

Abstract
Polyomaviruses are a family of small DNA viruses that are associated with a number of severe human diseases, particularly in immunocompromised individuals. The detailed virus-host interactions during lytic polyomavirus infection are not fully understood. Here we report the first nuclear proteomic study with BK polyomavirus (BKPyV) in a primary renal proximal tubule epithelial cell culture system using stable isotope labeling by amino acids in cell culture (SILAC) proteomic profiling coupled with LC-MS/MS. We demonstrated the feasibility of SILAC labeling in these primary cells and subsequently performed reciprocal labeling-infection experiments to identify proteins that are altered by BKPyV infection. Our analyses revealed specific proteins that are significantly up- or down-regulated in the infected nuclear proteome. The genes encoding many of these proteins were not identified in a previous microarray study, suggesting that differential regulation of these proteins may be independent of transcriptional control. Western blotting experiments verified the SILAC proteomic findings. Finally, pathway and network analyses indicated that the host cell DNA damage response signaling and DNA repair pathways are among the cellular processes most affected at the protein level during polyomavirus infection. Our study provides a comprehensive view of the host nuclear proteomic changes during polyomavirus lytic infection and suggests potential novel host factors required for a productive polyomavirus infection.
Keywords: BK polyomavirus, quantitative nuclear proteomics, SILAC, microarray
Graphical abstract
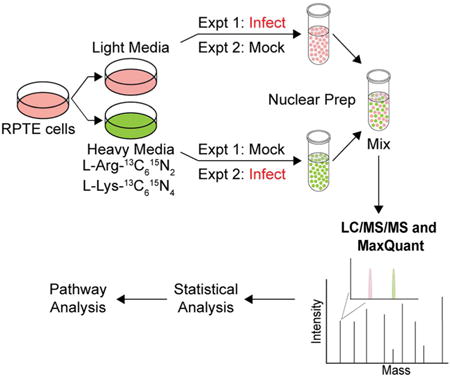
Introduction
Polyomaviruses are small double-stranded DNA (dsDNA) viruses with a ∼5kb genome. They belong to the family Polyomaviridate, which include several members associated with severe human disease.1 The first two identified human polyomaviruses, BK polyomavirus (BKPyV) and JC polyomavirus (JCPyV), were isolated in 1971.2, 3 These ubiquitous pathogens infect up to 90% of the adult population.4 BKPyV and JCPyV persist with occasional, subclinical replication in an immunocompetent host; however, as a consequence of immunosuppression, these viruses may reactivate and lead to disease. JCPyV is the causative agent of progressive multifocal leukoencephalopathy, a fatal demyelinating disease of the brain. JCPyV reactivation is most commonly associated with acquired immunodeficiency syndrome, and less frequently with other immunosuppressive conditions such as those found in organ transplant, certain autoimmune diseases, or patients treated with certain immunomodulatory monoclonal antibodies including multiple sclerosis and rheumatoid arthritis patients. 5-7 Similarly, immunosuppression during kidney transplant may result in BKPyV reactivation leading to polyomavirus associated nephropathy—potentially increasing the chance of graft rejection.8 BKPyV may also reactivate after bone marrow transplantation and is associated with hemorrhagic cystitis, a painful pathology of the bladder.9 In the past decade, a number of new polyomaviruses have been discovered,1 among which Merkel cell polyomavirus (MCPyV) has now been confirmed to cause Merkel cell carcinoma (MCC), an aggressive form of skin cancer. 10 Currently, there are no effective antiviral treatments and the lytic life cycle of these viruses is still poorly understood.
Due to its small genome size, polyomavirus has limited coding capacity. To overcome this limitation, polyomavirus relies heavily on host proteins to replicate. The polyomavirus large T Antigen (TAg) is a multifunction protein which governs many of the host interactions necessary for viral replication.11 For example, TAg directly interacts with replication protein A (RPA) to drive viral DNA synthesis.12 In addition to a direct role during viral DNA replication, TAg also interacts with multiple host proteins to create a cellular environment conducive to viral replication. TAg binds to the tumor suppressor protein retinoblastoma protein (pRb) and alleviates pRb suppression of E2F, an S phase transcription factor.13, 14 Liberated E2F drives S phase progression and stimulates the expression of host DNA synthesis proteins,15 which the virus may then appropriate. In tandem with these events, TAg from primate polyomaviruses inhibits host cell apoptosis by binding and inactivating p53, a tumor suppressor protein.16, 17 Recently, polyomavirus infection has also been shown to activate the host DNA damage responses (DDR) governed by both ataxia telangiectasia mutated (ATM) and ATM-Rad3 related (ATR), two phosphoinositide-3 kinase-like kinases (PIKKs), to facilitate viral DNA replication as well as to arrest the cells in the G2 phase for optimal viral progeny production.18-20
To identify additional host proteins important for polyomavirus infection, an early investigation performed a microarray analysis of BKPyV infection of primary cultures of renal proximal tubule epithelial (RPTE) cells.21 RPTE cells are the in vivo natural host cell targets of BKPyV lytic infection, and as such are a highly relevant primary tissue culture system to study BKPyV replication.22 The findings of this analysis indicated that genes associated with cell cycle regulation and apoptosis were primary targets of BKPyV host gene upregulation. Some genes involved in the DDR were also found to be up-regulated by BKPyV, while infection was found to down-regulate only four host genes at the level of transcription. Interestingly, there was no evidence observed to suggest an interaction of BKPyV with cellular innate immunity pathways, indicating that BKPyV may not elicit a strong innate immune response. Although microarray is a useful method for determining global changes at the transcript level, there are multiple potential additional layers of gene regulation that may not be reflected by changes in transcript abundance. For example, BKPyV TAg interacts with and stabilizes p53 in the host cell during infection,13, 19 but p53 was not identified as being upregulated by the microarray analysis.21 Therefore, investigating the regulatory changes caused by polyomavirus infection at the protein level may allow us to directly identify host protein factors that are essential for or inhibitory to polyomavirus replication.
Previous proteomic studies have been performed to investigate either proteins that interact with several polyomavirus tumor antigens,23 or proteomic changes in MCPyV-positive MCC tissues compared with MCPyV-negative tumor samples;24 no global analysis of host proteomic changes during lytic polyomavirus infection, however, has been reported. In this investigation we applied powerful quantitative analysis to determine global nuclear proteomic changes in primary RPTE cells lytically infected by BKPyV. From this approach we identified over 2000 proteins. Statistical analysis showed that 50 proteins were significantly up-regulated and 13 proteins were significantly down-regulated in BKPyV-infected cells. Pathway and network analysis of these differentially regulated proteins suggested that virus infection impacted multiple cellular functions including DDR signaling and DNA repair, cell cycle control, cellular movement, and DNA replication. These results revealed polyomavirus deregulation of host pathways that may be important mediators of viral infection.
Experimental Procedures
Cell culture, SILAC labeling, and viruses
RPTE cells (Lonza) were maintained for up to six passages in renal epithelial cell growth medium (REGM) as previously described.25 For SILAC labeling, custom MCDB 170 media (serum-, L-Lys-, L-Arg-free) were manufactured based on a previously described recipe (Caisson Laboratories,26). To produce heavy or light labeling media, either heavy amino acids (0.2 mM 13C6 15N2 L-lysine, and 0.3 mM 13C6 15N4 L-arginine, Thermo) or light amino acids (0.2 mM L-lysine, and 0.3 mM L-arginine, Thermo) were added to MCDB 170 supplemented with 0.5% dialyzed fetal bovine serum (FBS), 2 mg/L L-proline (Thermo) and SingleQuots™ Kit for REGM (Lonza). Cells were labeled for four doublings prior to infection. All cells were grown at 37°C with 5% CO2 in a humidified incubator.
BKPyV (Dunlop) was grown in Vero cells, purified, and titered using an infectious unit (IU) assay as previously described.27 Vero cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco) containing 10% FBS (HyClone), 100 U/ml penicillin, and 100 μg/ml streptomycin (Cambrex) as previously described.27
Infections
For SILAC labeling, 4 × 107 RPTE cells grown in light or heavy labeling media were infected with purified BKPyV at a multiplicity of infection (MOI) of 0.5 IU/cell. In repeat 1, the light amino acid-labeled RPTE cells were infected with BKPyV, while an equivalent number of heavy amino acid-labeled cells were mock-infected. In repeat 2, the light-labeled cells were mock infected and the heavy-labeled cells were BKPyV-infected. For infection, all RPTE cells were pre-chilled for 15 min at 4°C followed by incubation with viral inoculum diluted in respective labeling media for 1 h at 4°C. Infection was initiated by removing the viral inoculum, feeding with pre-warmed labeling media, and transferring the cells to 37°C for 3 days. For Western blotting confirmation experiments, RPTE cells were infected with crude viral lysates at an MOI of 0.5 IU/cell as described above.
