

Abstract
About 10% of inherited diseases are caused by nonsense mutations [Trends Mol Med 18 (2012) 688], and nonsense suppression drug therapy promoting translation through premature stop codons is an emerging therapeutic approach. Infantile neuronal ceroid lipofuscinosis (INCL), a childhood neurodegenerative disease, results from mutations in the CLN1 gene encoding the lysosomal enzyme, palmitoyl‐protein thioesterase 1 (PPT1) [Biochim Biophys Acta 1832 (2013) 1806, Hum Mutat (2012) 63, Biochim Biophys Acta 1832 (2013) 1881]. The nonsense mutation p.R151X is the most common disease‐causing CLN1 mutation Hum Mutat (2012) 63. In the novel Cln1 R151X mouse model of INCL, we found large, tissue‐specific variations in Cln1 R151X mRNA level and PPT1 residual enzyme activity. These tissue‐specific differences strongly influenced the read‐through efficiency of ataluren (PTC124), a well‐known nonsense suppression drug. A two‐day treatment with ataluren (10 mg/kg) increased PPT1 enzyme activity in the liver and muscle, but not in any other tissue examined. Our study identifies a new challenge/hurdle for read‐through drug therapy: variable efficiency of read‐through therapy in the different tissues/organs because of tissue‐specific variations in nonsense mutant transcript levels.
Keywords: infantile neuronal ceroid lipofuscinosis, Cln1R151X, nonsense mutation, read‐through drug, ataluren
Introduction
Nonsense mutations result in premature stop codons and are frequent causes of genetic diseases 1. mRNA transcripts containing premature stop codons are recognised and degraded by the nonsense‐mediated decay pathway to prevent the production and accumulation of truncated, non‐functional proteins within the cell 2. The so‐called read‐through drugs promote translation through premature stop codons, resulting full‐length proteins, which may be fully or partly functional, depending on the amino acid substitution for the stop codon. The first safe, specific and effective read‐through drug, PTC124, also known as ataluren, was described in a Nature Letter in 2007 3. The therapeutic effects of ataluren have been demonstrated in nonsense mutant mouse models of Duchene muscular dystrophy and cystic fibrosis 3, 4. Ataluren is an FDA‐approved investigational new drug, and it is currently in phase III clinical trials for cystic fibrosis (https://clinicaltrials.gov/ct2/show/NCT02139306) and for Duchenne muscular dystrophy (https://clinicaltrials.gov/ct2/show/NCT01826487).
Mutations in the CLN1 gene cause a rare fatal childhood neurodegenerative disorder, infantile neuronal ceroid lipofuscinosis (INCL), also known as infantile Batten disease 5, 6. CLN1 encodes a lysosomal enzyme, palmitoyl‐protein thioesterase 1 (PPT1), and the most frequent disease‐causing CLN1 mutation is the p.R151X nonsense mutation 6, 7. Previously, we have shown that ataluren (PTC124) significantly elevates PPT1 enzyme activity in a patient‐derived INCL lymphoblast cell line, harbouring the p.R151X mutation in CLN1 8. To test if read‐through drugs represent a potential treatment for INCL, we have generated the Cln1 R151X nonsense mutant mouse model of the disease. Cln1 R151X mice show a dramatic decrease in Cln1 mRNA level and CLN1 (PPT1) enzyme activity in the liver and brain, and display characteristic neuropathological and neurological features of the human disease 9. Treating Cln1 R151X mice with ataluren multiple times per day dose‐dependently increased PPT1 protein level and enzyme activity in the liver and brain 9.
In genetic diseases caused by nonsense mutations usually multiple organs/tissues are affected, and it is assumed that for a particular disease read‐through drug therapy will provide comparable beneficial effects in various target organs/tissues. Tissue‐specific variations in the level of nonsense mutation‐containing transcripts, however, may strongly influence the efficacy of read‐through drugs. Supporting this notion, up to twofold differences in nonsense‐mediated mRNA decay efficiency in various murine tissues have been shown 10.
