

Abstract
Peptoclostridium difficile (Clostridium difficile) is the major pathogen associated with infectious diarrhea in humans. Concomitant with the increased incidence of C. difficile infection worldwide, there is an increasing concern regarding this infection type. This study reports a draft assembly and detailed sequence analysis of C. difficile strain ZJCDC-S82. The de novo assembled genome was 4.19 Mb in size, which includes 4,013 protein-coding genes, 41 rRNA genes, and 84 tRNA genes. Along with the nuclear genome, we also assembled sequencing information for a single plasmid consisting of 11,930 nucleotides. Comparative genomic analysis of C. difficile ZJCDC-S82 and two other previously published strains, such as M120 and CD630, showed extensive similarity. Phylogenetic analysis revealed that genetic diversity among C. difficile strains was not influenced by geographic location. Evolutionary analysis suggested that four genes encoding surface proteins exhibited positive selection in C. difficile ZJCDC-S82. Codon usage analysis indicated that C. difficile ZJCDC-S82 had high codon usage bias toward A/U-ended codons. Furthermore, codon usage patterns in C. difficile ZJCDC-S82 were predominantly affected by mutation pressure. Our results provide detailed information pertaining to the C. difficile genome associated with a strain from mainland China. This analysis will facilitate the understanding of genomic diversity and evolution of C. difficile strains in this region.
Keywords: C. difficile ZJCDC-S82, genome sequencing, phylogenetic analysis, evolutionary analysis, codon usage
Introduction
Peptoclostridium difficile (Clostridium difficile) is a Gram-positive, spore-forming, anaerobic bacterium that commonly resides asymptomatically in the gut of healthy individuals.1,2 C. difficile infection (CDI) is now deemed one of the most important causes of infectious diarrhea. The risks associated with infection can be increased by antibiotic administration or prolonged periods of hospitalization.3 The clinical symptoms of CDI range from mild, self-limiting diarrhea to pseudomembranous colitis and life-threatening fulminant colitis.4,5 In addition, CDI has a high relapse rate due to the reactivation or reinfection of latent infections,4,5 which makes it difficult to cure completely. The morbidity and severity of CDI have been dramatically increasing in hospitals around the world. In particular, CDI outbreaks with polymerase chain reaction (PCR) ribotype 027 have emerged in Canada, the United States, and Western Europe since 2003, leading to increased colectomies and deaths.6–9 The lack of studies on CDI from mainland China contrasted with the relatively large number of reports from other Asian countries.10 The limited molecular epidemiological studies of C. difficile from China reported that PCR ribotypes 012 (ST54), 046 (ST35), and 017 (ST37) were the common strains.11 The three comprehensive Chinese studies of CDI have been conducted by far in Beijing,12 Shanghai,13 and Hangzhou11), respectively. As the data show, an epidemiological pattern in mainland China differed from other world regions. It would be important to disclose the specific molecular characteristics of PCR ribotypes 012 (ST54), 046 (ST35), and 017 (ST37) isolated from mainland China.
Given the importance of CDI in clinical epidemiology, extensive research focusing on genomic analysis was performed to gain insights into the pathogenicity of C. difficile.14–17 Since the release of the first complete genome sequence of C. difficile strain CD630,18 the number of whole-genome sequences of C. difficile strains in GenBank (http://www.ncbi.nlm.nih.gov/genome/) has rapidly increased to 262 as of May 2015. This is predominantly due to the widespread application of next-generation DNA-sequencing technology.15,16,19–22 Comparative genome analysis of different C. difficile strains revealed some genetic alterations that may account for C. difficile pathogenesis.14,23,24 For example, Forgetta et al sequenced the whole genome of 14 C. difficile isolates and identified 20 DNA markers that can predict disease severity caused by CDI.24 To ascertain the evolution of virulent strain C. difficile 027, Stabler et al performed a three-way genomic comparison of C. difficile strains displaying different levels of virulence.14 They reported several unique genes and genetic regions in the epidemic strain, which may explain the recent emergence of increased virulence associated with the pathogen.14 Furthermore, Darling et al analyzed the whole genome of C. difficile strain 5.3 and found several genes that may be associated with its virulence and pathogenicity.23
Although genome analysis has facilitated exploration into some of the underlying mechanisms associated with CDI in many countries, little is known regarding genomic information of C. difficile strains from mainland China. After performing an epidemiological survey in the Zhejiang region, we chose one of the dominant molecular types to facilitate whole-genome sequence analysis. We subsequently performed a detailed genome-wide analysis of the C. difficile strain ZJCDC-S82 that had been isolated from Zhejiang province. The analysis involved sequencing, de novo assembling, and annotating the whole draft genome of ZJCDC-S82. Next, we performed a three-way comparative genome analysis of C. difficile strains ZJCDC-S82, M120, and CD630. Phylogenetic analysis was also carried out using the genome sequences of ZJCDC-S82 and 10 additional published C. difficile strains. Finally, we performed evolutionary analysis and codon usage analysis of protein-coding genes from ZJCDC-S82.
Methods
Bacterial strain and DNA isolation
This study was approved by the Institutional Review Board of Zhejiang provincial Center for Disease Control and Prevention (ZJCDC) and Hangzhou First People’s Hospital (HFPH). The C. difficile strain ZJCDC-S82 was isolated from a C. difficile-positive stool specimen at ZJCDC, China. The clinical stool specimen was collected from an inpatient with severe diarrhea in the Department of Gastroenterology at HFPH. The stool specimen was treated with alcohol and then inoculated onto cefoxitin–cycloserine–fructose agar plates (Oxoid).8 After incubation for 48 hours at 37 °C in an anaerobic chamber with a GENbag anaer atmospheric generator (BioMérieux), the isolate was confirmed to be C. difficile using previously described assays.25 Genomic DNA was extracted using a DNA extraction kit (Qiagen, Inc.) according to the manufacturer’s instructions. DNA concentration was quantified using a NanoDrop™ spectrophotometer. DNA aliquots with concentrations of >100 ng/µL were required for library preparation prior to using the next-generation sequencing.