Cellular Fractionation
Cellular fractionation was carried out using a modified protocol.28 Briefly, RPTE cells were washed twice with cold PBS and lysed with a hypotonic buffer (20 mM HEPES pH 7.0, 10 mM KCl, 2 mM MgCl2, 0.5% Nonidet P40) with protease and phosphatase inhibitors (5 μg/ml PMSF, 5 μg/ml aprotinin, 5 μg/ml leupeptin, 50 mM NaF, and 0.2 mM Na3VO4) for 10 min on ice. The lysate was homogenized by 30 strokes in a tightly fitting Dounce homogenizer. Nuclei were pelleted at 1,500 g for 5 min. The supernatant was re-centrifuged at 15,000 g for 5 min and the resulting supernatant comprised the cytoplasmic fraction. The nuclei were further washed three times with the lysis buffer and finally extracted with the same buffer containing 0.5 M NaCl for 30 min on ice. The extracted fraction was centrifuged at 15,000 g for 10 min and the resulting supernatant comprised the nuclear fraction. Relatively pure nuclear fractions can be isolated using this method. However, residual cytoplasmic contamination may still be present; therefore the fractions were analyzed by Western blotting to determine the quality of the fractionation prior to mass spectrometry analysis.
SILAC sample preparation
All SILAC sample preparation, mass spectrometry (MS), and MS data processing were performed by MS Bioworks (http://www.msbioworks.com/). To determine label incorporation, the concentrations of proteins that were labeled with heavy labeling media were determined using Qubit fluorometry (Life Technologies). 20 μg of sample was processed by SDS-PAGE on a 4-12% Bis-Tris Novex mini-gel (Life Technologies) using the MOPS buffer system. The gel was stained with InstantBlue (Expedeon) and bands were excised at 50 and 100 kDa. Gel bands were processed using a robot (ProGest, DigiLab) as follows: the bands were washed with 25 mM ammonium bicarbonate followed by acetonitrile, reduced with 10 mM dithiothreitol at 60°C followed by alkylation with 50 mM iodoacetamide at room temperature, digested with trypsin (Promega) at 37°C for 4 h, quenched with formic acid, and the supernatant was analyzed directly without further processing. For the nuclear proteome SILAC analysis, 10 μg each of heavy and light media-labeled nuclear fractions were combined and processed by SDS-PAGE using a 4-12% Bis Tris NuPage mini-gel (Life Technologies). Calibration was performed with Thermo PageRuler broad range markers. The mobility region was excised into forty equal sized segments using a grid. Each gel segment was processed as described above.
Mass spectrometry and data processing
Each gel digest was analyzed by nano liquid chromatography with tandem MS (LC-MS/MS) with a Waters NanoAcquity HPLC system interfaced to a ThermoFisher Q Exactive mass spectrometer. Peptides were loaded on a trapping column and eluted over a 75 μm analytical column at 350nL/min; both columns were packed with Jupiter Proteo resin (Phenomenex). The mass spectrometer was operated in a data-dependent mode, with MS performed at 70,000 full width at half maximum (FWHM) resolution and MS/MS performed at 17,500 FWHM. The fifteen most abundant ions were selected for MS/MS.
For label incorporation, data were searched using a local copy of Mascot with the following parameters: Enzyme: Trypsin/P, Database: Swissprot Human (concatenated forward and reverse plus common contaminants), Fixed modifications: Carbamidomethyl (C), Variable modifications: Oxidation (M), Acetyl (N-term), Deamidation (N,Q), 13C6 15N2 (K), 13C6 15N4 (R),13C6 15N (P), Mass values: Monoisotopic, Peptide Mass Tolerance: 10 ppm, Fragment Mass Tolerance: 0.02 Da, Max Missed Cleavages: 2. Mascot DAT files were parsed into the Scaffold algorithm for validation, filtering and to create a non-redundant list per sample. Data were filtered using a 1% protein and peptide false discovery rate (FDR) and requiring at least two unique peptides per protein. Isotope incorporation was calculated using the following equation: .
For nuclear proteome SILAC analysis, data were processed with MaxQuant version 1.4.1.2 (Max Planck Institute for Biochemistry), which incorporates the Andromeda search engine, including recalibration of MS data, protein/peptide identification using the Andromeda database search engine, filtering of database search results at the 1% protein and peptide FDR, and calculation of SILAC heavy:light (H/L) ratios. The following Andromeda settings were used: Enzyme, Trypsin/P; Database, SwissProt Human; Fixed modification, Carbamidomethyl (C); Variable modifications, Oxidation (M), Acetyl (Protein N-term), Deamidation (N,Q), Pyro-Glu (N-term Q); Missed cleavages, 2; Multiplicity, 2; Labels, Lys8, Arg10; Unique peptides, 2. The median of the total ratio population is shifted to 1 to normalize the ratios between heavy and light partners.
Western blotting (WB) and antibodies
Total cell proteins were harvested, quantified, and immunoblotted as previously described.27 Nuclear and cytoplasmic fractions were quantified and analyzed similarly. The following antibodies and concentrations were used: Primary antibodies: anti-TAg pAb416 (29, 1:3,000), anti-p84 (Genetex, 1:10,000), anti-Grb2 (Cell Signaling, 1:1,000), anti-KIF22 (Cytoskeleton, 1:250), anti-PDCD4 (Cell signaling, 1:10,000), anti-p53 (Oncogene, 1:10,000), anti-GAPDH (Abcam, 1:10,000). For chemiluminescence-based WB, horseradish peroxidase-conjugated sheep anti-mouse and donkey anti-rabbit IgG secondary antibodies (Amersham) were used (1:2,000-10,000). For quantitative blots using the Odyssey infrared imaging system, the membrane was processed according to the manufacturer's instructions (LI-COR). Secondary goat anti-rabbit IRDye 680RD and goat anti-mouse IRDye 800CW antibodies were used at 1:10,000-1:20,000. The membrane was scanned using the Odyssey infrared imaging system and the relevant bands were quantified using the Odyssey software program.
Statistical and bioinformatic analyses
To compare between the two experiments, protein ratios within each replicate were converted to log2 and then a Z-score was calculated using the following formula:
A Z-score > 1.65σ or < -1.65σ indicates that the protein ratio between mock and infected is outside of the 90% confidence level and therefore was considered significantly up- or down-regulated. A list of proteins that are up- or down-regulated in both data sets was generated and the data were formatted and uploaded to the Ingenuity Pathway Analysis software for pathway and network analyses. For subcellular localization predictions, all proteins identified by more than one unique peptide were analyzed.
Results
Nuclear fractionation and SILAC labeling in primary RPTE cells
To begin examining nuclear proteomic changes during BKPyV infection, we first optimized the fractionation protocol to ensure that we were able to obtain clean nuclear fractions in RPTE cells. We tested several cellular fractionation protocols involving various methods for lysing cells, washing the nuclear pellets, and extracting nuclear proteins. A revised protocol based on Lin et al.28 was the most robust method with RPTE cells. The isolated nuclear and cytoplasmic fractions were immunoblotted for nuclear and cytoplasmic markers (Figure 1A). p84 is a nuclear matrix protein that has been widely used as a nuclear marker,30 whereas Grb2 (Growth factor receptor-bound protein 2) is a signal transduction adaptor protein and serves a cytoplasmic fraction marker.31 The immunoblots showed clear separation of the two markers (Figure 1A). BKPyV TAg is known to localize to the nucleus where it coordinates many aspects of virus replication;32 as expected, TAg co-purified with p84 in the nuclear fraction (Figure 1A), further validating our protocol.
Figure 1.
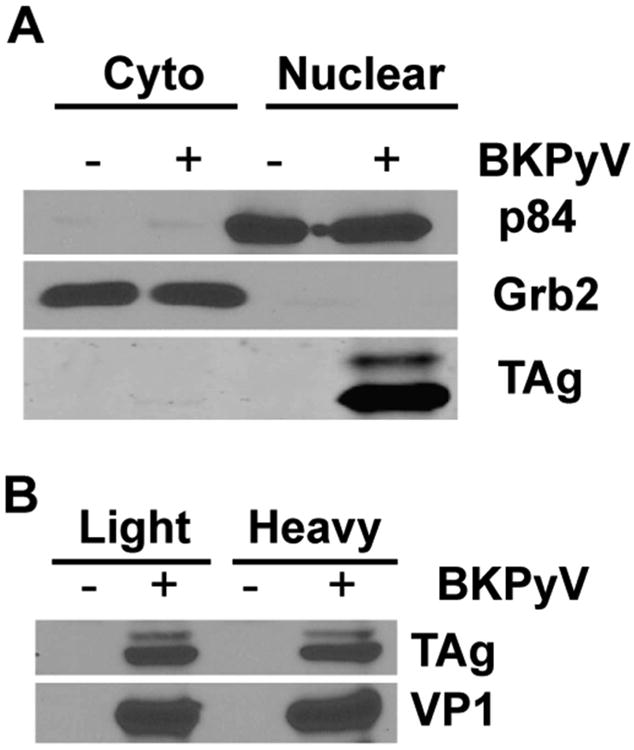
Confirmation of nuclear fractionation protocol and infection in SILAC-labeled RPTE cells. (A) RPTE cells were mock or infected with BKPyV at an MOI of 0.5 IU/cell. At 3 dpi, cytoplasmic and nuclear fractions were isolated as described in Experimental Procedures, and immunoblotted for p84 (nuclear marker), Grb2 (cytoplasmic marker), and TAg. (B) RPTE cells were grown in light or heavy labeling media and infected at an MOI of 0.5 IU/cell. Total cellular proteins were harvested at 2 dpi and immunoblotted for TAg and VP1.
