Immunofluorescence and electron microscopy immunodetection with gold particles localized these FABPs in the microvilli, cytosol, and nuclei of most enterocytes in the anterior intestinal mucosa. Nuclear localization was mostly in the interchromatin space outside the condensed chromatin clusters. Native PAGE binding assay of BODIPY-FL-labeled FAs demonstrated binding of BODIPY-FLC12 but not BODIPY-FLC5 to recombinant Fabp1b.1 and Fabp2. The binding of BODIPY-FLC12 to Fabp1b.1 was fully displaced by oleic acid. In vivo experiments demonstrated, for the first time, that intestinal absorption of dietary BODIPY-FLC12 was followed by colocalization of the labeled FA with Fabp1b and Fabp2 in the nuclei. These data suggest that dietary FAs complexed with FABPs are able to reach the enterocyte nucleus with the potential to modulate nuclear activity.
Keywords: BODIPY-labeled fatty acids, diet and dietary lipids, electron microscopy, fatty acid binding protein 1, fatty acid binding protein 2, fluorescence microscopy, gene expression, intestine, nucleus, zebrafish, Danio rerio
Fatty acid binding proteins (FABPs) were initially described as abundant intracellular cytosolic proteins with relatively low molecular masses (14–15 kDa) able to bind organic anions (1) and radioactively labeled FAs (2, 3). Together with cellular retinol and retinoic acid binding proteins, these abundant chaperone proteins are members of an ancient conserved multigene family of intracellular lipid binding proteins (4–6). The evolutionary relationships of vertebrate FABPs were clarified using phylogenetic and conserved synteny analyses (7, 8). They bind long-chain FAs (LCFAs) and other lipophilic compounds (9–12) and are believed to be implicated in FA intracellular uptake and transport, lipid metabolism regulation, protection from the harmful effects of nonesterified LCFAs, and modulation of cell growth and proliferation (13–17). Despite a low amino acid sequence identity among the various FABPs, these proteins fold in a highly conserved tertiary structure. A typical member of the family consists of 127–134 amino acid residues with 10 or 11 antiparallel β-strands folded into a barrel capped by two short α-helices (18). The ligand binding site is within the large water-filled interior (19, 20), but binding sites and interaction modes with some other lipophilic compounds may vary (12).
Duplication of genes and entire genomes is a major evolutionary force driving the increasing complexity of organisms (21), and the history of the FABP family has been shaped by duplication events (8). The molecular diversity of FABPs, together with overlapping and tissue-specific expression patterns, may facilitate redundancy, neofunctionalization, and subfunctionalization of biochemical properties. The precise in vivo function of each FABP is not yet clearly understood. Distinct developmental and tissue-specific expression patterns support the hypothesis of a subspecialization in ligand preferences and/or suggest diversity in uncharacterized functions.
It was demonstrated, using cell cultures, that FABPs may channel unesterified FAs and other lipophilic ligands into nuclei, potentially targeting them to transcription factors, and initiate nuclear receptor transcriptional activity (11, 22–33). To our knowledge, no in vivo data are currently available to support the hypothesis that exogenous FAs enter cell nuclei via their binding to FABPs. The dietary triacylglycerols are the major source of lipids in the intestinal lumen. Their hydrolysis releases large quantities of medium- to long-chain FAs absorbed by the enterocytes via complex mechanisms, involving both passive diffusion and protein-mediated transport (34–38). Once inside the cell, dietary FAs are reversibly bound to lipid binding proteins involved in the transport of FAs from the plasma membrane to cellular compartments (e.g., FABPs and acyl-CoA binding proteins). The intestinal mucosa and its enterocytes provide a very attractive system for evaluating the fate of exogenous FAs inside cells. FABP1 and FABP2 are the most strongly expressed FABP family members in the human small intestine (13, 14), and these proteins are found in abundance in absorptive cells (39, 40). In zebrafish, the anterior intestine is the major site of fat absorption (41–43), and different fabp genes were found expressed in the intestine including fabp1a, fabp1b.1, fabp1b.2, fabp2, fabp3, fabp6, fabp7a/fabp7b, fabp10a/fabp10b, and fabp11a (41, 44–47). The teleost ancestor experienced a whole-genome duplication event at the base of the teleost radiation (48) leading, in some cases, to the retention of pairs of duplicate genes (e.g., fabp1a/fabp1b). The resulting ohnologs may have retained their initial subfunctions, as well as acquiring new functions (i.e., neofunctionalization), and/or duplicate genes may be preserved by partitioning ancestral subfunctions between them (i.e., subfunctionalization). In addition, tandem gene duplication may arise as demonstrated with fabp1b.1 and fabp1b.2 found adjacent in the zebrafish genome (44).
Zebrafish were used to study the expression pattern and dietary regulation of homologs to human FABP1 and FABP2, which are significantly expressed in the anterior intestine of this vertebrate model. The next step was cellular and subcellular immunolocalization of Fabp1b and Fabp2 along the intestinal villi. Third, an assessment of the fate of dietary fluorescent (BODIPY)-labeled analogs of FAs revealed that dietary BODIPY-FLC12 (medium chain) but not BODIPY-FLC5 (short chain) was colocalized with Fabp1b and Fabp2 in enterocyte nuclei.
MATERIALS AND METHODS
Animals
Wild-type zebrafish (Danio rerio) were produced in our facilities according to the rules approved by the Ministère de l’Agriculture, de l’Agroalimentaire et de la Forêt, France, under permit number A33-522-6. All experiments were conducted in conformity with the 2010/63/EU directive on the protection of animals used for scientific purposes. Larvae were obtained by natural mating and raised in embryo water [90 µg/ml Instant Ocean (Aquarium Systems, Sarrebourg, France), 0.58 mM CaSO4, 2H2O, dissolved in reverse-osmosis purified water] at 28.5°C with an 11:13 light/dark photoperiod.
Feeding experiments and tissue sampling
Zebrafish adults were fed ad libitum with TetraMin flakes (Tetra GmbH, Melle, Germany) containing 48% protein, 8% lipid, 11% ash, 2% fibers, and 6% moisture. Larvae were fed on ZF Biolabs formulated diet (FD) flakes (Tres Cantos, Spain), dried chicken egg yolk (CEY) powder, or olive oil (OO).
Zebrafish eleutheroembryos at 5 days postfertilization (dpf) and larvae at 15 dpf, as well as adult males, were sampled for real-time quantitative PCR (qRT-PCR). On the day preceding the nutritional trials, larvae and adults were divided into two groups and transferred into containers with embryo water but no food. After ∼18 h (larvae) or 22 h (adults) food deprivation, one group of larvae or adults was fed for 2–2.5 h, while the second group continued to fast. After feeding, the eleutheroembryos/larvae were transferred into clean embryo water to eliminate any remaining food, and six pools of 25 eleutheroembryos (5 dpf) or 20 larvae (15 dpf) were immediately frozen in liquid nitrogen and stored at −80°C until RNA extraction. Two separate batches were used as sources of larvae experiments. The adult males were dissected, and six individual samples of liver and anterior intestine were isolated. The ventral white adipose tissue was carefully removed, as it was closely associated with the digestive tract on the ventral side and also with liver lobes at some points (49). Samples were immediately frozen in liquid nitrogen and stored at −80°C until RNA extraction for the RT-PCR and qRT-PCR experiments. Larvae and adult animals from the fasted group were sampled and treated in the same way as the fed animals. In addition to the liver and anterior intestine, six separate brain samples were collected, one ovarian, one testicular, and one heart sample were collected from a pool of six animals, and one kidney sample was collected from a pool of four animals. All samples, except the heart, kidney, and ovarian tissues, were collected from male organisms.
For whole-mount in situ hybridization, 15 dpf zebrafish larvae (i.e., at a fully exotrophic nutritional stage) were fasted for 24 h and were then fed for 3 h with FD, dried CEY, or OO. Approximately 15 larvae per treatment or sampling point were fixed in 4% paraformaldehyde (PFA) at 4°C overnight, rinsed three times in PBS (137 mM NaCl, 2.7 mM KCl, 0.02 M PO4), and dehydrated by successive washes with methanol (25%, 50%, 75%, 95%, and 100%) and stored at −20°C prior to analysis.
Dietary experiments with the green fluorescent BODIPY-FA analogs were performed using adult zebrafish. BODIPY [boron dipyrromethene (4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene)] and BODIPY-FL analogs (Life Technologies) were resuspended in methanol (1 µg/µl) and mixed well with a CEY emulsion (10 µl/ml), prepared from dried CEY mixed in water using an IKA Ultra-Turrax Homogenizer. The BODIPY493/503, BODIPY-FLC5 (D3834), or BODIPY-FLC12 (D3822)-CEY emulsion was maintained at 37°C for 10 min for the solvent to evaporate. BODIPY493/503 (D3922) was used as a negative control. Animals previously starved for 5 days were fed ad libitum (labeled emulsion/fish water = 1:600, v/v), and their food intake was checked visually. Samples were taken 3 h after feeding. Quantitative analysis of dietary BODIPY-FA analog fluorescence signal after intestinal absorption in zebrafish intestinal villi was determined using ImageJ free-processing software (National Institutes of Health, http://rsb.info.nih.gov/ij/) as previously described (50).
Extraction of total RNA, reverse transcription, and PCR analyses
Total zebrafish RNA was extracted using the NucleoSpin® RNA II extraction kit (Macherey-Nagel, Hoerdt, France) according to the manufacturer’s instructions. Total RNA was checked for integrity by ethidium bromide staining in agarose borax gel and quantified using a NanoDrop ND-1000 spectrophotometer (Nyxor Biotech, Paris, France). Reverse transcriptions were performed with 1 µg total RNAs using the StrataScriptTM qRT-PCR cDNA synthesis kit (Stratagene, Amsterdam, The Netherlands), with an oligo(dT) primer, according to the manufacturer’s instructions. The reaction mixture (20 µL) was incubated at 37°C for 30 min, 42°C for 1 h 30 min, and then 70°C for 10 min. Two reverse transcriptions and one reverse transcription negative (RTneg) control (i.e., without reverse transcriptase) were performed on each sample. Qualitative PCR was also carried out on 2 µl reverse transcription reaction product diluted 1:40 in a final volume of 20 µl containing 10 µl Brilliant® SYBR® Green qRT-PCR Master Mix and 0.25 µM of each primer. An aliquot of the PCR reactions was electrophoresed on 1.5% agarose gel containing ethidium bromide, and PCR products were visualized and photographed.
qRT-PCR
Transcripts were amplified using the MX 3000P qRT-PCR thermal cycler instrument (Stratagene). qRT-PCR amplification was carried out in a final volume of 20 µl, using 10 µl Brilliant® SYBR® Green qRT-PCR master mix (Stratagene), 0.25 µM oligodeoxyribonucleotide primers, and 3 µl diluted cDNA: 5 and 15 dpf zebrafish eleutheroembryos/larvae (1/160), anterior intestine (1/160), liver (1/80), and various adult tissues (1/40). Each run included a standard curve made up of seven to eight serial dilution points of a pool of cDNA from the samples to be quantified. Amplification was performed with systematic negative controls [nontemplate control containing no cDNA (NTC) and RTneg]. Each reverse transcription from the same sample was amplified in duplicate, and, when there were no significant differences in CT, the two reverse transcription products were pooled. Elongation factor 1α (eef1a1) and peptidylprolyl isomerase aa (ppiaa) were chosen as normalizing genes for qRT-PCR. The reference sequence, design details, size of the fragment produced, and Tm of the primer pairs used are described in supplementary Table 1. The qRT-PCR profiles contained an initial denaturation step at 95°C for 10 min, followed by 40 cycles: 30 s at 95°C, 30 s at the specific primer pair annealing Tm (see supplementary Table 1), and 10 s (30 s for eef1a1) at 72°C. After the amplification phase, 1 min incubation at 95°C and 30 s at 55°C was followed by a ramp up to 95°C, at 0.01°C/s, where data were collected in continuum to obtain a single product dissociation curve. The qRT-PCR product sizes were checked on 2% agarose gel, and all amplicons were sequenced to confirm specificity. No amplification was observed in RTneg controls, and no primer-dimer formation occurred in the NTC. qRT-PCR efficiency was 100 ± 2%, and the correlation coefficient was >0.995 for each run. Six independent samples (5 or 15 dpf eleutheroembryos/larvae and liver or anterior intestine from individual adults) were tested in triplicate each time. Four independent brain samples were tested in triplicate. Zebrafish heart (pooled from six animals), kidney (pooled from four animals), ovary, and testicle samples were subjected to a single determination in triplicate.