C. difficile typing
One reference strain with standard typing patterns (VPI 10463: ribotype 087) was used as a control for all typing tests described below. The isolate was subjected to PCR ribotyping using capillary gel electrophoresis with primer pairs as described previously.26 The 16S primer was labeled at the 5′ end with carboxyfluorescein. After PCR amplification, PCR fragments were analyzed in an ABI 3100 genetic analyzer (Applied Biosystems) with a 36-cm capillary loaded with a POP4 gel (Applied Biosystems). The size of each peak was determined by the use of the Genemapper ID-X software, version 1.3 (Applied Biosystems). The capillary sequencer-based PCR-ribotyping data were deposited in the WEBRIBO web site (https://webribo.ages.at/), and the results were analyzed and output automatically.
In addition, the isolate was subjected to multilocus sequence typing (MLST) as described elsewhere.27 Seven loci, consisting of adk, atpA, dxr, glyA, recA, sodA, and tpi genes, were chosen for PCR amplification and sequencing. PCR amplicons were detected using a 3730xl DNA analyzer (Applied Biosystems). Then, the data for C. difficile alleles and sequence types were deposited in the public C. difficile MLST database, which is available at http://pubmlst.org/cdifficile.
Genome sequencing and assembly
Genomic DNA was sequenced using two high-throughput sequencing platforms, Pacific Biosciences (PacBio) RS (http://www.pacificbiosciences.com/) and Ion Torrent Proton™ (http://www.lifetechnologies.com/). To assemble the whole-genome sequence, sequencing reads from both platforms were de novo assembled separately. PacBio RS reads were assembled using the hierarchical genome assembly process method (version 2.3.0, default setting).28 Ion Torrent Proton™ reads were trimmed, quality filtered, and further assembled using CLC Genomics Workbench (version 7.0, default setting).29 After that, assemblies from two previous assemblers were combined with the final assembly using the Mix software30 at the default setting. Finally, assembled contigs of ZJCDC-S82 were reordered according to the genome sequence of strain CD630 using Mauve 2.4.0.31
Genome annotation and comparison
The whole-genome sequence of C. difficile strain ZJCDC-S82 was annotated with the Rapid Annotations using Subsystems Technology (RAST) annotation system.32–34 In RAST, protein-coding genes were predicted by Glimmer 2.35 tRNA genes were identified using tRNAScan-SE2,36 and rRNA genes were annotated with Search_for_rnas script (Niels Larsen, unpublished results). The functional assignment of each gene was performed using FIGfams (release 70).32 The predicted genes of strain ZJCDC-S82 were compared against two strains with high-quality assembly and annotation, M120 and CD630, using the SEED viewer (http://seed-viewer.theseed.org). To identify conserved genes between genomes, we searched the homologous sequences using sequence identity (filtering threshold: 90%) and synteny information. In addition, genomic rearrangement patterns between strains ZJCDC-S82, M120, and CD630 were analyzed and visualized by Mauve.31
Phylogenetic analysis
To ascertain the phylogenetic relationship between ZJCDC-S82 and C. difficile strains from different global locations, we chose 10 additional C. difficile strains from the Ensembl genome database (http://bacteria.ensembl.org/index.html) according to their strain type and geographic locations. Detailed information for these strains is shown in Supplementary Table 1. The phylogenetic tree of C. difficile strains was generated from a concatenated alignment of 2,001 orthologous genes by MEGA version 6.0 using a maximum likelihood (ML) approach.37 The orthologous genes are those genes that are identified following the annotation of the strain CD630 genome where a homologous sequence was found in all the 11 genomes analyzed. This was performed using BLAT38 where 90% identity with the query sequence was required in the designation as an orthologous gene. The priori tree for the ML search was generated automatically by applying the Neighbor-Joining algorithm. Bootstrapping was performed with 1,000 replicates.
Evolutionary analysis
The ratio between the numbers of nonsynonymous substitutions (dN) and synonymous substitutions (dS) was used to estimate the selective pressure on protein-coding genes. Although a dN/dS value of <1.0 indicated negative (or purifying) selection, a dN/dS value of >1.0 suggested positive (or adaptive) selection. We calculated a dN/dS value for each protein-coding gene in ZJCDC-S82 by comparison with homologs from M120 and CD630, respectively. Multiple alignments of each gene were performed using CLC Genomics Workbench 7.0. Pairwise dN, dS, and dN/dS values were calculated using the codeml program from the PAML 4.8 package (setting: runmode = −2).39 The likelihood ratio test (LRT) was performed by comparing this model (M0) to the null model (setting: omega = 1).
Furthermore, we applied a pair of site models: M7 and M8 in the codeml program from the PAML 4.8 package39 to detect the particular amino acid sites under positive selection in two toxin genes (tcdA and tcdB). The null model (M7) uses distributions that do not admit positive selection, while the alternative model (M8) allows positive selection. The LRT statistic between the null model and the alternative model was compared with the χ2 distribution with two degrees of freedom. The Bayes empirical Bayes (BEB) approach was employed to detect amino acid sites with a posterior probability >95% of being under selection.40
Codon usage analysis
Most amino acids are encoded by more than one codon. Thus, there can be multiple codons for a given amino. Relative synonymous codon usage (RSCU) is a measurement that can be used to estimate the extent of nonrandom usage of synonymous codons for a specific amino acid.41 Synonymous codons with RSCU values close to 1 showed little codon usage bias for that amino acid. Other than RSCU, the effective number of codons (ENc) was used to quantify codon usage bias of a gene.42 ENc values range from 20 to 61. These values are independent of gene length and the number of amino acids. An ENc value of 20 implies a gene with extreme codon usage bias where only one codon was used for a particular amino acid, while a value of 61 showed no bias where all codons were used equally. We calculated RSCU values for each sensitive codon. ENc and GC3s for each protein-coding gene were generated using CodonW.43
Results
Sequencing and assembly statistics of C. difficile ZJCDC-S82
The sequencing statistics of both PacBio RS and Ion Torrent Proton™ are shown in Supplementary Table 2. A PacBio RS sequencing run resulted in 128,964 reads with an average read length of 2.2 kilobase pairs (bps; Supplementary Fig. 1), which corresponded to a 73-fold coverage of the genome. PacBio RS reads were assembled into 30 large contigs (longer than 5,000 bps; Supplementary Table 3). Additionally, an Ion Torrent Proton™ sequencing run generated 3,681,407 reads with an average read length of 223 bps before trimming and quality filtering (Supplementary Fig. 2). After filtering, Ion Torrent Proton™ reads were assembled into 259 contigs (Supplementary Table 3). Finally, the two separate assemblies were combined into the final assembly of ZJCDC-S82, which included 20 contigs in total (Supplementary Table 3). The assembled ZJCDC-S82 draft genome was deposited in the NCBI GenBank under the accession number JYNK00000000 (http://www.ncbi.nlm.nih.gov/nuccore/JYNK00000000).
