

Abstract
Malaria infection induces, alongside endothelial damage and obstruction hypoxia, a potent inflammatory response similar to that observed in other systemic diseases caused by bacteria and viruses. Accordingly, it is increasingly recognised that cerebral malaria (CM), the most severe and life threatening complication of Plasmodium falciparum infection, bears a number of similarities with sepsis, an often fatal condition associated with a misregulated inflammatory response triggered by systemic microbial infections.
Using a Plasmodium berghei ANKA mouse model, histology, immunohistochemistry and gene expression analysis, we showed that lipopolysaccharide S (LPS), at doses that normally induce inflammation tolerance, protects P. berghei infected mice against experimental CM (ECM). Vascular endothelial growth factor (VEGF) preserved blood vessel integrity, and the combination with LPS resulted in a strong synergistic effect. Treated mice did not develop ECM, showed a prolonged survival and failed to develop a significant inflammatory response and splenomegaly in spite of normal parasite loads. The protective role of VEGF was further confirmed by the observation that the treatment of P. berghei infected C57BL/6 and Balb/c mice with the VEGF receptor inhibitor axitinib exacerbates cerebral pathology and aggravates the course of infection. Infected mice treated with VEGF and LPS showed an induction of the anti-inflammatory genes Nrf2 and HO-1 and a suppression to basal levels of the genes IFN-γ and TNF-α. These results provide the rationale for developing new therapeutic approaches against CM and shed new light on how the inflammatory process can be modulated in the presence of systemic infectious diseases.
Keywords: Vascular endothelial growth factor, Angiogenesis, Cerebral malaria, Inflammation, Cytokines
Introduction
The pathology of Plasmodium falciparum malaria is the result of a combination of factors that involve the systemic activation of the inflammatory response and hypoxia from blood vessel obstruction, leading to endothelial damage. In cerebral malaria (CM) patients, the cerebral capillaries are damaged, lined with apoptotic cells and filled with parasitised erythrocytes, while the surrounding brain tissue shows monocyte infiltration and glial proliferation.1 The consequent disruption of the blood–brain barrier leads to cerebral oedema, coma and death.2,3 The adherence of large numbers of parasitised red blood cells (pRBCs) to endothelium of brain post-capillary venules would plug the vessels, leading to mechanical occlusion, impaired blood flow with resulting ischaemia and tissue hypoxia.4 The mechanisms behind vasoconstriction and vascular dysfunction in CM are not completely understood, although mediators such as carbon monoxide (CO), nitric oxide (NO), endothelins, growth factors and the angiopoietin–Tie2 axis play critical roles.4–6 To this end, very recently, it has been demonstrated that NO and CO suppress the development of severe forms of malaria associated with Plasmodium infection via a mechanism in which NO induces the expression of heme oxygenase-1 (HO-1) through activation of the nuclear factor erythroid 2 related factor 2 (Nrf2).7 These two genes are also targets of lipopolysaccharide S (LPS). It has been shown that LPS induces HO-1 expression via Nrf2 in both human monocytic cells and mouse brain endothelial cells.8,9 Moreover, there is strong evidence that hypoxia and inflammation cause secretion of vascular endothelial growth factor (VEGF), which then stimulates the release of NO and prostacyclin (PGI2) from endothelial cells,10,11 which then affect the main targets of these pathological events in the context of CM. Therefore, with normal vascular tonus restored, resistance to blood flow would decrease and normal shear stress would help washing out adhered cells.4
The rodent parasite Plasmodium berghei ANKA strain (PbA) induces in the brain of susceptible mice, pathological changes that are very similar to human CM.2,12 The utilisation of this experimental CM (ECM) model has provided a better understanding of the malaria pathology in the brain and supported the notion that the severity of the condition is linked to a dysregulation of the inflammation process.1,12 Compelling evidence implicates the cytokines IFN-γ and TNF-α in driving the inflammatory response leading to ECM.13–15 IFN-γ is required for up-regulating the expression of endothelial adhesion molecules, which bind to infected erythrocytes in the brain vessels, and for inducing the synthesis of macrophage derived TNF-α that in turn enhances the inflammatory response.16 Mice in which either the genes coding for IFN-γ and TNF-α or their receptors are disrupted fail to develop ECM.17,18 While activation of the inflammatory response is clearly necessary for developing ECM, several lines of evidence suggest that this alone may not be sufficient to fully explain experimental and human brain pathology. Mice in which the genes coding for the endothelial adhesion molecules ICAM-1, VCAM-1 and P-selectin have been disrupted do not develop ECM.19 Normally, these genes are highly induced during ECM and have been implicated in enhancing the binding of leukocytes, platelets and pRBCs to endothelial cells.19 In particular, the disruption of ICAM-1 and VCAM-1 would prevent the binding of platelets to endothelium, a process that has been shown to induce TGFβ1 mediated apoptosis of these cells.18,20,21 Furthermore, the observation that high levels of TNF and IFN-γ are detected in non-lethal cases of Plasmodium vivax infection casts doubts on the inflammation only hypothesis of CM.22 Accordingly, and even more convincingly, attempts to treat CM with anti-inflammatory agents such as anti-TNF monoclonal antibodies and dexamethasone, rather than ameliorating, exacerbated the course of malaria.23,24 Not all proinflammatory cytokines are equally relevant for the development of CM. The best documented evidence implicates IFN-γ, TNF-α and IL-12, whereas no evidence has been found for IL-6.25 In the case of IL-12 and/or IFN-γ, strong support for their involvement in the pathogenesis of CM comes from studies with knockout mice infected with P. berghei ANKA.26,27 In addition to systemic proinflammatory cytokine production, local cytokine release could contribute to organ specific pathophysiology, especially in the brain in the case of CM.28 The lack of a classical inflammatory response to the presence of parasitised erythrocytes in the brain microvasculature indicates anti-inflammatory cytokine involvement.19 However, the role of anti-inflammatory cytokines in the control of CM is less clear. Although data based on knockout mice indicate that IL-4 and IL-10 are not required for development of the disease,29 in models in which IL-12 and IFN-γ display a dominant pathological role, a protective effect has been demonstrated for IL-10. Tan et al.30 found that infected wild type mice showed an early Th1 response that shifted to a late Th2 response, whereas in infected IRF-1− / − animals, an early protective Th2 was founnd that avoids Th1 dependent pathological damage. Moreover, neutralisation of IL-10 in vivo has been demonstrated to increase the percentage of mice with CM in a CM resistant strain.26
Currently, >20% of children with CM are at risk of either dying or acquiring severe learning deficits in spite of being administered effective anti-parasitic treatment, thus highlighting the need to develop an effective support therapy. This must take into account the role of both the inflammatory response and the obstruction hypoxia mediated damage in shaping the pathology. Here, we report that the co-administration of sublethal doses of LPS, a potent suppressor of the inflammatory response, together with VEGF, a molecule known to produce the release of NO from endothelial cells as well as to exert a strong protective and regenerative activity on them, completely protects P. berghei infected mice from ECM, without interfering with parasite burden. Notably, VEGF-LPS treatment silences the inflammation process in all tissues and organs examined, and prevents the development of splenomegaly in spite of normal parasite loads. The treatment suppresses the induction of both TNF-α and IFN-γ and activates the transcription of the anti-inflammatory genes Nrf2 and HO-1, leading indirectly to the protection mechanism against ECM already described in the literature.7,31 The protective activity of VEGF is also supported by the observation that a recombinant form of this molecule increases the survival of P. berghei infected mice and prevents the development of canonical ECM symptoms. Accordingly, the VEGF inhibitor axitinib dramatically aggravates the course of infection in both ECM susceptible C57BL/6 and ECM resistant Balb/c mice.