To achieve SILAC labeling in any cells, the growth medium has to be supplemented with isotope-labeled amino acids (L-lysine and L-arginine) to allow for metabolic incorporation of these isotopes into proteins. We normally grow RPTE cells in the proprietary Renal Cell Growth Medium (REGM) with SingleQuot growth factor and cytokine supplements (Lonza). Because we could not obtain the composition of REGM in order to be able to add defined amino acids to our culture, we instead grew RPTE cells in the closely related MCDB170 mammary epithelial cell culture media, which we supplemented with either heavy amino acids (13C6 15N2 L-lysine and 13C6 15N4 L-arginine) or light amino acids (L-lysine and L-arginine). We confirmed that RPTE cells grown in either “heavy” or “light” medium could be infected by BKPyV as evidenced by expression of both TAg and capsid protein VP1 (Figure 1B). Finally, we determined that ∼98% labeling efficiency could be achieved in RPTE cells after four doublings of growth (see Experimental Procedures for a detailed protocol for labeling efficiency determination).
SILAC analyses of nuclear proteomes in BKPyV-infected RPTE cells
Since the nucleus is the major site for polyomavirus transcription, replication and assembly, we were particularly interested in the global changes that are caused by polyomavirus infection in the nucleus at the protein level. Having established that we could obtain efficient SILAC labeling, infection, and clean nuclear fractionation in RPTE cells, we set up experiments for total nuclear proteomic comparison analysis between mock and BKPyV-infected cells. RPTE cells were allowed to grow in the light or heavy labeling media for four doublings to reach sufficient isotope incorporation. We then infected the light-labeled cells with purified BKPyV and kept the heavy-labeled cells as mock infected control (Graphical Abstract, experiment 1). At 3 days post infection (dpi), nuclear fractions were isolated from both mock- and virus-infected cells, mixed, digested and subjected to LC-MS/MS for identification and quantification. We also performed a biological replicate of the experiment during which we switched the labels and infected the heavy-labeled cells with BKPyV. (Graphical Abstract, experiment 2). This will help ensure that any change we observe is not due to differential labeling of the cells. Overall, we identified a total of 2572 proteins from experiment 1 and 2411 proteins from experiment 2 by two or more unique peptides, with 2122 proteins identified in both data sets (74% overlap, Figure 2A). Localization prediction analyses revealed that among all the proteins identified, ∼32% of the proteins are predicted to be localized to the nucleus and ∼50% of the proteins are predicted to be cytoplasmic (Figure 2B). Apparent identification of cytoplasmic proteins in the nuclei-enriched fractions is not uncommon, as the localization prediction is not robust: a similar phenomenon has been observed in other nuclear proteomic analyses.33 There are also ∼3% and ∼8% of the total proteins that are predicted to localize to the plasma membrane and extracellular space, respectively.
Figure 2.
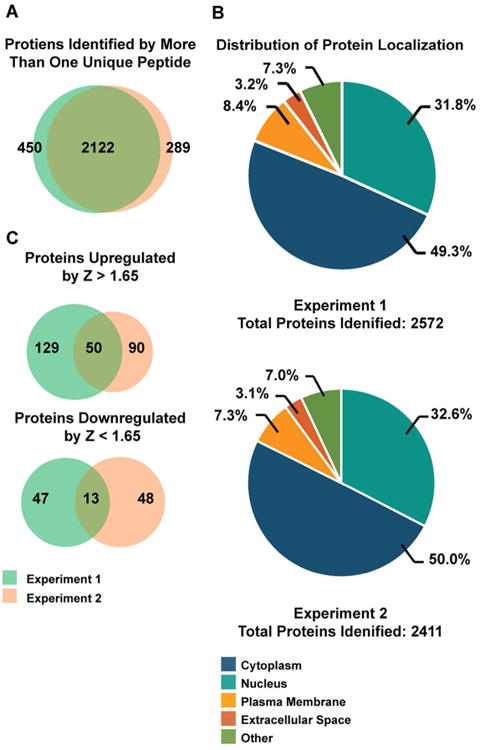
Distributions of proteins identified from two independent SILAC experiments. (A) Venn diagrams of total proteins identified from the two experiments using the opposite differential labeling protocols. (B) Sub-cellular localizations of total proteins identified were determined using the Ingenuity Pathway Analysis program. (C) Total number of proteins that are up-regulated (Z-score >1.65) or down-regulated (Z-score < -1.65) in the infected samples from the two labeling experiments.
To identify proteins that are significantly up- or down-regulated in BKPyV-infected nuclear fractions, we converted the Infected:Mock SILAC ratios to Z-scores and set a Z-score of 1.65 (90% confidence interval) as our cut-off. If the Infected:Mock Z-score is > 1.65 or < -1.65, we considered the protein up- or down-regulated in the infected sample, respectively. With this criterion, a total of 50 proteins were up-regulated whereas 13 proteins were down-regulated in BKPyV-infected nuclear fractions from both experiments (Figure 2C). A summary of these proteins and their Z-scores and Infected:Mock SILAC ratios are listed in Table 1.
Table 1.
List of proteins that are up- or down-regulated in BKPyV-infected samples identified from both SILAC nuclear proteomic analyses.
| Gene names | Accession Number | Protein Name | Mean Z-Score | Infected/Uninfected Ratio | Predicted Cellular Localization | Protein Type |
|---|---|---|---|---|---|---|
| Upregulated Proteins | ||||||
| ATAD2 | Q6PL18 | ATPase family, AAA domain containing 2 | 3.604 | 8.061 | Nucleus | Enzyme |
| AURKB | Q96GD4 | Aurora Kinase B | 3.222 | 7.351 | Nucleus | Kinase |
| CCNA2 | P20248 | Cyclin A2 | 3.531 | 8.585 | Nucleus | Other |
| CCNB1 | P14635 | Cyclin B1 | 4.532 | 14.949 | Cytoplasm | Kinase |
| CDK1 | P06493 | Cyclin-dependent Kinase 1 | 3.043 | 7.704 | Nucleus | Kinase |
| CENPF | P49454 | Centromere protein F, 350/400kDa | 3.014 | 6.421 | Nucleus | Other |
| CEP55 | Q53EZ4 | Centrosomal protein 55kDa | 3.953 | 9.579 | Cytoplasm | Other |
| DNMT1 | P26358 | DNA (cytosine-5-)-methyltransferase 1 | 3.214 | 6.431 | Nucleus | Enzyme |
| ECT2 | Q9H8V3 | Epithelial cell transforming 2 | 2.544 | 4.558 | Cytoplasm | Other |
| FANCD2 | Q9BXW9 | Fanconi anemia, complementation group D2 | 3.146 | 6.430 | Nucleus | Other |
| FEN1 | P39748 | Flap structure-specific endonuclease 1 | 4.264 | 11.486 | Nucleus | Enzyme |
| HELLS | Q9NRZ9 | Helicase, lymphoid-specific | 2.854 | 5.345 | Nucleus | Enzyme |
| HMGB1 | P09429 | High mobility group box 1 | 4.923 | 16.341 | Nucleus | Other |
| HMGB2 | P26583 | High mobility group box 2 | 4.120 | 11.174 | Nucleus | Transcription |
| HMGB3 | O15347 | High mobility group box 3 | 2.673 | 5.375 | Nucleus | Other |
| IQGAP3 | Q86VI3 | IQ motif containing GTPase activating protein 3 | 4.205 | 13.936 | Plasma Membrane | Other |
| KIF20A | O95235 | Kinesin family member 20A | 4.113 | 10.313 | Cytoplasm | Transporter |
| KIF20B | Q96Q89 | Kinesin family member 20B | 3.000 | 6.030 | Nucleus | Enzyme |
| KIF22 | Q14807 | Kinesin family member 22 | 3.704 | 8.280 | Nucleus | Other |
| KIF2C | Q99661 | Kinesin family member 2C | 3.553 | 7.674 | Nucleus | Other |
| KIF4A | O95239 | Kinesin family member 4A | 3.229 | 6.528 | Nucleus | Other |
| KIFC1 | Q9BW19 | Kinesin family member C1 | 3.826 | 9.350 | Nucleus | Enzyme |
| KPNA2 | P52292 | Karyopherin alpha 2 (RAG cohort 1, importin alpha 1) | 4.055 | 10.096 | Nucleus | Transporter |
| LASP1 | Q14847 | LIM and SH3 protein 1 | 2.642 | 4.721 | Cytoplasm | Transporter |
| MCM5 | P33992 | Minichromosome maintenance complex component 5 | 2.493 | 4.323 | Nucleus | Enzyme |
