

Abstract
Purpose of review
Extensive data indicate a role for reactive oxygen species (ROS) and redox signaling in vascular damage in hypertension. However, molecular mechanisms underlying these processes remain unclear, but oxidative post-translational modification of vascular proteins is critical. This review discusses how proteins are oxidatively modified and how redox signaling influences vascular smooth muscle cell growth and vascular remodeling in hypertension. We also highlight Nox5 as a novel vascular ROS-generating oxidase.
Recent findings
Oxidative stress in hypertension leads to oxidative imbalance that affects vascular cell function through redox signaling. Many Nox isoforms produce ROS in the vascular wall, and recent findings show that Nox5 may be important in humans. ROS regulate signaling by numerous processes including cysteine oxidative post-translational modification such as S-nitrosylation, S-glutathionylation and sulfydration. In vascular smooth muscle cells, this influences cellular responses to oxidative stimuli promoting changes from a contractile to a proliferative phenotype.
Summary
In hypertension, Nox-induced ROS production is increased, leading to perturbed redox signaling through oxidative modifications of vascular proteins. This influences mitogenic signaling and cell cycle regulation, leading to altered cell growth and vascular remodeling in hypertension.
Keywords: NADPH oxidase, oxidative post-translational modification, reactive oxygen species, vascular smooth muscle cells
INTRODUCTION
Reactive oxygen species (ROS) are produced in all cell types of the vasculature, including endothelial cells, smooth muscle cells, adventitial fibroblasts and perivascular adipocytes. In the cardiovascular system, the major ROS are superoxide
hydrogen peroxide (H2O2) and hydroxyl anion (OH-). Reactive nitrogen species, including nitric oxide (NO) and peroxynitrite (ONOO-), are also biologically important oxidants [1–3]. ROS regulate many cellular processes in the vasculature such as cell growth, contraction/dilation, migration, differentiation and cytoskeletal organization, important in maintaining vascular tone and integrity [4,5▪▪]. In stressed or pathological conditions, ROS-generating enzymes, such as nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (Nox), are activated leading to increased bioavailability of ROS (termed oxidative stress). When produced in excess, ROS interact with lipids and proteins leading to functional and structural changes of target molecules. This perturbation in oxidative balance promotes post-translational oxidative modification of lipids and proteins and impaired redox signaling [6,7]. Post-translational oxidative modification involves covalent changes of cysteine residues within redox-sensitive proteins, important processes that regulate protein structure and function. Redox-sensitive signaling is involved in endothelial dysfunction, arterial remodeling and vascular inflammation associated with hypertension and seems to play an important role in cardiovascular complications and target organ damage [8,9]. The present review discusses the role of ROS in vascular remodeling in hypertension and focuses on cellular and molecular mechanisms underlying ROS actions. We specifically highlight the potential role of the novel ROS-generating Nox isoform, Nox5 in the vasculature,; processes of oxidative post-translational modification and impact on redox signaling and vascular remodeling in hypertension and; targeting Nox/ROS as a potential therapeutic strategy.
Box 1.
no caption available
CHARACTERISTICS OF REACTIVE OXYGEN SPECIES IN BIOLOGICAL SYSTEMS
Most ROS and reactive nitrogen species are relatively unstable oxygen-centered or nitrogen-centered-free radicals, which contain unpaired electrons. Superoxide is water soluble, unstable and short-lived, whereas H2O2 is lipid soluble and more stable than
[10]. H2O2 is produced mainly from dismutation of
by superoxide dismutases and is scavenged by catalase and glutathione peroxide [11]. NO is enzymatically formed by nitric oxide synthase, which oxidizes L-arginine. NO acts as a second messenger with vasodilatory, anti-inflammatory and antiproliferative actions. In the presence of excess
NO is converted into the injurious oxidant, peroxynitrite. ROS are highly reactive and therefore they have short half-lives in biological systems. This makes it very challenging to measure ROS directly and accordingly most assays are indirect estimates of ROS abundance and reactivity.
PRODUCTION OF REACTIVE OXYGEN SPECIES IN THE VASCULAR WALL
All cell types of the vascular wall and endothelium have the capacity to produce ROS. Physiologically, ROS are generated in a regulated manner at low concentrations and function as signaling molecules modulating vascular function and structure. In pathophysiological conditions, increased activity/expression of ROS-generating enzymes, and/or decreased activation of antioxidant systems in the vasculature result in increased ROS bioavailability leading to oxidative stress.
ROS are produced as by-products by enzymatic reactions such as those catalyzed by cytochrome P450 enzymes, xanthine oxidase, uncoupling of eNOS, mitochondrial oxidases and glucose oxidase [12–14]. The Nox enzymes are ‘professional oxidases’, and have as their primary function, the production of ROS. They are the major enzymatic source of ROS in the vascular wall. NADPH oxidases are multisubunit enzymes that generate
by the one electron reduction of oxygen using NADPH as the electron donor
[15,16]. The major catalytic subunit is Nox (of which there are seven isoforms, Nox1-5, Duox1 and Duox2), which together with p22phox, are membrane-bound subunits. The cytosolic regulatory subunits include p40phox, p47phox and p67phox or homologues NoxO1 and NoxA1. Noxs are differentially regulated by the subunits and not all Noxs require subunits for activation. In human vascular cells, the major Nox isoforms are Nox1, Nox4 and Nox5. Nox2, the prototype Nox, is found primarily in phagocytic cells and may also be present in the vascular wall in pathological conditions, usually localized in macrophages and invading monocytes. Unlike phagocytic Nox, which is activated only upon stimulation and generates
in a burst-like manner extracellularly, vascular Nox may be constitutively active, preassembled and produces
intracellularly in a slow and sustained manner [16].