Materials and methods
Animals
Cln1 R151X mice were maintained on a mixed 129S6/SvEv x C57BL/6J genetic background, and hybrid 129S6/SvEv x C57BL/6J mice served as WT controls. Mice were housed in individually vented microisolator cages (4–5 mice/cage) with ad libitum access to food and water. Mice were fed with the Teklad Global 2918 diet (Harlan Laboratories, Indianapolis, IN, USA), and their drinking water was tap water. All procedures were carried out according to the guidelines of the Animal Welfare Act and NIH policies, and were approved by the Sanford Research Animal Care and Use Committee.
Sample processing and handling
For sample collection, mice were anesthetised followed by transcardial perfusion and vascular rinse using ice‐cold PBS. All tissue samples were collected and processed in a similar manner, and stored at −80°C no longer than 2 months before total RNA extraction or total protein isolation.
Nucleic acid extraction
Total RNA was extracted from all tissue samples with a Maxwell 16 LEV simplyRNA Cells Kit (Promega, Madison, WI, USA) using a Maxwell 16 Instrument (Promega), according to the manufacturer's instructions. Sample purities and yields were determined using a Nanodrop Spectrophotometer (Thermo Fischer Scientific, Waltham, MA, USA). All samples had A260/A280 values between 2.03 and 2.20. RNA integrity was assessed as previously described 8.
Reverse transcription
All samples, except muscle, were mass normalised using approximately 800 ng of total RNA for cDNA synthesis using a High Capacity cDNA Reverse Transcription Kit (Life Technologies, Carlsbad, CA, USA) according to the manufacturer's instructions in a 96‐well plate. Muscle samples were mass normalized using approximately 400 ng of total RNA. The reaction conditions were as follows: 25°C for 10 min., 37°C for 120 min., 85°C for 5 min. Samples were diluted with molecular grade water to 10 ng/μl following reverse transcription. Samples were assessed for DNA contamination using reactions with no reverse transcriptase added. All samples were DNA‐free and stored at −20°C until use.
Quantitative real‐time polymerase chain reaction
Quantitative real‐time polymerase chain reaction (qPCR) was performed for the target gene using TaqMan hydrolysis assays (Life Technologies) for Cln1 (Cat.# Mm00477078_m1). Quantitative real‐time polymerase chain reaction was performed for reference genes using TaqMan hydrolysis assays (Life Technologies) for Gapdh (Part.# Mm99999915_g1), Hgprt (Part.# 01318741_g1), B2m (Part.# Mm00437762_m1) and Gusb (Part.# Mm00446962_g1). Amplification was performed with 20 ng of cDNA in 10 μl reaction volumes for four technical replicates using Absolute Blue qPCR mix (Thermo Fischer Scientific) in 384 well plates (Roche Diagnostics, Indianapolis, IN, USA). Thermal cycling and fluorescence data collection were performed on a LightCycler 480 (Roche Diagnostics) using the following reaction conditions: 95°C for 15 min., followed by 40 cycles at 95°C for 15 sec., 60°C for 1 min.
qPCR data analysis
Raw fluorescence data were analysed as previously described 8 using REST‐MCS software 11, 12.
Protein isolation
Approximately 25–50 mg of each tissue sample was homogenised using an Ultra‐Turrax T8 homogenizer with a Dispergierstation T8.10 stand (Thermo Fischer Scientific) in 400 μl lysis buffer containing 50 mM Tris‐HCl, pH 7.4, 150 mM NaCl, 0.2% Triton X‐100 and 300 mM NP‐40, supplemented with 1:100 protease inhibitor cocktail and 1:1000 Phenylmethylsulfonyl fluoride (PMSF). This was followed by pulse sonication and 30 min. incubation on ice. Following a high‐speed centrifugation (12,000 × g at +4°C) for 10 min., the supernatant was collected and the total protein concentration was measured using the Pierce 660 nm Protein Assay (Thermo Fischer Scientific).