These findings add to our knowledge of the immunobiology of CM pathogenesis and provide the rationale for developing effective supporting therapy to reduce the risk of death and brain damage in CM.
Results
LPS and VEGF suppress the inflammatory response to PbA infection and prevents the development of CM
At low doses, LPS is known to elicit a complex orchestrated counter-regulatory response to inflammation known as ‘tolerance’ that involves the expression of HO-1 via the transcription factor Nrf2 as well as the induction of a number of antioxidant stress response genes including VEGF.8,9,32–34 When given to P. berghei Anka (PbA) infected C57BL/6 mice, LPS protected 100% of the animals from ECM and prolonged their survival until day +12 post-infection without affecting the parasite load (Fig. 1a). Immunohistochemistry analysis of brain sections from these mice showed a marked reduction of the CD3+ cell infiltrate (peculiar of CM pathology) and a similar decrease in the immunostaining of anti-cleaved caspase 3 antibodies compared to untreated C57BL/6 mice (Fig. 1b). LPS treatment induced from day +3 post-infection to the time of death a significant down-regulation to basal levels of both TNF-α and IFN-γ (p < 0.01). These expression changes were associated to a significant up-regulation of both HO-1 and Nrf2 (p < 0.01). The role of obstruction hypoxia and endothelial cell damage associated to brain pathology prompted us to also investigate whether the administration of recombinant VEGF could change the course of PbA infection. We investigated both ECM susceptible C57BL/6 and ECM resistant Balb/c mice treated with recombinant VEGF (200 ng/mouse daily) starting from day +1 post-infection over a period of 5 days. Under these experimental conditions, the treatment prolonged the survival of the C57BL/6 infected mice until day +10 post-infection without affecting the parasite load (Supplemental Fig. 1a). None of these animals developed the typical signs of ECM, such as a positive Woolley–White sign and convulsions. However, they showed ruffled fur, pale ears and footpad, and red discharge from the nose (which are typical signs of anaemia). Morphological analysis and immunohistochemistry of C57BL/6 brain sections revealed the presence of a moderate infiltrate of CD3 lymphocytes (mainly composed of CD8 rather than CD4; data not shown) from day +3 to end point (Supplemental Fig. 1b). Some apoptotic endothelial cells were also detected, particularly on the day when the animals died (Supplemental Fig. 1b, blue arrow). Important differences were detected when analysing the expression of inflammation markers in treated versus non-treated animals. VEGF induced a marked increase of both VEGFR1 and VEGFR2 that was associated with a down-regulation of the cytokines IL-23p19 (p < 0.05), TNF-α and IFN-γ (p < 0.01) (Table 1). On the contrary, VEGF treatment had little effect on the transcription of inflammation associated genes in Balb/c mice (Table 1).
Figure 1.
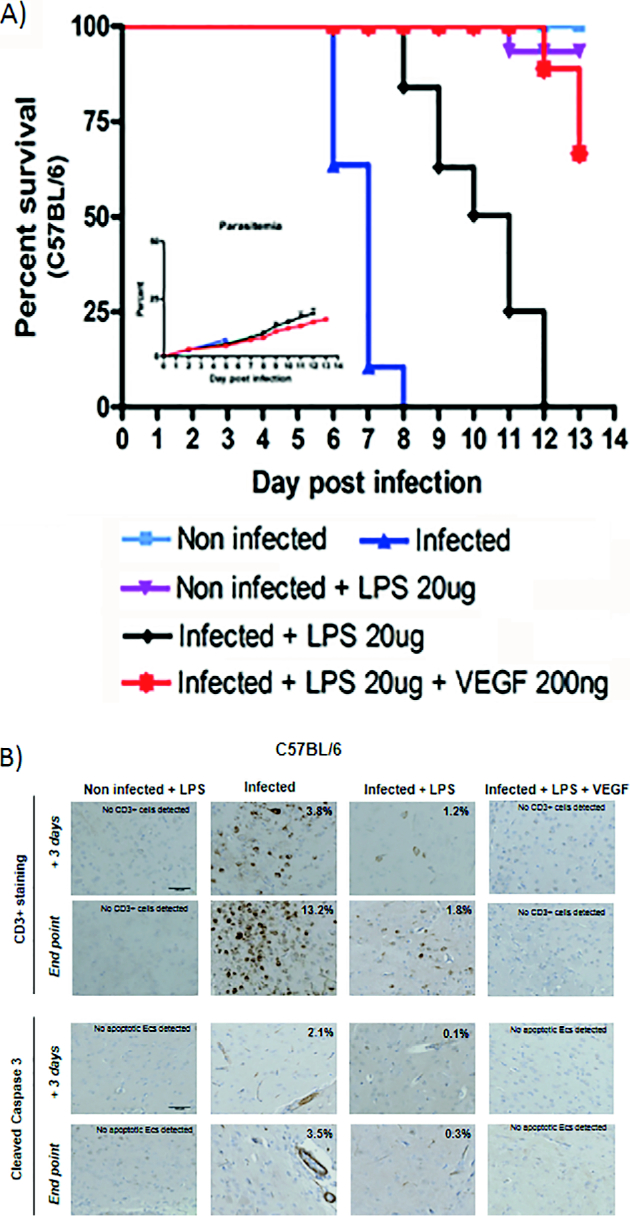
VEGF and LPS combination treatment on infected C57BL/6 mice a survival rate and parasitemia (inset) of infected C57BL/6 mice (dark blue filled triangle) treated with LPS alone (black filled diamonds) or with a combination of VEGF and LPS (red filled stars). The survival of not infected control groups either not treated (light blue filled squares) or treated with LPS (purple filled triangles) is also shown. b immunostaining of brain sections from infected, treated and control groups examined both at day +3 post-infection and at end point, stained with antibodies directed against the anti-CD3 lymphocyte antigen or the cleaved caspase 3 fragment. Percent of CD3+ cells and /or apoptotic endothelial cells (cleaved caspase 3) per field of view is indicated.