Vascular Nox is activated by many prohypertensive factors, including vasoactive agents, growth factors, cytokines, shear stress and mechanical forces [17,18]. Among these, angiotensin II (Ang II), through AT1 receptors, appears to be particularly important. Acute stimulation of vascular cells with Ang II causes increased Nox-derived ROS generation leading to activation of redox signaling pathways. Nox expression and activation are increased in cultured endothelial and vascular smooth muscle cells and in whole vessels in experimental and human hypertension [19–21]. Nox hyperactivation leads to excessive ROS generation that disrupts redox networks, normally regulated by antioxidant systems, resulting in oxidative stress, triggering molecular processes, which in the vasculature, contributes to vascular injury.
The biological significance of multiple Nox isoforms being expressed in the vascular wall is unclear, but their differential tissue distribution, cellular localization and subcellular compartmentalization probably play a major role in Nox-specific actions [22]. For example, whereas Ang II-activated Nox1 appears to be important in vascular smooth muscle cells from large arteries, especially in association with atherosclerosis, Nox4 and Nox5 may be more important in small resistance arteries, especially in humans [23,24]. Moreover, different Nox isoforms may generate different ROS. For example, Nox1, Nox2 and Nox5 generate
whereas Nox4 produces primarily H2O2 and may act as a vasodilator in some vascular beds. Nox isoforms in the cardiovascular system have recently been comprehensively reviewed [25–28] and only the most recent of the Nox family members, Nox5, is briefly discussed here.
VASCULAR NOX5
Nox5 (five splice variants: α, β, δ, γ, ε) is the most recently identified Nox and is unique: it is Ca2+-sensitive, it possesses a calmodulin-like domain with binding sites for Ca2+, the binding of which induces a conformational change leading to enhanced ROS formation, and it does not require any NADPH oxidase subunits for its activity [29–31,32▪▪,33–36]. Nox5 phosphorylation of serine/threonine (Ser475, Ser 490, Thr494 and Ser 498) enhances sensitivity to Ca2+ and facilitates ROS production at lower levels of Ca2+ [31]. Nox5 phosphorylation is regulated by protein kinase C (PKC), specifically PKCα. Nox5 is also regulated by other kinases, including c-Abl, Ca2+/calmodulin-dependent protein kinase II and MAP kinases [32▪▪,33–35]. Protein:protein interactions with the molecular chaperones Hsp90 and Hsp70 further regulate Nox5 expression and activity [35]. Nox5 was originally discovered in testes, spleen and lymph nodes, but more recently has been found in the vasculature, heart and kidney. In vascular cells, Nox5α and Nox5β are the major ROS-generating isoforms and are activated by thrombin, platelet derived growth factor, Ang II and endothelin-1. Although all Noxs are present in mice, rats and humans, the rodent genome does not contain the nox5 gene, making it challenging to study Nox5 in the experimental setting. The biological significance of vascular Nox5 is still unknown, although it has been implicated in cell proliferation, angiogenesis and migration [37–39]. In porcine aortic cells, Nox5-derived ROS is required for growth factor-induced potassium intermediate/small conductance calcium-activated channel, subfamily N, member 4 [40], important in vascular smooth muscle cell proliferation and migration in atherogenesis [40]. In pathological conditions, such as atherosclerosis, acute myocardial infarction, aneurysm and hypertension, vascular Nox5 expression is increased, implying a role for Nox5 in cardiovascular disease [41,42,43▪▪]. Macrophage and monocyte Nox5 have been shown to play a role in ROS production in atherosclerosis [44]. We recently demonstrated that in mice expressing human Nox5 in a podocyte-specific manner, renal function is markedly impaired and blood pressure is elevated [45], further suggesting a role for Nox5 in disease processes [45].
REDOX SIGNALING IN VASCULAR CELLS
ROS are important signaling molecules in vascular smooth muscle cells. In hypertension, perturbations in ROS signaling are associated with endothelial dysfunction, impaired vascular tone and arterial remodeling [46]. These processes are mediated by changes in redox state of ion channels (K+ channels and Ca2+ channels), cyclases (guanylate cyclase), kinases [mitogen-activated protein kinases (MAPK), Rho kinases and tyrosine kinases], phosphatases (protein tyrosine phosphatases), cytoskeletal proteins (actin and myosin) and activation of transcription factors [activator protein-1 (AP-1), nuclear factor κB (NFκB), and nuclear factor erythroid 2-related factor 2] [47–49].
The specific signaling effects of ROS are mediated by the covalent modification of specific cysteine residues in redox-sensitive proteins. These residues have unique features in that they contain a terminal thiol (-SH) functional group, which is electron-rich enabling different oxidation states, including S-nitrosylation (S-nitration; SNO), S-glutathionylation (RS-SG), sulfhydration (SSH), disulphide bonds (RS-SR’), sulfenylation (SOH), sulfinic acid (SO2H) and sulfonic acid (SO3H) [50]. Oxidative post-translational modifications influence target protein structure and function. The multiple types of oxidative post-translational modifications of myriad proteins translate into diverse cellular effects, which in vascular cells range from contraction to growth.
OXIDATIVE POST-TRANSLATIONAL MODIFICATION OF PROTEINS IN THE CARDIOVASCULAR SYSTEM
S-Nitrosylation
SNO of proteins in the cardiovascular system has been associated with protective effects. This has been demonstrated during ischemic preconditioning and has been attributed to SNO of ATP synthase at Cys294 [51]. The post-translational modification of Cys294 is altered in heart failure, and accordingly this Cys294 in the ATP synthase has been described as a redox switch [51]. In experimental models of hypertension, increased SNO modification of proteins was associated with impaired aortic relaxation [52]. A comprehensive dataset for modified SNO proteins in cardiac, endothelial and vascular smooth muscle cells has been compiled [50,53]. SNO-modified proteins are localized in many subcellular compartments and organelles and are involved in numerous cellular functions, especially those related to cell metabolism and cytoskeletal organization.