PPT1 enzyme assay
Palmitoyl‐protein thioesterase 1 enzyme activity was measured in a fluorogenic enzyme assay as previously described 8, 9, 13, 14, 15, 16. Total protein samples isolated from Cln1 WT/WT and Cln1 R151X/R151X tissues were used. Ten micrograms of total protein per well (in a black 96‐well plate) was incubated in a mixture containing 0.64 mM of the fluorogenic PPT1 substrate, 4‐methylumbelliferyl‐6‐thiopalmitoyl‐β‐glucoside (MU‐6S‐palm‐βGlc; Moscerdam Substrates, Oegstgeest, the Netherlands), 15 mM dithiothreitol (DTT) in 5.1 mg/ml bovine serum albumin and 0.02% Na‐azide, 0.375% Triton X‐100 in 2/1 chloroform/methanol, 0.1 U β‐glucosidase from almonds (Sigma–Aldrich, St. Louis, MO, USA) in double‐concentrated McIlvains phosphate/citric‐acid buffer, pH 4.0. The total protein reaction mixture was then incubated for 1 hr at 37°C. The reaction was stopped with the addition of 0.5 M NaHCO3/0.5 M Na2CO3 buffer, pH 10.7 with 0.025% Triton X‐100 and fluorescence was measured using a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA, USA). Relative enzyme activity was estimated using total fluorescence minus background.
Ataluren treatment
Cln1 R151X male mice were randomly assigned to either a treatment group or vehicle control group. Eight mice were treated with 10 mg/kg ataluren (Selleck Chemicals, Houston, TX, USA) dissolved in PBS containing DMSO (2%) and (2‐hydroxypropyl)‐β‐cyclodextrin (22%) (Cat.# H5784; Sigma–Aldrich) via intraperitoneal (i.p.) injections four times daily for two consecutive days. Eight control mice were treated with the vehicle of the drug. Immediately following the last injection on the second day, tissues were collected and stored at −80°C for further use.
Statistical analysis
The normality of all data sets was assessed using the Kolmogorov–Smirnov test and the Shapiro–Wilk test. All data were found to be normally distributed. Separate independent t‐tests were used to compare each tissue between control and ataluren‐treated mice. Levene's test for equality of variances was used to determine if variances of data sets being compared were equal. In instances where variances were found not to be similar (Levene's test statistic <0.05), unequal variances were assumed. All statistical analyses were done using IBM SPSS statistical software version 21 (IBM Corporation, Armonk, NY, USA) or GraphPad Prism software version 5.04 (GraphPad Software, Inc., La Jolla, CA, USA).
Results
To evaluate whether tissue‐specific differences in nonsense‐mediated decay affect the relative expression of Cln1 mRNA and PPT1 enzyme activity in the Cln1 R151X nonsense mutant mouse model of INCL, we compared multiple tissue types to each other. Cln1 mRNA expression varied widely among tissues collected from the central nervous system and periphery, with the testes showing the highest level of Cln1 expression (39% of wild‐type) and the kidney showing the lowest level of Cln1 expression (~4% of wild‐type) (Fig. 1). Residual PPT1 enzyme activity, measured in a fluorogenic enzyme assay, also varied widely among tissues, with the muscle showing the highest level of PPT1 activity (7% of wild‐type) and the cerebral cortex showing the lowest level of PPT1 activity (0.1% of wild‐type) (Fig. 2). Interestingly, the brain, which is primarily affected in INCL, showed the lowest level of PPT1 activity overall. However, there was no significant correlation between Cln1 mRNA expression and PPT1 enzyme activity in the tissues examined. This indicates that in addition to the strong effect of nonsense‐mediated mRNA decay on Cln1 mRNA abundance, multiple layers of regulation, at both the mRNA and protein levels, affect the tissue‐specific expression of enzymatically active PPT1.
