

Abstract
BACKGROUND
Corticotropin-independent macronodular adrenal hyperplasia may be an incidental finding or it may be identified during evaluation for Cushing’s syndrome. Reports of familial cases and the involvement of both adrenal glands suggest a genetic origin of this condition.
METHODS
We genotyped blood and tumor DNA obtained from 33 patients with corticotropin-independent macronodular adrenal hyperplasia (12 men and 21 women who were 30 to 73 years of age), using single-nucleotide polymorphism arrays, microsatellite markers, and whole-genome and Sanger sequencing. The effects of armadillo repeat containing 5 (ARMC5) inactivation and overexpression were tested in cell-culture models.
RESULTS
The most frequent somatic chromosome alteration was loss of heterozygosity at 16p (in 8 of 33 patients for whom data were available [24%]). The most frequent mutation identified by means of whole-genome sequencing was in ARMC5, located at 16p11.2. ARMC5 mutations were detected in tumors obtained from 18 of 33 patients (55%). In all cases, both alleles of ARMC5 carried mutations: one germline and the other somatic. In 4 patients with a germline ARMC5 mutation, different nodules from the affected adrenals harbored different secondary ARMC5 alterations. Transcriptome-based classification of corticotropin-independent macronodular adrenal hyperplasia indicated that ARMC5 mutations influenced gene expression, since all cases with mutations clustered together. ARMC5 inactivation decreased steroidogenesis in vitro, and its overexpression altered cell survival.
CONCLUSIONS
Some cases of corticotropin-independent macronodular adrenal hyperplasia appear to be genetic, most often with inactivating mutations of ARMC5, a putative tumor-suppressor gene. Genetic testing for this condition, which often has a long and insidious prediagnostic course, might result in earlier identification and better management. (Funded by Agence Nationale de la Recherche and others.)
Corticotropin-independent macronodular adrenal hyperplasia can lead to excess cortisol secretion and Cushing’s syndrome.1,2 Adrenocortical nodules in corticotropin-independent macronodular adrenal hyperplasia are, by definition, larger than 10 mm in diameter and frequently reach 30 to 40 mm in diameter. The condition is typically diagnosed in patients with Cushing’s syndrome who are between 40 and 60 years of age and who have suppressed levels of circulating corticotropin. Tumor growth and cortisol dysregulation appear to progress slowly in cases of corticotropin-independent macronodular adrenal hyperplasia, and the diagnosis is often made only after several years or decades of disease progression.3 Milder forms are commonly detected in patients with incidentally discovered adrenal tumors or hyperplasia. Aberrant receptor expression leading to unexpected cortisol responses to ligands that stimulate G-protein–coupled receptors has been reported.4–7 Treatment is most often surgical, but medical therapies targeting aberrant receptor expression have been effective in controlling excess cortisol in some cases.6
The bilateral nature of adrenal tumors in corticotropin-independent macronodular adrenal hyperplasia provides support for the hypothesis of a germline genetic predisposition. Reports of familial cases suggest the involvement of germline hereditary factors in the emergence of corticotropin-independent macronodular adrenal hyperplasia.1,8–11 The familial occurrence of the disease might be underrecognized because of the variation in disease severity. Corticotropin-independent macronodular adrenal hyperplasia has been reported in a small subgroup of patients with familial multiple tumor syndromes.1,11–13 In the McCune– Albright syndrome, mosaic mutations of the gene encoding G-protein subunit αs (GNAS) have been observed in young children with bilateral adrenocortical nodules.14 However, the vast majority of cases are not part of any known multiple tumor syndrome, and the genetic basis of this condition has not been established.
METHODS
STUDY OVERSIGHT
The first two authors and the last two authors vouch for the completeness and accuracy of the data and analyses. Written informed consent for the analysis of the tumor and leukocyte DNA was obtained from all patients. The study was approved by the institutional review board of Cochin Hospital.
STUDY PATIENTS
A total of 33 patients (21 women and 12 men; age range, 30 to 73 years) who had undergone surgery for corticotropin-independent macronodular adrenal hyperplasia were included in the study. They had various levels of corticotropin-independent hypercortisolism and underwent computed tomographic (CT) imaging and adrenalectomy.5,15 The diagnosis of corticotropin-independent macronodular adrenal hyperplasia was confirmed histologically. Detailed clinical phenotypes are described in Table 1 and in Table S1 in the Supplementary Appendix, available with the full text of this article at NEJM.org. Thirty-one patients were unrelated; 2 were siblings. The tumor samples were obtained prospectively by the Corticomedullosurrénale Tumeur Endocrine Network tumor bank.16
Table 1.