| MKI67 | P42695 | Marker of proliferation Ki-67 | 3.758 | 8.828 | Nucleus | Other |
| NCAPD3 | Q86XI2 | Non-SMC condensin II complex, subunit D3 | 3.234 | 7.368 | Nucleus | Other |
| NCAPG2 | P12004 | Non-SMC condensin II complex, subunit G2 | 4.010 | 10.492 | Nucleus | Other |
| PCNA | P53350 | Proliferating cell nuclear antigen | 6.434 | 36.623 | Nucleus | Enzyme |
| PLK1 | P28340 | Polo-like Kinase 1 | 3.109 | 6.614 | Nucleus | Kinase |
| POLD1 | O43663 | Polymerase (DNA directed), delta 1, catalytic subunit | 2.451 | 4.286 | Nucleus | Enzyme |
| PRC1 | Q9H0H5 | Protein regulator of cytokinesis 1 | 3.088 | 6.003 | Nucleus | Other |
| RACGAP 1 | P40938 | Rac GTPase activating protein 1 | 4.487 | 13.343 | Cytoplasm | Transporter |
| RFC3 | P35249 | Replication factor C (activator 1) 3, 38kDa | 2.263 | 3.868 | Nucleus | Enzyme |
| RFC4 | P40937 | Replication factor C (activator 1) 4, 37kDa | 2.487 | 4.314 | Nucleus | Other |
| RFC5 | P27694 | Replication factor C (activator 1) 5, 36.5kDa | 2.528 | 4.400 | Nucleus | Enzyme |
| RPA1 | P15927 | Replication protein A1, 70kDa | 4.730 | 14.564 | Nucleus | Other |
| RPA2 | P35244 | Replication protein A2, 32kDa | 3.732 | 8.426 | Nucleus | Other |
| RPA3 | Q8NEM2 | Replication protein A3, 14kDa | 4.394 | 12.104 | Nucleus | Other |
| SHCBP1 | O95347 | SHC SH2-domain binding protein 1 | 3.280 | 7.807 | Other | Other |
| SMC2 | Q9NTJ3 | Structural maintenance of chromosomes 2 | 3.540 | 7.645 | Nucleus | Transporter |
| SMC4 | P53814 | Structural maintenance of chromosomes 4 | 4.267 | 11.264 | Nucleus | Transporter |
| SMTN | Q5JUR7 | Smoothelin | 1.706 | 2.835 | Extracellular Space | Other |
| TEX30 | P42166 | Testis expressed 30 | 2.189 | 3.740 | Other | Other |
| TMPO | P11388 | Thymopoietin | 2.508 | 4.352 | Nucleus | Other |
| TOP2A | P04637 | Topoisomerase (DNA) II alpha 170kDa | 3.916 | 10.066 | Nucleus | Enzyme |
| TP53 | Q9ULW0 | Tumor protein p53 | 4.979 | 16.672 | Nucleus | Transcription Regulator |
| TPX2 | Q99986 | TPX2, microtubule-associated | 2.791 | 5.554 | Nucleus | Other |
| VRK1 | O75717 | Vaccinia related Kinase 1 | 1.844 | 3.053 | Nucleus | Kinase |
| WDHD1 | O75717 | WD repeat and HMG-box DNA binding protein 1 | 3.186 | 8.224 | Nucleus | Other |
| Downregulated Proteins | ||||||
| AQP1 | P29972 | Aquaporin 1 (Colton blood group) ATPase, H+ | -1.828 | 0.422 | Plasma Membrane | Transporter |
| ATP6AP1 | Q15904 | transporting, lysosomal accessory protein 1 | -3.006 | 0.224 | Cytoplasm | Transporter |
| ATP6V0A 1 | Q93050 | ATPase, H+ transporting, lysosomal V0 subunit a1 | -3.168 | 0.205 | Cytoplasm | Transporter |
| ATP6V0 D1 | P61421 | ATPase, H+ transporting, lysosomal 38kDa, V0 subunit d1 | -2.575 | 0.283 | Cytoplasm | Transporter |
| ATP6V1 D | Q9Y5K8 | ATPase, H+ transporting, lysosomal 34kDa, V1 subunit D | -1.939 | 0.398 | Cytoplasm | Transporter |
| CPM | P14384 | CarboxyPeptidase M | -1.995 | 0.391 | Plasma Membrane | Peptidase |
| FLOT1 | O75955 | Flotillin 1 | -2.930 | 0.233 | Plasma Membrane | Other |
| FLOT2 | Q14254 | fFotillin 2 | -3.053 | 0.218 | Plasma Membrane | Other |
| NT5E | P21589 | 5′-nucleotidase, ecto (CD73) | -4.208 | 0.118 | Plasma Membrane | Phosphatase |
| PDCD4 | Q53EL6 | Programmed cell death 4 (neoplastic transformation inhibitor) | -2.030 | 0.380 | Nucleus | Other |
| PLAU | P00749 | Plasminogen activator, uroKinase | -1.807 | 0.427 | Extracellular Space | Peptidase |
| TCIRG1 | Q13488 | T-cell, immune regulator 1, ATPase, H+ transporting, lysosomal V0 subunit A3 | -3.515 | 0.170 | Plasma Membrane | Enzyme |
| TMEM50 A | O95807 | Transmembrane protein 50A | -2.194 | 0.346 | Plasma Membrane | Other |
Validation of SILAC results by Western blotting
To validate the SILAC results, we performed Western blotting analyses on select proteins that were identified to be up- or down-regulated during BKPyV infection (Figure 3). Cytoplasmic and nuclear fractions were harvested from mock or BKPyV-infected RPTE cells grown in standard REGM medium and immunoblotted for KIF22, PDCD4, p53, and TAg (as an infection marker). p84 and GAPDH were used as a nuclear and cytoplasmic marker, respectively. BKPyV TAg is known to bind to p5313 and BKPyV infection has been shown to increase total p53 levels in the cell.19 Both SILAC and Western blotting confirmed these previous findings with p53 and demonstrated an up-regulation of p53 in the nuclear fraction in the infected sample. KIF22 is a kinesin-like protein that is involved in spindle formation and chromosome movement during mitosis.34, 35 It was identified by SILAC as up-regulated ∼8 fold in BKPyV-infected samples (Table 1) and the Western blotting result showed a similar trend (Figure 3). Programmed cell death protein 4 (PDCD4) is a nuclear/cytoplasmic-shuttling protein and is a tumor suppressor protein involved in apoptosis.36, 37 Both the SILAC and Western blotting displayed an ∼2 fold reduction of this protein in BKPyV-infected nuclear fraction. Overall, the Western blotting data agreed with the SILAC proteomic results.
Figure 3.
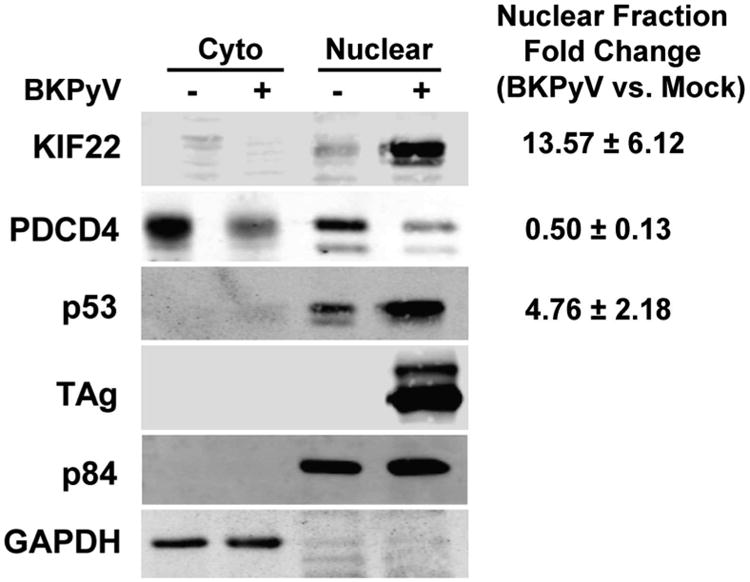
Western blotting validation of SILAC results. RPTEs were infected with BKPyV at an MOI of 0.5 IU/cell. At 3 dpi the nuclear and cytoplasmic fractions of the RPTEs were isolated and subjected to Western blotting analysis. Three protein targets were chosen from significantly up-regulated (p53 and KIF22) or down-regulated (PDCD4) proteins found in both SILAC experiments. p84 and GAPDH serve as fractionation markers for the nuclear and cytoplasmic fractions, respectively. Representative blots and quantitation using the Odyssey system (mean ± standard deviation) from three independent experiments were shown.
Comparison with the microarray data
To identify protein changes that occur at the post-transcriptional level, we compared our proteomic results to a previous microarray study21 (Table 2). Of the total 63 proteins identified by SILAC as up- or down-regulated upon BKPyV infection, 29 were previously reported by the microarray study as having significant differences in RNA expression between mock and infected samples (Table 2). All of the 29 proteins were upregulated in BKPyV-infected cells and the general trend is consistent between the SILAC and the microarray studies. Some of these proteins had a higher level of increase in the SILAC datasets, suggesting additional post-transcriptional regulation. The remaining 34 proteins identified by SILAC were not reported by the microarray study. These include all the proteins that were found down-regulated in the BKPyV-infected cells by the SILAC study. These results showcased the power of the SILAC proteomic approach at identifying host gene expression changes that occur at multiple levels.