Figure 1.
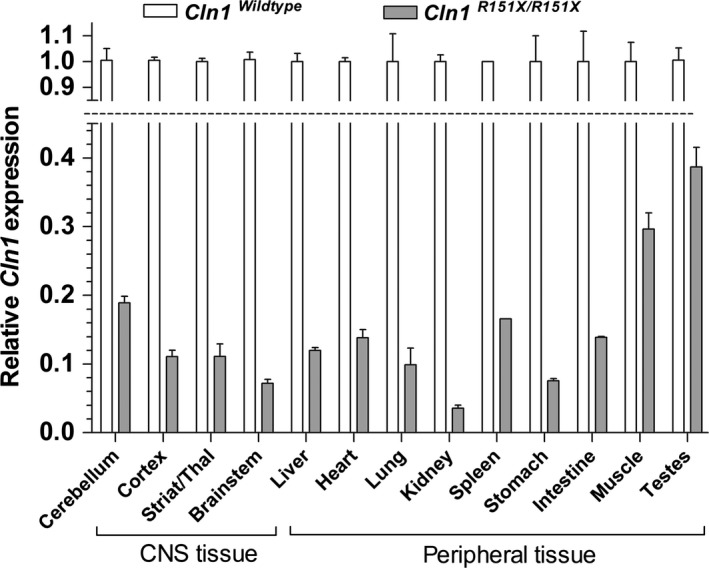
Tissue‐specific variations of Cln1 mRNA level in Cln1 R151X mice. Endogenous Cln1 mRNA levels were measured by quantitative real‐time PCR in the brain (cerebellum, cortex, striatum/thalamus, brainstem) and peripheral tissues (liver, heart, lung, kidney, spleen, stomach, intestine, muscle, testes) from 5‐month‐old wild‐type (white bars) and Cln1 R151X (grey bars) male mice. The relative expression of Cln1 was normalized with four reference genes (B2m, Gapdh, Gusb, Hgprt). Columns and bars represent mean ± S.E.M. (three wild‐type and three Cln1 R151X mice; four technical replicates were used for each mouse).
Figure 2.
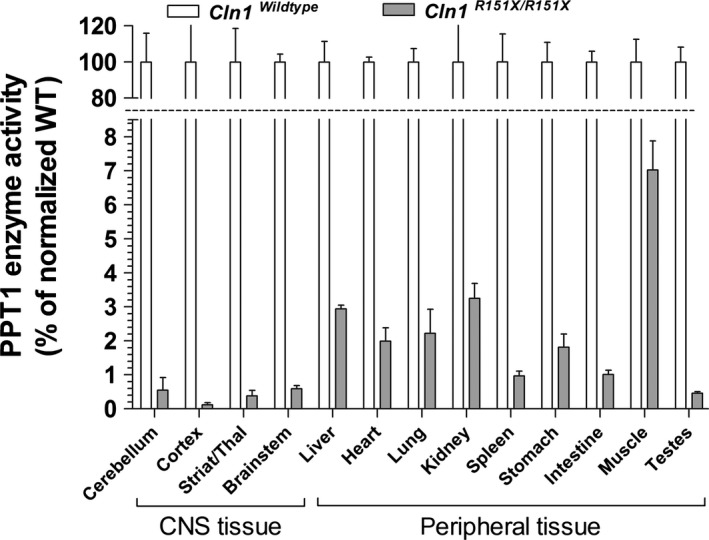
Tissue‐specific variations of PPT1 enzyme activity in Cln1 R151X mice. Endogenous PPT1 enzyme activity was measured in the brain (cerebellum, cortex, striatum/thalamus, brainstem) and peripheral tissues (liver, heart, lung, kidney, spleen, stomach, intestine, muscle, testes) from 5‐month‐old wild‐type (white bars) and Cln1 R151X (grey bars) male mice. The brain in particular has low levels of PPT1 activity compared to almost all peripheral tissues tested. Columns and bars represent mean ± S.E.M. (three wild‐type and three Cln1 R151X mice; four technical replicates were used for each mouse).