Whole-mount in situ hybridization
Whole-mount in situ hybridization was carried out on zebrafish larvae, sampled, and fixed in 4% PFA, as described previously. A clone purchased from the German Resource Center for Genome Research, Berlin, Germany (IMAGE: 7225625), corresponding to GenBank database dbEST zebrafish clone gb BC095259.1, was used to generate the fabp1b RNA probe. The sense (ZF-fabp1b F1) 5-CAAGACTATTGTGAACAGAGA-3 and antisense (ZF-fabp1b R1) 5-TGAGATTGAGAACACTTTAATG-3 primers were designed from this clone and used for probe synthesis, as previously described (43), except that the primer annealing temperature (Tm) in the thermal profile was 55°C for fabp1b in the first PCR amplification. For the fabp2 RNA probe, the ZF-FB clone (41) was used, corresponding to a 203 bp PCR product of GenBank database dbEST zebrafish clone gbAJ132590 after amplification with the sense (oligo ZFA1) 5-CTGTCATCATCATGACCTTCAACGG-3 and antisense (oligo ZF A3) 5-CCGCACACTGGAAATTAACTTTAC-3 primers, subcloned using the pGEM-T Easy vector kit (Promega, France). The second PCR was performed with the two pGEM-T Easy vector cDNAs, using the T7 and SP6 universal primers, as previously described (43). As the fragments were sense oriented in the vector, the PCR template for the fabp1b.1 sense probe was produced using T7 universal primer, while the PCR template for the antisense probe was produced using SP6 universal primer and the inverse for the fabp2 probes. Both antisense and sense digoxigenin (DIG)-labeled RNA probes were synthesized using the DIG RNA labeling kit (SP6/T7) (Roche Diagnostics, Meylan, France), following the manufacturer’s instructions. The 211 bp fabp1b probe was able to hybridize to fabp1b.1 and fabp1b.2 transcript variants. The fabp2 probe was 203 bp long. The protocol for in situ hybridization was as previously described (51, 52), except that prehybridization and hybridization were conducted at 60°C. The posthybridization stringent baths were at hybridization temperature, except for the last two baths in 0.2× SSC Tween at 57°C (30 min each). In contrast, the hybridization buffer contained 50% formamide, and the animals were incubated in preabsorbed sheep anti-DIG-AP Fab (Roche Diagnostics) fragments at 1:5,000 dilution at 4°C overnight. The antibody was rinsed in six PBS-Tween baths for 30 min each time.
Amino acid sequence analyses
Deduced protein sequences were extracted from the UniProt (53) database. Sequences were aligned (supplementary Fig. 1) using the ClustalW2 program (54).
Recombinant Fabp1b.1 and Fabp2 production and purification
Zebrafish fabp1b.1 and fabp2 full coding cDNA sequences (supplementary Table 1) were cloned in the pET5a vector (Promega). pET5a-Fabp1b.1 and pET5a-Fabp2 constructs were transformed into BL21 (DE3) Star Escherichia coli strain (Novagen). Bacterial cells containing the relevant expression plasmid were cultured in 2× yeast extract and tryptone media at 30°C for Fabp1b.1 and 37°C for Fabp2 for ∼4 h before Fabp synthesis was induced by adding 0.4 mM isopropyl β-d-1-thiogalactopyranoside and incubating for a further 4 h. After centrifugation at 5,000 g for 10 min, the cells were collected and resuspended in cell lysis buffer (30 mM Tris-HCl, 1 mM EDTA, 1 mM DTT, pH 8.3) and ruptured by sonication. Cell debris was removed by centrifugation at 15,000 g at 4°C for 30 min, and the supernatant was processed in two ammonium sulfate precipitation steps. (NH4)2SO4 was first added to a final concentration of 30% with stirring at room temperature for 2 h, followed by centrifugation at 15,000 g for 15 min. The supernatant fraction was treated with (NH4)2SO4 to a final concentration of 50% and centrifuged at 15,000 g for 15 min. The latter supernatant fraction (∼40 ml) was dialyzed against 4 liters of 30 mM Tris-HCl, pH 8.3, at room temperature for 12 h, changing the buffer every 4 h, and finally concentrated by ultrafiltration. The concentrated fraction (∼10 ml) was loaded onto a SephadexG50 fine gel filtration column (141 cm × 1.9 cm). Chromatography was performed at 4°C, using 30 mM Tris-HCl, pH 8.3, as the mobile phase at a flow rate of 15 ml/h. Fabp fractions were analyzed by SDS-PAGE, MALDI-TOF, and MALDI-TOF-TOF MS to determine protein purity.
Antibody production and characterization
Polyclonal antisera against recombinant zebrafish Fabp1b.1 and Fabp2 were obtained by injecting New Zealand white rabbits with 0.8 mg purified recombinant proteins dissolved in 0.4 ml 30 mM Tris-HCl, pH 8.3, together with an equal amount of Freund’s complete adjuvant. One month later, a booster injection contained 0.4 mg purified protein in Freund’s incomplete adjuvant. Bleed tests were carried out at days 0, 20, and 40, for antibody title response measurement, and a final blood sample was collected at day 68. Blood was allowed to clot and retract at 37°C for 1 h, followed by 16 h at 4°C, and the collected serum was stored at −20°C. Crude polyclonal antisera were purified by affinity column chromatography at room temperature after linking highly purified Fabp1b or Fabp2 to cyanogen bromide-activated Sepharose (Sigma-Aldrich), according to the manufacturer’s instructions. The eluate was washed by adding 0.1 M glycine and 0.15 M NaCl, pH 8.2, until its A280 nm reached zero. The antibodies were desorbed using 0.1 M glycine and 0.5 M NaCl, pH 2.6. The fractions collected were immediately spiked with 1:10 (v/v) Tris-HCl 1 M, pH 8.3, to neutralize the pH and stored at −20°C until use. Antiserum titration was performed by ELISA. Plates were coated with 100 ng highly purified Fabp1b.1 or Fabp2. All the following steps were conducted in PBS-Tween buffer. Plates were blocked with 5% BSA at room temperature for 1 h. Serial dilutions (1:200 to 1:102,400) of each antiserum were incubated for 2 h at room temperature. Plates were extensively washed, incubated with Alexa-488 goat anti-rabbit IgG antibody (Molecular Probes), and, finally, extensively rewashed. The signal was measured in a Varioskan Flash spectrofluorometer (Thermo Scientific). Antibody specificity was assayed by Western immunoblotting. Following SDS-PAGE, total proteins extracted from 5 dpf zebrafish eleutheroembryos were transferred to Hybond-C membrane (GE Healthcare Amersham). Membranes were blocked with 0.1% BSA and 2% glycine in PBS-Tween, and then incubated in the presence of crude anti-Fabp1b.1 (1:10,000) or anti-Fabp2 (1:1,000) antisera or purified polyclonal antibodies at room temperature for 2 h. Membranes were washed and incubated with affinity purified anti-rabbit IgG-alkaline phosphatase produced in goat (1:30,000; Sigma-Aldrich), at room temperature for 1 h and then washed again. Alkaline phosphatase activity was developed with 0.33 mg/ml nitro-blue tetrazolium and 0.165 mg/ml 5-bromo-4-chloro-3′-indolyphosphate in developing buffer (100 mM Tris-HCl, pH 9.5, 0.1 M NaCl, and 5 mM MgCl2). Proteins extracted from 5 dpf zebrafish eleutheroembryos exhibited one immunoreactive band at the right predicted relative molecular mass of Fabp1b.1 and Fabp2, thus demonstrating the monospecificity of each antibody (supplementary Fig. 2).
Immunofluorescence labeling analyses
Immunofluorescence was measured on dissected zebrafish digestive cavity and anterior intestinal fragments. Tissues were fixed with 4% PFA in PBS at 4°C overnight and embedded in graded alcohol/paraplast and toluene/paraplast series, and then, finally, in paraplast. Sections 10 µm thick were then used for immunodetection and confocal microscopy. The sections were blocked in 5% normal goat serum, 0.1% BSA, and 0.05% Tween-20 in PBS (PBST) at room temperature for 20 min. Sections were incubated with rabbit preimmune serum, or purified polyclonal anti-Fabp1b.1 (1:2,000), or anti-Fabp2 (1:200) primary antibodies in 1% normal goat serum and 0.1% BSA in PBST at 37°C for 2 h. After extensive washing, 4 × 5 min with PBS, the sections were incubated with secondary Alexa 488- or Alexa 594-conjugated goat anti-rabbit IgG (H+L) (Life Technology) at 1:1,000 dilution in PBS at room temperature for 1 h. The slides were then immersed in a solution of To-Pro 3 iodide (TP3) or 4’,6’-diamidino-2-phenylindole (DAPI) (1:4,000) to label the nuclei red or blue. Following several washes in PBS, samples were mounted with Gold ProLong antifade mounting medium (Life Technology), sealed with nail polish, and stored in a dark place at 4°C prior to analysis. Control sections were treated without the primary antibody. Sections were viewed using an Olympus FV-300 scanning laser confocal microscope.
Electron microscope immunocytochemistry
Fragments of anterior intestine were fixed in 1% glutaraldehyde and 4% formaldehyde in 0.1 M PBS, pH 7.4, for 12 h, changing the solution three times. After washing in the same buffer, the samples were embedded in LR white resin (Sigma-Aldrich). Ultrathin sections were treated successively with glycine 0.02 M and PBS, blocked with 1% BSA for 5 min, treated with primary antibody diluted in PBS containing 1% BSA for 1 h, washed with PBS, and then incubated with the 10 nm protein A-colloidal-gold complex (Sigma-Aldrich) in PBS containing 1% BSA for 1 h. All the incubation samples consisted of a 30 µl drop of solution at 25°C. Controls were prepared by floating sections on PBS to replace primary antibodies. Slices were contrasted with 5% uranyl acetate for 5 min and examined under a Zeiss 6EM902 electron microscope. Subnuclear immunogold particle density was determined using the area and particle tools in ImageJ free-processing software.