Characteristics of the Patients.*
| Patient No. | Sex | Age | Corticotropin Level† | Plasma Cortisol Level after 1 mg of Dexamethasone‡ | Urinary Cortisol Level§ | Cushing’s Syndrome | Adrenal Weight | ||
|---|---|---|---|---|---|---|---|---|---|
| Right | Left | Total | |||||||
| y | pg/ml | μg/dl | μg/24 hr | g | |||||
| Patients with mutated ARMC5 | |||||||||
| 1 | F | 34 | <5 | 23.6 | 373 | Clinical | 40 | 26 | 66 |
| 2 | F | 41 | <5 | NA | NA | Clinical | 30 | 94 | 124 |
| 3 | M | 64 | 6 | 27.2 | 866 | Clinical | 39 | 118 | 157 |
| 4 | F | 40 | <5 | 18.0 | 233 | Clinical | NA | NA | NA |
| 5 | F | 30 | <5 | 26.1 | 458 | Clinical | 52 | 67 | 119 |
| 6 | F | 48 | <5 | 27.4 | 118 | Clinical | 34 | 50 | 84 |
| 7 | F | 51 | <10 | 22.4 | 248 | Clinical | 45 | 64 | 109 |
| 8 | M | 45 | <10 | 36.7 | 1463 | Clinical | 48 | 71 | 119 |
| 9 | F | 70 | <5 | 16.0 | 122 | Clinical | 21 | 60 | 81 |
| 10 | M | 41 | <5 | 29.0 | 394 | Clinical | 65 | 121 | 186 |
| 11 | M | 46 | <5 | 18.0 | 204 | Clinical | 34 | 46 | 80 |
| 12 | M | 55 | <5 | 3.6 | 135 | Subclinical | 42 | 30 | 72 |
| 13 | M | 63 | 8 | 22.9 | 302 | Clinical | 35 | 50 | 85 |
| 14 | M | 53 | <5 | 25.1 | 293 | Clinical | 65 | 77 | 142 |
| 15 | M | 56 | <5 | NA | 107 | Clinical | 56 | 110 | 166 |
| 16 | M | 51 | <5 | NA | NA | Clinical | NA | NA | NA |
| 17 | F | 52 | <5 | 8.9 | 37 | Subclinical | 24 | 29 | 53 |
| 18 | F | 73 | <5 | 15.0 | NA | Subclinical | 21 | 34 | 55 |
| Total¶ | 9 F, 9 M | 51 (27–74) | 6 (<5 to 9) | 21.3 (4.7 to 38) | 357 (0 to 1089) | Clinical, 15; subclinical, 3 | 41 (13 to 69) | 65 (3 to 128)‖ | 106 (25 to 188)‖ |
| Patients with nonmutated ARMC5 | |||||||||
| Total | 12 F, 3 M | 52 (31 to 74) | 8 (<5 to 18) | 14.6 (0 to 32.5) | 362 (0 to 1466) | Clinical, 11; subclinical, 3 | 27 (0 to 75) | 38 (0 to 88) | 55 (0 to 144) |
NA denotes not available.
The normal corticotropin level is 10 to 60 pg per milliliter.
The normal plasma cortisol level is <1.8 μg per deciliter.
The normal urinary cortisol level is <90 μg per 24 hours.
Quantitative variables are expressed as means with 95% confidence intervals, and qualitative variables are expressed as numbers of patients.
P<0.05.
GENOTYPING AND SEQUENCING
Genomic DNA was isolated as previously described from 41 adrenal nodules and 25 leukocyte samples obtained from the 33 patients.17,18 The DNA from 34 nodules and 18 leukocyte samples was hybridized to single-nucleotide polymorphism (SNP) arrays (Affymetrix SNP 6.0). Somatic loss of heterozygosity and copy-number variants were identified as previously described19 (see the Methods section and Fig. S1 in the Supplementary Appendix). Loss of heterozygosity was further investigated in 7 nodules and their paired leukocytes by means of microsatellite analysis.
DNA from five adrenal nodules and their paired leukocyte samples were sequenced by Complete Genomics20 with the use of the Cancer Sequencing Service pipeline, version 2.0.2.26. Subsequent filtering is detailed in the Methods section and Figure S2 in the Supplementary Appendix. Sanger sequencing of armadillo repeat containing 5 (ARMC5) is described in the Methods section in the Supplementary Appendix.
The transcriptomes of 10 adrenal samples from patients with corticotropin-independent macronodular adrenal hyperplasia were studied as previously described21 (see the Methods section in the Supplementary Appendix).
CELL CULTURE AND TRANSFECTION
Human HeLa cells and adrenocortical cancer cells (H295R) were cultured, transfected, and stimulated with forskolin as previously described.22,23 The small interfering RNA (siRNA) targeting ARMC5 and the control siRNA used are described in the Methods section in the Supplementary Appendix. ARMC5 expression vector containing a FLAG tag (Origen RC226267) was used for mutagenesis with Agilent Technologies kit 200521.
WESTERN BLOTTING, IMMUNOSTAINING, AND MESSENGER RNA ANALYSIS
Preparations of whole-cell or tissue lysates, Western blotting, immunohistochemical analysis, and immunofluorescence were performed as previously described23,24 (see the Methods section in the Supplementary Appendix).