Table 1.
Fold change expression of inflammation markers in the brain of VEGF treated mice
| VEGF | VEGFR1 | VEGFR2 | S1Pr1 | IL-6 | IL-23p19 | TNF-α | IFN-γ | HO-1 | Nrf2 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Mouse strain C57BL/6 | ||||||||||
| Infected | 7.43 | 2.80 | 4.76 | 2.30 | 3.80 | 20.60 | 1.30 | 2.64 | 1.10 | 1.30 |
| 7.50 | 2.23 | 7.10 | 1.60 | 8.30 | 21.10 | 4.30 | 9.70 | 0.40 | 0.88 | |
| Infected+VEGF | 7.35 | 0.50* | 1.77* | 5.10 | 1.20* | 25.44* | 0.89 | 1.02** | 1.76 | 3.76* |
| 8.11 | 2.05 | 9.07** | 1.40 | 0.20** | 5.43* | 2.10** | 2.43** | 1.20* | 0.90 | |
| Mouse strain Balb/c | ||||||||||
| Infected | 1.55 | 0.89 | 0.85 | 5.36 | 0.58 | 0.27 | 0.49 | 0.80 | 3.48 | 2.79 |
| 0.85 | 0.61 | 0.16 | 3.32 | 0.72 | 1.14 | 0.50 | 1.13 | 6.22 | 6.25 | |
| Infected+VEGF | 2.73* | 1.32 | 0.60 | 3.60 | 0.09 | 0.50 | 0.80 | 0.40 | 4.30 | 2.70 |
| 1.99 | 1.0** | 2.37** | 4.60 | 0.20 | 0.90 | 0.30* | 0.68 | 6.70 | 7.10 | |
Expression of inflammation markers in brain tissue of C57BL/6 and Balb/c infected versus treated mice (VEGF 200 μg), +3 days post-infection (blue) and at the end point (red). Each value represents the mean of the fold changes obtained from three biological replicates. Induction relative to the non-infected control (fold change) was calculated using the comparative C t method (ΔΔC t). Values obtained from non-treated versus treated mice were compared using a one sample t test to access statistical significance: * (0.01 ≤ p < 0.05) and ** (p < 0.01).
The VEGF inhibitor axitinib exacerbates ECM pathology
To better understand the role of VEGF, we investigated disease progression, brain pathology and inflammatory gene expression in both C57BL/6 and Balb/c mice treated with axitinib.35 This is a potent small molecule inhibitor of the VEGF receptor tyrosine kinase that induces a significant reduction in the VEGF response to inflammation and hypoxia.35 Typically, untreated PbA infected C57BL/6 mice showed signs of convulsions, ataxia, coma and a positive Woolley–White sign and died of ECM within 7 days post-infection. All axitinib treated C57BL/6 mice died 1 or 2 days earlier than the untreated control group, showing severe signs of ECM (Fig. 2a). Treatment of Balb/c mice with axitinib did not induce ECM, but these mice also died earlier than untreated control starting from day +8 post-infection (survival: day 8: 98%, day 9: 80%, day 10: 50%) (Fig. 2b). Brain samples from C57BL/6 and Balb/c mice were collected in a replica experiment and processed for histological examination at day +3 post-infection and at the time of death (end point). Brain sections from C57BL/6 mice stained with hematoxylin and eosin (H&E) showed a considerable monocyte infiltrate and accumulation of malaria pigment that progressively increased from day +3 to the time of death (Fig. 3, top panel). Immunohistochemistry of brain samples revealed that the monocyte infiltrate was composed of CD3 positive cells (CD4+ and CD8+ mixed subpopulations) with a higher number of CD4, calculated as percent of area of the positive signal (infected C57BL/6: 4.3%, C57BL/6 treated with axitinib: 12.4%; Balb/c infected: 2.3%, Balb/c treated with axitinib: 4%), versus CD8 (infected C57BL/6: 2.8%, C57BL/6 treated with axitinib: 2.9%; Balb/c infected: 0.5%, Balb/c treated with axitinib: 2.4%), in both mouse strains (Fig. 3, middle panel). To evaluate the endothelial cell apoptosis, we used an antibody directed against the cleaved form of caspase 3.36,37 The immunohistochemistry showed an increased staining of brain parenchyma and endothelial cells in axitinib treated C57BL/6 mice (Fig. 3, bottom panel), which is in line with the worsening of disease progression in axitinib treated mice.
Figure 2.