Table 2.
Comparison of differentially regulated proteins identified in this study to the previous microarray study.
| Gene names | Accession Number | Protein Name | Mean Z-Score | Infected/Uninfected Ratio | Microarray |
|---|---|---|---|---|---|
| Upregulated Proteins | |||||
| ATAD2 | Q6PL18 | ATPase family, AAA domain containing 2 | 3.604 | 8.061 | 6.32 |
| AURKB | Q96GD4 | Aurora kinase B | 3.222 | 7.351 | 5.35 |
| CCNA2 | P20248 | Cyclin A2 | 3.531 | 8.585 | 7.06 |
| CCNB1 | P14635 | Cyclin B1 | 4.532 | 14.949 | 2.99 |
| CDK1 | P06493 | Cyclin-dependent kinase 1 | 3.043 | 7.704 | N/A |
| CENPF | P49454 | Centromere protein F, 350/400kDa | 3.014 | 6.421 | 2.99 |
| CEP55 | Q53EZ4 | Centrosomal protein 55kDa | 3.953 | 9.579 | 6.11 |
| DNMT1 | P26358 | DNA (cytosine-5-)-methyltransferase 1 | 3.214 | 6.431 | N/A |
| ECT2 | Q9H8V3 | Epithelial cell transforming 2 | 2.544 | 4.558 | N/A |
| FANCD2 | Q9BXW9 | Fanconi anemia, complementation group D2 | 3.146 | 6.430 | 2.33 |
| FEN1 | P39748 | Flap structure-specific endonuclease 1 | 4.264 | 11.486 | 4.99 |
| HELLS | Q9NRZ9 | Helicase, lymphoid-specific | 2.854 | 5.345 | 4.86 |
| HMGB1 | P09429 | High mobility group box 1 | 4.923 | 16.341 | N/A |
| HMGB2 | P26583 | High mobility group box 2 | 4.120 | 11.174 | 3.86 |
| HMGB3 | O15347 | High mobility group box 3 | 2.673 | 5.375 | N/A |
| IQGAP3 | Q86VI3 | IQ motif containing GTPase activating protein 3 | 4.205 | 13.936 | 3.73 |
| KIF20A | O95235 | Kinesin family member 20A | 4.113 | 10.313 | 3.76 |
| KIF20B | Q96Q89 | Kinesin family member 20B | 3.000 | 6.030 | N/A |
| KIF22 | Q14807 | Kinesin family member 22 | 3.704 | 8.280 | N/A |
| KIF2C | Q99661 | Kinesin family member 2C | 3.553 | 7.674 | 4.44 |
| KIF4A | O95239 | Kinesin family member 4A | 3.229 | 6.528 | 4.63 |
| KIFC1 | Q9BW19 | Kinesin family member C1 | 3.826 | 9.350 | N/A |
| KPNA2 | P52292 | Karyopherin alpha 2 (RAG cohort 1, importin alpha 1) | 4.055 | 10.096 | N/A |
| LASP1 | Q14847 | LIM and SH3 protein 1 | 2.642 | 4.721 | N/A |
| MCM5 | P33992 | Minichromosome maintenance complex component 5 | 2.493 | 4.323 | 3.51 |
| MKI67 | P42695 | Marker of proliferation Ki-67 | 3.758 | 8.828 | 4.2 |
| NCAPD3 | Q86XI2 | Non-SMC condensin II complex, subunit D3 | 3.234 | 7.368 | N/A |
| NCAPG2 | P12004 | Non-SMC condensin II complex, subunit G2 | 4.010 | 10.492 | 5.66 |
| PCNA | P53350 | Proliferating cell nuclear antigen | 6.434 | 36.623 | 3.27 |
| PLK1 | P28340 | Polo-like kinase 1 | 3.109 | 6.614 | 2.6 |
| POLD1 | O43663 | Polymerase (DNA directed), delta 1, catalytic subunit | 2.451 | 4.286 | N/A |
| PRC1 | Q9H0H5 | Protein regulator of cytokinesis 1 | 3.088 | 6.003 | 4.29 |
| RACGAP1 | P40938 | Rac GTPase activating protein 1 | 4.487 | 13.343 | 2.62 |
| RFC3 | P35249 | Replication factor C (activator 1) 3, 38kDa | 2.263 | 3.868 | 6.11 |
| RFC4 | P40937 | Replication factor C (activator 1) 4, 37kDa | 2.487 | 4.314 | 2.91 |
| RFC5 | P27694 | Replication factor C (activator 1) 5, 36.5kDa | 2.528 | 4.400 | 3.18 |
| RPA1 | P15927 | Replication protein A1, 70kDa | 4.730 | 14.564 | N/A |
| RPA2 | P35244 | Replication protein A2, 32kDa | 3.732 | 8.426 | N/A |
| RPA3 | Q8NEM2 | Replication protein A3, 14kDa | 4.394 | 12.104 | N/A |
| SHCBP1 | O95347 | SHC SH2-domain binding protein 1 | 3.280 | 7.807 | 8.28 |
| SMC2 | Q9NTJ3 | Structural maintenance of chromosomes 2 | 3.540 | 7.645 | N/A |
| SMC4 | P53814 | Structural maintenance of chromosomes 4 | 4.267 | 11.264 | N/A |
| SMTN | Q5JUR7 | Smoothelin | 1.706 | 2.835 | N/A |
| TEX30 | P42166 | Testis expressed 30 | 2.189 | 3.740 | N/A |
| TMPO | P11388 | Thymopoietin | 2.508 | 4.352 | 3.46 |
| TOP2A | P04637 | Topoisomerase (DNA) II alpha 170kDa | 3.916 | 10.066 | 8.75 |
| TP53 | Q9ULW0 | Tumor protein p53 | 4.979 | 16.672 | N/A |
| TPX2 | Q99986 | TPX2, microtubule-associated | 2.791 | 5.554 | 3.89 |
| VRK1 | O75717 | Vaccinia related kinase 1 | 1.844 | 3.053 | 2.73 |
| WDHD1 | O75717 | WD repeat and HMG-box DNA binding protein 1 | 3.186 | 8.224 | N/A |
| Downregulated Proteins | |||||
| AQP1 | P29972 | Aquaporin 1 (Colton blood group) | -1.828 | 0.422 | N/A |
| ATP6AP1 | Q15904 | ATPase, H+ transporting, lysosomal accessory protein 1 | -3.006 | 0.224 | N/A |
| ATP6V0A1 | Q93050 | ATPase, H+ transporting, lysosomal V0 subunit a1 | -3.168 | 0.205 | N/A |
| ATP6V0D1 | P61421 | ATPase, H+ transporting, lysosomal 38kDa, V0 subunit d1 | -2.575 | 0.283 | N/A |
| ATP6V1D | Q9Y5K8 | ATPase, H+ transporting, lysosomal 34kDa, V1 subunit D | -1.939 | 0.398 | N/A |
| CPM | P14384 | Carboxypeptidase M | -1.995 | 0.391 | N/A |
| FLOT1 | O75955 | Flotillin 1 | -2.930 | 0.233 | N/A |
| FLOT2 | Q14254 | Flotillin 2 | -3.053 | 0.218 | N/A |
| NT5E | P21589 | 5′-nucleotidase, ecto (CD73) | -4.208 | 0.118 | N/A |
| PDCD4 | Q53EL6 | Programmed cell death 4 (neoplastic transformation inhibitor) | -2.030 | 0.380 | N/A |
| PLAU | P00749 | Plasminogen activator, urokinase | -1.807 | 0.427 | N/A |
| TCIRG1 | Q13488 | T-cell, immune regulator 1, ATPase, H+ transporting, lysosomal V0 subunit A3 | -3.515 | 0.170 | N/A |
| TMEM50A | O95807 | Transmembrane protein 50A | -2.194 | 0.346 | N/A |
Bioinformatic pathway and network analyses
To identify cellular pathways that are enriched in our datasets, we subjected differentially regulated proteins between mock- and BKPyV-infected cells to the Ingenuity Pathway Analysis (IPA, www.ingenuity.com) with the “Canonical Pathway” analysis function (Figure 4). This function identifies pathways that contain differentially regulated proteins with the most significant p-values (the gold line shows the –log(p-value)). Among the top pathways identified, there is an enrichment of pathways involved in DNA damage signaling, DNA damage repair, and cell cycle checkpoint control functions.
Figure 4.