Total RNA was extracted from the cell lines, and the expression levels of target genes were determined by means of real-time polymerase chain reaction (PCR) as previously described23 (see the Methods section in the Supplementary Appendix). Cortisol concentrations in culture medium were assayed as previously described.15
RESULTS
GENOMEWIDE GENOTYPING AND SEQUENCING
To search for gene alterations with the potential to cause corticotropin-independent macronodular adrenal hyperplasia, we used SNP arrays for genomewide screening of chromosomal alterations in 34 tumor specimens obtained from 26 patients with corticotropin-independent macronodular adrenal hyperplasia who had undergone surgery. Recurrent somatic chromosomal alterations in nodules from the patients were rare (Fig. S3 and Tables S2, S3, and S4 in the Supplementary Appendix), except at 16p (Fig. 1). A copy-neutral loss of heterozygosity was identified in 10 of 34 tumor specimens (29%) obtained from 7 of the 26 patients (27%). In addition, somatic loss of heterozygosity in 16p11. was detected with the use of microsatellite markers in 1 of 7 other patients (Table S5 in the Supplementary Appendix); thus, loss of heterozygosity was detected at 16p in 8 of 33 patients (24%) with corticotropin-independent macronodular adrenal hyperplasia.
Figure 1. Chromosomal Alterations in Nodules Identified by Means of Single-Nucleotide Polymorphism (SNP) Arrays.
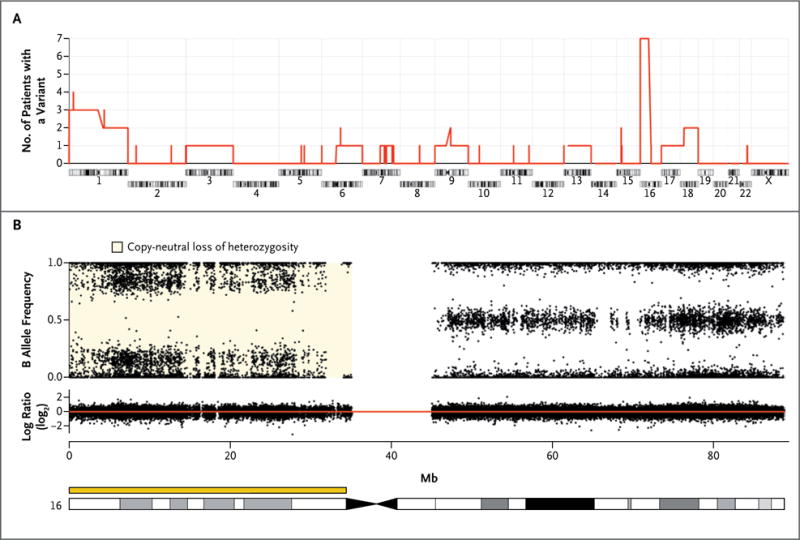
Panel A shows the number of patients with corticotropin-independent macronodular adrenal hyperplasia who had a chromosomal alteration (gain, loss, or copy-neutral loss of heterozygosity) in at least one nodule. The most common event, the copy-neutral loss of heterozygosity in 16p, was detected in 7 of 26 patients. Panel B shows chromosome 16 from a nodule obtained from Patient 5 with a copy-neutral loss of heterozygosity in 16p. The upper part of the panel shows genotypes of the SNPs expressed as the B allele frequency. The lower part of the panel shows the DNA copy number expressed on a base-2 log scale (log ratio), with the red line corresponding to two copies of DNA.
Whole-genome sequencing in five paired tumor and leukocyte DNA samples identified somatic mutations affecting the coding sequence of 85 genes and structural variants affecting the coding sequence of 12 genes (Tables S6, S7, and S8 in the Supplementary Appendix). Only 1 gene, ARMC5, which maps to 16p11.2, was modified in more than one tumor sample; tumor samples obtained from three patients had a mutation. The somatic mutations of ARMC5 included two frameshift mutations and one missense mutation. These mutations were confirmed by means of Sanger sequencing.
Direct sequencing of tumor DNA identified ARMC5 mutations in 18 of the 33 patients (55%). A total of 26 tumor specimens obtained from these 18 patients were analyzed. All tumors tested had 2 genetic alterations in the ARMC5 locus: 2 mutations in 16 specimens, 1 mutation with loss of heterozygosity at 16p (loss of the ARMC5 nonmutated allele) in 9 specimens, and 1 mutation plus a microdeletion (1.3 Mb) in 1 specimen (Fig. 2A). The 28 mutations identified included 6 nonsense, 10 frameshift, 8 missense, and 4 more complex mutations (Fig. S4 in the Supplementary Appendix). None of these mutations were detected in the 186 control leukocyte DNA samples that were sequenced in the laboratory or in several thousand other controls from the exome variant server, hosted by the National Heart, Lung, and Blood Institute (http://evs.gs.washington.edu/EVS), with the exception of the p.R267X mutation, which was detected in 1 of 6297 controls (Table S9 in the Supplementary Appendix). Western blot analysis showed that the level of ARMC5 protein was decreased in the majority of patients with corticotropin-independent macronodular adrenal hyperplasia who had an ARMC5 mutation, and especially in those with nonsense mutations leading to a premature stop codon combined with a somatic loss of the nonmutated allele (Fig. 2B).
Figure 2. ARMC5 Alterations in Leukocyte and Tumor DNA.