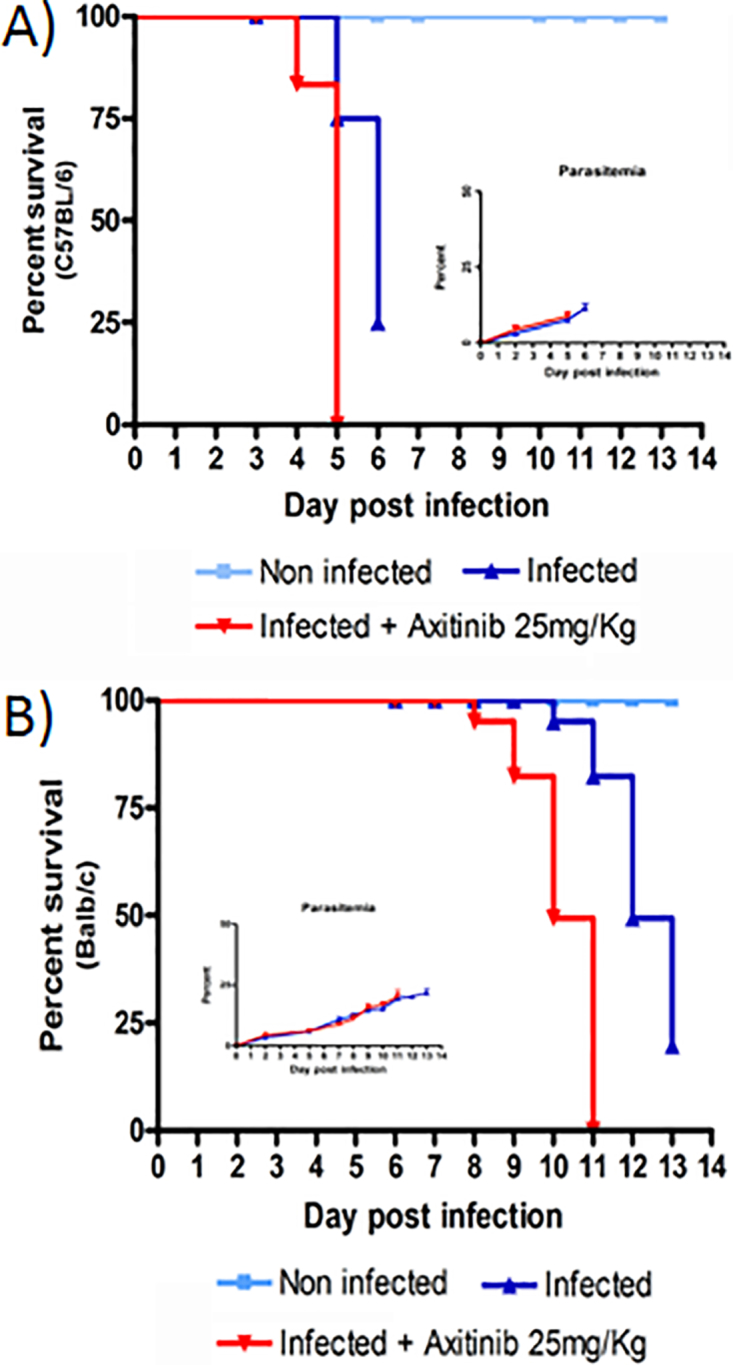
Effect of axitinib treatment on P. berghei infected mice. Survival rate and parasitemia (inset) of C57BL/6 a and Balb/c b of P. berghei infected mice either not treated (blue filled triangle) or treated (red filled triangle) with axitinib. Treated animals received from day +1 post-infection a daily dose of axitinib (25 mg/kg i.p.) over a period of 5 days. The parasitemia was determined by Giemsa staining, and microscopic examination of thin blood smears from individual mice was taken at regular interval from day 2 to end point (time of culling).
Figure 3.

Morphological analysis of brain sections from infected C57BL/6 and Balb/c mice treated with VEGF inhibitorC57BL/6 and Balb/c mice [non-infected, infected and infected plus treatment ( + axitinib)] rain sections are shown. The specimens were collected at day +3 post-infection and at end point, stained with H&E, or processed for immunohistochemistry with anti-CD3 and cleaved caspase 3 antibodies. Blue arrows show cleaved caspase 3 reactive endothelial cells. Scale bars are indicated. Percent of CD3+ cells and/or apoptotic endothelial cells (cleaved caspase 3) per field of view is indicated.
Analysis of cytokine and inflammatory markers in axitinib treated animals
The brains of PbA infected C57BL/6 mice showed consistent high levels of IL-23p19 and a progressive up-regulation of TNF-α and IFN-γ from day +3 to the onset of ECM together with basal levels of both Nrf2 and HO-1. In these mice, axitinib treatment strongly suppressed the expression of VEGF and both its receptors VEGFR1 and VEGFR2. We also observed that axitinib significantly down-regulated the expression of a number of cytokines including IL-12p40, IL-6 and IL-23p19 (p < 0.01) while leaving the induction of TNF-α and IFN-γ unaffected (Table 2 and Supplemental Table 1). When analysing circulating cytokines, we observed that axitinib treated mice showed significantly higher level of IFN-γ (Supplemental Table 2). Balb/c mice responded to PbA infection with a marked up-regulation in the brain of both HO-1 and Nrf2 as shown in previous observations.31 In these mice, axitinib treatment significantly inhibited this response (p < 0.01), in agreement with its worsening effect on disease progression (Table 2).
Table 2.