Molecular typing of C. difficile ZJCDC-S82
While a panel of data from capillary gel electrophoresis-based PCR ribotyping was entered into the web-based database, a close matched ribotype corresponding to each submitted isolate was identified and output automatically. The result showed that C. difficile ZJCDC-S82 was PCR ribotype 012. In addition, after seven PCR amplicons were sequenced respectively, the gene sequences for the ZJCDC-S82 were deposited in the MLST database and aligned automatically. The result indicated that C. difficile ZJCDC-S82 was ST54.
General genome features
Our draft genome assembly of C. difficile strain ZJCDC-S82 consisted of a circular nuclear genome, which was ~4,190,365 bps in length with a guanine-cytosine (GC) content of 29.1%. In total, we identified 4,032 protein-coding genes (4,013 genes in the nuclear genome and 19 genes in the plasmid). Among them, 1,161 genes were hypothetical protein-coding genes. We also predicted 41 rRNA gene loci, consisting of 5S, 16S, and 23S rRNA genes and 84 tRNA genes, representing all 20 amino acids. In addition, 11 genes were predicted as possibly missing by the RAST annotation pipeline. Sequences of these possibly missing genes have long gaps when comparing genes in neighboring genomes. The general features of the C. difficile ZJCDC-S82 genome are summarized in Table 1. The overall functional profile of genes in C. difficile ZJCDC-S82 is shown in Figure 1A, and the schematic circular representation of the ZJCDC-S82 draft genome is shown in Figure 1B.
Table 1.
General features of the ZJCDC-S82 genome.
| GENETIC ELEMENT | SIZE (bp) | NO. OF CONTIGS | N50 (bp) | GC CONTENT (%) | NO. OF CODING SEQUENCES | NO. OF rRNA GENES | NO. OF tRNA GENES |
|---|---|---|---|---|---|---|---|
| Chromosome | 4190365 | 19 | 598695 | 29.1 | 4013 | 41 | 84 |
| Plasmid | 11930 | 1 | 11930 | 26.4 | 19 | 0 | 0 |
Figure 1.

General features of the C. difficile strain ZJCDC-S82 genome. (A) Genes connected to subsystems and their distribution in different categories with the genome of ZJCDC-S82. The results were obtained using SEED viewer (http://rast.nmpdr.org). (B) Circular representations of the C. difficile strain ZJCDC-S82 chromosome. From the outside: circles 1 and 2 show the position of all genes transcribed in a clockwise and counterclockwise direction, circle 3 shows RNA genes (cyan, rRNA genes; red, tRNA genes), circle 4 shows GC content (plotted using a 10-kb window), and circle 5 shows GC deviation (plotted using a 10-kb window; orange, >0%; red, <0%).
C. difficile ZJCDC-S82 plasmid genome
In addition to the 4.19 Mb chromosome, C. difficile ZJCDC-S82 was also found to carry a plasmid, which was 11,930 bps in length with a GC content of 26.4%. To our knowledge, plasmids have been found in several different strains.18,44–46 The sequence of the C. difficile ZJCDC-S82 plasmid (pZJCDC-S82) was compared against the NCBI nucleotide database using blastn. The sequence demonstrating the highest identity to pZJCDC-S82 was the C. difficile 630 plasmid. Through sequence analysis, we identified that pZJCDC-S82 carried sequences with homology to genes that encoded a transcriptional regulator, RNA polymerase sigma-70 factor, Cys-tRNA(Pro) deacylase YbaK, DNA invertase, and an outer membrane lipoprotein-sorting protein (which may influence the function of nuclear genes).
Whole-genome comparison
The genome of the C. difficile strain ZJCDC-S82 was compared with the genomes of two C. difficile strains, CD630 and M120. For consistency of comparison, the protein-coding sequences in the genome of strains CD630 and M120 were also reanalyzed using RAST. The predicted subsystem features of genes in these three genomes are shown in Supplementary Table 4. We found that 2,895 Coding sequences (CDSs) have >90% sequence identity between them (Fig. 2A). Gene ontology analysis showed that these genes were mainly involved in encoding cofactors, vitamins, prosthetic groups, pigments, protein metabolism proteins, RNA metabolism proteins, amino acids and derivatives, carbohydrates, cell wall and capsule proteins, nucleosides, and nucleotides. In addition, 717 CDSs were identified as unique to strain ZJCDC-S82 but were not located in strains CD630 and M120 (Supplementary Table 5). These included CDS with homology to genes that encoded for proteins involved in or associated with DNA metabolism, phage, prophage, trans-posable elements, plasmids, respiration, virulence, disease, and defense. Similarly, there were 610 CDSs and 480 CDSs that were unique to strains CD630 and M120, respectively (Fig. 2A). In order to perform genome comparison of these three strains, the whole genome was progressively divided into 14 locally collinear blocks by Mauve (Fig. 2B). We found that the majority of the genomes were conserved in all three strains, although there were some strain-specific regions.
Figure 2.

Comparative genome analysis results of C. difficile strains ZJCDC-S82, M120, and CD630. (A) Unique and shared CDSs of C. difficile strains ZJCDC-S82, M120, and CD630. The Venn diagram shows the number of unique and shared genes between the three strains. (B) Genome alignment of C. difficile strains ZJCDC-S82, M120, and CD630. Pairwise comparison of the three genomes as visualized using Mauve is shown.
Phylogenetic relationships among strains
In the last decade, C. difficile has become the most important cause of infectious diarrhea worldwide. However, the global spread pattern remains unknown. To explore this issue, we collected publicly available genomes of 10 additional worldwide strains (Supplementary Table 1) and performed phylogenetic analysis in association with our sequenced strain, ZJCDC-S82. The phylogenetic relationship between these strains was constructed based on a concatenated alignment of 2,001 orthologous genes using an ML approach. We observed that the ZJCDC-S82 strain was most closely related to the common ancestor of five strains, CD630, E14, R20291, QCD-66c26, and CD196 (Fig. 3). These strains have PCR ribotypes 012 (CD630), 014 (E14), and 027 (CD196, QCD-66c26, and R20291; Fig. 3). Strains with the same PCR ribotypes are more likely to group together in the phylogenetic tree (Fig. 3). However, the three strains isolated from Ireland (E13, T20, and M120) did not group together in the phylogenetic tree (Fig. 3). This result suggested that the PCR ribotype of C. difficile strains, rather than the geographic location, was more related to its genetic diversity.