BODIPY-FL FAs binding to recombinant Fabps
Stock 2 mM solutions of BODIPY-FLC5 or BODIPY-FLC12 in ethanol were diluted in PBS prior to mixing and incubating with purified proteins for a few minutes. Prior to binding assays, the proteins were delipidated in batch using hydroxyalkoxypropyl-dextran (Sigma-Aldrich) chromatography, equilibrated with 50 mM Tris-HCl, pH 7.4, at 37°C for 45 min. The proteins were then diluted at the working concentration with 30 mM Tris HCl, pH 8.3, sample buffer. Five microliters of 20 µM BODIPY-FLC5 or BODIPY-FLC12 were mixed with 20 µl 5.6 µM recombinant zebrafish Fabp1b.1, 12 µl 4.3 µM tropomyosin A (TPMA) from Echinococcus granulosus (GenBank: AAB65799.4), or 6.6 µl 7.5 µM BSA. A 12 µl sample of 20 µM BODIPY-FLC5 or BODIPY-FLC12 was mixed with 20 µl 30 µM recombinant zebrafish Fabp2. The binding of fluorescent carbon chain BODIPY FAs to recombinant Fabps was then evaluated by an in-gel fluorescence imaging method derived from the one previously described for visualizing BODIPY-arachidonic acid-tagged cellular proteins (55, 56). A dye-free 4× loading sample buffer containing 200 mM Tris-HCl (pH 6.8), 20% glycerol, and 8% SDS was then added to each sample. Samples were then loaded on a 1.5 mm thick, 15% native polyacrylamide gel and electrophoresed using a running buffer containing 0.05 M Tris-HCl and 2.6 M glycine. After in-gel fluorescence imaging (G:Box, Syngene), gels were stained with Coomassie blue R250 to check for protein loading and electrophoretic migration. All manipulations were done in the dark.
Fluorescence emission spectra of BODIPY-FLC5 or BODIPY-FLC12 in the presence and absence of Fabp1b.1 were determined at 25°C using a Cary Eclipse fluorescence spectrophotometer (Agilent Technologies, Santa Clara). BODIPY-FLC5 or BODIPY-FLC12 2 mM stock solutions in ethanol were diluted in PBS prior to mixing and incubating for 2 min with purified FABP1b.1. The protein sample was previously dissolved with 30 mM Tris HCl, pH 8.3, buffer. The emission spectrum was measured from 500 to 600 nm while the excitation wavelength was set at 488 nm.
Displacement experiments
Fabp1b.1 was dissolved in 30 mM Tris HCl, pH 8.3, sample buffer (5.6 µM, 22.5 µl) and incubated with BODIPY-FLC12 (20 µM, 7.5 µl) at room temperature for 10 min. The solution was then mixed with 3 µl unlabeled FA taken from a 5 mM stock solution in ethanol and supplemented with 10 µl loading sample buffer used for loading on the native polyacrylamide gel. Under these conditions, the unlabeled FAs were in ∼100-fold molar excess (i.e., 0.35 mM) over the labeled FA and protein used. The following competitor ligands assayed were oleic acid (C18:1w9), linoleic acid (C18:2w6), and DHA (C22:6w3). The samples were mixed and allowed to equilibrate for 20 min at room temperature before loading 29 µl into the native polyacrylamide gel.
Tandem MS
Stained PAGE bands were faded, excised, and in-gel digested with trypsin in the appropriate buffer. The peptides resulting from the proteolytic digestion were identified by peptide mass fingerprinting and collision-induced dissociation using a 4800 MALDI-TOF-TOF mass spectrometer (Applied Biosystems). The MS data were analyzed using Mascot software (Matrix Science Ltd., London, United Kingdom) and searching the NCBInr and ZFIN zebrafish databases.
Statistics
Variations in transcription levels (normalized by reference genes) were analyzed for statistical significance using REST-384-version 2 software for multiple reference genes (http://rest.gene-quantification.info/) and/or a nonparametric Mann-Whitney test (GraphPad InStat v3.5, GraphPad Software). The significance level was set at 0.05. Data are presented as mean ± SD. The same test was used to check for variations in normalized ef1α/ppiaa values.
RESULTS
FABP1 and FABP2 homologs in zebrafish
Tandem duplicated fabp1b.1 and fabp1b.2 on zebrafish chromosome 8 and fabp2 on chromosome 1 (http://zfin.org/) generated proteins of the predicted molecular mass of 14,115 Da and 128 amino acids long, 14,461 Da and 128 amino acids long, and 15,090 Da and 132 amino acids long, respectively. Human FABP1 exhibited 54.33% and 41.73% identity with Fabp1b.1 and Fabp1b.2, respectively (supplementary Fig. 1). Zebrafish Fabp2 was identical in size and exhibited 66.67% identity with its human homolog (supplementary Fig. 1). Zebrafish Fabp1b.1 and Fabp1b.2 exhibited 21.09% and 15.62% identity with zebrafish Fabp2, while these two proteins showed 50.78% identity between them.
Tissue-specific distribution of zebrafish fabp1b.1, fabp1b.2, and fabp2 transcripts
Qualitative RT-PCR detected fabp1b.1 and fabp1b.2 transcripts in total RNA extracted from the anterior intestine in adult zebrafish, irrespective of the animals’ nutritional status (Fig. 1A). qRT-PCR indicated similarly high levels of transcripts for fabp1b.1 and fabp2 in the anterior intestine, by comparison with normalizing genes eef1a1 (Fig. 1B, D) and ppiaa (data not shown). The fabp1b.2 mRNA levels were around 3-fold lower (Fig. 1C, data not shown). The normalized level of fabp1b.2 transcripts was very high in brain (Fig. 1C, data not shown). Trace amounts of fabp2 transcripts were also expressed in adult brain while fabp1b.1 transcripts were not detected. By contrast, fabp1b.1 transcripts were detected in the liver, together with very small amounts of fabp2 mRNA, while fabp1b.2 transcripts were not detected (Fig. 1; data not shown). Zebrafish fabp1b.2 transcripts were also detected in ovaries, testicles, kidneys, and heart, and trace amounts of fabp2 transcripts were found in kidneys.
Fig. 1.

The fabp1b.1, fabp1b.2, and fabp2 transcript expression in various adult zebrafish tissues. A: RT-PCR products of fabp1b.1and fabp1b.1 transcripts resolved by electrophoresis on agarose ethidium bromide gels. The eef1a1 transcripts were analyzed to assess qualitative integrity of mRNAs. Lane 1: size standard. Lanes 2 to 7: RT-PCR products generated from total RNAs and extracted from dissected brain (b) (lanes 2 and 3), anterior intestine (ai) (lanes 4 and 5), and liver (l) (lanes 6 and 7) after a period of over 24 h fasting (lanes 2, 4, 6) or 22 h fasting followed by 2 h feeding (lanes 3, 5, 7). Lane 8: A negative control lacking reverse transcriptase generated no RT-PCR products. B–D: qRT-PCR analysis of fabp1b.1 (B), fabp1b.2 (C), and fabp2 (D) transcript expression was performed on fed animals after dissecting various tissues. Six independent liver or anterior intestine and four brain samples from individual adults were tested in triplicate in each condition. Zebrafish heart (h; pool from six animals), kidney (k; pool from four animals), ovary (o), and testicle (t) samples were subjected to a single determination in triplicate. The eef1a1 transcript levels were determined and used as a normalizing gene. The results shown are the number of copies of fabp1b.1, fabp1b.2, and fabp2 transcripts divided by the number of copies of eef1a1. nd, no amplification product detected.
Regulating fabp1b.1 and fabp2 transcript levels by the nutritional status and lipid composition of the diet and cephalocaudal patterning of their expression in gut
The aim of the experimental work was to determine whether the fabp1b.1 and fabp2 transcript levels were regulated after feeding and/or according to the lipid composition of the diet. Suitable primer sets were designed on the basis of the cDNA sequences previously obtained to be used for qRT-PCR and primers for synthesizing specific probes for whole-mount, in situ hybridization. The modulation, if any, of transcript levels after feeding and by dietary lipid composition was evaluated by using commercially and in-house formulated synthetic larval diets and the techniques described previously.
The first step was to study whether regulation of fabp1b.1 and fabp2 transcript levels occurred in the zebrafish adult intestine and liver after feeding. As indicated in Fig. 2, transcript levels of fabp2 (P = 0.0087) were upregulated by 1.59-fold and fabp1b.1 by 1.36-fold (not significant) in the anterior intestine when the reference gene used was eef1a1. REST software, using both ppiaa and eef1a1as reference transcripts, gave P = 0.021 for fabp2. This upregulation was not observed in the liver (Fig. 2). A similar study was then conducted at early developmental nutritional stages (i.e., first feeding at 5 dpf) and at the start of the exogenous nutritional phase of larval development (i.e., 15 dpf) (Fig. 3). There were no significant differences in fabp1b.1 and fabp2 transcript levels between nourished and unnourished 5 dpf eleutheroembryos when normalized with the two reference genes, while a slight upregulation of fabp1b.1 was detected when using ppiaa only (×1.15-fold, P = 0.026). Transcripts levels of fabp1b.1 and fabp2 were also significantly upregulated after feeding in whole 15 dpf zebrafish larvae homogenates, irrespective of the reference gene used (Fig. 3). The fabp1b.1 transcript levels were 1.41 (P = 0.026) and 1.54 (P = 0.0022) higher, respectively, when eef1a1 and ppiaa were used as reference transcripts. REST software using the two reference transcripts at the same time gave a P value of 0.001. The fabp2 transcripts levels were 1.59 (P = 0.026) and 1.71 (P = 0.0022) higher, respectively, when eef1a1 and ppiaa were used as reference transcripts. REST software using the two reference transcripts at the same time gave a P value of 0.004. The effect of diet composition on the level of fabp1b (fabp1b.1 + fabp1b.2) and fabp2 transcript levels was then studied by whole-mount in situ hybridization (Fig. 4). An upregulation of fabp1b and fabp2 hybridization signals between starved and fed conditions was detected, and the signal was apparently modulated according to the lipid composition of the diet. Larvae (15 dpf) nourished with FD experienced a high induction of the signal after feeding, while larvae fed with dried CEY or OO were in an intermediate state (Fig. 4).
Fig. 2.
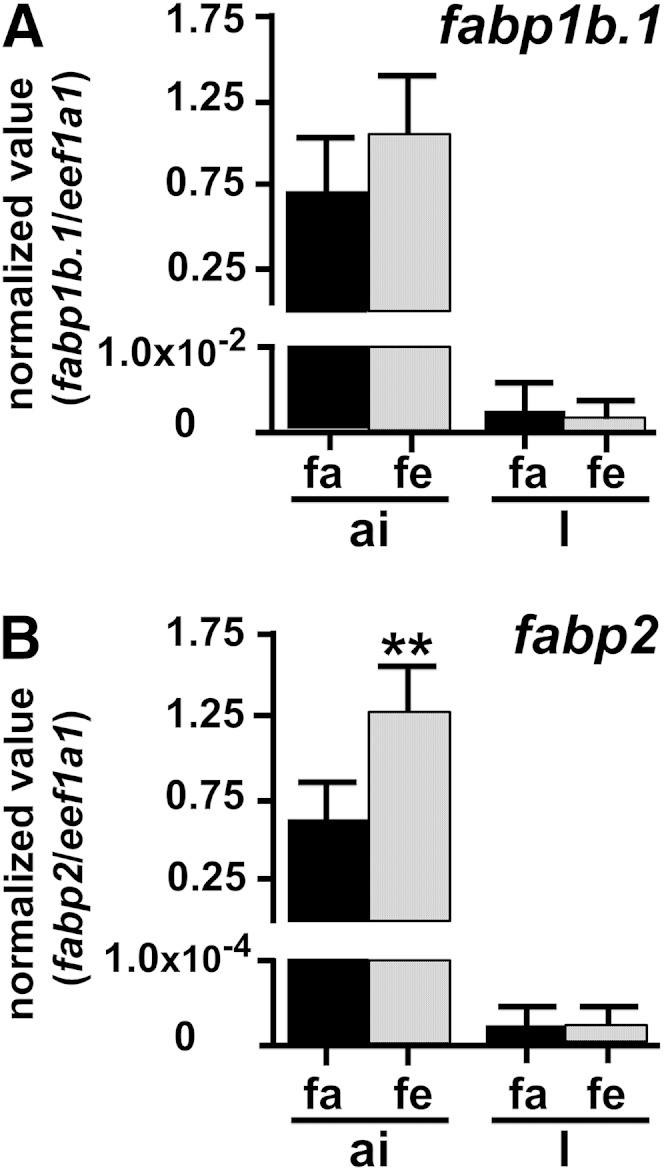
qRT-PCR analysis of fabp1b.1 and fabp2 transcript expression in the dissected anterior intestine (ai) and liver (l) of adult zebrafish. A: fabp1b.1. B: fabp2. After a period of over 24 h fasting (fasted group, fa) or 22 h fasting followed by 2 h feeding (fed group, fe), organs from 6 individuals were sampled and analyzed independently, in triplicate. The number of eef1a1 transcripts was also quantified and used to express the results as a number of copies of fabp1b.1 or fabp2 divided by the number of copies of the normalizing gene. Very similar results were obtained with ppiaa, used as another normalizing gene (data not shown). Data are means ± SD. ** P < 0.01, significant difference between fa and fe groups for the organ examined using the nonparametric Mann-Whitney test.