Panel A shows the two ARMC5 alterations detected in the tumor DNA obtained from 18 patients. The analysis of leukocyte DNA (left column) shows the germline alteration (blue box). Each tumor under “Somatic Variants in Different Tumors” is shown as a box with a blue rectangle (showing the alteration in leukocyte DNA) and a yellow rectangle (showing the alteration that is detected only in the tumor DNA). Germline DNA from 4 patients was not available (bottom right). Panel B shows Western blot analysis of ARMC5 protein in adrenocortical human cell line H295R transfected with control small interfering RNA (siRNA) or ARMC5 siRNA (left), 3 normal adrenal glands (middle), and 17 samples obtained from patients with corticotropin-independent macronodular adrenal hyperplasia (right): 7 without an ARMC5 alteration and 10 with various types of ARMC5 alterations. LOH denotes loss of heterozygosity, LT left tumor, and RT right tumor.
ARMC5 was analyzed in leukocyte DNA obtained from 14 of the 18 patients with ARMC5 mutations. A germline ARMC5 alteration was detected in all 14 patients: a mutation in 13 patients and a microdeletion (1.3 Mb) in 1 patient (Fig. 2A, and Fig. S5 in the Supplementary Appendix). This observation suggests that ARMC5 might be a tumor-suppressor gene that leads to the development of a tumor when a primary inactivating alteration on one allele in the germ-line is present and a somatic secondary event affecting the second allele occurs. This could be directly observed by sequencing for two nodules obtained from 2 patients with two ARMC5 mutations close enough to be amplified together; the sequences of the PCR products from these two nodules showed that the two mutations were present on two different alleles (Fig. S6 in the Supplementary Appendix).
In four patients with an ARMC5 germline mutation, two or more nodules from one or both adrenal glands were analyzed. In each case, the same germline ARMC5 mutation was associated with different, nodule-specific, somatic ARMC5 alterations (Fig. 2A and 3). This observation suggests that in addition to the germline inactivating mutation, there were different somatic ARMC5 second hits in the two adrenals of each patient, one for each nodule. In contrast, in the inter-nodular regions in four other patients with corticotropin-independent macronodular adrenal hyperplasia, no somatic second hit could be detected; only the germline ARMC5 mutation was observed (Fig. S7 in the Supplementary Appendix).
Figure 3. Analysis of Multiple Nodules Obtained from the Same Patient.
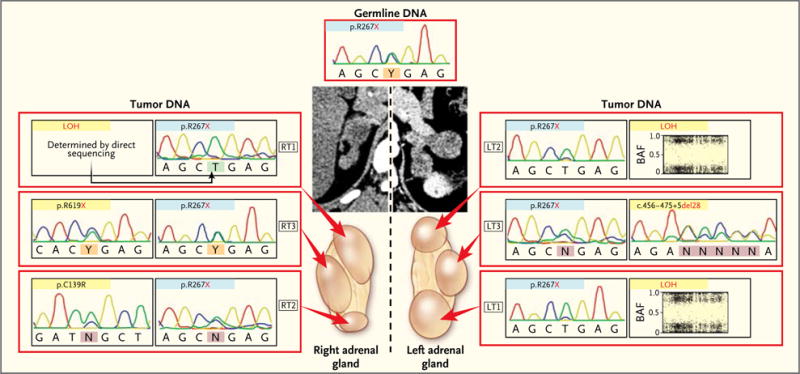
Computed tomographic scans and the various nodules present on both adrenal glands in Patient 5 are shown. Each nodule showed the germline defect (blue). A second alteration (yellow) differed between the two adrenals.
Familial screening for ARMC5 mutations was subsequently performed in 11 first-degree relatives of 7 index patients. None of these relatives had known Cushing’s syndrome or any known adrenal tumor. A germline ARMC5 alteration was detected in 6 of the 11 relatives, who were younger siblings or children of the index patients. Subsequent CT scanning revealed adrenal nodular hyperplasia with bilateral nodules ranging from 8 mm to 2.5 cm in 5 of these 6 relatives. Abnormal responses to dexamethasone were observed in 3 of the 6 relatives, and an increased 24-hour urinary cortisol excretion was detected in 1. There were no abnormalities in adrenal secretion or CT studies in any of the 5 relatives with nonmutated ARMC5 alleles.
TRANSCRIPTOME ANALYSIS
The function of ARMC5 is not known. To assess the functional consequences of ARMC5 inactivation, we analyzed the transcriptome of 10 tumor specimens obtained from patients with corticotropin-independent macronodular adrenal hyperplasia (5 with ARMC5 mutations and 5 with nonmutated ARMC5). Sorting of these tumors without awareness of their mutation status (unsupervised hierarchical clustering) resulted in two distinct groups: one containing the 5 mutated tumors and the other containing the 5 nonmutated tumors (P = 0.008 by Fisher’s exact test) (Fig. S8 in the Supplementary Appendix). Several hundred genes are differentially expressed between the two groups. The transcriptome of mutated tumors was significantly enriched in genes related to RNA processing (Tables S10 and S11 in the Supplementary Appendix). This clearly indicates that ARMC5 mutations have a large effect on gene expression and identify a subgroup of patients with corticotropin-independent macronodular adrenal hyperplasia.