Expression of inflammation markers in the brain of Anti-VEGF treated mice
| VEGF | VEGFR1 | VEGFR2 | S1Pr1 | IL-6 | IL-23p19 | TNF-α | IFN-γ | HO-1 | Nrf2 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Mouse strain C57BL/6 | ||||||||||
| Infected | 7.43 | 2.80 | 4.76 | 2.30 | 3.80 | 20.60 | 1.30 | 2.64 | 1.10 | 1.30 |
| 7.50 | 2.23 | 7.10 | 1.60 | 8.30 | 21.10 | 4.30 | 9.70 | 0.40 | 0.88 | |
| Infected+axitinib | 0.08* | 0.30** | 0.30** | 1.10** | 1.90** | 0.85** | 0.16* | 0.74** | 0.17* | 0.16** |
| 0.10* | 0.10* | 0.16** | 1.70 | 0.16** | 0.08** | 3.87 | 10.40 | 0.15** | 0.05** | |
| Mouse strain Balb/c | ||||||||||
| Infected | 1.55 | 0.89 | 0.85 | 5.36 | 0.58 | 0.27 | 0.49 | 0.80 | 3.48 | 2.79 |
| 0.85 | 0.61 | 0.16 | 3.32 | 0.72 | 1.14 | 0.50 | 1.13 | 6.22 | 6.25 | |
| Infected+axitinib | 0.61** | 0.38* | 0.14* | 0.66** | 0.06 | 0.36 | 1.41** | 1.14 | 0.99* | 0.78* |
| 0.47** | 0.34 | 0.75* | 0.41** | 0.20* | 0.96 | 1.29 | 1.47 | 1.04** | 2.27** | |
Expression of inflammation markers in brain tissue of C57BL/6 and Balb/c infected versus treated mice (axitinib 25 mg/kg), +3 days post-infection (blue) and at the end point (red). Each value represents the mean of the fold changes obtained from three biological replicates. Induction relative to the non-infected control (fold change) was calculated using the comparative C t method (ΔΔC t). Values obtained from non-treated versus treated mice were compared using a one sample t test to access statistical significance: * (0.01 ≤ p < 0.05) and ** (p < 0.01).
LPS and VEGF act synergistically to completely silence the inflammatory response to PbA infection
The exogenous addition of recombinant VEGF to LPS further improved the course of PbA infection in C57BL/6 mice without affecting the parasite load. Not only did none of the treated mice not develop ECM but also their survival was significantly prolonged. This improvement was mirrored by a complete absence of brain pathology. Very few if any cells were found to react against antibodies directed to CD3 lymphocyte antigen and to cleaved caspase 3 (Fig. 1b). VEGF-LPS treatment also induced changes in the inflammation response to parasite infection that were striking when compared to those induced by VEGF or LPS alone. In the brain, the genes S1Pr1 (a molecule involved in endothelial remodelling), Nrf2 and HO-1 were significantly up-regulated by ∼10-, 25- and 9-fold respectively (p < 0.01), while the expression of TNF-α and IFN-γ was reduced at the level of non-infected control mice (p < 0.01) (Table 3 and Supplemental Table 3). Most surprisingly, the spleens of LPS-VEGF treated mice were very similar in terms of morphology (size, colour and appearance) to those of non-infected controls (Supplemental Fig. 2). We therefore performed an expression analysis of inflammation markers of the internal organs (liver, spleen and lung) from treated and untreated mice (Table 4). This investigation revealed that LPS-VEGF treatment completely abolished the inflammatory response in all organs analysed. In the liver, PbA infection induced an important activation of the inflammatory response at day +3 post-infection, up-regulating the expression of both TNF-α and IFN-γ by ∼50- and 30-fold respectively. The LPS-VEGF treatment reversed this response to baseline levels while up-regulating the Nrf2 gene (p < 0.01). Notably, by the time the non-treated mice developed ECM, the liver had down-regulated the inflammatory response, thus suggesting the presence of a potent autoregulatory pathway in this organ. The spleens of untreated C57BL/6 mice showed a dramatic inflammatory response to PbA infection: the development of ECM coincided with a strong up-regulation of VEGF, VEGFR1, VEGFR2, TNF-α and IFN-γ. Here, LPS-VEGF treatment completely silenced this response to baseline level while significantly up-regulating the expression of the Nrf2 gene close to 40-fold (p < 0.01). Unlike the brain, we did not observe the induction of HO-1 gene neither in the liver nor in the spleen. The lung was not affected by the inflammatory response elicited by PbA infection.
Table 3.
Expression of inflammation markers in the brain of LPS-VEGF treated mice
| Mouse strain C57BL/6 | VEGF | VEGFR1 | VEGFR2 | S1Pr1 | IL-6 | IL-23p19 | TNF-α | IFN-γ | HO-1 | Nrf2 |
|---|---|---|---|---|---|---|---|---|---|---|
| Infected | 7.43 | 2.80 | 4.76 | 2.30 | 3.80 | 20.60 | 1.30 | 2.64 | 1.10 | 1.30 |
| 7.50 | 2.23 | 7.10 | 1.60 | 8.30 | 21.10 | 4.30 | 9.70 | 0.40 | 0.88 | |
| Infected+LPS | 7.66 | 5.15* | 1.98* | 1.80 | 5.60 | 23.50 | 0.98 | 0.70* | 1.70* | 0.60 |
| 4.28** | 3.16 | 2.95** | 2.20 | 0.30* | 7.90** | 1.00** | 0.84** | 3.42* | 3.70* | |
| Infected+LPS+VEGF | 1.25** | 1.20* | 1.74** | 12.60** | 4.00 | 40.57 | 0.80 | 0.95 | 3.30** | 23.01** |
| 5.46* | 1.90 | 7.30 | 10.30** | 0.50** | 4.64** | 1.40* | 0.77** | 9.10** | 25.21** | |
| Non-infected+LPS | 0.60** | 4.50* | 0.90** | 0.75* | 3.70 | 6.30* | 0.80* | 0.50* | 2.00** | 3.91* |
| 0.97** | 0.93* | 1.33** | 2.40 | 0.40** | 10.20 | 2.92* | 1.50* | 3.10** | 3.81* |
Expression of inflammation markers in brain tissue of C57BL/6 infected and treated mice (LPS+VEGF), +3 days post-infection (blue) and at the end point (red). Each value is the mean of the fold changes obtained from three biological replicates. Induction relative to the non-infected control (fold change) was calculated using the comparative C t method (ΔΔC t). Values obtained from non-treated versus treated mice were compared using a one sample t test to access statistical significance: * (0.01 ≤ p < 0.05) and ** (p < 0.01).
Table 4.