Figure 3.

Phylogenetic tree based on genome sequences of 11 C. difficile strains.
Evolutionary analysis
In order to evaluate whether or not certain genes had evolved differently in C. difficile strain ZJCDC-S82, we aligned homologous gene sequences from strains ZJCDC-S82, M120, and CD630 and calculated the ratios of dN/dS. As shown in Figure 4, the dN/dS values in pairwise comparisons between the three genomes had a left skewed distribution with a peak around 0.03 and the shape of the curves had a similar appearance. Approximately 99% of the genes had dN/dS values below 1.0, suggesting that the genes had evolved under purifying selection. There are 11 genes with dN/dS values more than 1.0, which suggested that they may undergo positive selection. Four genes from ZJCDC-S82 and M120 with dN/dS values >1.0 were identified as encoding a fema-like peptidoglycan biosynthesis protein, a lipoprotein, and 50S ribosomal proteins. Seven genes from ZJCDC-S82 and CD630 were identified as encoding hypothetical proteins and membrane proteins (Table 2). But, dN/dS values of these 11 genes show no significant deviation to 1 using LRT.
Figure 4.
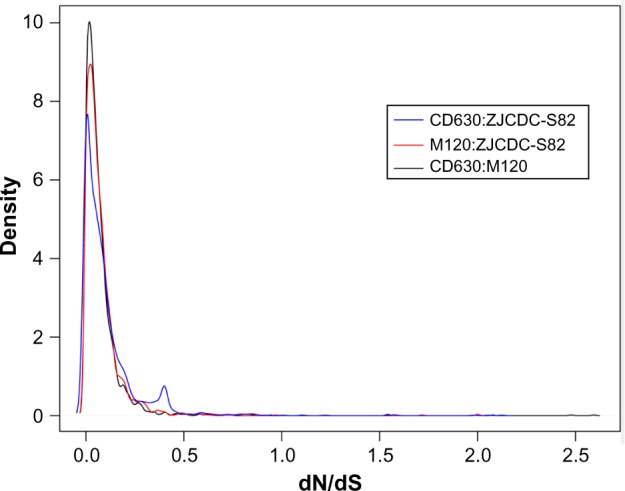
The distribution of dN/dS estimates for genes with ortholog relationships between C. difficile strains ZJCDC-S82, M120, and CD630.
Table 2.
Positively selected genes in C. difficile strain ZJCDC-S82.
| GENEa | dN/dS | P-VALUE | ANNOTATION | GENOMES COMPAREDb |
|---|---|---|---|---|
| CD0410 | 2.12 | n.s | Hypothetical protein | ZJCDC-S82:CD630 |
| CD2317 | 2.06 | n.s | Hypothetical protein | ZJCDC-S82:CD630 |
| CD3205 | 1.98 | n.s | Hypothetical protein | ZJCDC-S82:CD630 |
| CD2768 | 1.58 | n.s | Membrane protein | ZJCDC-S82:CD630 |
| CD0855 | 1.57 | n.s | Hypothetical protein | ZJCDC-S82:CD630 |
| CD1509 | 1.22 | n.s | Hypothetical protein | ZJCDC-S82:CD630 |
| CD3922 | 1.11 | n.s | Membrane protein | ZJCDC-S82:CD630 |
| CD0789 | 2.00 | n.s | 50S ribosomal protein L21 | ZJCDC-S82:M120 |
| CD3380 | 2.00 | n.s | 50S ribosomal protein L36 | ZJCDC-S82:M120 |
| CD3544 | 1.72 | n.s | Fema-like peptidoglycan biosynthesis protein | ZJCDC-S82:M120 |
| CD3135 | 1.11 | n.s | Lipoprotein | ZJCDC-S82:M120 |
Notes:
Gene name of C. difficile strain ZJCDC-S82.
CD630 – C. difficile strain CD630; M120 – C. difficile strain M120; ZJCDC-S82 – C. difficile strain ZJCDC-S82.
Abbreviation: n.s, nonsignificant.
To detect potential genetic regions that might be associated with the virulence of this strain, we also performed site-specific evolutionary analysis on tcdA and tcdB genes by comparing orthologous genes in strains ZJCDC-S82, M120, and CD630 using site-specific model M7 versus M8. LRT results between the likelihood scores of the null (M7) and alternative (M8) models for each gene are shown in Table 3. The test for site model in tcdA gene was significant (2ΔlnL = 12.24, df = 2, P < 0.01), indicating the presence of amino acid sites under positive selection (2032G, 2180N, 2219G, 2361L, 2428N, and 2581N). However, the test for site model in tcdB gene was nonsignificant (2ΔlnL = 3.20, df = 2, P > 0.05), in which no amino acid sites were under positive selection.
Table 3.
Toxin genes (tcdA and tcdB) LRTs for PAML M7 and M8 site models.
| GENE | MODEL | lnLnulla | lnLalternativea | 2ΔlnLb | P-VALUE | POSITIVELY SELECTED SITESd | |
|---|---|---|---|---|---|---|---|
| tcdA | M7 vs M8 | −12224.17 | −12218.05 | 12.24 | P < 0.01 | 2032G(0.966) | |
| 2180N(0.968) | |||||||
| 2219G(0.968) | |||||||
| 2361L(0.967) | |||||||
| 2428N(0.966) | |||||||
| 2581N(0.964) | |||||||
| tcdB | M7 vs M8 | −11112.81 | −11111.21 | 3.20 | n.s. | none | |
Notes:
log-likelihood scores.
LRT to detect positive selection.
Positively selected sites: posterior probabilities >0.95 in the BEB analyses.
Abbreviation: n.s, nonsignificant.