Sulfydration
H2S is produced by cystathionine γ-lyase and has been identified as a vasodilator. Mice lacking cystathionine are hypertensive [54]. H2S can modify downstream proteins by sulfydration (SSH), which is an important Cys modification. H2S produced in response to endoplasmic reticulum (ER) stress sulfydrates protein tyrosine phosphatase 1B (PTP1B), and possibly other protein tyrosine phosphatases [55]. Sulfydration of PTP1B reduces its activity leading to decreased PTP1B-induced dephosphorylation of protein kinases, such as protein kinase-like ER kinase and MAPK, key proteins that regulate vascular smooth muscle cell function [55].
S-glutathionylation
Another form of oxidized post-translational modification of proteins is S-glutathionylation (RS-SG), which is reversible [56,57]. It is formed by a reaction of glutathione (oxidized form, GSSG) or S-nitrosoglutathione (GSNO) with free thiol. Many proteins in cardiovascular cells are able to undergo S-glutathionylation, which when exposed to an oxidative milieu, such as in hypertension and aging, is increased [56,57]. S-glutathionylation of endothelial NOS (eNOS) has recently been identified as an important novel mechanism of eNOS regulation, processes that are altered in cardiovascular disease [58,59].
Sulfenylation
Sulfenylation has been associated with injurious oxidative damage. Sulfenic acid is very reactive, unstable and hence is short-lived. Sulfenic acid can be converted into other oxidized post-translational modifications, which may be reversible or irreversible [49].
OXIDATION OF CYTOSKELETAL PROTEINS IN VASCULAR SMOOTH MUSCLE CELLS
Of the many proteins that undergo oxidative changes is actin, a major protein involved in cytoskeletal organization in vascular cells [60]. Actin is highly redox-sensitive and when cells are exposed to oxidative stress actin is among the most prominent proteins to become oxidized [60]. Stimulation of vascular cells with H2O2 causes cytoskeletal disorganization and morphological changes. In the heart, actin oxidation is associated with impaired contractility [60,61]. In vascular smooth muscle cells, interleukin-22 stimulation leads to carbonylation of actin, as well as α-enolase, heat shock cognate 71kDa protein and mitochondrial 60kDa heat shock protein, proteins associated with cell stress and growth [62].
Growing evidence indicates that oxidized proteins may function collaboratively with proteins that undergo other post-translational modifications such as phosphorylation, acetylation and ubiquitination [49]. This networking between proteins impacts on downstream signaling that determines the final biological cellular response, such as cell growth.
REDOX REGULATION OF VASCULAR CELL GROWTH: IMPLICATIONS IN HYPERTENSION
Vascular smooth muscle cells are intrinsically contractile in nature and exhibit very low rates of proliferation. However, in pathological conditions associated with vascular injury, such as in hypertension, vascular smooth muscle cells proliferate, undergo hypertrophy, dedifferentiate and migrate [63]. These processes are regulated in large part through ROS which influence redox-sensitive mitogenic signaling and cell cycle progression through oxidative post-translational modification of proteins, including protein tyrosine phosphatases, protein kinases, cytoskeletal proteins and transcription factors [63–65] (Fig. 1).
FIGURE 1.
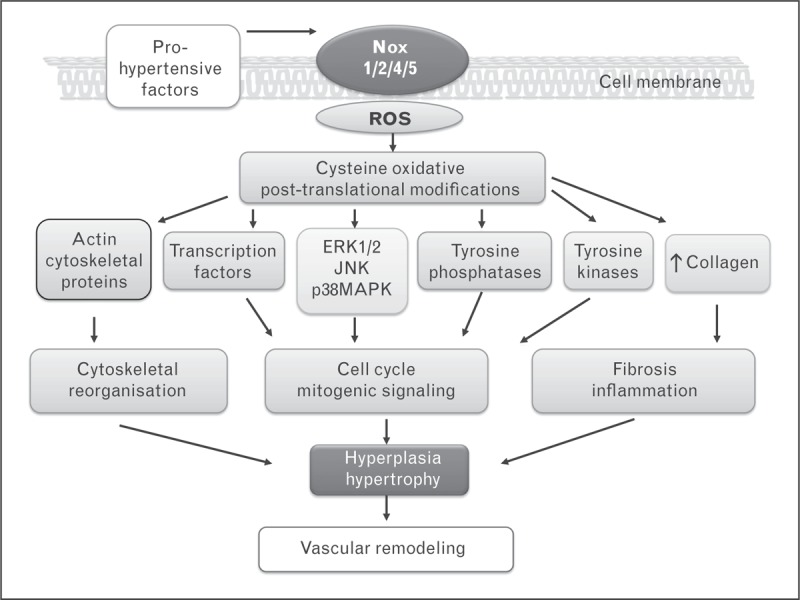
Diagram demonstrating mechanisms whereby increased Nox-induced ROS generation regulates redox signaling and vascular smooth muscle cell function in hypertension. Prohypertensive factors, such as activation of the renin angiotensin system, growth factors, sympathetic nervous system activation and proinflammatory mediators, induce activation of Nox isoforms, leading to increased ROS production. Changes in intracellular redox status cause oxidative modification of proteins important in proliferative and profibrotic signaling. Increased vascular smooth muscle cell growth, cytoskeletal reorganization and fibrosis contribute to vascular remodeling in hypertension. ROS, reactive oxygen species.
Despite both
and H2O2 inducing vascular smooth muscle cell proliferation, their growth signaling pathways are different. Whereas
primarily activates extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase, H2O2 increases expression of p38 mitogen-activated protein kinase [66–68]. Mitogenic signaling pathways in vascular smooth muscle cells that are sensitive to oxidant levels also directly influence expression and activity of cyclins and cyclin-dependent kinases, which regulate cell cycle progression [69]. Cdk 4, Cdk 6, Cdk 2 and Cdk 1 are activated by binding with their regulatory subunits cyclin D1, cyclin E, cyclin A and cyclin B, respectively. The kinase activities of cyclin/Cdk complexes are negatively regulated by Cdk inhibitor proteins p21, p27, p16 and p15.