After finding tissue‐specific variations in Cln1 mRNA level and PPT1 enzyme activity, we treated Cln1 R151X mice with the read‐through drug, ataluren (10 mg/kg), injecting the drug intraperitoneally four times a day for two consecutive days. Immediately following the last injection on the second day, tissues were collected, and PPT1 enzyme activity was measured. This proof‐of‐concept trial was performed to identify tissue‐specific variations in the efficacy of read‐through therapy in the Cln1 R151X mouse model of INCL. Ataluren treatment did not result in any significant increase in PPT1 enzyme activity in the different brain regions examined (cerebellum, cortex, striatum/thalamus and brainstem) (Fig. 3). Among the five peripheral tissues analysed from ataluren‐treated Cln1 R151X mice, only the liver and skeletal muscle showed a slight but statistically significant increase in PPT1 enzyme activity as compared to the same tissues from control, vehicle‐treated mice (Fig. 3). The liver of ataluren‐treated and control (vehicle‐treated) Cln1 R151X mice had 5.46% and 4.48% PPT1 activity, respectively (unpaired t‐test, P = 0.004). Skeletal muscle from ataluren‐treated Cln1 R151X mice showed 8.72% PPT1 activity compared to 7.59% in the control group (unpaired t‐test, P = 0.009). Ataluren treatment did not increase PPT1 enzyme activity in the heart, lung and kidney (Fig. 3). Although the ~1% increase in PPT1 enzyme activity in the liver and muscle after a short‐term, 2‐day treatment, seems negligible, it has previously been demonstrated that restoring PPT1 activity to only 4–5% of the normal level results in significant therapeutic benefits 17.
Figure 3.
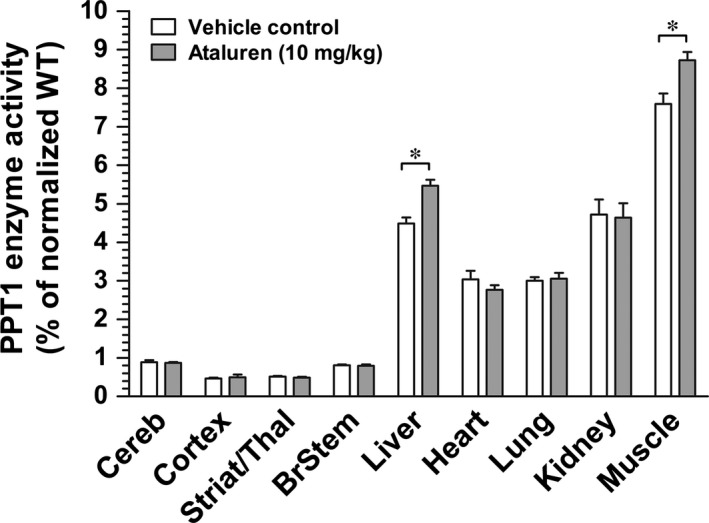
Tissue‐dependent effectiveness of the read‐through drug, ataluren (PTC124), in Cln1 R151X mice. Two‐month old Cln1 R151X male mice were treated with ataluren (10 mg/kg, i.p.) four times a day for 2 days. Control mice were treated with the vehicle of the drug. Immediately following the last injection on the second day, tissues were collected, and PPT1 enzyme activity was measured in the brain (cerebellum, cortex, striatum/thalamus, brainstem) and peripheral tissues (liver, heart, lung, kidney, skeletal muscle). Ataluren only increased PPT1 activity in the liver and muscle. Columns and bars represent mean ± S.E.M. (eight wild‐type and eight Cln1 R151X mice). Statistical significance was determined by unpaired t‐test.: *p=0.004 for liver, *p=0.009 for skeletal muscle.