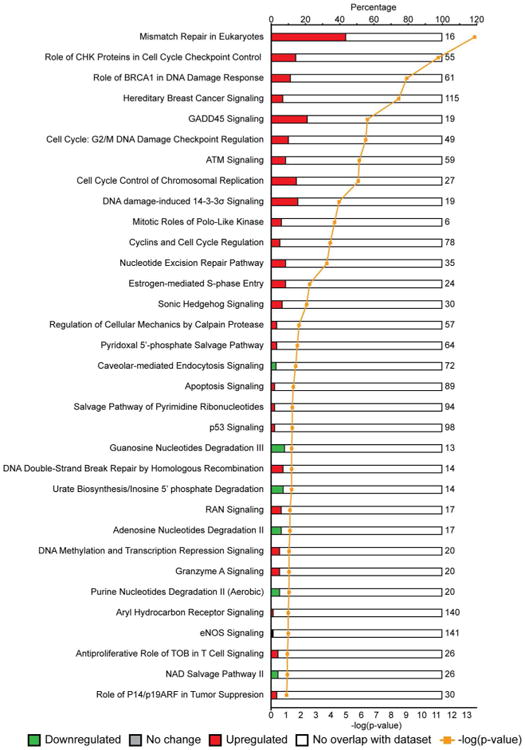
Canonical pathway analysis of differentially regulated proteins. Top canonical pathways identified by Ingenuity Pathway Analysis (IPA) of the significantly up and down regulated proteins recovered in both experiments are listed. The red and green bars represent proteins up- and down-regulated respectively as a percent (top x-axis) of the total proteins present in that pathway (to the right of the empty bars, determined by IPA). P-value is defined as the likelihood of each ratio occurring due to random chance and the gold line displays the -log(p-value) (bottom x-axis). Therefore the greater the -log(p-value) is, the more significant the pathway is represented in the SILAC datasets.
We next aimed to understand the potential interactions among all the differentially regulated proteins. To achieve this, we used the “Network” function of the IPA software. This analysis takes into consideration direct protein-protein interactions that overlap with the most differentially regulated proteins found in our SILAC datasets. The top two networks that were identified using this approach are displayed in Figure 5. In both networks, cellular functions including cell cycle control, cellular movement, cellular assembly and organization, DNA replication, DNA recombination, and DNA repair are overrepresented in the datasets, which is consistent with the pathways analysis. Some of the proteins present in these networks, such as RPA, are well-known essential factors for polyomavirus replication.38
Figure 5.
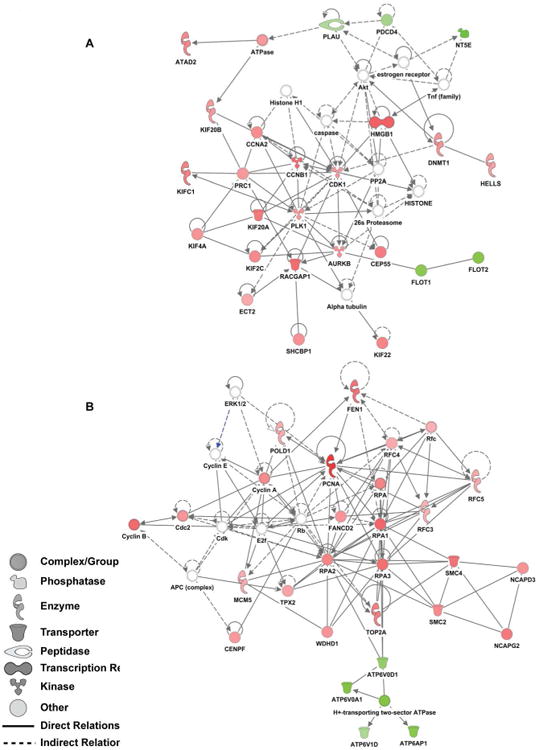
Graphical representation of the top two networks determined by IPA analysis. Both network A and B include interactions of the cell cycle, cellular movement, cellular assembly and organization, DNA replication, DNA recombination, and DNA repair. Red and green colored symbols represent up- and down-regulated proteins in the BKPyV-infected samples, respectively. Uncolored symbols are proteins that were not identified in the dataset, but rather likely interact with other identified proteins within the network.
Discussion
In this study we applied SILAC coupled with LC-MS/MS to analyze nuclear proteomic changes upon BKPyV infection of primary RPTE cells. Previously, proteomics studies were performed either to examine polyomavirus tumor antigen-interacting proteins23 or to analyze proteomic changes in Merkel cell carcinoma tumor samples24. To our knowledge, our study is the first analysis to examine global nuclear changes at the protein level during lytic polyomavirus infection in primary cells. Using this powerful technique, we identified over 2000 proteins from both biological repeats. With stringent statistical cut-off, we found that 50 proteins are significantly up-regulated and 13 proteins are down-regulated in BKPyV infected cells. Because of our reciprocal labeling design, we can also conclude that these changes are not due to differential labeling of the cells, but reflect bona fide changes that are caused by lytic BKPyV infection.
Our nuclear proteomic analyses identified a high percentage of proteins that are predicted to have cytoplasmic localization. We do not think this directly reflects cytoplasmic contamination in our nuclear fractions for several reasons: First, many proteins can localize to both the cytoplasm and the nucleus even though the predominant prediction is cytoplasmic or nuclear. PDCD4 represents such an example. Second, BKPyV infection itself may result in a change in the localization of certain proteins. Finally, the presence of a high percentage of non-nuclear proteins in nuclear fraction has been reported in other nuclear proteomic studies,33, 39, 40 suggesting that the localization prediction methods of many proteins can be inaccurate.
The low levels of protein overlap between the reciprocal SILAC labeling experiments (Figure 2) may be the result of either the stringency of the cutoff for significantly up- or down-regulated hits (lowering the cutoff threshold results in a much higher proportion of overlap between the datasets) or differences in labeling. Additionally, because only proteins identified by more than one unique peptide in both datasets were analyzed, significantly down-regulated proteins may be under-represented as their levels might be too low to be detected by mass spectrometry in both experiments.
Our SILAC studies revealed the differential regulation of a number of canonical pathways and networks that are involved in DNA damage signaling (such as ATM signaling and checkpoint cell cycle control signaling) and DNA repair (such as mismatch repair and nucleotide excision repair) (Figure 4 and 5). This is consistent with the expanding literature demonstrating the importance of the DDR during polyomavirus infection.18-20, 41, 42 It is thought that polyomaviruses hijack components of the DDR pathways to maintain replication fidelity or to arrest the cell cycle to maximize viral yield. Our proteomic results revealed the differential regulation of several previously reported important players for polyomavirus replication such as RPA43 and FANCD2,44 as well as many other uncharacterized proteins in DDR signaling and repair pathways. The detailed molecular functions of these proteins and pathways during viral replication remain to be determined.
We think that much of the protein upregulation during polyomavirus infection is likely due to E2F-mediated transcription, as the viral TAg is known to bind to pRb, thus relieving the inhibition of pRb on E2F.45 Alternatively, the upregulation could be derived from post-transcriptional regulation such as protein stabilization in the case of p53.46-48 Among all the proteins that were found to be significantly different between mock and infected samples, the levels of RNAs representing over half of them were not previously reported to be different by microarray analysis (Table 2). This demonstrates the importance of using various approaches to probe global changes caused by viral infections. The discrepancy between the proteomic and microarray analyses in our studies likely reflects differential regulation occurring independent of transcriptional control, as well as differences in the two methods with regards to sensitivity in detecting changes in the levels of individual genes and proteins. It would be interesting to investigate the mechanism of differential regulation of these proteins during viral infection. These studies will provide knowledge on how polyomaviruses modulate various cellular proteins and will lead to novel insight into how these cellular proteins function in normal cells.
Among the proteins that were down-regulated by BKPyV infection, IPA network analysis revealed a cluster of vacuolar ATPase subunits, including v-ATPase a3 (TCIRG1) (Figure 5B). These proteins are normally involved in acidification of the autolysosome49 and can also contribute to autophagy.50 Autophagy plays a number of antiviral roles including antigen presentation and activating cellular immunity (reviewed in 51). Other human tumor viruses, including Epstein-Barr virus (EBV) and Kaposi's sarcoma-associated herpesvirus (KSHV), have been shown to modify autophagy to enhance infection and prevent senescence.52, 53 Conversely, stimulating autophagy restricts herpes simplex virus 1 (HSV-1) and human papillomavirus (HPV) infection. 54, 55 These data suggest that limiting autophagy may be an important step in viral replication. Consistent with this, MCPyV encodes a miRNA, which interferes with AMBRA1, an autophagy gene.56 Interestingly, another report in renal proximal tubule cells demonstrates that inhibition of autophagy by targeting TCIRG1 led to defects in cytokinesis, rapid aneuploidy, and enhanced cell survival.57 Therefore, the down-regulation of these autophagy-related proteins during BKPyV infection may have further implications related to immune avoidance, cell survival, and host chromosomal instability caused by infection.