In hypertension, vascular smooth muscle cells are stimulated to divide in response to mitogenic factors. ROS induces variable growth-related responses, including increased proliferation, apoptosis, transient cell cycle arrest, permanent senescence and cell death, depending mainly on the relative dose of exposure. Mitogenic stimulation initiates re-entry into the cell cycle. They exit the G1 phase and enter the S phase. Cyclin D1/Cdk 4, cyclin E/Cdk 2 and cyclin A/Cdk 2 complexes hyperphosphorylate the pRb in the late G1 phase, which then triggers entry into S phase [69,70]. Cyclin D1 controls the G0−G1 transition and is highly sensitive to oxidative stress. It is the only cyclin that can drive terminally differentiated cells back into the cell cycle. Transcription of cyclin D1 is upregulated by growth factors, including Ang II, through ERK1/2 and downstream redox-sensitive transcription factors, such as AP-1 and NFκB, which are highly redox-sensitive [70]. These effects are modulated by microRNA-365 [71].
Oxidants also regulate G1, S and G2 phases of the cell cycle. Peroxides induce a G1 checkpoint response that is attenuated by antioxidants. H2O2 suppresses S phase entry by inhibiting cyclin E/Cdk2 activity, by upregulating p21 and p53 and through downregulation of cyclin A expression [72].
REACTIVE OXYGEN SPECIES AND VASCULAR REMODELING IN HYPERTENSION
ROS-induced change of vascular smooth muscle cells to a proliferative phenotype contributes to vascular hypertrophy and remodeling in hypertension, characterized by reduced vascular lumen, increased media thickness, increased stiffness and reduced distensibility [73,74]. At the molecular and cellular levels, remodeling involves changes in cytoskeletal organization, altered growth/apoptosis, senescence and rearrangement of vascular smooth muscle cells, processes that are highly sensitive to alterations in the intracellular redox milieu. Remodeling is also influenced by changes in extracellular matrix protein composition and reorganization of proteoglycans, collagens and fibronectin [74]. Targeting ROS to reduce vascular smooth muscle cell proliferation, prevent vascular cell dedifferentiation and inhibit fibrosis may be an interesting approach to ameliorate arterial remodeling in hypertension [47,75–77].
TARGETING REACTIVE OXYGEN SPECIES AS A THERAPEUTIC STRATEGY IN CARDIOVASCULAR DISEASE
Considering the important role of oxidative damage associated with vascular injury, strategies to reduce ROS bioavailability should have vasoprotective effects in cardiovascular disease. Two major approaches to reduce oxidative stress have been explored including antioxidants to scavenge ROS, and Nox inhibitors to block ROS generation.
Antioxidants
Data from experimental studies have demonstrated that antioxidants improve endothelial function, promote regression of vascular remodeling and reduce blood pressure in hypertension [78,79]. These phenomena are associated with decreased MAP kinase signaling, decreased activation of transcription factors and reduced vascular smooth muscle cell proliferation, inflammation and fibrosis [78–82]. However, clinical findings have been inconsistent and results from antioxidant clinical trials have mainly been negative, showing little cardiovascular benefit of antioxidant vitamins and carotene [83–86]. Possible reasons for these disappointing results from antioxidant trials have been discussed in detail elsewhere [83–86], but a number of points should be highlighted: patients who entered into the trials already had long-standing cardiovascular disease wherein irreversible oxidative damage may have already occurred and hence scavenging of ROS already formed may have little benefit; inappropriate antioxidants may have been used, as antioxidant vitamins themselves can act as oxidants thereby promoting oxidative stress; antioxidant-dosing regimens and duration of therapy may have been suboptimal to effectively scavenge ROS in vivo; orally administered antioxidants may be inaccessible to the source of free radicals, particularly if ROS are generated in intracellular compartments and organelles. This may be especially pertinent to water soluble vitamins, such as vitamin C, which may not cross the cell membrane to scavenge intracellular ROS and; antioxidants do not inhibit the production of ROS; they scavenge free radicals once they are formed. Finally, in none of the large antioxidant clinical trials was it ever proven that patients did indeed have evidence of oxidative stress.
Nox inhibitors
Theoretically, compounds that block ROS generation to reduce an oxidative load should be more efficacious than nonspecific antioxidant ROS scavengers. This is based on experimental evidence in which it has been shown that inhibition of Nox-mediated ROS generation, using pharmacological and gene-targeted strategies, leads to regression of vascular remodeling, improved endothelial function and lowering of blood pressure [87,88]. A number of pharmacological agents have now been developed as NADPH oxidase/Nox inhibitors, including nonspecific and isoform-specific compounds.
Classical NADPH oxidase inhibitors, apocynin and diphenyleneiodinium (DPI), are nonspecific and may act as ROS scavengers. Apocynin has intrinsic antioxidant activity and DPI acts as a general flavoprotein inhibitor [76,77,89]. Hence, because of the nonspecific nature of these agents, they should not be used as selective Nox inhibitors.
Recently, new isoform-specific Nox inhibitors have been characterized from rational drug delivery. These include the small molecule inhibitors GKT137831 and GKT136901 (GenKyoTex), which variably inhibit Nox1, Nox4 and Nox5; 2-actylphenothiazine (ML171) (Scripps Research Institute), which inhibits Nox1; VAS2870 and VAS3947 (Vasopharm GmbH), which inhibit mainly Nox2 and to a lesser extent Nox4; S17834 (Servier), which inhibits Nox2 and Nox4; and Fulvene-5, which inhibits Nox2 and Nox4 [75–77]. Biological peptidic inhibitors of NADPH oxidase have been developed by the Pagano group [90▪▪,91], including NOX2ds-tat, which prevents p47phox binding to Nox2, thereby preventing assembly of the active Nox2 oxidase, and NOXA1ds, a Nox1 inhibitor, which inhibits binding of NOXA1 to NOX1.