IMMUNOHISTOCHEMICAL ANALYSIS AND CELL CULTURE AND TRANSFECTION
Immunohistochemical analysis of adrenal samples and two cell lines (H295R and HeLa) showed that ARMC5 is mostly located in the cytoplasm (Fig. S9 in the Supplementary Appendix). In patients with corticotropin-independent macronodular adrenal hyperplasia and an ARMC5 nonsense mutation, a decreased level of ARMC5 and steroidogenic enzyme CYP11A1 immunostaining were observed, findings that are consistent with the reduced expression of steroidogenic enzymes in the transcriptome (Table S11 in the Supplementary Appendix).
We investigated the role of ARMC5 in steroidogenesis in vitro. ARMC5 inactivation by siRNA in the H295R cell line reduced messenger RNA (mRNA) levels of genes encoding steroidogenic enzymes CYP17A1 and CYP21A2 and reduced mRNA levels of the gene encoding adrenal transcription factor NR5A1 (also called steroidogenic factor 1) and the melanocortin 2 receptor MC2R (Fig. 4A). In contrast, ARMC5 inactivation had no effect on the mRNA level of the gene encoding transcription factor NR0B1 (DAX-1). Cortisol synthesis was reduced in H295R cells after ARMC5 inactivation (Fig. 4B).
Figure 4. Analysis of the Role of ARMC5.
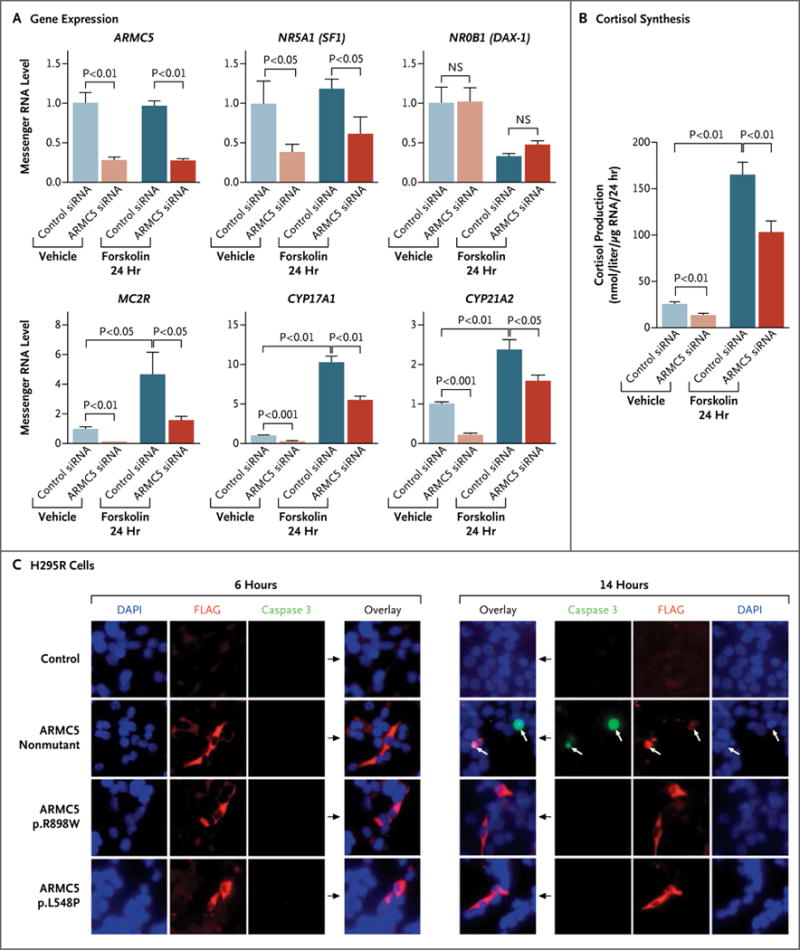
Panels A and B show the effects of ARMC5 inactivation by siRNA on the expression of ARMC5, NR5A1, NR0B1, MC2R, CYP17A1, and CYP21A2 and on basal and forskolin-stimulated cortisol synthesis in adrenocortical H295R cells. NS denotes not significant. Panel C shows immunofluorescence staining of transfected H295R cells with different ARMC5-FLAG constructs (a nonmutated construct and two missense mutants: p.R898W and p.L548P). After 6 hours, the cells expressing the ARMC5 constructs had normal morphologic features. After 14 hours, the cells expressing the nonmutant ARMC5 construct (white arrows) were apoptotic (coexpression of the cleaved caspase 3 [green stain] and altered cell morphology with condensed nuclei), and the cells expressing the missense ARMC5 constructs (p.R898W and p.L548P) were not apoptotic. Red staining shows FLAG antibodies (ARMC5 constructs), and blue staining shows 4′,6-diamidino-2-phenylindole (DAPI) (cell nuclei).
We next studied the role of ARMC5 in cell survival. In transient transfection experiments, the expression of nonmutated ARMC5 in H295R and HeLa cells resulted in cell death. No viable transfected cells could be detected after 14 hours, as shown by costaining of cleaved caspase 3 and ARMC5-FLAG (Fig. 4C, and Fig. S10 in the Supplementary Appendix). Moreover, apoptosis was confirmed by the cleavage of two markers of apoptosis: poly(ADP-ribose) polymerase and lamin A and C (Fig. S11 in the Supplementary Appendix). In contrast, this effect was not observed with the two germline missense mutations p.R898W and p.L548P (Fig. 4C, and Fig. S10 and S11 in the Supplementary Appendix).