Fig. 3.

qRT-PCR analysis of the expression of fabp1b.1 and fabp2 transcripts in whole, 5 dpf zebrafish eleutheroembryos and 15 dpf larvae homogenates. A, B: fabp1b.1. C, D: fabp2. Animals were sampled after a period of over 18 h fasting (fasted group, fa) or 18 h fasting followed by 2–2.5 h feeding with FD (fed group, fe). At each age, six independent pools of 25 (5 dpf) or 20 (15 dpf) animals were used to quantify fabp1b.1 and fabp2 transcript levels in triplicate. The eef1a1 and ppiaa were also quantified and used as normalizing genes. The results shown are the number of copies of the fabp1b.1 or fabp2 transcripts divided by the number of copies of eef1a1 (A, C) or ppiaa (B, D). Data are means ± SD. Means were compared in reference to the fasted group using the nonparametric Mann-Whitney test. Asterisks indicate significant differences (* P < 0.05; *** P < 0.005) between the fasted and fed groups, for the transcript and age concerned.
Fig. 4.
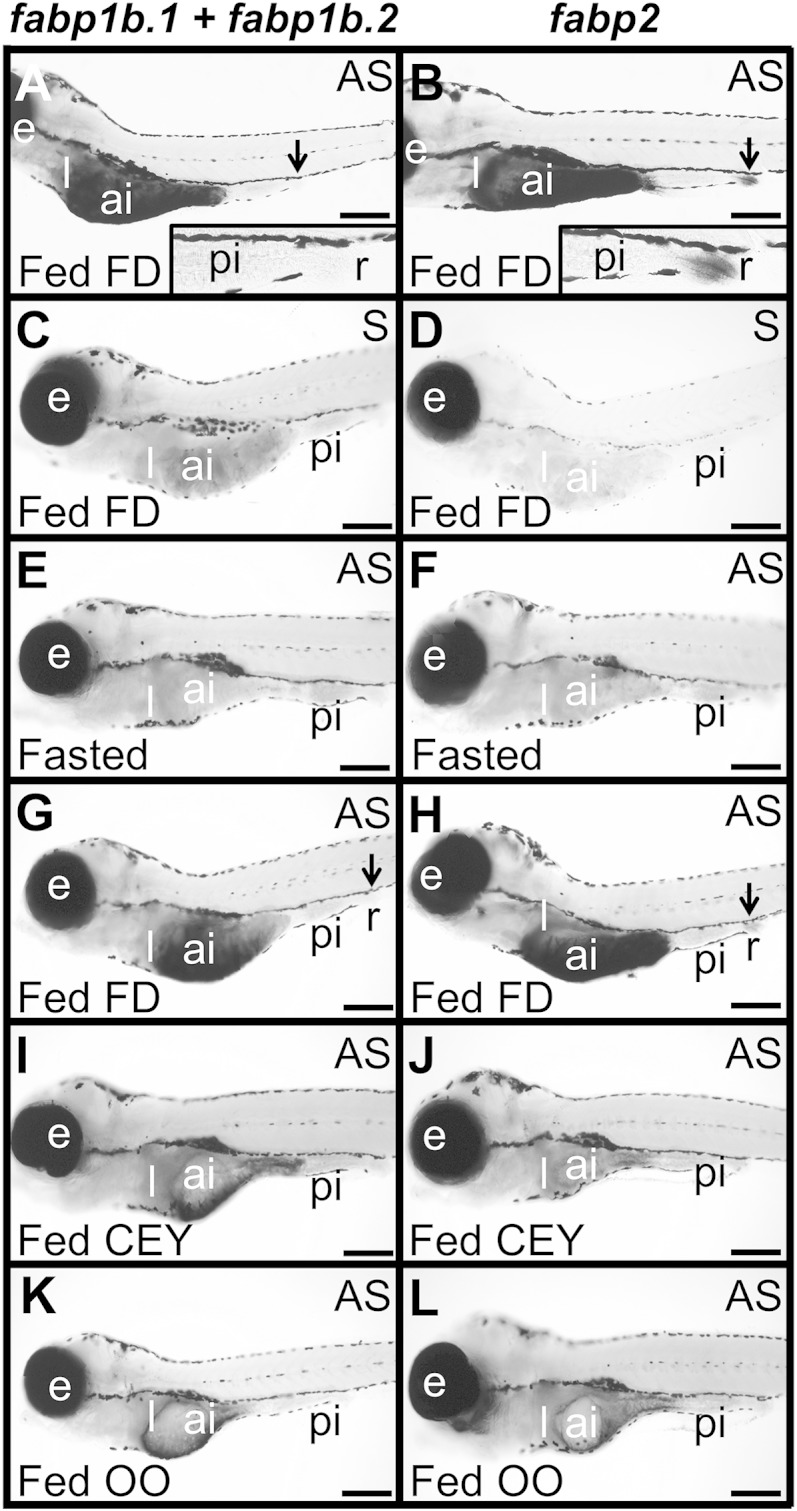
The effects of feeding and diet formulation on fabp1b (fabp1b.1 plus fabp1b.2) and fabp2 mRNA transcript hybridization signal in zebrafish larvae were evaluated by whole-mount in situ hybridization. A, C, E, G, I, K: fabp1b. B, D, F, H, J, L: fabp2. Larvae [8 dpf (A, B) or 15 dpf (C–L)] were fasted for 24 h and were then fed (A–D, G–L) or not (E, F) for 3 h with FD (A–D, G, H), dried CEY (I, J), or OO (K, L). The larvae were then collected and hybridized with sense (S) (C) or antisense (AS) (A, E, G, I, K) fabp1b or with sense (D) or antisense (B, F, H, J, L) fabp2 riboprobes, respectively. The fabp1b antisense probe was able to hybridize to fabp1b.1 and fabp1b.2 transcript variants. Representative larvae are shown in lateral view with the anterior part to the left. The specificity of the riboprobes used was demonstrated by a differential expression pattern between fabp1b and fabp2 transcripts in the rectum (r) (indicated by a black arrow in panels A, B, G, H) (see also supplementary Fig. 3). High transcript levels were found for these genes in the anterior intestine (ai), while no mRNAs were detected in the posterior intestine (pi) (A, B, G, H). Under the hybridization conditions used, high level of fabp2 transcript was detected in the rectum (r) (B) at 8 dpf and, to a lesser extent, at 15 dpf (H), while no hybridization signal was retrieved with fabp1b (A, G). Inserts in A and B are enlargements of the most posterior part of the gut including the rectal area. The fabp1b and fabp2 hybridization signal in the anterior intestine rose significantly in fed larvae nourished with FD (G, H) compared with fasting larvae (E, F). A weaker signal was measured when larvae were nourished with dried CEY (I, J) or OO (K, L). The staining hybridization reaction was stopped after 2 h (i.e., before appearance of the signal in the starved animals) to facilitate comparison between starved and fed states. A longer staining hybridization reaction led to a dark saturated signal in both starved and nourished larvae (data not shown), as these genes were strongly expressed in the anterior intestine, irrespective of the animals’ nutritional status. No staining signal was detected using control sense probes (C, D). e, eye; l, liver. Scale bar represents 250 µm in A, B and 300 µm in C–L.
A differential expression pattern of fabp genes along the anterior-posterior axis of the gut was demonstrated by whole-mount in situ hybridization (Fig. 4; supplementary Fig. 3). The fabp1b (fabp1b.1 plus fabp1b.2) and fabp2 transcripts were strongly expressed in the anterior intestine and its rostral expansion, and no mRNAs could be detected in the posterior intestine. A significant fabp2 transcript level was detected in the rectum, but no hybridization signal with fabp1b.
Localization of Fabp1b and Fabp2 in adult zebrafish intestine
Histological sections of the digestive cavity were stained with monospecific polyclonal antibodies raised against recombinant zebrafish Fabp1b.1 or Fabp2 (Fig. 5). It is important to notice that the antibody raised against Fabp1b.1 may also detect an Fabp1b.2 signal, due to a significant sequence identity between the two; from now on, we will refer to the signal generated by Fabp1b. Fabp1b and Fabp2 were immunodetected at high levels in the anterior intestine. A lower-level Fabp1b signal was detected in the liver (Fig. 5A and data not shown). No Fabp1b (Fig. 5A) or Fabp2 (data not shown) labeling signal was found in the posterior intestine, testicles, white adipose tissue, or infiltrated pancreatic islets. Higher magnification revealed homogenous cytoplasmic Fabp1b and Fabp2 expression in the intestinal absorptive cells. Expression was detected through the cytoplasm, including the microvilli, and inside the nuclei of some enterocytes mostly outside condensed chromatin clusters.
Fig. 5.

Immunofluorescence localization of Fabp1b and Fabp2 in the digestive cavity of zebrafish. The histological sections were stained with monospecific polyclonal antibodies developed against recombinant zebrafish Fabp1b.1 (A, B, E) or Fabp2 (C, D, F) proteins, with Alexa 488-conjugated secondary antibody (green). The sections were counterstained with TP3 to label the nuclei red. A: High levels of Fabp1b were immunodetected in the anterior intestine (ai) and lower levels in the liver (l) while no signal was detected in the posterior intestine (pi), testicles (t), adipose tissue (at), or pancreatic islets (p). B: In the intestinal mucosa, Fabp1b was detected inside the intestinal villi (iv) while the lamina propria (lp) was devoid of any signal. C, D: Immuno-fluorescent staining for Fabp2 was similar to that of Fabp1b, with high levels in iv. E, F: Higher magnification revealed Fabp1b and Fabp2 expression in the enterocytes (e), including the microvilli (mv), while goblet cell (gc) and infiltrated cells from lymph and blood vessels (lbv) were devoid of any immunofluorescent signal. Nuclear localization of Fabp1b and Fabp2 was highlighted by the yellow coloring in the nuclei of most enterocytes in the intestinal mucosa (B, D, E, F). TP3 nuclear staining demonstrated that cells with fully condensed chromatin appeared homogeneously red and did not exhibit any nuclear Fabp signal. The nuclei located in the supra-basal position of a large number of enterocytes exhibited immunofluorescent labeling outside condensed chromatin clusters. White arrow: nucleus (n) with condensed chromatin clusters, immunolabeled with Fabp antibodies outside the clusters. White arrowhead: nucleus with condensed chromatin clusters, not immunolabeled with Fabp1b antibody. Inserts in E, F are enlarged views of the region surrounded by dashed lines in the main pictures. il, intestinal lumen. Scale bar represents 75 µm in A, B, C, D; 20 µm in E, F.