Discussion
Although nonsense‐mediated decay is an important process by which cells degrade mRNA transcripts containing premature stop codons to prevent the catastrophic damage of truncated proteins, very little is currently known about tissue‐specific nonsense‐mediated decay and how this could affect genetic disease pathology and therapeutic outcome 2. In the Cln1 R151X nonsense mutant mouse model of INCL, we found large, tissue‐specific variations in Cln1 R151X mRNA level, indicating tissue‐specific differences in nonsense‐mediated decay. Tissue‐specific variations in Cln1 gene expression, however, might also contribute to the observed differences. Based on what is known about the nonsense‐mediated decay pathway and the degradation of truncated proteins within the cell, it is reasonable to conclude that if larger pools of Cln1 R151X mRNA transcript are present, the more PPT1 enzyme activity will be measured because more transcripts are available for natural read‐through. This hypothesis is not supported, however, when the tissue‐specific Cln1 mRNA levels and PPT1 enzyme activities are compared. Although skeletal muscle has both the highest levels of Cln1 mRNA expression and PPT1 enzyme activity, kidney has the lowest Cln1 mRNA level, yet the second highest level of PPT1 enzyme activity. Complete analysis of Cln1 mRNA level as it relates to PPT1 enzyme activity in the same tissue yielded no correlation. The lack of correlation could be because of tissue‐specific differences in the level of Cln1 mRNA synthesis and rate of PPT1 protein turnover. Since premature stop codons frequently induce alternative splicing 18, 19, another possible explanation is that the Cln1 R151X mutation‐induced altered splicing is different in the various tissues. The known tissue‐specific differences in the unfolded protein response 20 in combination with the recently identified negative regulation of unfolded protein response by NMD 21 could also contribute to the observed lack of correlation between Cln1 mRNA level and PPT1 enzyme activity in tissues of Cln1 R151X mice.
Treatment of Cln1 R151X mice with the read‐through drug, ataluren (10 mg/kg), resulted in a slight but statistically significant increase in PPT1 enzyme activity in the liver and muscle only. Although the ~1% increase in PPT1 enzyme activity in the liver and muscle after a short‐term, 2‐day treatment, seems negligible, it has previously been demonstrated that restoring PPT1 activity to only 4–5% of the normal level results in significant therapeutic benefits 17. The current experiment involved administering ataluren for only two days. The lack of increased PPT1 enzyme activity in the heart, lung, brain and kidney may suggest that more time is required for ataluren to achieve optimal concentrations in some tissues. In a previous study, we have shown that much higher dose of ataluren (100 mg/kg) is required to increase PPT1 enzyme activity within the brain 9. With this higher dose of ataluren (100 mg/kg), however, the therapeutic effect in the liver was lost 9, indicating that above a certain tissue concentration ataluren does not promote translation through the premature stop codon.
In summary, our study identified a new challenge/hurdle for read‐through drug therapy: variable efficiency of read‐through therapy in the different tissues/organs because of tissue‐specific variations in nonsense mutant transcript levels. This means that a read‐through drug at any given dose cannot be therapeutically effective in all tissues. Accordingly, it will be important to consider which organ is affected the most in a particular genetic disease and to determine the optimal dose of read‐through drug therapy for that organ.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported in part by Hayden's Hope, the Beat Batten Foundation, Sanford Health and the National Institutes of Health (NS044310).