DISCUSSION
The detection of ARMC5 mutations in more than half the patients in this series who underwent surgery for corticotropin-independent macro nodular adrenal hyperplasia suggests a common genetic cause of the condition. A combined genomic approach showed that ARMC5 genetic alterations conform to the two-hit model for a likely tumor-suppressor gene. Indeed, patients with a germ-line ARMC5-inactivating mutation had nodule-specific secondary somatic mutations inactivating ARMC5. The occurrence of various somatic mutations within a single adrenal suggests polyclonal tumorigenesis related to a predisposition arising from a single germline mutation. The consistent occurrence of ARMC5 mutations in these adrenal nodules clearly suggests a major role of ARMC5 inactivation in their development.
Inactivation of ARMC5 was associated with a slow process of apparent dedifferentiation of adrenocortical cells and growth of bilateral masses in the patients with corticotropin-independent macronodular adrenal hyperplasia, along with reduced expression of steroidogenic enzymes and MC2R and abnormal cortisol production; these findings are consistent with previous expression-profile studies.25,26 In our patients, the weight of the adrenals in patients in whom ARMC5 was mutated was 4 to 24 times that of a normal adrenal. It is therefore likely that, despite the reduced secretory capacity of each cell, the overall production of cortisol was increased because of the large adrenal mass. This phenomenon may largely explain why a severe form of clinical Cushing’s syndrome develops only in patients with very large adrenal nodules and why the disease often has an insidious course.
Most patients with an ARMC5 mutation who were evaluated for aberrant membrane-receptor expression had positive upright and metoclopramide tests.27 In contrast, the three patients in this cohort and three additional index patients with a food-dependent Cushing’s syndrome, which is characterized by aberrant adrenal sensitivity to gastric inhibitory polypeptide,4,7 did not have any ARMC5 mutations (Table S1 in the Supplementary Appendix). Thus, ARMC5 inactivation may be associated with a particular type of aberrant receptor expression. Furthermore, ARMC5 mutations were associated with a specific transcriptome profile in the adrenal nodules.
As noted above, the function of ARMC5 is unknown. It encodes a protein that contains an armadillo repeat domain, suggesting that protein–protein interactions may be important for its function. β-catenin, which also contains armadillo repeats, is another gene that is frequently mutated in various cancers, including adrenocortical tumors.28
The present study also shows that corticotropin-independent macronodular adrenal hyperplasia is frequently genetic. A limitation of previous studies that were based on clinical screening may be the substantial variation in disease severity both within a given pedigree and among index case patients. For example, the siblings of two of the index patients in the present series had a severe form of adrenal Cushing’s syndrome, but most relatives of the seven other index patients with an ARMC5 mutation who agreed to be evaluated had adrenal nodules either without Cushing’s syndrome or with only subclinical evidence of it. The slow progression of the disease and the advanced age at diagnosis make it difficult to study older generations in the families of index patients. In the current study, we were unable to evaluate any older relatives of the patients in whom mutations were identified. We speculate that mortality among some of the older relatives who were not examined may have been affected by cardiometabolic consequences of undiagnosed Cushing’s syndrome. An alternative hypothesis is that secondary genetic or environmental factors might modulate the effects of germline ARMC5 mutations. Such genetic secondary factors have been observed in another form of bilateral adrenal tumors leading to Cushing’s syndrome (primary pigmented nodular adrenocortical disease) in patients with Carney complex due to PRKAR1A mutations.29
In conclusion, we identified ARMC5 mutations in a substantial proportion of patients with corticotropin-independent macronodular adrenal hyperplasia. In affected patients, recovery from the effects of excess cortisol after definitive therapy for Cushing’s syndrome is often only partial because of years of exposure to increased cortisol levels. This study provides information that may be helpful in developing diagnostic tests that will lead to earlier diagnosis and management of this disease.
Supplementary Material
Acknowledgments
Supported in part by grants from Agence Nationale de la Recherche (ANR-10-Blan-1136), Corticomedullosurrénale Tumeur Endocrine Network (Programme Hospitalier de Recherche Clinique grant AOM95201), Assistance Publique–Hôpitaux de Paris (Clinical Research Center Grant Genhyper P061006), Institut National du Cancer (Recherche Translationelle 2009-RT-02), the Seventh Framework Program of the European Commission (F2-2010-259735), INSERM (Contrat d’Interface, to Dr. Assié), the Conny-Maeva Charitable Foundation, and the intramural program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development.
We thank Drs. J. Chelly and M. Delpech of the cell bank of Cochin Hospital and Dr. B. Terris of the tumor bank of Cochin Hospital for their help in sample collection; Dr. E. Clauser of the oncogenetic unit of Cochin Hospital for help in microsatellite analysis; Drs. J. Guibourdenche and E. Clauser of the hormone biology unit of Cochin Hospital for cortisol assays; Drs. F. Tissier and Pierre Colin for pathological analysis; Anne Audebourg for technical assistance; J. Metral and A. de Reynies of the Cartes d’Identité des Tumeurs program of Ligue Nationale contre le Cancer for help in genomics studies and fruitful discussions; Dr. P. Nietschke of the bioinformatics platforms of Paris Descartes University for helpful discussions; all the members of the Genomics and Signaling of Endocrine Tumors team and of the genomic platform of Cochin Institute for their help in these studies; and the patients and their families, as well as the physicians and staff involved in patient care, for their active participation.