Fold change expression of inflammation markers in internal organs of LPS-VEGF treated mice
| Mouse strain C57BL/6 | VEGF | VEGFR1 | VEGFR2 | S1Pr1 | TNF-α | IFN-γ | HO-1 | Nrf2 |
|---|---|---|---|---|---|---|---|---|
| Liver | ||||||||
| Infected | 132.01 | 55.40 | 72.50 | 12.00 | 54.29 | 31.77 | 0.01 | 1.00 |
| 27.11 | 10.23 | 17.38 | 8.99 | 0.60 | 0.79 | 0.30 | 1.45 | |
| Infected+LPS+VEGF | 8.55** | 3.28** | 4.00** | 1.24** | 3.07** | 0.77** | 1.00* | 67.50** |
| 9.81** | 2.15** | 5.21** | 1.04** | 0.56 | 0.34* | 1.30* | 4.11** | |
| Spleen | ||||||||
| Infected | 920.11 | 288.74 | 625.19 | 2.00 | 30.93 | 14.99 | 0.13 | 1.00 |
| 790.88 | 177.50 | 548.60 | 1.00 | 238.38 | 223.73 | 0.79 | 0.80 | |
| Infected+LPS+VEGF | 225.20** | 98.15** | 119.12** | 6.00 | 8.45** | 3.90* | 0.87** | 39.07** |
| 42.15** | 20.11** | 24.04** | 13.00** | 1.50** | 0.69** | 1.30 | 1.46 | |
| Lung | ||||||||
| Infected | 3.11 | 1.25 | 1.00 | 1.00 | 1.00 | 1.10 | 0.90 | 0.80 |
| 0.55 | 0.22 | 0.10 | 0.24 | 0.10 | 0.12 | 0.47 | 0.20 | |
| Infected+LPS+VEGF | 0.32** | 0.18** | 0.10** | 1.10 | 1.20 | 0.28 | 1.16 | 2.10 |
| 0.51 | 0.24 | 0.20 | 2.80** | 0.30 | 0.10 | 1.12** | 6.60** | |
Fold changes of gene expression in liver, spleen and lung of C57BL/6 infected versus treated mice (LPS-VEGF), +3 days post-infection (blue) and at the end point (red). Each value represents the mean of the fold changes obtained from three biological replicates. Induction relative to the non-infected control (fold change) was calculated using the comparative C t method (ΔΔC t). Values obtained from non-treated versus treated mice were compared using a one sample t test to access statistical significance: * (0.01 ≤ p < 0.05) and ** (p < 0.01).
Discussion
The pathology of CM involves the binding of infected erythrocytes to the venular endothelium38 and the concomitant activation of a strong inflammatory response characterised by lymphocyte infiltration of the brain, up-regulation of the cytokines TNF-α and IFN-γ, and endothelial cell damage.3 Efforts to develop supportive therapy to decrease morbidity and mortality of malaria must take into account such a dual nature of the pathogenesis of CM. We show here that the administration of LPS, at doses that normally induce a state of tolerance to further inflammatory stimuli,32 changed significantly the course of PbA infection in ECM susceptible C57BL/6 mice. The treated animals showed a prolonged survival compared to untreated controls and failed to develop signs of ECM. Morphological analysis and immunohistochemistry revealed a marked reduction of the cellular infiltrate in the brains of LPS treated and a substantial decrease of reactivity to CD3 and activated caspase 3 antibodies, in agreement with the anticipated effect of LPS at low doses. Gene expression analysis revealed that LPS had a profound inhibitory effect on the induction of inflammatory cytokines TNF-α and IFN-γ while up-regulating Nrf2 and HO-1 to levels comparable to those observed in ECM resistant Balb/c mice. However, it was already known that LPS induces the expression of HO-1 via Nrf2 in mouse brain endothelial cells.9
Recently, Jeney and colleagues have shown that NO suppresses the pathogenesis of ECM also via a mechanism involving Nrf2, induction of HO-1 and CO production via heme catabolism by HO-1.7
We hypothesised that VEGF could also exert a protective role against ECM. This originated from the notion that the protective activities on brain vessels of CO and NO, two molecules known to prevent the development of ECM, mirror those mediated by VEGF in response to tissue injury and resolution of inflammation.39–41 Indeed, the relationship linking NO and CO with VEGF is quite intricate. It has been demonstrated that VEGF produces release of NO from endothelial cells by increasing endothelial NO synthetase expression.11 Different activators of HO-1 induce VEGF expression.42,43 This is mediated by the activity of HO-1 that produces CO starting from free heme.42,44 In turn, VEGF up-regulates the expression of HO-1 in endothelial cells, thereby establishing a positive feedback circuit.44 Accordingly, endothelial cells from HO-1 knockout mice do not respond optimally to VEGF stimulation.45 NO and NO donors are also potent inducers of VEGF synthesis and potentiate its effect on endothelial cells.46 Here, we show that treatment of C57BL/6 mice with axitinib, a potent inhibitor of VEGF receptors 1, 2, and 3, CD117 (cKIT), and platelet derived growth factor receptor (PDGFR), aggravates brain pathology dramatically and decreases the survival of the mice. At day 5 post-infection, treated animals showed an important CD3 lymphocyte infiltrate of the brain and widespread apoptosis of both parenchyma and endothelial cells. In ECM resistant Balb/c mice, axitinib significantly reduced the accelerated mortality in 100% of the treated animals. This was associated with sign of brain pathology such as lymphocyte infiltrate and cell apoptosis that were never observed in PbA infected Balb/c mice. The notion that axitinib acted on the VEGF receptor pathway is corroborated by the protective effect of VEGF treatment. Compared to untreated control group, VEGF treated mice did not develop ECM and showed moderate signs of brain inflammation and damage in terms of lymphocyte infiltrate and cell apoptosis respectively and a decreased up-regulation of both TNF-α and IFN-γ.