Codon usage in C. difficile ZJCDC-S82
We performed codon usage analysis using 4,032 protein-coding genes in C. difficile ZJCDC-S82. The observed number for each codon is shown in Table 4. Among these codons, AAA (68.9%) and CGG (0.3%) were the most prominent and least prominent codons, respectively. The overall GC content of protein- coding genes in C. difficile ZJCDC-S82 was 29.7%, while the mean value of the GC3s was 15.3%. Clearly, codons with the third-position nucleotide of A or U were much more frequently used than those ending with G or C, as observed in most previous studies.47,48 This might be due to the structure of AU-rich segments, which may give rise to the higher codon bias.
Table 4.
Codon usage for the C. difficile strain ZJCDC-S82 genome.
| CODON | COUNT | RSCU | CODON | COUNT | RSCU |
|---|---|---|---|---|---|
| GCU(A) | 25956 | 1.68 | CCU(P) | 11631 | 1.49 |
| GCC(A) | 3204 | 0.21 | CCC(P) | 940 | 0.12 |
| GCA(A) | 30118 | 1.95 | CCA(P) | 17659 | 2.26 |
| GCG(A) | 2356 | 0.15 | CCG(P) | 1066 | 0.14 |
| UGU(C) | 12217 | 1.68 | CAA(Q) | 21948 | 1.59 |
| UGC(C) | 2296 | 0.32 | CAG(Q) | 5597 | 0.41 |
| GAU(D) | 53559 | 1.64 | CGU(R) | 2912 | 0.47 |
| GAC(D) | 11932 | 0.36 | CGC(R) | 612 | 0.1 |
| GAA(E) | 66979 | 1.58 | CGA(R) | 1320 | 0.21 |
| GAG(E) | 17683 | 0.42 | CGG(R) | 289 | 0.05 |
| UUU(F) | 42831 | 1.73 | AGA(R) | 28026 | 4.52 |
| UUC(F) | 6766 | 0.27 | AGG(R) | 4031 | 0.65 |
| GGU(G) | 27083 | 1.5 | UCU(S) | 21829 | 1.74 |
| GGC(G) | 4279 | 0.24 | UCC(S) | 2177 | 0.17 |
| GGA(G) | 34321 | 1.91 | UCA(S) | 21418 | 1.7 |
| GGG(G) | 6319 | 0.35 | UCG(S) | 1624 | 0.13 |
| CAU(H) | 13007 | 1.65 | AGU(S) | 23118 | 1.84 |
| CAC(H) | 2749 | 0.35 | AGC(S) | 5323 | 0.42 |
| AUU(I) | 38103 | 1 | ACU(T) | 24691 | 1.74 |
| AUC(I) | 6539 | 0.17 | ACC(T) | 2701 | 0.19 |
| AUA(I) | 69662 | 1.83 | ACA(T) | 26932 | 1.89 |
| AAA(K) | 79658 | 1.51 | ACG(T) | 2583 | 0.18 |
| AAG(K) | 25995 | 0.49 | GUU(V) | 30086 | 1.57 |
| UUA(L) | 53307 | 3.06 | GUC(V) | 3465 | 0.18 |
| UUG(L) | 13573 | 0.78 | GUA(V) | 35899 | 1.87 |
| CUU(L) | 21357 | 1.22 | GUG(V) | 7162 | 0.37 |
| CUC(L) | 1446 | 0.08 | UGG(W) | 7235 | 1 |
| CUA(L) | 12061 | 0.69 | UAU(Y) | 38643 | 1.64 |
| CUG(L) | 2941 | 0.17 | UAC(Y) | 8586 | 0.36 |
| AUG(M) | 30486 | 1 | UAA(*) | 2702 | 1.99 |
| AAU(N) | 60832 | 1.67 | UAG(*) | 1029 | 0.76 |
| AAC(N) | 12095 | 0.33 | UGA(*) | 339 | 0.25 |
In addition, we performed RSCU analysis to identify the general patterns of synonymous codon usage in C. difficile ZJCDC-S82. About one-third of the codons (19 of 64) were abundantly used, and the frequently used codons were always A/U-ended, denoted using bold letters in Table 4. Previous studies have shown that the RSCU of NCG codons is related to the level of DNA methylation.49,50 In C. difficile ZJCDC-S82, four NCG codons (ACG, UCG, CCG, and GCG) had very low RSCU values (0.18, 0.13, 0.14, and 0.15, respectively). This result suggested that high methylation level might result in the mutation from methylated C to T, which causes the highly enriched U/A-ending codons in C. difficile ZJCDC-S82. In relation to stop codons, UAA was the most widely used with an RSCU value of 1.99, and UGA was the least commonly used stop codon with an RSCU value of 0.25.
The ENc-plot analysis was used to evaluate the correlation between codon usage and GC3s. This was an effective method to study the key factor in influencing the usage bias of synonymous codons. The ENc values varied from 22.55 to 61.00 for all genes in C. difficile ZJCDC-S82, with an average value of 36.37. As shown in Figure 5, most of the points were located near the standard curve (expected ENc-plot curve), while only a small number of points lay on the curve itself. These results indicated that mutation bias was a major factor, although not a unique factor, in shaping the codon bias. Some other factors, such as translational selection, also accounted for biased codon usage in C. difficile ZJCDC-S82.
Figure 5.
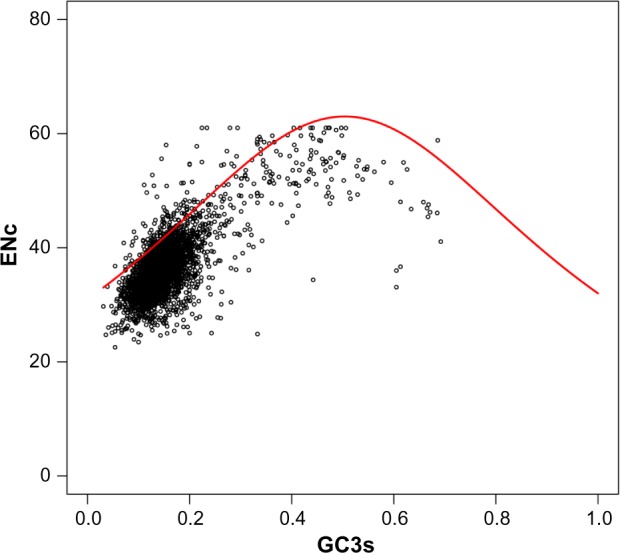
ENc of each CDS plotted against GC3s. ENc shows the effective number of codons, and GC3s shows the GC content on the synonymously variable third position of the sense codon. The red points denote total CDSs; the red solid line represents the expected ENc.