Of the Nox inhibitors that have been registered in the patent literature [76,77,89,90▪▪], only one has progressed through to clinical trials, specifically GKT137831, which has entered into a phase 2 trial in diabetic nephropathy (www.genkyotex.com). Clinical outcomes of this trial should shed light on the role of Nox as a therapeutic target in oxidative stress-related diseases. Further clinical studies are still needed to confirm the clinical utility of Nox inhibitors, but these drugs may hold some promise in patients with Nox/ROS-associated diseases.
CONCLUSION
In hypertension, dysregulation of ROS-generating enzymes, including the novel NADPH oxidase, Nox5, results in oxidative stress, which contributes to vascular damage, through multiple processes including activation of transcription factors, stimulation of mitogenic signaling pathways and modulation of cell cycle progression. Fundamental to these phenomena is cysteine oxidative post-translational modification of vascular proteins, which determines final cellular responses to oxidative stimuli. The field of oxidative proteomics [92▪▪] in the vascular system is still immature and the vascular oxidative proteome in hypertension has yet to be elucidated, but increasing evidence indicates that oxidation of vital cytoskeletal proteins, kinases and phosphatases in vascular smooth muscle cells is critical in phenotype switches in pathological conditions. Although inconclusive at present, strategies to regulate ROS bioavailability by decreasing production and/or by increasing radical scavenging may regress vascular remodeling, prevent vascular damage and reduce hypertension and its associated end-organ damage. Targeting oxidative stress with novel Nox inhibitors and other ROS modulators may be an attractive therapeutic strategy to ameliorate endothelial dysfunction and vascular damage in hypertension and associated diseases. Outcomes of current clinical studies evaluating cardiovascular and renal effects of Nox inhibitors will shed light on the clinical utility of this approach and will also inform on the role of Nox/ROS in human disease.
Acknowledgements
None.
Financial support and sponsorship
Studies performed by the authors were supported by grants 57786 and 44018 from the Canadian Institutes of Health Research and by grants from the British Heart Foundation (29762, 30099).
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES
- 1. Brown DI, Griendling KK. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ Res 2015; 116:531–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Montezano AC, Touyz RM. Reactive oxygen species, vascular Noxs, and hypertension: focus on translational and clinical research. Antioxid Redox Signal 2014; 20:164–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Al Ghouleh I, Khoo NK, Knaus UG, et al. Oxidases and peroxidases in cardiovascular and lung disease: new concepts in reactive oxygen species signalling. Free Radic Biol Med 2011; 51:1271–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Salabei JK, Hill BG. Autophagic regulation of smooth muscle cell biology. Redox Biol 2015; 4:97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5▪▪. MacKay CE, Knock GA. Control of vascular smooth muscle function by Src-family kinases and reactive oxygen species in health and disease. J Physiol 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]; An excellent overview linking molecular mechanism whereby ROS and redox signaling network with the Src family kinases.
- 6. Lee SE, Park YS. Role of lipid peroxidation-derived α, β-unsaturated aldehydes in vascular dysfunction. Oxid Med Cell Longev 2013; 2013:629028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sinha N, Dabla PK. Oxidative stress and antioxidants in hypertension: a current review. Curr Hypertens Rev 2015; May 29. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 8. Lee MY, Griendling KK. Redox signaling, vascular function, and hypertension. Antioxid Redox Signal 2008; 10:1045–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fridovich I. Superoxide anion radical, superoxide dismutases, and related matters. J Biol Chem 1997; 272:18515–18517. [DOI] [PubMed] [Google Scholar]
- 10. Fukai T, Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal 2011; 15:1583–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cantu-Medellin N, Kelley EE. Xanthine oxidoreductase-catalyzed reactive species generation: a process in critical need of reevaluation. Redox Biol 2013; 1:353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Montezano AC, Touyz RM. Reactive oxygen species and endothelial function: role of nitric oxide synthase uncoupling and Nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic Clin Pharmacol Toxicol 2012; 110:87–94. [DOI] [PubMed] [Google Scholar]
- 13. Dikalov SI, Ungvari Z. Role of mitochondrial oxidative stress in hypertension. Am J Physiol Heart Circ Physiol 2013; 305:H1417–H1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Laurindo FR, Araujo TL, Abrahão TB. Nox NADPH oxidases and the endoplasmic reticulum. Antioxid Redox Signal 2014; 20:2755–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brandes RP, Weissmann N, Schröder K. Nox family NADPH oxidases in mechano-transduction: mechanisms and consequences. Antioxid Redox Signal 2014; 20:887–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Montezano AC, Nguyen Dinh Cat A, Rios FJ, Touyz RM. Angiotensin II and vascular injury. Curr Hypertens Rep 2014; 16:431. [DOI] [PubMed] [Google Scholar]
- 17. Raaz U, Toh R, Maegdefessel L, et al. Hemodynamic regulation of reactive oxygen species: implications for vascular diseases. Antioxid Redox Signal 2014; 20:914–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Briones AM, Tabet F, Callera GE, et al. Differential regulation of Nox1, Nox2 and Nox4 in vascular smooth muscle cells from WKY and SHR. J Am Soc Hypertens 2011; 5:137–153. [DOI] [PubMed] [Google Scholar]