The addition of VEGF to LPS treatment had a strong synergistic effect that manifested itself with a complete silencing of the inflammatory response to PbA infection. CD3 lymphocytes were absent in the brain parenchyma of treated C57BL/6 mice in repeated examinations. Similarly, sections stained with antibodies directed activated caspase 3 did not show any reactivity. Expression analysis revealed that the combination treatment was extremely effective in up-regulating the expression of the regulatory genes Nrf2 and HO-1 to levels much higher than those observed in ECM resistant Balb/c mice or when treating the mice with either LPS or VEGF alone. Accordingly, VEGF-LPS treated mice showed a prolonged survival and did not develop ECM. When analysing the internal organs at the time of death, we noticed that the spleens of VEGF-LPS treated mice could not be distinguished in term of size, colour and weight from those of non-infected controls. This morphological observation was in agreement with the effect of VEGF-LPS treatment on the expression of inflammation markers. In the spleen of untreated PbA infected animals, we observed a progressive very strong up-regulation of VEGF, VEGFR1, VEGFR2, TNF-α and IFN-γ respectively. LPS-VEGF treatment completely silenced TNF-α and IFN-γ response to baseline level while up-regulating the expression of the Nrf2 gene, thus shedding light for the first time on the mechanism leading to splenomegaly, a condition observed in human malaria as well as in several acute and chronic infectious diseases.
The synergy achieved by combining LPS and VEGF reflects their respective activities on the two pathogenic mechanisms leading to CM: an unregulated activation of the inflammation response and obstruction–hypoxia endothelial cell damage. On one hand, LPS induces antioxidant genes, thereby protecting endothelial cells from injury while down-regulating the inflammatory response47; on the other hand, VEGF induces endothelial cell proliferation and restores blood vessel integrity. These observations all together significantly add to our understanding of the factors regulating the inflammatory response to plasmodium infection and provide the rationale to develop novel effective supportive therapies to treat CM and, in general, to protect the endothelium in the presence of life threatening inflammatory processes.
Methods
Animals
C57BL/6 and Balb/c females (age 4–6 weeks) were used. The animal work carried out at Perugia University have been performed according to the D.L 27 January 1992, no. 116, Italian legislation, and approved under Ethics Committee license no. PR 0161.
Parasite and infection procedure
The parasite P. berghei ANKA clone 2.34 was originally obtained from The University of Perugia, Dipartimento di Medicina Sperimentale, Italy. Serial passages of P. berghei were carried out by i.p. inoculation of naïve C57BL/6 mice with 105 pRBCs to obtain parasitised erythrocytes stocks. In this study, C57BL/6 or Balb/c mice were infected by i.p. injection of 105 infected erythrocytes at day 0. Parasitemia was first measured 3 days post-infection followed by a daily measurement until the day of death. A drop of blood was collected by venesection of the tail of the mouse and transferred onto the edge of a microscope slide. The blood was drawn evenly across a second slide to make a thin blood film and allowed to dry at room temperature before staining with Giemsa stain. Five fields of ∼200 cells each were counted, and the parasitemia was calculated as the percentage of the total RBCs containing parasites.
Treatment regimen
Starting from day 1 post-infection, the mice were treated daily with axitinib (25 mg/kg) (Tocris Bioscience, #4350) or mouse vascular endothelial growth factor-164 (mVEGF164) (200 ng/mouse) (Cell Signaling, #5211) over a period of 5 days. LPS from Escherichia coli 055:B5 (LPS) (20 μg/mouse) (Sigma-Aldrich, UK, #L6529) was injected i.p. route from Escherichia coli 055:B5 5 days before infection.
Parameters used to assess the development of CM
The onset of cerebral complications was determined by monitoring a number of clinical signs, such as ruffled fur, hunching, wobbly gait, limb paralysis, convulsion and death. Moreover, a drop in body temperature < 34°C was also used to confirm the onset of cerebral complications. Mice were monitored daily from the day of the infection. At the end of the experiment, mice underwent deep anaesthesia for blood sampling by cardiac puncture. Afterwards, they were euthanised and organ collection (brain, spleen, lung, and liver) took place.
Cytokine and chemokine measurements
Circulating cytokine/chemokine levels were measured using the BD Cytometric Bead Array (CBA) Mouse Th1/Th2/Th17 Cytokine kit (R&D System, USA, #560485) on 50 μL of plasma obtained from blood samples collected from either infected, treated or wild type littermates at each of the different time points. The assay was performed in triplicate. For all the soluble markers included in the panels, the detection limit of CBA was 20 pg mL− 1.
Histopathology and immunohistochemistry
Formalin fixed samples of brain, liver and spleen collected from infected, treated and wild type littermates were embedded in paraffin blocks. Thin tissue sections (5 μm thick) were obtained and routinely processed for histopathological examination (H&E stain) and for immunohistochemistry using antibodies to detect CD3 (anti-CD3 antibody, Abcam ab5690/antigen retrieval: citrate buffer 10 min; MW, dilution: 1:100; 4°C overnight) and cleaved caspase 3 positive cells (cleaved caspase 3 antibody, Cell Signaling #9661, antigen retrieval: citrate buffer 10 min; MW, dilution: 1:100; 4°C overnight). All the antibodies were visualised using the Vectastain Elite ABC kit (Vector Laboratories, #PK-6101) and DAB Peroxidase Substrate kit (Vector Laboratories, #SK-4100). Additional serial tissue sections were also processed for indirect immunoperoxidase staining following the procedures described elsewhere.48
Percent of area of the signal [CD3+ cells and apoptotic endothelial cells (cleaved caspase 3)] has been calculated using Fiji ImageJ. The RGB image was thresholded by signal intensity.
Microphotographs of organs were taken using a Canon PowerShot A520 with a close-up lens 250D (5.7 zoom).
mRNA transcript quantification
Total RNA was extracted using Trizol (Life Technologies) and reverse transcribed using the SuperScript III First Strand Synthesis System kit (Life Technologies). Transcript abundance was measured using an Applied Biosystems Thermocycler and Fast SYBER Green Master Mix (Life Technologies).
GAPDH was used as housekeeping reference gene in a qPCR reaction. Induction relative to the non-infected control (fold change) was calculated using the comparative C t method (ΔΔC t).