Discussion
Over the last decade, genomic features of C. difficile strains from various host species and geographic regions have been a topic of considerable interest. Even though the genomes of large-scale strains have been sequenced, no C. difficile isolates from mainland China were analyzed. Using molecular epidemiological data from Zhejiang province (data not published) and References 10–12, PCR ribotype 012 (ST54) was shown to be one of the dominant types demonstrating similar antibiotic profiles in Zhejiang province and other regions. The ribotype 012 strain contained both tcdA and tcdB (A+B+) and did not contain the binary toxin genes cdtA and cdtB, and exhibited an 18-bp deletion in the tcdC gene. The results of molecular typing showed that the strain ZJCDC-S82 was PCR ribotypes 012 and ST54 with multidrug resistance but was sensitive to both vancomycin and metronidazole. The strain PCR ribotype 012 was equal to ST54 by MLST, having less genetic diversity than other ribotypes.27 We speculated that PCR ribotype 012 might be related to local prevalence in hospitalized patients. Therefore, this strain, ZJCDC-S82, was chosen to perform subsequent genomic analysis in order to disclose the molecular characteristics of PCR ribotype 012 in this region. To the best of our knowledge, C. difficile ZJCDC-S82 is the first C. difficile strain in mainland China to have its whole genome sequenced and assembled. We also performed phylogenetic analysis, evolutionary analysis, and codon usage analysis on this strain.
Phylogenetic analysis and comparative genomic analysis revealed that the strain ZJCDC-S82 had low geographic similarity, but high genome stability. The phylogenetic tree showed that the genome sequences derived from C. difficile in remote geographic regions did not show phylogenetic segregation. Phylogenetic analysis ambiguously indicated the genetic clustering of the strains according to their geographic origins. Practically, this may suggest that the exchange of C. difficile among the host populations was frequent and that, therefore, the bacteria have evolved more or less dependently. Furthermore, we observed 2,001 orthologous genes in all 11 genomes, differing substantially from observations in previous studies, which estimated the presence of <1,000 genes.21,51–53 The different number of orthologous genes was likely caused by the use of different methodologies and analysis techniques in these studies.
Evolutionary analysis revealed some genes that possibly experienced positive selection in C. difficile ZJCDC-S82. In total, we identified 11 genes that might have demonstrated positive selection. The number of positively selected genes seemed very low for such a diverse species. This finding is consistent with a previous study that demonstrated that the number of genes under positive selection was comparatively small for isolated C. difficile strains.21 It appeared from this analysis that the positively selected genes were surface proteins, including membrane proteins, a fema-like peptidoglycan biosynthesis protein, and a lipoprotein. These proteins are likely candidates in facilitating diversification selection driven by the host immune system. Moreover, it is quite possible that many more positively selected genes exist, but these are part of the bacterial accessory gene pool and were therefore not detected. Consequently, our results showed that host immune selection might play an important role in C. difficile evolution. Site-specific evolutionary analysis on two genes that are related to C. difficile virulence, tcdA and tcdB genes, revealed that several sites in tcdA genes experienced positive selection in ZJCDC-S82 strain. But, there is no signal of positive selection for amino acids in tcdB genes. This suggested that tcdA gene, rather than tcdB gene, might be associated with the pathogenicity of C. difficile ZJCDC-S82 strain. Although our analysis presented several potential sites that might be related to strain pathogenicity, more thorough analyses are needed to explain the detailed mechanism of its pathogenicity and virulence.
Codon usage bias is an important evolutionary feature in most genomes and has been widely reported in many genome analyses. In general, biased usage of synonymous codons, in relation to translational efficiency, is balanced between genetic drift, mutation pressure, and natural selection. We observed that the most abundant codons in the C. difficile ZJCDC-S82 genome were codons that ended with A or U. This is very similar to the codon usage pattern observed in other organisms, such as Onchocerca volvulus, Bombyx mori, and Plasmodium falciparum.48,54,55 In addition, the characteristics of synonymous codon usage patterns and ENc-plot analysis revealed that the most important determinant of codon usage patterns was mutational bias in C. difficile ZJCDC-S82. In contrast, natural selection might have little influence in shaping the codon usage patterns in C. difficile ZJCDC-S82.
Conclusion
In this article, we initially sequenced the genome of C. difficile strain ZJCDC-S82 isolated from mainland China using the PacBio RS and Ion Torrent Proton™ sequencing platforms. The C. difficile strain ZJCDC-S82 was one of the dominant types (PCR ribotype 012) isolated from C. difficile-positive stool specimens in Zhejiang province. The C. difficile ZJCDC-S82 draft genome consisted of a nuclear genome of ~4.19 Mb and a plasmid of 11,930 bps containing 4,013 and 19 CDSs, respectively. The ML trees of 11 C. difficile strains suggested that the genetic diversity of C. difficile strains was not influenced by geographic location. Evolutionary analysis confirmed that the number of positively selected genes was very low for this C. difficile strain. The ENc-plot analysis showed that mutation bias was the predominant factor influencing the usage bias of synonymous codons. Our results provided extensive genomic information in relation to a C. difficile strain from mainland China. This study will aid our understanding of global genomic diversity associated with C. difficile.
Supplementary Materials
Supplementary Table 1. Genomics information of the 11 C. difficile strains used in this study.
Supplementary Table 2. Statistics of sequencing information.
Supplementary Table 3. Statistics of the genomic assembly of C. difficile strain ZJCDC-S82.
Supplementary Table 4. The predicted subsystem features in the genomes of C. difficile strains ZJCDC-S82, M120, and CD630.
Supplementary Table 5. Unique genes of C. difficile strain ZJCDC-S82.
Supplementary Figure 1. Reads from PacBio platform length distribution.
Supplementary Figure 2. Reads from Proton platform length distribution.
Acknowledgments
We would like to thank the sequencing and informatics teams of the NJDG Bioinformatics Department for their technical assistance.
Footnotes
ACADEMIC EDITOR: Jike Cui, Associate Editor
PEER REVIEW: Four peer reviewers contributed to the peer review report. Reviewers’ reports totaled 2133 words, excluding any confidential comments to the academic editor.