Our proteomic study also established the feasibility of applying SILAC and mass spectrometry analyses to primary RPTE cells and to infection studies. This adds to the growing list of examples applying such proteomic studies to examining global protein changes that occur upon various viral infections.33, 58-60 With proper adaptation, the SILAC and mass spectrometry approach can be combined with other sub-cellular enrichment methods for proteomic studies during infection in the future. For example, it would be interesting to determine which host proteins interact with the viral TAg during replication as previous TAg-interactome studies were performed with TAg expression alone and not in the context of infection.23
Conclusions
We have applied the powerful SILAC to identify nuclear proteomic changes during a lytic polyomavirus infection in relevant primary cells. Our studies led to the identification of several important cellular pathways such as DDR signaling and DNA repair pathways that are altered by polyomavirus at the protein level. This study forms the basis for future detailed molecular characterizations of essential host factors that are crucial for the establishment of a productive viral infection.
Acknowledgments
We thank members of the Jiang, Lefkowitz, and Imperiale labs for help and discussion with this work. We thank Dr. David Crossman and UAB Heflin Center for Genomic Science for assistance with the IPA software. This work was supported by the UAB Department of Microbiology start-up fund, UAB Faculty Development Grant Program (Office of the Provost), a UAB Cancer Center Pilot Program Project grant, a YSB UAB Cancer Center New Faculty Development Award, and an American Heart Association Scientist Development Grant 15SDG25680061 to M.J., grant award UL1TR00165 from the National Center for Advancing Translational Sciences of the National Institutes of Health (NIH) to the UAB Center for Clinical and Translational Sciences under, and NIH grant AI060584 awarded to M.J.I.
References
- 1.DeCaprio JA, Garcea RL. A cornucopia of human polyomaviruses. Nat Rev Microbiol. 2013;11:264–76. doi: 10.1038/nrmicro2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gardner SD, Field AM, Coleman DV, Hulme B. New human papovavirus (B.K.) isolated from urine after renal transplantation. Lancet. 1971;1:1253–7. doi: 10.1016/s0140-6736(71)91776-4. [DOI] [PubMed] [Google Scholar]
- 3.Padgett BL, Walker DL, ZuRhein GM, Eckroade RJ, Dessel BH. Cultivation of papova-like virus from human brain with progressive multifocal leucoencephalopathy. Lancet. 1971;1:1257–60. doi: 10.1016/s0140-6736(71)91777-6. [DOI] [PubMed] [Google Scholar]
- 4.Egli A, Infanti L, Dumoulin A, Buser A, Samaridis J, Stebler C, Gosert R, Hirsch HH. Prevalence of polyomavirus BK and JC infection and replication in 400 healthy blood donors. J Infect Dis. 2009;199:837–46. doi: 10.1086/597126. [DOI] [PubMed] [Google Scholar]
- 5.Astrom KE, Mancall EL, Richardson EP., Jr Progressive multifocal leuko-encephalopathy; a hitherto unrecognized complication of chronic lymphatic leukaemia and Hodgkin's disease. Brain. 1958;81:93–111. doi: 10.1093/brain/81.1.93. [DOI] [PubMed] [Google Scholar]
- 6.Ferenczy MW, Marshall LJ, Nelson CD, Atwood WJ, Nath A, Khalili K, Major EO. Molecular biology, epidemiology, and pathogenesis of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin Microbiol Rev. 2012;25:471–506. doi: 10.1128/CMR.05031-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calabrese LH, Molloy E, Berger J. Sorting out the risks in progressive multifocal leukoencephalopathy. Nat Rev Rheumatol. 2015;11:119–23. doi: 10.1038/nrrheum.2014.167. [DOI] [PubMed] [Google Scholar]
- 8.Suwelack B, Malyar V, Koch M, Sester M, Sommerer C. The influence of immunosuppressive agents on BK virus risk following kidney transplantation, and implications for choice of regimen. Transplant Rev (Orlando) 2012;26:201–11. doi: 10.1016/j.trre.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 9.Dropulic LK, Jones RJ. Polyomavirus BK infection in blood and marrow transplant recipients. Bone Marrow Transplant. 2008;41:11–8. doi: 10.1038/sj.bmt.1705886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096–100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Topalis D, Andrei G, Snoeck R. The large tumor antigen: a “Swiss Army knife” protein possessing the functions required for the polyomavirus life cycle. Antiviral Res. 2013;97:122–36. doi: 10.1016/j.antiviral.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 12.Melendy T, Stillman B. An interaction between replication protein A and SV40 T antigen appears essential for primosome assembly during SV40 DNA replication. J Biol Chem. 1993;268:3389–95. [PubMed] [Google Scholar]
- 13.Harris KF, Christensen JB, Imperiale MJ. BK virus large T antigen: interactions with the retinoblastoma family of tumor suppressor proteins and effects on cellular growth control. J Virol. 1996;70:2378–86. doi: 10.1128/jvi.70.4.2378-2386.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeCaprio JA, Ludlow JW, Figge J, Shew JY, Huang CM, Lee WH, Marsilio E, Paucha E, Livingston DM. SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell. 1988;54:275–83. doi: 10.1016/0092-8674(88)90559-4. [DOI] [PubMed] [Google Scholar]
- 15.Bracken AP, Ciro M, Cocito A, Helin K. E2F target genes: unraveling the biology. Trends Biochem Sci. 2004;29:409–17. doi: 10.1016/j.tibs.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 16.Sheppard HM, Corneillie SI, Espiritu C, Gatti A, Liu X. New insights into the mechanism of inhibition of p53 by simian virus 40 large T antigen. Mol Cell Biol. 1999;19:2746–53. doi: 10.1128/mcb.19.4.2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Segawa K, Minowa A, Sugasawa K, Takano T, Hanaoka F. Abrogation of p53-mediated transactivation by SV40 large T antigen. Oncogene. 1993;8:543–8. [PubMed] [Google Scholar]
- 18.Sowd GA, Li NY, Fanning E. ATM and ATR activities maintain replication fork integrity during SV40 chromatin replication. PLoS Pathog. 2013;9:e1003283. doi: 10.1371/journal.ppat.1003283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang M, Zhao L, Gamez M, Imperiale MJ. Roles of ATM and ATR-mediated DNA damage responses during lytic BK polyomavirus infection. PLoS Pathog. 2012;8:e1002898. doi: 10.1371/journal.ppat.1002898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orba Y, Suzuki T, Makino Y, Kubota K, Tanaka S, Kimura T, Sawa H. Large T antigen promotes JC virus replication in G2-arrested cells by inducing ATM- and ATR-mediated G2 checkpoint signaling. J Biol Chem. 2010;285:1544–54. doi: 10.1074/jbc.M109.064311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abend JR, Low JA, Imperiale MJ. Global effects of BKV infection on gene expression in human primary kidney epithelial cells. Virology. 2010;397:73–9. doi: 10.1016/j.virol.2009.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Low J, Humes HD, Szczypka M, Imperiale M. BKV and SV40 infection of human kidney tubular epithelial cells in vitro. Virology. 2004;323:182–8. doi: 10.1016/j.virol.2004.03.027. [DOI] [PubMed] [Google Scholar]
- 23.Rozenblatt-Rosen O, Deo RC, Padi M, Adelmant G, Calderwood MA, Rolland T, Grace M, Dricot A, Askenazi M, Tavares M, Pevzner SJ, Abderazzaq F, Byrdsong D, Carvunis AR, Chen AA, Cheng J, Correll M, Duarte M, Fan C, Feltkamp MC, Ficarro SB, Franchi R, Garg BK, Gulbahce N, Hao T, Holthaus AM, James R, Korkhin A, Litovchick L, Mar JC, Pak TR, Rabello S, Rubio R, Shen Y, Singh S, Spangle JM, Tasan M, Wanamaker S, Webber JT, Roecklein-Canfield J, Johannsen E, Barabasi AL, Beroukhim R, Kieff E, Cusick ME, Hill DE, Munger K, Marto JA, Quackenbush J, Roth FP, DeCaprio JA, Vidal M. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature. 2012;487:491–5. doi: 10.1038/nature11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shao Q, Byrum SD, Moreland LE, Mackintosh SG, Kannan A, Lin Z, Morgan M, Stack BC, Jr, Cornelius LA, Tackett AJ, Gao L. A Proteomic Study of Human Merkel Cell Carcinoma. J Proteomics Bioinform. 2013;6:275–282. doi: 10.4172/jpb.1000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abend JR, Low JA, Imperiale MJ. Inhibitory effect of gamma interferon on BK virus gene expression and replication. J Virol. 2007;81:272–9. doi: 10.1128/JVI.01571-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hammond SL, Ham RG, Stampfer MR. Serum-free growth of human mammary epithelial cells: rapid clonal growth in defined medium and extended serial passage with pituitary extract. Proc Natl Acad Sci U S A. 1984;81:5435–9. doi: 10.1073/pnas.81.17.5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang M, Abend JR, Tsai B, Imperiale MJ. Early events during BK virus entry and disassembly. J Virol. 2009;83:1350–8. doi: 10.1128/JVI.02169-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin SY, Makino K, Xia W, Matin A, Wen Y, Kwong KY, Bourguignon L, Hung MC. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol. 2001;3:802–8. doi: 10.1038/ncb0901-802. [DOI] [PubMed] [Google Scholar]