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
References
- 1.Hsiao HP, Kirschner LS, Bourdeau I, et al. Clinical and genetic heterogeneity, overlap with other tumor syndromes, and atypical glucocorticoid hormone secretion in adrenocorticotropin-independent macronodular adrenal hyperplasia compared with other adrenocortical tumors. J Clin Endocrinol Metab. 2009;94:2930–7. doi: 10.1210/jc.2009-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lacroix A. ACTH-independent macronodular adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab. 2009;23:245–59. doi: 10.1016/j.beem.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 3.Swain JM, Grant CS, Schlinkert RT, et al. Corticotropin-independent macronodular adrenal hyperplasia: a clinicopathologic correlation. Arch Surg. 1998;133:541–5. doi: 10.1001/archsurg.133.5.541. [DOI] [PubMed] [Google Scholar]
- 4.Reznik Y, Allali-Zerah V, Chayvialle JA, et al. Food-dependent Cushing’s syndrome mediated by aberrant adrenal sensitivity to gastric inhibitory polypeptide. N Engl J Med. 1992;327:981–6. doi: 10.1056/NEJM199210013271403. [DOI] [PubMed] [Google Scholar]
- 5.Libé R, Coste J, Guignat L, et al. Aberrant cortisol regulations in bilateral macronodular adrenal hyperplasia: a frequent finding in a prospective study of 32 patients with overt or subclinical Cushing’s syndrome. Eur J Endocrinol. 2010;163:129–38. doi: 10.1530/EJE-10-0195. [DOI] [PubMed] [Google Scholar]
- 6.Lacroix A, Bourdeau I, Lampron A, Mazzuco TL, Tremblay J, Hamet P. Aberrant G-protein coupled receptor expression in relation to adrenocortical overfunction. Clin Endocrinol (Oxf) 2010;73:1–15. doi: 10.1111/j.1365-2265.2009.03689.x. [DOI] [PubMed] [Google Scholar]
- 7.Lacroix A, Bolté E, Tremblay J, et al. Gastric inhibitory polypeptide-dependent cortisol hypersecretion — a new cause of Cushing’s syndrome. N Engl J Med. 1992;327:974–80. doi: 10.1056/NEJM199210013271402. [DOI] [PubMed] [Google Scholar]
- 8.Findlay JC, Sheeler LR, Engeland WC, Aron DC. Familial adrenocorticotropin-independent Cushing’s syndrome with bilateral macronodular adrenal hyperplasia. J Clin Endocrinol Metab. 1993;76:189–91. doi: 10.1210/jcem.76.1.8380604. [DOI] [PubMed] [Google Scholar]
- 9.Gagliardi L, Hotu C, Casey G, et al. Familial vasopressin-sensitive ACTH-independent macronodular adrenal hyperplasia (VPs-AIMAH): clinical studies of three kindreds. Clin Endocrinol (Oxf) 2009;70:883–91. doi: 10.1111/j.1365-2265.2008.03471.x. [DOI] [PubMed] [Google Scholar]
- 10.Vezzosi D, Cartier D, Régnier C, et al. Familial adrenocorticotropin-independent macronodular adrenal hyperplasia with aberrant serotonin and vasopressin adrenal receptors. Eur J Endocrinol. 2007;156:21–31. doi: 10.1530/eje.1.02324. [DOI] [PubMed] [Google Scholar]
- 11.Matyakhina L, Freedman RJ, Bourdeau I, et al. Hereditary leiomyomatosis associated with bilateral, massive, macronodular adrenocortical disease and atypical Cushing syndrome: a clinical and molecular genetic investigation. J Clin Endocrinol Metab. 2005;90:3773–9. doi: 10.1210/jc.2004-2377. [DOI] [PubMed] [Google Scholar]
- 12.Gaujoux S, Pinson S, Gimenez-Roqueplo AP, et al. Inactivation of the APC gene is constant in adrenocortical tumors from patients with familial adenomatous polyposis but not frequent in sporadic adrenocortical cancers. Clin Cancer Res. 2010;16:5133–41. doi: 10.1158/1078-0432.CCR-10-1497. [DOI] [PubMed] [Google Scholar]
- 13.Yoshida M, Hiroi M, Imai T, et al. A case of ACTH-independent macronodular adrenal hyperplasia associated with multiple endocrine neoplasia type 1. Endocr J. 2011;58:269–77. doi: 10.1507/endocrj.k10e-218. [DOI] [PubMed] [Google Scholar]
- 14.Carney JA, Young WF, Stratakis CA. Primary bimorphic adrenocortical disease: cause of hypercortisolism in McCune-Albright syndrome. Am J Surg Pathol. 2011;35:1311–26. doi: 10.1097/PAS.0b013e31821ec4ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bertherat J, Contesse V, Louiset E, et al. In vivo and in vitro screening for illegitimate receptors in adrenocorticotropin-independent macronodular adrenal hyperplasia causing Cushing’s syndrome: identification of two cases of gonadotropin/gastric inhibitory polypeptide-dependent hypercortisolism. J Clin Endocrinol Metab. 2005;90:1302–10. doi: 10.1210/jc.2004-1256. [DOI] [PubMed] [Google Scholar]