FUNDING: This work was supported by the National High-Tech Research and Development Program of China (863 Program; 2014AA021402), the Program for Major Science and Technology Medicine and Health Care in Zhejiang (WKJ-ZJ-1507), the Program for the National Natural Science Foundation of China (81471998), and Provincal Major Science and Technology Special Projects of Zhejiang (2013C03045-1). The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE). Provenance: the authors were invited to submit this paper.
Author Contributions
Constructed and sequenced the genomic libraries: YL, CH. Isolated the C. difficile ZJCDC-S82 strain and extracted genomic DNA: JY. Conducted the contig assembly, the associated analysis, data deposition, and interpretation: WF, WG, ZC, HL, XW. Conceived and designed the study: DJ, YL, XW. Drafted the manuscript: YL, CH, DJ. All the authors read and approved the final manuscript.
REFERENCES
- 1.Mackenzie JS, Jeggo M, Daszak PS, et al. One Health: The Human-Animal-Environment Interfaces in Emerging Infectious Diseases. Heidelberg: Springer; 2013. [Google Scholar]
- 2.Rupnik M, Wilcox MH, Gerding DN. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol. 2009;7(7):526–36. doi: 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- 3.Bartlett JG. Historical perspectives on studies of Clostridium difficile and C. difficile infection. Clin Infect Dis. 2008;46(suppl 1):S4–11. doi: 10.1086/521865. [DOI] [PubMed] [Google Scholar]
- 4.Bartlett JG. Clostridium difficile: history of its role as an enteric pathogen and the current state of knowledge about the organism. Clin Infect Dis. 1994;18(suppl 4):S265–72. doi: 10.1093/clinids/18.supplement_4.s265. [DOI] [PubMed] [Google Scholar]
- 5.Kato H, Ito Y, van den Berg RJ, et al. First isolation of Clostridium difficile 027 in Japan. Euro Surveill. 2007;12(1) doi: 10.2807/esw.12.02.03110-en. E070111070113. [DOI] [PubMed] [Google Scholar]
- 6.Gilca R, Fortin E, Rocher I, et al. Surveillance des diarrhées associées à Clostridium difficile au Québec: bilan du 16 août 2009 au 5 décembre 2009. Institut National de Santé Publique du Québec; Québec, Canada: http://www.inspq.qc.ca/pdf/publications/745_Cdifficile_bilan2004-2007.pdf. [Google Scholar]
- 7.Kuijper E, Coignard B, Tüll P. Emergence of Clostridium difficile-associated disease in North America and Europe. Clin Microbiol Infect. 2006;12(s6):2–18. doi: 10.1111/j.1469-0691.2006.01580.x. [DOI] [PubMed] [Google Scholar]
- 8.Loo VG, Poirier L, Miller MA, et al. A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality. N Engl J Med. 2005;353(23):2442–9. doi: 10.1056/NEJMoa051639. [DOI] [PubMed] [Google Scholar]
- 9.Warny M, Pepin J, Fang A, et al. Toxin production by an emerging strain of Clostridium difficile associated with outbreaks of severe disease in North America and Europe. Lancet. 2005;366(9491):1079–84. doi: 10.1016/S0140-6736(05)67420-X. [DOI] [PubMed] [Google Scholar]
- 10.Collins DA, Hawkey PM, Riley TV. Epidemiology of Clostridium difficile infection in Asia. Antimicrob Resist Infect Control. 2013;2(1):21. doi: 10.1186/2047-2994-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y-B, Gu S-L, Wei Z-Q, et al. Molecular epidemiology of Clostridium difficile in a tertiary hospital of China. J Med Microbiol. 2014;63(4):562–9. doi: 10.1099/jmm.0.068668-0. [DOI] [PubMed] [Google Scholar]
- 12.Yan Q, Zhang J, Chen C, et al. Multilocus sequence typing (MLST) analysis of 104 Clostridium difficile strains isolated from China. Epidemiol Infect. 2013;141(1):195–9. doi: 10.1017/S0950268812000453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang H, Wu S, Wang M, et al. Clostridium difficile infections in a Shanghai hospital: antimicrobial resistance, toxin profiles and ribotypes. Int J Antimicrob Agents. 2009;33(4):339–42. doi: 10.1016/j.ijantimicag.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 14.Stabler RA, He M, Dawson L, et al. Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol. 2009;10(9):R102. doi: 10.1186/gb-2009-10-9-r102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dingle KE, Elliott B, Robinson E, et al. Evolutionary history of the Clostridium difficile pathogenicity locus. Genome Biol Evol. 2014;6(1):36–52. doi: 10.1093/gbe/evt204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eyre DW, Cule ML, Wilson DJ, et al. Diverse sources of C. difficile infection identified on whole-genome sequencing. N Engl J Med. 2013;369(13):1195–205. doi: 10.1056/NEJMoa1216064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mac Aogáin M, Moloney G, Kilkenny S, et al. Whole-genome sequencing improves discrimination of relapse from reinfection and identifies transmission events among patients with recurrent Clostridium difficile infections. J Hosp Infect. 2015;90(2):108–16. doi: 10.1016/j.jhin.2015.01.021. [DOI] [PubMed] [Google Scholar]
- 18.Sebaihia M, Wren BW, Mullany P, et al. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet. 2006;38(7):779–86. doi: 10.1038/ng1830. [DOI] [PubMed] [Google Scholar]
- 19.Pettit LJ, Browne HP, Yu L, et al. Functional genomics reveals that Clostridium difficile Spo0A coordinates sporulation, virulence and metabolism. BMC Genomics. 2014;15(1):160. doi: 10.1186/1471-2164-15-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kurka H, Ehrenreich A, Ludwig W, et al. Sequence similarity of Clostridium difficile strains by analysis of conserved genes and genome content is reflected by their ribotype affiliation. PLoS One. 2014;9(1):e86535. doi: 10.1371/journal.pone.0086535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He M, Sebaihia M, Lawley TD, et al. Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc Natl Acad Sci U S A. 2010;107(16):7527–32. doi: 10.1073/pnas.0914322107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He M, Miyajima F, Roberts P, et al. Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nat Genet. 2013;45(1):109–13. doi: 10.1038/ng.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Darling AE, Worden P, Chapman TA, et al. The genome of Clostridium difficile 5.3. Gut Pathog. 2014;6(1):4. doi: 10.1186/1757-4749-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forgetta V, Oughton MT, Marquis P, et al. Fourteen-genome comparison identifies DNA markers for severe-disease-associated strains of Clostridium difficile. J Clin Microbiol. 2011;49(6):2230–8. doi: 10.1128/JCM.00391-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McDonald LC, Killgore GE, Thompson A, et al. An epidemic, toxin gene–variant strain of Clostridium difficile. N Engl J Med. 2005;353(23):2433–41. doi: 10.1056/NEJMoa051590. [DOI] [PubMed] [Google Scholar]