Immunogold labeling electron microscopy was used for cellular and subcellular localization of Fabp1b (Fig. 6; supplementary Fig. 4) and Fabp2 (supplementary Fig. 5) in enterocytes. Gold particles were observed inside and on the surface of the microvilli, as well as in the terminal web. Immunogold particles were also abundant in the cytosol, outside the endoplasmic reticulum membrane, in close proximity to the plasma membrane, and inside the nucleus. No immunogold particles were detected inside oil globules, mitochondria, intercellular space, goblet cells, or lamina propria. Fabp1b nuclear immunogold particles were found in the interchromatin space with a labeling efficiency 4.34 times higher than in nuclear membrane-associated condensed chromatin and condensed chromatin clusters. The same was true of Fabp2, with 3.01-fold more labeling in the interchromatin space by comparison with the condensed chromatin areas.
Fig. 6.

Immunocytochemical labeling of Fabp1b in zebrafish enterocytes. Fabp1b antigenic sites, revealed using anti-Fabp1b/secondary IgG coupled to 10 nm protein A-colloidal-gold complex, registered as dark spots on the micrographs. A–C: Localization of antigen sites in the microvilli (mv), cytosol, and nucleus (n), respectively. Inserts in A and C are controls without the primary antibody. Nuclear immunogold particles were found in the interchromatin space (i.e., euchromatin), with a labeling efficiency (gold particle number/arbitrary surface area unit) of 2.04, while membrane-associated condensed chromatin (mc) and condensed chromatin clusters (ccc) (i.e., heterochromatin) were mostly unlabeled, with a labeling efficiency of 0.47 (ratio = 4.34). c, cytosol; er, endoplasmic reticulum; gc, goblet cell; ics, interchromatin space; il, intestinal lumen; is, intercellular space; nm, nuclear membrane; np, nuclear pore (indicated by a black arrow); m, mitochondria; og, oil globule; pm, plasma membrane; tw, terminal web. Scale bar represents 0.5 µm.
Native PAGE binding assay of BODIPY-FL-labeled FAs to recombinant Fabp1b.1 and Fabp2
A native PAGE binding assay was used to check that recombinant Fabp1b.1 or Fabp2 were able to specifically bind BODIPY-labeled FAs (Fig. 7). TPMA, used as a negative control, was unable to bind the labeled FAs. We demonstrated that neither Fabp1b.1 nor Fabp2 were able to bind BODIPY-FLC5 to any significant extend, but both of them avidly bound BODIPY-FLC12. While a very heavily labeled band was observed with BODIPY-FLC12 (Fig. 7B, D), only a very faintly labeled band was observed with Fabp1b.1 and BODIPY-FLC5 (Fig. 7B, lane 5). Adding SDS to a final concentration of 2% in the loaded sample improved the PAGE protein resolution without a drastic change in the quantity of BODIPY-FLC12 fluorescence bound to FABPs (supplementary Fig. 6; data not shown). The inability of BODIPY-FLC5 to bind to Fabp1b.1 was confirmed by very similar fluorescence emission spectra of this labeled compound in the presence or absence of Fabp1b.1 (supplementary Fig. 7A). The binding of BODIPY-FLC12 by Fabp1b.1 was supported by the increase in fluorescence quantum yield to the quantum yield obtained with BODIPY-FLC12 in buffer alone (supplementary Fig. 7B). Native PAGE of purified recombinant zebrafish Fabp2 revealed two protein bands (Fig. 7C). In both bands extracted from native PAGE, MALDI-TOF-TOF mass spectrometer analysis recovered Fabp2. The lower molecular mass Fabp2 bound BODIPY-FLC12, while the Fabp2 multimer upper band was unable to do so (Fig. 7D). BSA was used as a positive control. Under these experimental conditions, BSA was able to bind to both BODIPY-FL-labeled FAs (Fig. 7B, lanes 8 and 9). However, the intensity of the labeled bands was much lower than that observed with BODIPY-FLC12 binding to Fabp1b.1 or Fabp2. By increasing the amount of BSA loaded into the gel, our in-gel fluorescence imaging method confirmed that this protein was able to bind BODIPY-FLC12 (supplementary Fig. 8).
Fig. 7.

Native PAGE binding assay of BODIPY-FL-labeled FAs to recombinant Fabp1b.1 and Fabp2. Representative native PAGE of purified proteins, labeled with BODIPY-FL FAs or unlabeled, was stained with Coomassie blue (A, C) and detected for fluorescence (B, D). Only parts of the gels stained with Coomassie blue and contained the migrated proteins have been shown. A, B: Lane 1, 41.8 ng BODIPY-FLC12; lane 2, 1.66 µg TPMA plus 32 ng BODIPY-FLC5; lane 3, 1.66 µg TPMA plus 41.8 ng BODIPY-FLC12; lane 4, 1.66 µg TPMA; lane 5, 1.58 µg Fabp1b.1 plus 32 ng BODIPY-FLC5; lane 6, 1.58 µg Fabp1b.1 plus 41.8 ng BODIPY-FLC12; lane 7, 1.58 µg Fabp1b.1; lane 8, 3.28 µg BSA plus 32 ng BODIPY FL-C5; lane 9, 3.28 µg BSA plus 41.8 ng BODIPY-FLC12; lane 10, 3.28 µg BSA; lane 11, Fabp2 plus 76.8 ng BODIPY-FLC5; lane 12, Fabp2 plus 100 ng BODIPY-FLC12; lane 13, Fabp2. TPMA from E. granulosus (GenBank: AAB65799.4) and BSA fraction V (Sigma-Aldrich) were used for comparison. C, D: Lane 1, 9.05 µg Fabp2 + 76.8 ng BODIPY-FLC5; lane 2, 9.05 µg Fabp2 + 57.8 ng BODIPY-FLC5; lane 3, 9.05 µg Fabp2; lane 4, 9.05 µg Fabp2 + 146.5 ng BODIPY-FLC12; lane 5, no sample; lane 6, 9.05 µg Fabp2 + 100 ng BODIPY-FLC12; lane 7, 9.05 µg Fabp2 + 43.8 ng BODIPY-FLC12; lane 8, 4.52 µg Fabp2 + 146.5 ng BODIPY-FLC12; lane 9, 4.52 µg Fabp2 + 119.2 ng BODIPY-FLC12; lane 10, 4.52 µg Fabp2 + 75.3 ng BODIPY-FLC12; lane 11, no sample; lane 12, 9.05 µg Fabp2; lane 13, 4.52 µg Fabp2. Labeled BODIPY-FLC12 proteins observed in (B, D) are Fabp1b.1, Fabp2, and, to a lesser extent, BSA (see also supplementary Fig. 8 for a higher amount of BSA loaded). Fabp2* is an Fabp multimer able to enter the polyacrylamide gel but unable to bind BODIPY-FL-labeled FAs.
We then demonstrated that oleic acid was able to fully abolish the binding of BODIPY-FL C12 to Fabp1b.1 (Fig. 8). DHA had partial displacement activity under these experimental conditions, while linoleic acid was ineffective.
Fig. 8.
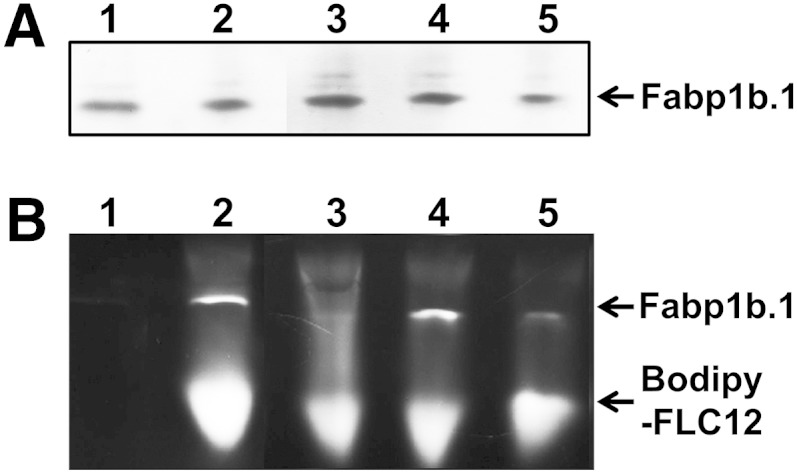
Displacement of BODIPY-FLC12 to recombinant Fabp1b.1. A: Representative native PAGE of purified recombinant Fabp1b.1 labeled with BODIPY-FLC12 and displaced by 100-fold molar excess of unlabeled FAs or not. Lane 1, Fabp1b; lane 2, Fabp1b.1 plus BODIPY-FLC12; lane 3, Fabp1b.1 plus BODIPY-FLC12 and oleic acid (C18:1w9); lane 4, Fabp1b.1 plus BODIPY-FLC12 and linoleic acid (C18:2w6); lane 5, Fabp1b.1 plus BODIPY-FLC12 and DHA (C22:6w3). B: Labeled Fabp1b.1 bands were detected for fluorescence, and full displacement of BODIPY-FLC12 to Fabp1b.1 was observed in the presence of oleic acid (lane 3). DHA exhibited partial displacement activity (lane 5), while linoleic acid was ineffective (lane 4). Lanes 3 to 5 come from the same gel with the lanes moved.
Feeding with BODIPY-labeled FAs
To investigate the involvement of Fabp1b and Fabp2 in the targeting of dietary FAs to nuclei, adult zebrafish were fed with BODIPY-FLC5 (Fig. 9) or BODIPY-FLC12 (Fig. 10; supplementary Fig. 9) mixed with dried CEY. Both fluorescent FAs were taken up by the enterocyte, and a wide cytoplasmic distribution was observed. These experiments also showed that nuclear incorporation of FA analogs depended on the ligand carbon chain length. A strong BODIPY-FLC5 signal was located in the basal region of the enterocyte and surrounding the nuclei, while a robust BODIPY-FLC12 signal was also observed inside the nuclei. Fluorescence microscopy images demonstrated that the BODIPY-FLC12 signal associated with the nuclear fraction was 18.05 ± 1.76% of the total fluorescence detected in the villi (7 villi analyzed which included 225 enterocyte nuclei). Some enterocyte nuclei were heavily stained while other ones with more condensed chromatin had low BODIPY-FLC12 intranuclear labeling (Fig. 10; supplementary Fig. 9). The labeled enterocyte nuclei were 13.17 ± 2.04% of the total surface area analyzed demonstrating a significant nuclear fraction enrichment of BODIPY-FLC12 label (P < 0.0006). Because both fluorescent FAs and Fabp1b and Fabp2 have a homogenous cytoplasmic distribution, it was difficult to assign a colocalization. However, an accumulation of both labels was observed in the basal region of the enterocyte, and colocalization of BODIPY-FLC12 with Fabp1b and Fabp2 was observed inside the enterocyte nuclei, in the interchromatin space between the condensed chromatin areas (Fig. 10).
Fig. 9.

The intestinal absorption of dietary BODIPY-FLC5 and simultaneous localization of Fabp1b and Fabp2 in zebrafish intestinal villi. Histological sections were counterstained with DAPI to label the nuclei blue (A–D). The sections were stained with monospecific polyclonal antibodies developed against recombinant zebrafish Fabp1b.1 (E–G) or Fabp2 (H) proteins with Alexa 594-conjugated secondary antibody (red). Adult animals were previously fed with BODIPY493/503 mixed with CEY powder and used as a negative control (see Materials and Methods for details) (I) or BODIPY-FLC5 mixed with CEY powder (J–L), and fluorescence was detected on histological sections in green fluorescent protein channel (green). (N–P) Merging between nuclei labeling, immunofluorescence signal of Fabps, and BODIPY493/503 or BODIPY-FLC5 signals are presented for each corresponding column of the figure. The apex of the villi is positioned at the bottom of each image. C, G, K, and O are enlargements of the indicated area on B, F, J, and M, respectively. No BODIPY-FLC5 signal was detected inside the nuclei and the intranuclear overlap between Fabp and DAPI labels is shown as pink spots (white arrows in N, O, P). e, enterocyte; lbv, lymph and blood vessels; mv, microvilli. Scale bar represents 20 µm, except 5 µm in C, G, K, O.