References
- 1. Bidou L, Allamand V, Rousset JP, et al Sense from nonsense: therapies for premature stop codon diseases. Trends Mol Med. 2012; 18: 679–88. [DOI] [PubMed] [Google Scholar]
- 2. Miller JN, Pearce DA. Nonsense‐mediated decay in genetic disease: friend or foe? Mutat Res, Rev Mutat Res. 2014; 762: 52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Welch EM, Barton ER, Zhuo J, et al PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007; 447: 87–91. [DOI] [PubMed] [Google Scholar]
- 4. Du M, Liu X, Welch EM, et al PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR‐G542X nonsense allele in a CF mouse model. Proc Natl Acad Sci USA. 2008; 105: 2064–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schulz A, Kohlschutter A, Mink J, et al NCL diseases ‐ clinical perspectives. Biochim Biophys Acta. 2013; 1832: 1801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kousi M, Lehesjoki AE, Mole SE. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum Mutat. 2012; 33: 42–63. [DOI] [PubMed] [Google Scholar]
- 7. Kollmann K, Uusi‐Rauva K, Scifo E, et al Cell biology and function of neuronal ceroid lipofuscinosis‐related proteins. Biochim Biophys Acta. 2013; 1832: 1866–81. [DOI] [PubMed] [Google Scholar]
- 8. Miller JN, Chan CH. Pearce DA. The role of nonsense‐mediated decay in neuronal ceroid lipofuscinosis. Hum Mol Genet. 2013; 22: 2273–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Miller JN, Kovacs AD, Pearce DA. The novel Cln1 R151X mouse model of infantile neuronal ceroid lipofuscinosis (INCL) for testing nonsense suppression therapy. Hum Mol Genet. 2015; 24: 185–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zetoune AB, Fontaniere S, Magnin D, et al Comparison of nonsense‐mediated mRNA decay efficiency in various murine tissues. BMC Genet. 2008; 9: 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pfaffl MW. A new mathematical model for relative quantification in real‐time RT‐PCR. Nucleic Acids Res. 2001; 29: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group‐wise comparison and statistical analysis of relative expression results in real‐time PCR. Nucleic Acids Res. 2002; 30: e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Voznyi YV, Keulemans JL, Mancini GM, et al A new simple enzyme assay for pre‐ and postnatal diagnosis of infantile neuronal ceroid lipofuscinosis (INCL) and its variants. J Med Genet. 1999; 36: 471–4. [PMC free article] [PubMed] [Google Scholar]
- 14. van Diggelen OP, Keulemans JL, Winchester B, et al A rapid fluorogenic palmitoyl‐protein thioesterase assay: pre‐ and postnatal diagnosis of INCL. Mol Genet Metab. 1999; 66: 240–4. [DOI] [PubMed] [Google Scholar]
- 15. van Diggelen OP, Thobois S, Tilikete C, et al Adult neuronal ceroid lipofuscinosis with palmitoyl‐protein thioesterase deficiency: first adult‐onset patients of a childhood disease. Ann Neurol. 2001; 50: 269–72. [DOI] [PubMed] [Google Scholar]
- 16. Miller JN, Pearce DA. A, novel c.776_777insA mutation in CLN1 leads to infantile neuronal ceroid lipofuscinosis. J Child Neurol. 2013; 28: 1106–11. [DOI] [PubMed] [Google Scholar]
- 17. Griffey MA, Wozniak D, Wong M. CNS‐directed AAV2‐mediated gene therapy ameliorates functional deficits in a murine model of infantile neuronal ceroid lipofuscinosis. Mol Ther. 2006; 13: 538–47. [DOI] [PubMed] [Google Scholar]
- 18. Buhler M, Muhlemann O. Alternative splicing induced by nonsense mutations in the immunoglobulin mu VDJ exon is independent of truncation of the open reading frame. RNA. 2005; 11: 139–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang J, Chang YF, Hamilton JI, et al Nonsense‐associated altered splicing: a frame‐dependent response distinct from nonsense‐mediated decay. Mol Cell. 2002; 10: 951–7. [DOI] [PubMed] [Google Scholar]
- 20. Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012; 197: 857–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Karam R, Lou CH, Kroeger H, et al The unfolded protein response is shaped by the NMD pathway. EMBO Rep. 2015; 16: 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]