- 26.Indra A, Huhulescu S, Schneeweis M, et al. Characterization of Clostridium difficile isolates using capillary gel electrophoresis-based PCR ribotyping. J Med Microbiol. 2008;57(pt 11):1377–82. doi: 10.1099/jmm.0.47714-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Griffiths D, Fawley W, Kachrimanidou M, et al. Multilocus sequence typing of Clostridium difficile. J Clin Microbiol. 2010;48(3):770–8. doi: 10.1128/JCM.01796-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chin C-S, Alexander DH, Marks P, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10(6):563–9. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 29.Miller JR, Koren S, Sutton G. Assembly algorithms for next-generation sequencing data. Genomics. 2010;95(6):315–27. doi: 10.1016/j.ygeno.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soueidan H, Maurier F, Groppi A, et al. Finishing bacterial genome assemblies with Mix. BMC Bioinformatics. 2013;14(suppl 15):S16. doi: 10.1186/1471-2105-14-S15-S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rissman AI, Mau B, Biehl BS, et al. Reordering contigs of draft genomes using the Mauve aligner. Bioinformatics. 2009;25(16):2071–3. doi: 10.1093/bioinformatics/btp356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aziz RK, Bartels D, Best AA, et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9(1):75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Overbeek R, Olson R, Pusch GD, et al. The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST) Nucleic Acids Res. 2014;42(D1):D206–14. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brettin T, Davis JJ, Disz T, et al. RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci Rep. 2015;5:8365. doi: 10.1038/srep08365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Delcher AL, Harmon D, Kasif S, et al. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999;27(23):4636–41. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):0955–64. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tamura K, Stecher G, Peterson D, et al. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kent WJ. BLAT – the BLAST-like alignment tool. Genome Res. 2002;12(4):656–64. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–91. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 40.Yang Z, Wong WS, Nielsen R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005;22(4):1107–18. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
- 41.harp PM, Li W-H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J Mol Evol. 1986;24:1–2. 28–38. doi: 10.1007/BF02099948. [DOI] [PubMed] [Google Scholar]
- 42.Wright F. The ‘effective number of codons’ used in a gene. Gene. 1990;87(1):23–9. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- 43.Peden JF. PhD thesis. Nottingham: University of Nottingham; 1999. Analysis of codon usage. [Google Scholar]
- 44.Clabots C, Lee S, Gerding D, et al. Clostridium difficile plasmid isolation as an epidemiologic tool. Eur J Clin Microbiol Infect Dis. 1988;7(2):312–5. doi: 10.1007/BF01963112. [DOI] [PubMed] [Google Scholar]
- 45.Clabots C, Peterson L, Gerding D. Characterization of a nosocomial Clostridium difficile outbreak by using plasmid profile typing and clindamycin susceptibility testing. J Infect Dis. 1988;158(4):731–6. doi: 10.1093/infdis/158.4.731. [DOI] [PubMed] [Google Scholar]
- 46.Muldrow L, Archibold E, Nunez-Montiel O, et al. Survey of the extrachromosomal gene pool of Clostridium difficile. J Clin Microbiol. 1982;16(4):637–40. doi: 10.1128/jcm.16.4.637-640.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peixoto L, Fernandez V, Musto H. The effect of expression levels on codon usage in Plasmodium falciparum. Parasitology. 2004;128(03):245–51. doi: 10.1017/s0031182003004517. [DOI] [PubMed] [Google Scholar]
- 48.Jia X, Liu S, Zheng H, et al. Non-uniqueness of factors constraint on the codon usage in Bombyx mori. BMC Genomics. 2015;16(1):356. doi: 10.1186/s12864-015-1596-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sterky F, Bhalerao RR, Unneberg P, et al. A Populus EST resource for plant functional genomics. Proc Natl Acad Sci U S A. 2004;101(38):13951–6. doi: 10.1073/pnas.0401641101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gonzalez-Ibeas D, Blanca J, Roig C, et al. MELOGEN: an EST database for melon functional genomics. BMC Genomics. 2007;8(1):306. doi: 10.1186/1471-2164-8-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Janvilisri T, Scaria J, Thompson AD, et al. Microarray identification of Clostridium difficile core components and divergent regions associated with host origin. J Bacteriol. 2009;191(12):3881–91. doi: 10.1128/JB.00222-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scaria J, Ponnala L, Janvilisri T, et al. Analysis of ultra low genome conservation in Clostridium difficile. PLoS One. 2010;5(12):e15147. doi: 10.1371/journal.pone.0015147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stabler R, Gerding D, Songer J, et al. Comparative phylogenomics of Clostridium difficile reveals clade specificity and microevolution of hypervirulent strains. J Bacteriol. 2006;188(20):7297–305. doi: 10.1128/JB.00664-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saul A, Battistutta D. Codon usage in Plasmodium falciparum. Mol Biochem Parasitol. 1988;27(1):35–42. doi: 10.1016/0166-6851(88)90022-9. [DOI] [PubMed] [Google Scholar]
- 55.Milhon JL, Tracy JW. Updated codon usage in Schistosoma. Exp Parasitol. 1995;80(2):353–6. doi: 10.1006/expr.1995.1046. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Genomics information of the 11 C. difficile strains used in this study.
Supplementary Table 2. Statistics of sequencing information.
Supplementary Table 3. Statistics of the genomic assembly of C. difficile strain ZJCDC-S82.
Supplementary Table 4. The predicted subsystem features in the genomes of C. difficile strains ZJCDC-S82, M120, and CD630.
Supplementary Table 5. Unique genes of C. difficile strain ZJCDC-S82.
Supplementary Figure 1. Reads from PacBio platform length distribution.
Supplementary Figure 2. Reads from Proton platform length distribution.