Fig. 10.

Intestinal absorption of dietary BODIPY-FLC12 and its colocalization with Fabp1b and Fabp2 in zebrafish enterocytes. A–C: Histological sections were counterstained with DAPI to label the nuclei blue. D, E: The sections were stained with monospecific polyclonal antibodies developed against recombinant zebrafish Fabp1b.1 or Fabp2 proteins with Alexa 594-conjugated secondary antibody (red). F, H: Adult animals were previously fed with BODIPY-FLC12 mixed with CEY powder (see Materials and Methods for details), and fluorescence was detected in the green fluorescent protein channel (green). I, J: Merged view of labeled nuclei, Fabp immunofluorescence signal, and BODIPY-FLC12 signals are presented in the left and right columns of the figure. The apex of the villi is positioned at the bottom in each image. B, G are enlargements of the indicated large area from A, I, respectively. Inserts in A, F are enlargements of the indicated small area focused on an enterocyte nucleus with a strong BODIPY-FLC12 signal. The selected nucleus has been surrounded by a yellow line after using the threshold and wand tool options of ImageJ software (see supplementary Fig. 9 for an enlarged view of these panels). Yellow intracytoplasmic granulation indicates an overlap between Fabp1b and BODIPY-FLC12 (I, J, red arrow in G). Some nuclei with more condensed chromatin had low BODIPY-FLC12 intranuclear labeling while the pink granules indicate intranuclear Fabp1b (white arrowhead in G). White and red asterisks indicate goblet cell and putative red blood cell nuclei, respectively, free of any Fabp1b and BODIPY-FLC12 labeling (B, G). Intranuclear overlapping was observed between Fabp, BODIPY-FLC12, and nucleus labels (white arrows in G, J). e, enterocyte; lbv, lymph and blood vessels; mv, microvilli. Scale bar represents 20 µm.
DISCUSSION
The primary role of all FABP family members is to bind FAs and other hydrophobic ligands, such as lipophilic drugs and xenobiotics, controlling their uptake and intracellular transport (i.e., FABPs act as chaperones) (13–17, 57). The cephalocaudal patterning of FABP transcript expression in the mammalian intestine was conserved through evolution. In mice, FABP1 is highly expressed in the proximal two-thirds of the small intestine (58), FABP2 is expressed in the distal jejunum, decreasing gradually toward both the proximal duodenum and the midcolon (59), while FABP6 transcripts encoding ileal lipid binding protein were located in the distal one-third of the small intestine (60). The molecular patterning of the intestine is remarkably conserved in zebrafish, where the fabp2 expression domain is restricted to the anterior intestine (41), while fabp6 transcripts are found in the posterior intestine (61). Functional cis-acting regulatory sequences may be conserved between zebrafish and mammals, driving intestine-specific FABP2/fabp2 expression (62). Axial patterning of FABP expression has been attributed to ligand and absorption specificities along the digestive track (58), but, in other cases, overlapping FABP expression sites and ligand binding specificities may represent a mechanism for ensuring functional redundancy. Studies directly comparing FABP1- and FABP2-null mice have revealed markedly different phenotypes, indicating that the encoded proteins indeed have different functions in the intestinal lipid metabolism and whole body energy homeostasis (13, 63, 64).
The first step in using the zebrafish model was to determine the expression pattern and dietary regulation of homologs to human FABP1 and FABP2, which were significantly expressed in their anterior intestine. Different fabp genes were expressed in the zebrafish intestine (41, 44–47), including high transcript levels of fabp1b.1 and fabp2 and trace amounts of fabp1a and fabp1b.2 (44, 45, 47, 65). By carefully removing the ventral white adipose tissue closely associated with the digestive tract and dissecting the anterior intestine, which is the major site of fat absorption (41–43), we retrieved similarly high transcript levels of fabp1b.1 and fabp2 and smaller amounts of fabp1b.2, irrespective of the animals’ nutritional status. In addition, fabp1b.1 and fabp1b.2 transcripts were found in liver and brain, respectively, supporting the hypothesis that these tandem-duplicated genes have a differential, tissue-specific function. The relatively high levels of fabp1b.2 in brain may be attributable to ligand preferences because it is known that polyunsaturated FAs are highly abundant in the central nervous system (66, 67). Whole-mount in situ hybridization demonstrated high transcript levels of fabp1b (fabp1b.1 plus fabp1b.2) and fabp2 in the anterior intestine, but no mRNAs were detected in the posterior intestine. As previously described (41), a significant level of fabp2 transcripts was detected in the rectum while no hybridization signal with fabp1b, thus highlighting the differential expression pattern of these genes. Levels of fabp1b.1 and fabp2 transcripts were upregulated in the anterior intestine after feeding, both at the larval stages and in adults. As previously reported (45, 47), these levels were modulated according to the lipid composition of the diet. Lipid-mediated regulation of gene transcription via the activation of nuclear receptors to modify the expression of genes coding for proteins involved in the lipid metabolism triggered the proliferation of intestinal cells and modulated the expression of molecular signals that may affect homeostasis (26, 36).
The second step of our study was to produce monospecific polyclonal antibodies against purified recombinant proteins from the two most highly expressed fabp’s in the zebrafish anterior intestine (i.e., fabp1b.1 and fabp2). These tools were used for the cellular and subcellular immunolocalization of Fabp1b and Fabp2 in the digestive cavity and demonstrated a very high level of these FABPs in enterocytes in the anterior intestine. Given the very intense immune staining signal observed for both Fabps in the zebrafish intestinal villi, it is very likely that these proteins are present simultaneously in the same enterocytes. Immunofluorescence and immunocytochemical labeling methods revealed Fabps in the microvilli and cytosol, as previously demonstrated with human enterocytes from jejunal mucosa (39), as well as in the nuclei of most enterocytes in the anterior intestine. No staining signal was observed inside mitochondria or oil globules. Nuclei of cells present in the intestinal mucosa and with fully condensed chromatin, as well as cells from lymph and blood vessels, were devoid of any Fabp immunofluorescent signal. Electron microcopy immunodetection with gold particles demonstrated that enterocyte nuclear localization was mostly in the interchromatin space outside the condensed chromatin clusters. The nucleus is compartmentalized into chromosome territories and interchromatin spaces (68, 69). Zebrafish enterocyte nuclei revealed the conventional pattern with dense chromatin/heterochromatin enriched at the nuclear periphery and around the nucleoli, whereas less condensed/euchromatin regions extended toward the nuclear interior, together with dispersed interior clumps of condensed chromatin/heterochromatin. The spatial organization of eukaryotic genomes in the cell nucleus is linked to their transcriptional regulation (70, 71). However, it has been difficult to find general rules on the involvement of nuclear organization in transcriptional regulation. It remain to be determined whether Fabp1b and Fabp2 immunolabeling, found almost exclusively in less condensed/euchromatin regions, was located in transcriptionally permissive areas.
The third step was to evaluate whether fluorescent FA analogs were able to bind to recombinant zebrafish Fabp1b.1 and Fabp2. We demonstrated that neither recombinant Fabp1b.1 nor Fabp2 were able to bind significantly to BODIPY-FLC5 (short chain), but both of them bound avidly to BODIPY-FLC12 (medium chain). Our in-gel binding assay showed that FABP2 in an oligomerization state was unable to bind to the labeled FA analog. Fluorescent short- to long-chain BODIPY-labeled FAs were used in cell culture to assess the cellular uptake and nuclear localization of unesterified FAs (72–75). These fluorescent FAs analogs, which have a fluorophore linked to the terminal (omega) carbon atom furthest from the carboxylate moiety, were also esterified into complex lipids after adding to cells/animals. Human FABP1 had high affinity for BODIPY-FLC12, in the same range as LCFAs (74). FABP1 expression significantly enhanced the uptake of medium-chain BODIPY-FLC12 and differentially targeted BODIPY-FLC12 into nuclei, in marked contrast to the lack of effect of FABP1 expression on the intracellular distribution of short-chain BODIPY-FLC5 (73). The findings demonstrated that oleic acid was the only FA tested able to fully abolish the binding of BODIPY-FLC12 to zebrafish Fabp1b.1. This is interesting, as oleate binding causes a conformational change in rat FABP1 (76) and increases the nuclear localization of FABP1 and FABP2 (23, 33). Oleic acid may be involved in the transcriptional regulation of gene expression (77, 78), and the presence of either FABP1 or FABP2 magnifies the transcriptional activation mediated by PPARα (33).
The last step was to assess the fate of dietary fluorescent (BODIPY) FAs at the intestinal level, as the digestive physiology of zebrafish may be addressed using these labeled analogs (79). In vertebrates, the absorption and transport of short- to medium-chain FAs occurs differently in enterocytes, as LCFAs are usually reesterified inside the cell. There is evidence indicating that both FABP1 and FABP2 participate in the cellular uptake and transport of unesterified FAs and the lipid metabolism (16, 80, 81). However, some authors report that they play a role in lipid sensing but not in direct dietary FA absorption (13, 62, 82, 83). Cell culture models have proved very useful for mechanistic investigations into the role of FABPs in modulating nuclear receptors and gene transcription (26). For example, it was demonstrated that FABP1 interacted with PPARα (24, 27), and FABP4 with PPARγ (22), respectively, in cultured primary hepatocytes, HepG2 cells, and 3T3-L1 adipocytes. A physical contact between FABP1 and PPARα occurs during this activation, so it has been suggested that FABP1 is a coactivator in PPAR-mediated gene control (27). It has also been demonstrated that FABP5 served as coactivator in the nucleus of transfected cells for PPARβ/δ-mediated gene transcription control (25). FABP5 shuttles FAs directly to the nucleus where it may deliver the ligand to PPARβ/δ, thus enabling its activation (31, 32). In addition, DHA binding to FABP7 triggers the accumulation of FABP7 in the nucleus and modulates cell migration (28). In transfected COS-7 cells, nuclear accumulation of FABP1 and FABP2 is unlikely to be mediated by increased nuclear transport. FABP accumulation in the nucleus is due to a reduced rate of nuclear egress in the presence of the ligand and the dependent interaction of FABP with PPARα (33). The results reported in this manuscript indicate that dietary BODIPY-FLC12 but not BODIPY-FLC5 may be detected in enterocyte nuclei after feeding and colocalized with Fabp1b and Fabp2 at the nuclear level. To our knowledge, this is the first demonstration that exogenous unesterified FAs reach the nucleus in an in vivo context. The interaction between FA-transporter FABPs and FA-activated, nuclear-receptor PPARs may promote nuclear localization of their ligands, thus constituting a mechanism whereby, following uptake by the cell, dietary FAs become signaling molecules for conveying messages to the nucleus. It cannot be completely ruled out that these FABPs may also play a role in FA retention in and/or release from the nuclei.
In summary, we demonstrated that fabp1b.1 and fabp2 transcript levels in the zebrafish anterior intestine were upregulated after feeding and modulated according to diet formulation. We provided also evidence that BODIPY-FLC12 was able to bind to recombinant Fabp1b.1 and Fabp2. The interaction of BODIPY-FLC12 with Fabp1b.1 was fully displaced by unlabeled oleic acid. In addition to their cytosolic localization, Fabp1b and Fabp2 were also found in enterocyte nuclei. In vivo experiments demonstrated that, following intestinal absorption of dietary fluorescent BODIPY-FLC12, the labeled FA analog was colocalized with Fabp1b and Fabp2 in the nucleus. It appears likely that dietary FAs complexed with FABPs may promote gene transcription regulation, either indirectly or via interaction with nuclear receptors.