- 19. Konior A, Schramm A, Czesnikiewicz-Guzik M, Guzik TJ. NADPH oxidases in vascular pathology. Antioxid Redox Signal 2014; 20:2794–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Montezano AC, Dulak-Lis M, Tsiropoulou S, et al. Oxidative stress and human hypertension: vascular mechanisms, biomarkers, and novel therapies. Can J Cardiol 2015; 31:631–641. [DOI] [PubMed] [Google Scholar]
- 21. Kaludercic N, Deshwal S, Di Lisa F. Reactive oxygen species and redox compartmentalization. Front Physiol 2014; 5:285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barman SA, Chen F, Su Y, et al. NADPH oxidase 4 is expressed in pulmonary artery adventitia and contributes to hypertensive vascular remodeling. Arterioscler Thromb Vasc Biol 2014; 34:1704–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Montezano AC, Burger D, Paravicini TM, et al. Nicotinamide adenine dinucleotide phosphate reduced oxidase 5 (Nox5) regulation by angiotensin II and endothelin-1 is mediated via calcium/calmodulin-dependent, rac-1-independent pathways in human endothelial cells. Circ Res 2010; 106:1363–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Montezano AC, Touyz RM. Oxidative stress, Noxs, and hypertension: experimental evidence and clinical controversies. Ann Med 2012; 44:S2–S16. [DOI] [PubMed] [Google Scholar]
- 25. Rivera J, Sobey CG, Walduck AK, Drummond GR. Nox isoforms in vascular pathophysiology: insights from transgenic and knockout mouse models. Redox Rep 2010; 15:50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. San Martin A, Griendling KK. NADPH oxidases: progress and opportunities. Antioxid Redox Signal 2014; 20:2692–2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Santos CX, Nabeebaccus AA, Shah AM, et al. Endoplasmic reticulum stress and Nox-mediated reactive oxygen species signaling in the peripheral vasculature: potential role in hypertension. Antioxid Redox Signal 2014; 20:121–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pandey D, Fulton DJ. Molecular regulation of NADPH oxidase 5 via the MAPK pathway. Am J Physiol Heart Circ Physiol 2011; 300:H1336–H1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Qian J, Chen F, Kovalenkov Y, et al. Nitric oxide reduces NADPH oxidase 5 (Nox5) activity by reversible S-nitrosylation. Free Radic Biol Med 2012; 52:1806–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hahn NE, Meischl C, Kawahara T, et al. NOX5 expression is increased in intramyocardial blood vessels and cardiomyocytes after acute myocardial infarction in humans. Am J Pathol 2012; 180:2222–2229. [DOI] [PubMed] [Google Scholar]
- 31. Guzik TJ, Chen W, Gongora MC, et al. Calcium dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J Am Coll Cardiol 2008; 52:1803–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32▪▪. Chen F, Yu Y, Haigh S, et al. Regulation of NADPH oxidase 5 by protein kinase C isoforms. Plos One 2014; 9:e88405. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study advances the field of Nox5 biology by demonstrating mechanisms whereby kinases, specifically PKC, regulate activity of Nox5.
- 33. El Jamali A, Valente AJ, Lechleiter JD, et al. Novel redox-dependent regulation of NOX5 by the tyrosine kinase c-Abl. Free Radic Biol Med 2008; 44:868–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen F, Pandey D, Chadli A, et al. Hsp90 regulates NADPH oxidase activity and is necessary for superoxide but not hydrogen peroxide production. Antioxid Redox Signal 2011; 14:2107–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bedard K, Jaquet V, Krause KH. NOX5: from basic biology to signaling and disease. Free Radic Biol Med 2012; 52:725–734. [DOI] [PubMed] [Google Scholar]
- 36. Serrander L, Jaquet V, Bedard K, et al. NOX5 is expressed at the plasma membrane and generates superoxide in response to protein kinase C activation. Biochimie 2007; 89:1159–1167. [DOI] [PubMed] [Google Scholar]
- 37. Greco TM, Hodara R, Parastatidis I, et al. Identification of S-nitrosylation motifs by site specific mapping of the S-nitrosocysteine proteome in human vascular smooth muscle cells. Proc Natl Acad Sci USA 2006; 103:7420–7425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pandey D, Patel A, Patel V, et al. Expression and functional significance of NADPH oxidase 5 (Nox5) and its splice variants in human blood vessels. Am J Physiol Heart Circ Physiol 2012; 302:H1919–H1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gole1 Hope K A, Tharp1 Darla L, Bowles Douglas K. Upregulation of intermediate-conductance Ca2+- activated K+ channels (KCNN4) in porcine coronary smooth muscle requires NADPH oxidase 5 (NOX5). Plos One 2014; 9:e105337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fulton DJ. Nox5 and the regulation of cellular function. Antioxid Redox Signal 2009; 11:2443–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yu P, Han W, Villar VA, et al. Unique role of NADPH oxidase 5 in oxidative stress in human renal proximal tubule cells. Redox Biol 2014; 2:570–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Manea A, Manea SA, Gan AM, et al. Human monocytes and macrophages express NADPH oxidase 5; a potential source of reactive oxygen species in atherosclerosis. Biochem Biophys Res Commun 2015; 461:172–179. [DOI] [PubMed] [Google Scholar]
- 43▪▪. Holterman CE, Thibodeau JF, Towaij C, et al. Nephropathy and elevated BP in mice with podocyte-specific NADPH oxidase 5 expression. J Am Soc Nephrol 2014; 25:784–797. [DOI] [PMC free article] [PubMed] [Google Scholar]; The first study to show that renal (podocyte) Nox5 plays an important role in renal dysfunction and hypertension. This study was performed in mice expressing human Nox5 in a podocyte-specific manner.