- 29.Harlow E, Crawford LV, Pim DC, Williamson NM. Monoclonal antibodies specific for simian virus 40 tumor antigens. J Virol. 1981;39:861–9. doi: 10.1128/jvi.39.3.861-869.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hartman KG, Vitolo MI, Pierce AD, Fox JM, Shapiro P, Martin SS, Wilder PT, Weber DJ. Complex formation between S100B protein and the p90 ribosomal S6 kinase (RSK) in malignant melanoma is calcium-dependent and inhibits extracellular signal-regulated kinase (ERK)-mediated phosphorylation of RSK. J Biol Chem. 2014;289:12886–95. doi: 10.1074/jbc.M114.561613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ueda Y, Koya T, Yoneda-Kato N, Kato JY. Small mitochondrial ARF (smARF) is located in both the nucleus and cytoplasm, induces cell death, and activates p53 in mouse fibroblasts. FEBS Lett. 2008;582:1459–64. doi: 10.1016/j.febslet.2008.03.032. [DOI] [PubMed] [Google Scholar]
- 32.Jiang M, Entezami P, Gamez M, Stamminger T, Imperiale MJ. Functional reorganization of promyelocytic leukemia nuclear bodies during BK virus infection. MBio. 2011;2:e00281–10. doi: 10.1128/mBio.00281-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kroeker AL, Ezzati P, Coombs KM, Halayko AJ. Influenza A infection of primary human airway epithelial cells up-regulates proteins related to purine metabolism and ubiquitin-related signaling. J Proteome Res. 2013;12:3139–51. doi: 10.1021/pr400464p. [DOI] [PubMed] [Google Scholar]
- 34.Funabiki H, Murray AW. The Xenopus chromokinesin Xkid is essential for metaphase chromosome alignment and must be degraded to allow anaphase chromosome movement. Cell. 2000;102:411–24. doi: 10.1016/s0092-8674(00)00047-7. [DOI] [PubMed] [Google Scholar]
- 35.Antonio C, Ferby I, Wilhelm H, Jones M, Karsenti E, Nebreda AR, Vernos I. Xkid, a chromokinesin required for chromosome alignment on the metaphase plate. Cell. 2000;102:425–35. doi: 10.1016/s0092-8674(00)00048-9. [DOI] [PubMed] [Google Scholar]
- 36.Shibahara K, Asano M, Ishida Y, Aoki T, Koike T, Honjo T. Isolation of a novel mouse gene MA-3 that is induced upon programmed cell death. Gene. 1995;166:297–301. doi: 10.1016/0378-1119(95)00607-9. [DOI] [PubMed] [Google Scholar]
- 37.Cmarik JL, Min H, Hegamyer G, Zhan S, Kulesz-Martin M, Yoshinaga H, Matsuhashi S, Colburn NH. Differentially expressed protein Pdcd4 inhibits tumor promoter-induced neoplastic transformation. Proc Natl Acad Sci U S A. 1999;96:14037–42. doi: 10.1073/pnas.96.24.14037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fanning E, Zhao K. SV40 DNA replication: from the A gene to a nanomachine. Virology. 2009;384:352–9. doi: 10.1016/j.virol.2008.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lietzen N, Ohman T, Rintahaka J, Julkunen I, Aittokallio T, Matikainen S, Nyman TA. Quantitative subcellular proteome and secretome profiling of influenza A virus-infected human primary macrophages. PLoS Pathog. 2011;7:e1001340. doi: 10.1371/journal.ppat.1001340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Forbus J, Spratt H, Wiktorowicz J, Wu Z, Boldogh I, Denner L, Kurosky A, Brasier RC, Luxon B, Brasier AR. Functional analysis of the nuclear proteome of human A549 alveolar epithelial cells by HPLC-high resolution 2-D gel electrophoresis. Proteomics. 2006;6:2656–72. doi: 10.1002/pmic.200500652. [DOI] [PubMed] [Google Scholar]
- 41.Zhao X, Madden-Fuentes RJ, Lou BX, Pipas JM, Gerhardt J, Rigell CJ, Fanning E. Ataxia telangiectasia-mutated damage-signaling kinase- and proteasome-dependent destruction of Mre11-Rad50-Nbs1 subunits in Simian virus 40-infected primate cells. J Virol. 2008;82:5316–28. doi: 10.1128/JVI.02677-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi Y, Dodson GE, Shaikh S, Rundell K, Tibbetts RS. Ataxia-telangiectasia-mutated (ATM) is a T-antigen kinase that controls SV40 viral replication in vivo. J Biol Chem. 2005;280:40195–200. doi: 10.1074/jbc.C500400200. [DOI] [PubMed] [Google Scholar]
- 43.Taneja P, Boche I, Hartmann H, Nasheuer HP, Grosse F, Fanning E, Weisshart K. Different activities of the largest subunit of replication protein A cooperate during SV40 DNA replication. FEBS Lett. 2007;581:3973–8. doi: 10.1016/j.febslet.2007.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boichuk S, Hu L, Hein J, Gjoerup OV. Multiple DNA damage signaling and repair pathways deregulated by simian virus 40 large T antigen. J Virol. 2010;84:8007–20. doi: 10.1128/JVI.00334-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chellappan S, Kraus VB, Kroger B, Munger K, Howley PM, Phelps WC, Nevins JR. Adenovirus E1A, simian virus 40 tumor antigen, and human papillomavirus E7 protein share the capacity to disrupt the interaction between transcription factor E2F and the retinoblastoma gene product. Proc Natl Acad Sci U S A. 1992;89:4549–53. doi: 10.1073/pnas.89.10.4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kress M, May E, Cassingena R, May P. Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. J Virol. 1979;31:472–83. doi: 10.1128/jvi.31.2.472-483.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature. 1979;278:261–3. doi: 10.1038/278261a0. [DOI] [PubMed] [Google Scholar]
- 48.Linzer DI, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell. 1979;17:43–52. doi: 10.1016/0092-8674(79)90293-9. [DOI] [PubMed] [Google Scholar]
- 49.Hinton A, Bond S, Forgac M. V-ATPase functions in normal and disease processes. Pflugers Arch. 2009;457:589–98. doi: 10.1007/s00424-007-0382-4. [DOI] [PubMed] [Google Scholar]
- 50.Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, Uchiyama Y, Westaway D, Cuervo AM, Nixon RA. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–58. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chiramel AI, Brady NR, Bartenschlager R. Divergent roles of autophagy in virus infection. Cells. 2013;2:83–104. doi: 10.3390/cells2010083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Granato M, Santarelli R, Farina A, Gonnella R, Lotti LV, Faggioni A, Cirone M. Epstein-barr virus blocks the autophagic flux and appropriates the autophagic machinery to enhance viral replication. J Virol. 2014;88:12715–26. doi: 10.1128/JVI.02199-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leidal AM, Cyr DP, Hill RJ, Lee PW, McCormick C. Subversion of autophagy by Kaposi's sarcoma-associated herpesvirus impairs oncogene-induced senescence. Cell Host Microbe. 2012;11:167–80. doi: 10.1016/j.chom.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 54.Yakoub AM, Shukla D. Autophagy stimulation abrogates herpes simplex virus-1 infection. Sci Rep. 2015;5:9730. doi: 10.1038/srep09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Griffin LM, Cicchini L, Pyeon D. Human papillomavirus infection is inhibited by host autophagy in primary human keratinocytes. Virology. 2013;437:12–9. doi: 10.1016/j.virol.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee S, Paulson KG, Murchison EP, Afanasiev OK, Alkan C, Leonard JH, Byrd DR, Hannon GJ, Nghiem P. Identification and validation of a novel mature microRNA encoded by the Merkel cell polyomavirus in human Merkel cell carcinomas. J Clin Virol. 2011;52:272–5. doi: 10.1016/j.jcv.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Belaid A, Cerezo M, Chargui A, Corcelle-Termeau E, Pedeutour F, Giuliano S, Ilie M, Rubera I, Tauc M, Barale S, Bertolotto C, Brest P, Vouret-Craviari V, Klionsky DJ, Carle GF, Hofman P, Mograbi B. Autophagy plays a critical role in the degradation of active RHOA, the control of cell cytokinesis, and genomic stability. Cancer Res. 2013;73:4311–22. doi: 10.1158/0008-5472.CAN-12-4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kroeker AL, Ezzati P, Halayko AJ, Coombs KM. Response of primary human airway epithelial cells to influenza infection: a quantitative proteomic study. J Proteome Res. 2012;11:4132–46. doi: 10.1021/pr300239r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berard AR, Coombs KM, Severini A. Quantification of the host response proteome after herpes simplex virus type 1 infection. J Proteome Res. 2015;14:2121–42. doi: 10.1021/pr5012284. [DOI] [PubMed] [Google Scholar]
- 60.Berard AR, Cortens JP, Krokhin O, Wilkins JA, Severini A, Coombs KM. Quantification of the host response proteome after mammalian reovirus T1L infection. PLoS One. 2012;7:e51939. doi: 10.1371/journal.pone.0051939. [DOI] [PMC free article] [PubMed] [Google Scholar]