Supplementary Material
Acknowledgments
The authors thank Antonio Osuna Carillo de Albornoz and Juan de Dios Bueno Pérez from the Centro de Instrumentación Científica, Universidad de Granada, Spain, for their help in electron microscopy studies, and the Unidad de Bioquímica y Proteómica Analítica, Institut Pasteur de Montevideo, for MALDI-TOF-TOF MS data.
Footnotes
Abbreviations:
- CEY
- chicken egg yolk
- DAPI
- 4’,6’-diamidino-2-phenylindole
- DIG
- digoxigenin
- dpf
- days postfertilization
- eef1a1
- elongation factor 1α FABP, fatty acid binding protein
- FD
- formulated diet
- LCFA
- long-chain fatty acid
- OO
- olive oil
- ppiaa
- peptidylprolyl isomerase aa
- qRT-PCR
- real-time quantitative RT-PCR
- RTneg
- reverse transcription negative (i.e., performed without reverse transcriptase)
- TPMA
- tropomyosin A
- TP3
- To-Pro 3 iodide
This work was supported by the ECOS-Sud program (U09B03) (A.E. and P.J.B.) and the Ministère de l’Enseignement Supérieur et de la Recherche, France. The authors declare no conflicts of interest.
The online version of this article (available at http://www.jlr.org) contains a supplement.
REFERENCES
- 1.Levi A. J., Gatmaitan Z., and Arias I. M.. 1969. Two hepatic cytoplasmic protein fractions, Y and Z, and their possible role in the hepatic uptake of bilirubin, sulfobromophthalein, and other anions. J. Clin. Invest. 48: 2156–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ockner R. K., Manning J. A., Poppenhausen R. B., and Ho W. K.. 1972. A binding protein for fatty acids in cytosol of intestinal mucosa, liver, myocardium, and other tissues. Science. 177: 56–58. [DOI] [PubMed] [Google Scholar]
- 3.Ockner R. K., and Manning J. A.. 1974. Fatty acid-binding protein in small intestine. Identification, isolation, and evidence for its role in cellular fatty acid transport. J. Clin. Invest. 54: 326–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banaszak L., Winter N., Xu Z., Bernlohr D. A., Cowan S., and Jones T. A.. 1994. Lipid-binding proteins: a family of fatty acid and retinoid transport proteins. Adv. Protein Chem. 45: 89–151. [DOI] [PubMed] [Google Scholar]
- 5.Esteves A., and Ehrlich R.. 2006. Invertebrate intracellular fatty acid binding proteins. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 142: 262–274. [DOI] [PubMed] [Google Scholar]
- 6.Zheng Y., Blair D., and Bradley J. E.. 2013. Phyletic distribution of fatty acid-binding protein genes. PLoS One. 8: e77636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agulleiro M. J., André M., Morais S., Cerdà J., and Babin P. J.. 2007. High transcript level of fatty acid-binding protein 11 but not of vitellogenin receptor during ovarian follicle atresia of a teleost fish (Solea senegalensis). Biol. Reprod. 77: 504–516. [DOI] [PubMed] [Google Scholar]
- 8.Babin P. J. 2009. Molecular evolution of vertebrate fatty acid-binding proteins. In Fatty Acid-Binding Proteins. A. Esteves, editor. Transworld Research Network, Trivandrum, Kerala, India. Chapter 2, 17–29. [Google Scholar]
- 9.Richieri G. V., Ogata R. T., Zimmerman A. W., Veerkamp J. H., and Kleinfeld A. M.. 2000. Fatty acid binding proteins from different tissues show distinct patterns of fatty acid interactions. Biochemistry. 39: 7197–7204. [DOI] [PubMed] [Google Scholar]
- 10.Velkov T., Horne J., Laguerre A., Jones E., Scanlon M. J., and Porter C. J.. 2007. Examination of the role of intestinal fatty acid-binding protein in drug absorption using a parallel artificial membrane permeability assay. Chem. Biol. 14: 453–465. [DOI] [PubMed] [Google Scholar]
- 11.Kaczocha M., Vivieca S., Sun J., Glaser S. T., and Deutsch D. G.. 2012. Fatty acid-binding proteins transport N-acylethanolamines to nuclear receptors and are targets of endocannabinoid transport inhibitors. J. Biol. Chem. 287: 3415–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patil R., Laguerre A., Wielens J., Headey S. J., Williams M. L., Hughes M. L., Mohanty B., Porter C. J., and Scanlon M. J.. 2014. Characterization of two distinct modes of drug binding to human intestinal fatty acid binding protein. ACS Chem. Biol. 9: 2526–2534. [DOI] [PubMed] [Google Scholar]
- 13.Furuhashi M., and Hotamisligil G. S.. 2008. Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat. Rev. Drug Discov. 7: 489–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Storch J., and Corsico B.. 2008. The emerging functions and mechanisms of mammalian fatty acid-binding proteins. Annu. Rev. Nutr. 28: 73–95. [DOI] [PubMed] [Google Scholar]
- 15.Storch J., and McDermott L.. 2009. Structural and functional analysis of fatty acid-binding proteins. J. Lipid Res. 50: S126–S131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Storch J., and Thumser A. E.. 2010. Tissue-specific functions in the fatty acid-binding protein family. J. Biol. Chem. 285: 32679–32683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glatz J. F. 2015. Lipids and lipid binding proteins: a perfect match. Prostaglandins Leukot. Essent. Fatty Acids. 93: 45–49. [DOI] [PubMed] [Google Scholar]
- 18.Sacchettini J. C., Scapin G., Gopaul D., and Gordon J. I.. 1992. Refinement of the structure of Escherichia coli-derived rat intestinal fatty acid binding protein with bound oleate to 1.75-A resolution. Correlation with the structures of the apoprotein and the protein with bound palmitate. J. Biol. Chem. 267: 23534–23545. [PubMed] [Google Scholar]
- 19.LaLonde J. M., Bernlohr D. A., and Banaszak L. J.. 1994. The up-and-down beta-barrel proteins. FASEB J. 8: 1240–1247. [DOI] [PubMed] [Google Scholar]
- 20.Gillilan R. E., Ayers S. D., and Noy N.. 2007. Structural basis for activation of fatty acid-binding protein 4. J. Mol. Biol. 372: 1246–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohno S. 1970. Evolution by Gene and Duplication. Springer-Verlag, Berlin. [Google Scholar]
- 22.Helledie T., Antonius M., Sorensen R. V., Hertzel A. V., Bernlohr D. A., Kølvraa S., Kristiansen K., and Mandrup S.. 2000. Lipid-binding proteins modulate ligand-dependent trans-activation by peroxisome proliferator-activated receptors and localize to the nucleus as well as the cytoplasm. J. Lipid Res. 41: 1740–1751. [PubMed] [Google Scholar]
- 23.Lawrence J. W., Kroll D. J., and Eacho P. I.. 2000. Ligand-dependent interaction of hepatic fatty acid-binding protein with the nucleus. J. Lipid Res. 41: 1390–1401. [PubMed] [Google Scholar]
- 24.Wolfrum C., Borrmann C. M., Borchers T., and Spener F.. 2001. Fatty acids and hypolipidemic drugs regulate peroxisome proliferator-activated receptors alpha- and gamma-mediated gene expression via liver fatty acid binding protein: a signaling path to the nucleus. Proc. Natl. Acad. Sci. USA. 98: 2323–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan N. S., Shaw N. S., Vinckenbosch N., Liu P., Yasmin R., Desvergne B., Wahli W., and Noy N.. 2002. Selective cooperation between fatty acid binding proteins and peroxisome proliferator-activated receptors in regulating transcription. Mol. Cell. Biol. 22: 5114–5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schroeder F., Petrescu A. D., Huang H., Atshaves B. P., McIntosh A. L., Martin G. G., Hostetler H. A., Vespa A., Landrock D., Landrock K. K., et al. 2008. Role of fatty acid binding proteins and long-chain fatty acids in modulating nuclear receptors and gene transcription. Lipids. 43: 1–17. [DOI] [PubMed] [Google Scholar]
- 27.Hostetler H. A., McIntosh A. L., Atsha B. P., Storey S. M., Payne H. R., Kier A. B., and Schroeder F.. 2009. L-FABP directly interacts with PPARα in cultured primary hepatocytes. J. Lipid Res. 50: 1663–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mita R., Beaulieu M. J., Field C., and Godbout R.. 2010. Brain fatty acid-binding protein and omega-3/omega-6 fatty acids: mechanistic insight into malignant glioma cell migration. J. Biol. Chem. 285: 37005–37015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Velkov T. 2013. Interactions between human liver fatty acid binding protein and peroxisome proliferator activated receptor selective drugs. PPAR Res. 2013: 938401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McIntosh A. L., Petrescu A. D., Hostetler H. A., Kier A. B., and Schroeder F.. 2013. Liver-type fatty acid binding protein interacts with hepatocyte nuclear factor 4α. FEBS Lett. 587: 3787–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Armstrong E. H., Goswami D., Griffin P. R., Noy N., and Ortlund E. A.. 2014. Structural basis for ligand regulation of the fatty acid-binding protein 5, peroxisome proliferator-activated receptor β/δ (FABP5-PPARβ/δ) signaling pathway. J. Biol. Chem. 289: 14941–14954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu S., Levi L., Casadesus G., Kunos G., and Noy N.. 2014. Fatty acid-binding protein 5 (FABP5) regulates cognitive function both by decreasing anandamide levels and by activating the nuclear receptor peroxisome proliferator-activated receptor β/δ (PPARβ/δ) in the brain. J. Biol. Chem. 289: 12748–12758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hughes M. L., Liu B., Halls M. L., Wagstaff K. M., Patil R., Velkov T., Jans D. A., Bunnett N. W., Scanlon M. J., and Porter C. J.. 2015. Fatty acid-binding proteins 1 and 2 differentially modulate the activation of peroxisome proliferator-activated receptor α in a ligand-selective manner. J. Biol. Chem. 290: 13895–13906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stremmel W. 1988. Uptake of fatty acids by jejunal mucosal cells is mediated by a fatty acid binding membrane protein. J. Clin. Invest. 82: 2001–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trotter P. J., Ho S. Y., and Storch J.. 1996. Fatty acid uptake by Caco-2 human intestinal cells. J. Lipid Res. 37: 336–346. [PubMed] [Google Scholar]
- 36.Niot I., Poirier H., Trang Tran T. T. P., and Besnard P.. 2009. Intestinal absorption of long-chain fatty acids: evidence and uncertainties. Prog. Lipid Res. 48: 101–115. [DOI] [PubMed] [Google Scholar]
- 37.Schwenk R. W., Holloway G. P., Luiken J. J., Bonen A., and Glatz J. F.. 2010. Fatty acid transport across the cell membrane: regulation by fatty acid transporters. Prostaglandins Leukot. Essent. Fatty Acids. 82: 149–154. [DOI] [PubMed] [Google Scholar]
- 38.Abumrad N. A., and Davidson N. O.. 2012. Role of the gut in lipid homeostasis. Physiol. Rev. 92: 1061–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Levy E., Ménard D., Delvin E., Montoudis A., Beaulieu J. F., Mailhot G., Dubé N., Sinnett D., Seidman E., and Bendayan M.. 2009. Localization, function and regulation of the two intestinal fatty acid-binding protein types. Histochem. Cell Biol. 132: 351–367. [DOI] [PubMed] [Google Scholar]
- 40.Alpers D. H., Bass N. M., Eagle M. J., and DeSchyver-Kecskemeti K.. 2000. Intestinal fatty acid binding protein may favor differential apical fatty acid binding in the intestine. Biochim. Biophys. Acta. 1483: 352–362. [DOI] [PubMed] [Google Scholar]