- 44. Madamanchi NR, Runge MS. Redox signaling in cardiovascular health and disease. Free Radic Biol Med 2013; 61:473–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chatterjee S, Fujiwara K, Perez NG, et al. Mechanosignaling in the vasculature: emerging concepts in sensing, transduction and physiological responses. Am J Physiol Heart Circ Physiol 2015; 308: (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Thomas SR, Witting PK, Drummond GR. Redox control of endothelial function and dysfunction: molecular mechanisms and therapeutic opportunities. Antioxid Redox Signal 2008; 10:1713–1765. [DOI] [PubMed] [Google Scholar]
- 47. Androwiki AC, Camargo Lde L, Sartoretto S, et al. Protein disulfide isomerase expression increases in resistance arteries during hypertension development. Effects on Nox1 NADPH oxidase signaling. Front Chem 2015; 3:24–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chung HS, Wang S-B, Venkatraman V, et al. Cysteine oxidative posttranslational modifications emerging regulation in the cardiovascular system. Circ Res 2013; 112:382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang SB, Foster DB, Rucker J, et al. Redox regulation of mitochondrial ATP synthase: implications for cardiac resynchronization therapy. Circ Res 2011; 109:750–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Choi H, Allahdadi KJ, Tostes RC, Webb RC. Augmented S-nitrosylation contributes to impaired relaxation in angiotensin II hypertensive mouse aorta: role of thioredoxinreductase. J Hypertens 2011; 29:2359–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang G, Wu L, Jiang B, et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science 2008; 322:587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Murray CI, Kane LA, Uhrigshardt H, et al. Site mapping of in vitro S-nitrosation in cardiac mitochondria: implications for cardioprotection. Mol Cell Proteomics 2011; 10: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang X, Ling S, Zhao D, et al. Redox regulation of actin by thioredoxin-1 is mediated by the interaction of the proteins via cysteine 62. Antioxid Redox Signal 2010; 13:565–573. [DOI] [PubMed] [Google Scholar]
- 54. Krishnan N, Fu C, Pappin DJ, Tonks NK. H2S-induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci Signal 2011; 4:ra86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dalle-Donne I, Giustarini D, Colombo R, et al. S-glutathionylation in human platelets by a thiol-disulfide exchange independent mechanism. Free Radic Biol Med 2005; 38:1501–1510. [DOI] [PubMed] [Google Scholar]
- 56. Oelze M, Kröller-Schön S, Steven S, et al. Glutathione peroxidase-1 deficiency potentiates dysregulatory modifications of endothelial nitric oxide synthase and vascular dysfunction in aging. Hypertension 2014; 63:390–396. [DOI] [PubMed] [Google Scholar]
- 57. Zweier JL, Chen CA, Druhan LJ. S-glutathionylation reshapes our understanding of endothelial nitric oxide synthase uncoupling and nitric oxide/reactive oxygen species-mediated signaling. Antioxid Redox Signal 2011; 14:1769–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mieyal JJ, Gallogly MM, Qanungo S, et al. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid Redox Signal 2008; 10:1941–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dalle-Donne I, Rossi R, Milzani A, et al. The actin cytoskeleton response to oxidants: from small heat shock protein phosphorylation to changes in the redox state of actin itself. Free Radic Biol Med 2001; 31:1624–1632. [DOI] [PubMed] [Google Scholar]
- 60. Canton M, Menazza S, Sheeran FL, et al. Oxidation of myofibrillar proteins in human heart failure. J Am Coll Cardiol 2011; 57:300–309. [DOI] [PubMed] [Google Scholar]
- 61. Bansal G, Das D, Hsieh CY, et al. IL-22 activates oxidant signaling in pulmonary vascular smooth muscle cells. Cell Signal 2013; 25:2727–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Savoia C, Burger D, Nishigaki N, et al. Angiotensin II and the vascular phenotype in hypertension. Expert Rev Mol Med 2011; 13:e11. [DOI] [PubMed] [Google Scholar]
- 63. Rao GN, Berk BC. Active oxygen species stimulate vascular smooth muscle cell growth and proto-oncogene expression. Circ Res 1992; 70:593–599. [DOI] [PubMed] [Google Scholar]
- 64. Tabet F, Schiffrin EL, Touyz RM. Mitogen-activated protein kinase activation by hydrogen peroxide is mediated through tyrosine kinase-dependent, protein kinase C-independent pathways in vascular smooth muscle cells: upregulation in spontaneously hypertensive rats. J Hypertens 2005; 23:2005–2012. [DOI] [PubMed] [Google Scholar]
- 65. Knock GA, Ward JP. Redox regulation of protein kinases as a modulator of vascular function. Antioxid Redox Signal 2011; 15:1531–1547. [DOI] [PubMed] [Google Scholar]
- 66. Touyz RM, Yao G, Viel E, et al. Angiotensin II and endothelin-1 regulate MAP kinases through different redox-dependent mechanisms in human vascular smooth muscle cells. J Hypertens 2004; 22:1141–1149. [DOI] [PubMed] [Google Scholar]
- 67. Touyz RM, Cruzado M, Tabet F, et al. Redox-dependent MAP kinase signaling by Ang II in vascular smooth muscle cells: role of receptor tyrosine kinase transactivation. Can J Physiol Pharmacol 2003; 81:159–167. [DOI] [PubMed] [Google Scholar]