- 16.Gicquel C, Bertagna X, Gaston V, et al. Molecular markers and long-term recurrences in a large cohort of patients with sporadic adrenocortical tumors. Cancer Res. 2001;61:6762–7. [PubMed] [Google Scholar]
- 17.Libé R, Fratticci A, Coste J, et al. Phosphodiesterase 11A (PDE11A) and genetic predisposition to adrenocortical tumors. Clin Cancer Res. 2008;14:4016–24. doi: 10.1158/1078-0432.CCR-08-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Libè R, Groussin L, Tissier F, et al. Somatic TP53 mutations are relatively rare among adrenocortical cancers with the frequent 17p13 loss of heterozygosity. Clin Cancer Res. 2007;13:844–50. doi: 10.1158/1078-0432.CCR-06-2085. [DOI] [PubMed] [Google Scholar]
- 19.Assié G, LaFramboise T, Platzer P, Bertherat J, Stratakis CA, Eng C. SNP arrays in heterogeneous tissue: highly accurate collection of both germline and somatic genetic information from unpaired single tumor samples. Am J Hum Genet. 2008;82:903–15. doi: 10.1016/j.ajhg.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drmanac R, Sparks AB, Callow MJ, et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science. 2010;327:78–81. doi: 10.1126/science.1181498. [DOI] [PubMed] [Google Scholar]
- 21.de Reyniès A, Assié G, Rickman DS, et al. Gene expression profiling reveals a new classification of adrenocortical tumors and identifies molecular predictors of malignancy and survival. J Clin Oncol. 2009;27:1108–15. doi: 10.1200/JCO.2008.18.5678. [DOI] [PubMed] [Google Scholar]
- 22.Groussin L, Massias JF, Bertagna X, Bertherat J. Loss of expression of the ubiquitous transcription factor cAMP response element-binding protein (CREB) and compensatory overexpression of the activator CREMtau in the human adrenocortical cancer cell line H295R. J Clin Endocrinol Metab. 2000;85:345–54. doi: 10.1210/jcem.85.1.6307. [DOI] [PubMed] [Google Scholar]
- 23.Ragazzon B, Cazabat L, Rizk-Rabin M, et al. Inactivation of the Carney complex gene 1 (protein kinase A regulatory subunit 1A) inhibits SMAD3 expression and TGF beta-stimulated apoptosis in adrenocortical cells. Cancer Res. 2009;69:7278–84. doi: 10.1158/0008-5472.CAN-09-1601. [DOI] [PubMed] [Google Scholar]
- 24.Rizk-Rabin M, Assie G, Rene-Corail F, et al. Differential expression of parathyroid hormone-related protein in adrenocortical tumors: autocrine/paracrine effects on the growth and signaling pathways in H295R cells. Cancer Epidemiol Biomarkers Prev. 2008;17:2275–85. doi: 10.1158/1055-9965.EPI-07-2924. [DOI] [PubMed] [Google Scholar]
- 25.Assie G, Louiset E, Sturm N, et al. Systematic analysis of G protein-coupled receptor gene expression in adrenocorticotropin-independent macronodular adrenocortical hyperplasia identifies novel targets for pharmacological control of adrenal Cushing’s syndrome. J Clin Endocrinol Metab. 2010;95(10):E253–E262. doi: 10.1210/jc.2009-2281. [DOI] [PubMed] [Google Scholar]
- 26.Antonini SR, Baldacchino V, Tremblay J, Hamet P, Lacroix A. Expression of ACTH receptor pathway genes in glucose-dependent insulinotrophic peptide (GIP)-dependent Cushing’s syndrome. Clin Endocrinol (Oxf) 2006;64:29–36. doi: 10.1111/j.1365-2265.2005.02411.x. [DOI] [PubMed] [Google Scholar]
- 27.Bourdeau I, D’Amour P, Hamet P, Boutin JM, Lacroix A. Aberrant membrane hormone receptors in incidentally discovered bilateral macronodular adrenal hyperplasia with subclinical Cushing’s syndrome. J Clin Endocrinol Metab. 2001;86:5534–40. doi: 10.1210/jcem.86.11.8062. [DOI] [PubMed] [Google Scholar]
- 28.Tissier F, Cavard C, Groussin L, et al. Mutations of beta-catenin in adrenocortical tumors: activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res. 2005;65:7622–7. doi: 10.1158/0008-5472.CAN-05-0593. [DOI] [PubMed] [Google Scholar]
- 29.Libé R, Horvath A, Vezzosi D, et al. Frequent phosphodiesterase 11A gene (PDE11A) defects in patients with Carney complex (CNC) caused by PRKAR1A mutations: PDE11A may contribute to adrenal and testicular tumors in CNC as a modifier of the phenotype. J Clin Endocrinol Metab. 2011;96(1):E208–E214. doi: 10.1210/jc.2010-1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.