- 41.André M., Ando S., Ballagny C., Durliat M., Poupard G., Briançon C., and Babin P. J.. 2000. Intestinal fatty acid binding protein gene expression reveals the cephalocaudal patterning during zebrafish gut morphogenesis. Int. J. Dev. Biol. 44: 249–252. [PubMed] [Google Scholar]
- 42.Marza E., Barthe C., André M., Villeneuve L., Hélou C., and Babin P. J.. 2005. Developmental expression and nutritional regulation of a zebrafish gene homologous to mammalian microsomal triglyceride transfer protein large subunit. Dev. Dyn. 232: 506–518. [DOI] [PubMed] [Google Scholar]
- 43.Morais S., Knoll-Gellida A., André M., Barthe C., and Babin P. J.. 2007. Conserved expression of alternative splicing variants of peroxisomal acyl-CoA oxidase 1 in vertebrates and developmental and nutritional regulation in fish. Physiol. Genomics. 28: 239–252. [DOI] [PubMed] [Google Scholar]
- 44.Karanth S., Denovan-Wright E. M., Thisse C., Thisse B., and Wright J. M.. 2009. Tandem duplication of the fabp1b gene and subsequent divergence of the tissue-specific distribution of fabp1b.1 and fabp1b.2 transcripts in zebrafish (Danio rerio). Genome. 52: 985–992. [DOI] [PubMed] [Google Scholar]
- 45.Karanth S., Lall S. P., Denovan-Wright E. M., and Wright J. M.. 2009. Differential transcriptional modulation of duplicated fatty acid-binding protein genes by dietary fatty acids in zebrafish (Danio rerio): evidence for subfunctionalization or neofunctionalization of duplicated genes. BMC Evol. Biol. 9: 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Venkatachalam A. B., Thisse C., Thisse B., and Wright J. M.. 2009. Differential tissue-specific distribution of transcripts for the duplicated fatty acid-binding protein 10 (fabp10) genes in embryos, larvae and adult zebrafish (Danio rerio). FEBS J. 276: 6787–6797. [DOI] [PubMed] [Google Scholar]
- 47.Venkatachalam A. B., Sawler D. L., and Wright J. M.. 2013. Tissue-specific transcriptional modulation of fatty acid-binding protein genes, fabp2, fabp3 and fabp6, by fatty acids and the peroxisome proliferator, clofibrate, in zebrafish (Danio rerio). Gene. 520: 14–21. [DOI] [PubMed] [Google Scholar]
- 48.Braasch I., and Postlethwait J. H.. 2012. Fish polyploidy and the teleost genome duplication. In Polyploidy and Genome Evolution. P. S. Soltis and D. E. Soltis, editors. Springer, Berlin. 341–383. [Google Scholar]
- 49.Tingaud-Sequeira A., Knoll-Gellida A., André M., and Babin P. J.. 2012. Vitellogenin expression in white adipose tissue in female teleost fish. Biol. Reprod. 86: 38. [DOI] [PubMed] [Google Scholar]
- 50.Tingaud-Sequeira A., Ouadah N., and Babin P. J.. 2011. Zebrafish obesogenic test: a tool for screening molecules that target adiposity. J. Lipid Res. 52: 1765–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cotto E., André M., Forgue J., Fleury H. J., and Babin P. J.. 2005. Molecular characterization, phylogenetic relationships, and developmental expression patterns of prion genes in zebrafish (Danio rerio). FEBS J. 272: 500–513. [DOI] [PubMed] [Google Scholar]
- 52.Thisse C., and Thisse B.. 2008. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 3: 59–69. [DOI] [PubMed] [Google Scholar]
- 53. The UniProt Consortium. 2014. Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res. 42: D191–D198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., et al. 2007. Clustal W and Clustal X version 2.0. Bioinformatics. 23: 2947–2948. [DOI] [PubMed] [Google Scholar]
- 55.Higdon A. N., Dranka B. P., Hill B. G., Oh J. Y., Johnson M. S., Landar A., and Darley-Usmar V. M.. 2009. Methods for imaging and detecting modification of proteins by reactive lipid species. Free Radic. Biol. Med. 47: 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cummins T. D., Higdon A. N., Kramer P. A., Chacko B. K., Riggs D. W., Salabei J. K., Dell’Italia L. J., Zhang J., Darley-Usmar V. M., and Hill B. G.. 2013. Utilization of fluorescent probes for the quantification and identification of subcellular proteomes and biological processes regulated by lipid peroxidation products. Free Radic. Biol. Med. 59: 56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang G., Bonkovsky H. L., de Lemos A., and Burczynski F. J.. 2015. Recent insights into the biological functions of liver fatty acid binding protein 1. J. Lipid Res. 56: 2238–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Agellon L. B., Toth M. J., and Thomson A. B. R.. 2002. Intracellular lipid binding proteins of the small intestine. Mol. Cell. Biochem. 239: 79–82. [PubMed] [Google Scholar]
- 59.Cohn S. M., Simon T. C., Roth K. A., Birkenmeier E. H., and Gordon J. I.. 1992. Use of transgenic mice to map cis-acting elements in the intestinal fatty acid binding protein gene (Fabpi) that control its cell lineage-specific and regional patterns of expression along the duodenal-colonic and crypt-villus axes of the gut epithelium. J. Cell Biol. 119: 27–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crossman M. W., Hauft S. M., and Gordon J. I.. 1994. The mouse ileal lipid-binding protein gene: a model for studying axial patterning during gut morphogenesis. J. Cell Biol. 126: 1547–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alves-Costa F. A., Denovan-Wright E. M., Thisse C., Thisse B., and Wright J. M.. 2008. Spatio-temporal distribution of fatty acid-binding protein 6 (fabp6) gene transcripts in the developing and adult zebrafish (Danio rerio). FEBS J. 275: 3325–3334. [DOI] [PubMed] [Google Scholar]
- 62.Her G. M., Yeh Y. H., and Wu J. L.. 2004. Functional conserved elements mediate intestinal-type fatty acid binding protein (I-FABP) expression in the gut epithelia of zebrafish larvae. Dev. Dyn. 230: 734–742. [DOI] [PubMed] [Google Scholar]
- 63.Haunerland N. H., and Spener F.. 2004. Fatty acid-binding proteins–insights from genetic manipulations. Prog. Lipid Res. 43: 328–349. [DOI] [PubMed] [Google Scholar]
- 64.Gajda A. M., and Storch J.. 2015. Enterocyte fatty acid-binding proteins (FABPs): different functions of liver and intestinal FABPs in the intestine. Prostaglandins Leukot. Essent. Fatty Acids. 93: 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Venkatachalam A. B., Lall S. P., Denovan-Wright E. M., and Wright J. M.. 2012. Tissue-specific differential induction of duplicated fatty acid-binding protein genes by the peroxisome proliferator, clofibrate, in zebrafish (Danio rerio). BMC Evol. Biol. 12: 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Domenichiello A. F., Kitson A. P., and Bazinet R. P.. 2015. Is docosahexaenoic acid synthesis from α-linolenic acid sufficient to supply the adult brain? Prog. Lipid Res. 59: 54–66. [DOI] [PubMed] [Google Scholar]
- 67.Liu J. J., Green P., John Mann J., Rapoport S. I., and Sublette M. E.. 2015. Pathways of polyunsaturated fatty acid utilization: implications for brain function in neuropsychiatric health and disease. Brain Res. 1597: 220–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cremer T., and Cremer M.. 2010. Chromosome territories. Cold Spring Harb. Perspect. Biol. 2: a003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rouquette J., Cremer C., Cremer T., and Fakan S.. 2010. Functional nuclear architecture studied by microscopy: present and future. Int. Rev. Cell Mol. Biol. 282: 1–90. [DOI] [PubMed] [Google Scholar]
- 70.Fedorova E., and Zink D.. 2008. Nuclear architecture and gene regulation. Biochim. Biophys. Acta. 1783: 2174–2184. [DOI] [PubMed] [Google Scholar]
- 71.Jost K. L., Bertulat B., and Cardoso M. C.. 2012. Heterochromatin and gene positioning: inside, outside, any side? Chromosoma. 121: 555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang H., Starodub O., McIntosh A., Kier A. B., and Schroeder F.. 2002. Liver fatty acid-binding protein targets fatty acids to the nucleus. Real time confocal and multiphoton fluorescence imaging in living cells. J. Biol. Chem. 277: 29139–29151. [DOI] [PubMed] [Google Scholar]
- 73.Huang H., Starodub O., McIntosh A., Atshaves B. P., Woldegiorgis G., Kier A. B., and Schroeder F.. 2004. Liver fatty acid-binding protein colocalizes with peroxisome proliferator activated receptor alpha and enhances ligand distribution to nuclei of living cells. Biochemistry. 43: 2484–2500. [DOI] [PubMed] [Google Scholar]
- 74.Thumser A. E., and Storch J.. 2007. Characterization of a BODIPY-labeled fluorescent fatty acid analogue. Binding to fatty acid-binding proteins, intracellular localization, and metabolism. Mol. Cell. Biochem. 299: 67–73. [DOI] [PubMed] [Google Scholar]
- 75.Hostetler H. A., Balanarasimha M., Huang H., Kelzer M. S., Kaliappan A., Kier A. B., and Schroeder F.. 2010. Glucose regulates fatty acid binding protein interaction with lipids and peroxisome proliferator-activated receptor α. J. Lipid Res. 51: 3103–3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Honma Y., Niimi M., Uchiumi T., Takahashi Y., and Odani S.. 1994. Evidence for conformational change of fatty acid-binding protein accompanying binding of hydrophobic ligands. J. Biochem. 116: 1025–1029. [DOI] [PubMed] [Google Scholar]
- 77.Leng S., Lu S., Yao Y., Kan Z., Morris G. S., Stair B. R., Cherny M. A., and Black D. D.. 2007. Hepatocyte nuclear factor-4 mediates apolipoprotein A-IV transcriptional regulation by fatty acid in newborn swine enterocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 293: G475–G483. [DOI] [PubMed] [Google Scholar]
- 78.van Deursen D., van Leeuwen M., Akdogan D., Adams H., Jansen H., and Verhoeven A. J.. 2009. Activation of hepatic lipase expression by oleic acid: possible involvement of USF1. Nutrients. 1: 133–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Semova I., Carten J. D., Stombaugh J., Mackey L. C., Knight R., Farber S. A., and Rawls J. F.. 2012. Microbiota regulate intestinal absorption and metabolism of fatty acids in the zebrafish. Cell Host Microbe. 12: 277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Montoudis A., Seidman E., Boudreau F., Beaulieu J. F., Menard D., Elchebly M., Mailhot G., Sane A. T., Lambert M., Delvin E., et al. 2008. Intestinal fatty acid binding protein regulates mitochondrion beta-oxidation and cholesterol uptake. J. Lipid Res. 49: 961–972. [DOI] [PubMed] [Google Scholar]
- 81.Lagakos W. S., Gajda A. M., Agellon L., Binas B., Choi V., Mandap B., Russnak T., Zhou Y. X., and Storch J.. 2011. Different functions of intestinal and liver-type fatty acid-binding proteins in intestine and in whole body energy homeostasis. Am. J. Physiol. Gastrointest. Liver Physiol. 300: G803–G814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Darimont C., Gradoux N., Persohn E., Cumin F., and De Pover A.. 2000. Effects of intestinal fatty acid-binding protein overexpression on fatty acid metabolism in Caco-2 cells. J. Lipid Res. 41: 84–92. [PubMed] [Google Scholar]
- 83.Vassileva G., Huwyler L., Poirier K., Agellon L. B., and Toth M. J.. 2000. The intestinal fatty acid binding protein is not essential for dietary fat absorption in mice. FASEB J. 14: 2040–2046. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.