- 68. Zhang X, Liu L, Chen C, et al. Interferon regulatory factor-1 together with reactive oxygen species promotes the acceleration of cell cycle progression by up-regulating the cyclin E and CDK2 genes during high glucose-induced proliferation of vascular smooth muscle cells. Cardiovasc Diabetol 2013; 12:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kim TH, Oh S, Kim SS. Recombinant human prothrombin kringle-2 induces bovine capillary endothelial cell cycle arrest at G0-G1 phase through inhibition of cyclin D1/CDK4 complex: modulation of reactive oxygen species generation and up-regulation of cyclin-dependent kinase inhibitors. Angiogenesis 2005; 8:307–314. [DOI] [PubMed] [Google Scholar]
- 70. Kim MH, Ham O, Lee SY, et al. MicroRNA-365 inhibits the proliferation of vascular smooth muscle cells by targeting cyclin D1. J Cell Biochem 2014; 115:1752–1761. [DOI] [PubMed] [Google Scholar]
- 71. Barnouin K, Dubuisson ML, Child ES, et al. H2O2 induces a transient multiphase cell cycle arrest in mouse fibroblasts through modulating cyclin D and p21Cip1 expression. J Biol Chem 2002; 277:13761–13770. [DOI] [PubMed] [Google Scholar]
- 72. Rizzoni D, Agabiti Rosei E. Small artery remodeling in hypertension and diabetes. Curr Hypertens Rep 2006; 8:90–95. [DOI] [PubMed] [Google Scholar]
- 73. Schiffrin EL. Remodeling of resistance arteries in essential hypertension and effects of antihypertensive treatment. Am J Hypertens 2004; 17 (12 Pt 1):1192–1200. [DOI] [PubMed] [Google Scholar]
- 74. Liu B, Luo XJ, Yang ZB, et al. Inhibition of NOX/VPO1 pathway and inflammatory reaction by trimethoxystilbene in prevention of cardiovascular remodeling in hypoxia-induced pulmonary hypertensive rats. J Cardiovasc Pharmacol 2014; 63:567–576. [DOI] [PubMed] [Google Scholar]
- 75. Altenhöfer S, Radermacher KA, Kleikers PW, et al. Evolution of NADPH oxidase inhibitors: selectivity and mechanisms for target engagement. Antioxid Redox Signal 2014; Feb 26. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Drummond GR, Sobey CG. Endothelial NADPH oxidases: which NOX to target in vascular disease? Trends Endocrinol Metab 2014; 25:452–463. [DOI] [PubMed] [Google Scholar]
- 77. Sorce S, Krause KH, Jaquet V. Targeting NOX enzymes in the central nervous system: therapeutic opportunities. Cell Mol Life Sci 2012; 69:2387–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sato Y, Ikeda M, Ito T, et al. Ascorbic acid levels and neutrophil superoxide production in blood of pre, early and late hypertensive stroke-prone spontaneously hypertensive rats. Clin Exp Hypertens 2011; 33:397–403. [DOI] [PubMed] [Google Scholar]
- 79. Chen X, Touyz RM, Park JB, Schiffrin EL. Antioxidant effects of vitamins C and E are associated with altered activation of vascular NADPH oxidase and superoxide dismutase in stroke-prone SHR. Hypertension 2001; 38 (3 Pt 2):606–611. [DOI] [PubMed] [Google Scholar]
- 80. Scioli MG, Bielli A, Agostinelli S, et al. Antioxidant treatment prevents serum deprivation- and TNF-α-induced endothelial dysfunction through the inhibition of NADPH oxidase 4 and the restoration of β-oxidation. J Vasc Res 2014; 51:327–337.Kyaw M. [DOI] [PubMed] [Google Scholar]
- 81. Yoshizumi M, Tsuchiya K, Izawa Y, et al. Atheroprotective effects of antioxidants through inhibition of mitogen-activated protein kinases. Acta Pharmacol Sin 2004; 25:977–985. [PubMed] [Google Scholar]
- 82. Ashor AW, Siervo M, Lara J, et al. Effect of vitamin C and vitamin E supplementation on endothelial function: a systematic review and meta-analysis of randomised controlled trials. Br J Nutr 2015; 113:1182–1194. [DOI] [PubMed] [Google Scholar]
- 83. Desai CK, Huang J, Lokhandwala A, et al. Antioxidants, inflammation and cardiovascular disease. World J Cardiol 2014; 6:462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Tousoulis D, Psaltopoulou T, Androulakis E, et al. Oxidative stress and early atherosclerosis: novel antioxidant treatment. Cardiovasc Drugs Ther 2015; 29:75–88. [DOI] [PubMed] [Google Scholar]
- 85. Mangge H, Becker K, Fuchs D, Gostner JM. The role of vitamin supplementation in the prevention of cardiovascular disease events. Clin Cardiol 2014; 37:576–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Paganini-Hill A, Kawas CH, Corrada MM. Antioxidant vitamin intake and mortality: the Leisure World Cohort Study. Am J Epidemiol 2015; 181:120–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Bruder-Nascimento T, Callera GE, Montezano AC, et al. Vascular injury in diabetic db/db mice is ameliorated by atorvastatin: role of Rac1/2-sensitive Nox-dependent pathways. Clin Sci (Lond) 2015; 128:411–423. [DOI] [PubMed] [Google Scholar]
- 88. Sabuhi R, Asghar M, Hussain T. Inhibition of NAD(P)H oxidase potentiates AT2 receptor agonist-induced natriuresis in Sprague-Dawley rats. Am J Physiol Renal Physiol 2010; 299:F815–F820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Heumüller S, Wind S, Barbosa-Sicard E, et al. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension 2008; 51:211–217. [DOI] [PubMed] [Google Scholar]
- 90▪▪. Cifuentes-Pagano E, Meijles DN, Pagano PJ. The quest for selective Nox inhibitors and therapeutics: challenges, triumphs and pitfalls. Antioxid Redox Signal 2014; 20:2741–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]; A comprehensive update and critical review of Nox inhibitors, with a discussion about novel approaches to inhibit Nox isoforms.
- 91. Cifuentes-Pagano E, Csanyi G, Pagano PJ. NADPH oxidase inhibitors: a decade of discovery from Nox2ds to HTS. Cell Mol Life Sci 2012; 69:2315–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92▪▪. Go YM, Chandler JD, Jones DP. The cysteine proteome. Free Radic Biol Med 2015; 84:227–245. [DOI] [PMC free article] [PubMed] [Google Scholar]; An outstanding and comprehensive overview of oxidative modifications of proteins.