The specific mouse primer sequences (5′-3′) used in this work to profile the cytokines and other immune related factors were as follows: GAPDH,49 ATGACATCAAGAAGGTGGTG (forward) and CATACCAGGAAATGAGCTTG (reverse); VEGF,50,51 CTGTGCAGGCTGCTGTAACG (forward) and GTTCCCGAAACCCTGAGGAG (reverse); VEGFR1,52 CGGAAGGAAGACAGCTCATC (forward) and CTTCACGCGACAGGTGTAGA (reverse); VEGFR2,52 GGCGGTGGTGACAGTATCTT (forward) and TCTCCGGCAAGCTCAAT (reverse); S1Pr1,53 AACTTTGCGAGTGAGCTGGT (forward) and CTAGAGGGCGAGGTTGAGTG (reverse); IL-6,54 AAGAGTTGTGCAATGGCAATTCT (forward) and AAGTGCATCATCGTTGTTCATACA (reverse); IL-23p19,53,55 TGCTGGATTGCAGAGCAGTAA (forward) and CTGGAGGAGTTGGCTGAGTC (reverse); TNF-α,56 CCCAGGGACCTCTCTCTAATC (forward) and ATGGGCTACAGGCTTGTCACT (reverse); IFN-γ,57 CTAA TTATTCGGTAACTGACTTGA (forward) and ACAGTTCAGCCATCACTTGGA (reverse); IDO,27 GGCTTCTTCCTCGTCTCTCTATTG (forward) and TGACGCTCTACTGCACTGGATAC (reverse); HO-1,58,59 GGTGATGGCTTCCTTGTACC (forward) and AGTGAGGCCCATACCAGAAG (reverse); Nrf2,60,61 TCTCCTCGCTGGAAAAAGAA (forward) and AATGTGCTGGCTGTGCTTTA (reverse); IL-1Ra,62 CCTCGGGATGGAAATCTGCT (forward) and CCAGATTCTGAAGGCTTGCAT (reverse); IL-18,27 TTCCATGCTTTCTGGACTCCTG (forward) and TGCTGGAGGTTGCAGAAGATG (reverse); IL-12p35,53 CATCGATGAGCTGATGCAGT (forward) and CAGATAGCCCATCACCCTGT (reverse); IL-12p40,53 TGGAAGCACGGCAGCAGAATAAAT (forward) and TGCGCTGGATTCGAACAAAGAACT (reverse); IL-10,63 ATTTGAATTCCCTGGGTGAGAAG (forward) and CACAGGGGAGAAATCGATGACA (reverse); IL-17,64 CTCCAGAAGGCCCTCAGACTAC (forward) and AGCTTTCCCTCCGCATTGACACAG (reverse); MCP-1,62 CTTCTGGGCCTGCTGTTCA (forward) and CCAGCCTACTCATTGGGATCA (reverse); IL-2,62 CCTGAGCAGGATGGAGAATTACA (forward) and TCCAGAACATGCCGCAGAG (reverse); IL-4,62 ACAGGAGAAGGGACGCCAT (forward) and GAAGCCCTACAGACGAGCTCA (reverse); TGFβ1,62 TGACGTCACTGGAGTTGTACGG (forward) and GGTTCATGTCATGGATGGTGC (reverse).
Statistical analysis
All statistical analyses were performed using GraphPad Prism 4.0 (GraphPad Software Inc.). Analysis of variance (ANOVA) was followed by Sidak's multiple comparison test. Values of p < 0.05 were considered significant, and all data are displayed as mean ± SD of results from single representative experiment performed in duplicate and/or in triplicate. Each experiment was repeated at least twice to confirm reproducibility.
Fold changes obtained from real time PCR data for each of the genes selected from individuals in different conditions were compared. More specifically, the pairings for comparison were done as follows (condition A versus condition B):
- - Not treated (infected) versus
- ○ infected+axitinib,
- ○ infected+VEGF,
- ○ non infected+LPS,
- ○ infected+LPS,
- ○ infected+LPS+VEGF littermates (C57BL/6 or Balb/c)
- infected+axitinib versus infected+LPS+VEGF
- infected+VEGF versus infected+LPS+VEGF
Each condition A versus condition B pairing was explored with biological triplicates. In order to detect the genes for whom the difference of expression between conditions was significant, the following procedure was adopted individually onto each selected gene (by means of an Excel spreadsheet):
-
(i)
the difference between condition related expressions was computed as follows: diff = log2(expressioncondA/expressioncondB) so that no difference between expressions (i.e. expressioncondA/expressioncondB = 1) resulted in diff = 0;
-
(ii)
to test for statistical significance of expression difference, a one sample t test was constructed, the null hypothesis being H0: μdiff = 0, with a sample formed of the three diff values obtained from the three biological replicates. Rejection of the null hypothesis (p < 0.05) was used to identify significance
-
(iii)
genes exhibiting significant differences were labelled with * (0.01 ≤ p < 0.05) and ** (p < 0.01)
Disclaimer Statements
Contributors
M.C. conceived the study, performed all the experiments, and analysed and interpreted the data. A.C. conceived and directed the experiments, interpreted the data, and wrote the manuscript. T.D. performed statistical analysis on gene expression. M.C. and A.C. discussed the results and commented on the manuscript.
Funding
The research leading to these results has received funding from the European Union Seventh Framework Programme (FP7/2007-2013) under the Marie Curie Co-Funding of Regional, National and International Programme I-MOVE (grant agreement no. 267232).
Conflicts of interest
The authors declare no competing financial interests.
Ethics approval
All animal studies and the protocols were approved under Ethics Committee License no. PR 0161.
References
- 1.de Souza JB, Riley EM. Cerebral malaria: the contribution of studies in animal models to our understanding of immunopathogenesis. Microbes Infect. 2002;4(3):291–300. [DOI] [PubMed] [Google Scholar]
- 2.Mishra SK, Newton CR. Diagnosis and management of the neurological complications of falciparum malaria. Nat. Rev. Neurol. 2009;5(4):189–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller LH, Ackerman HC, Su XZ, Wellems TE. Malaria biology and disease pathogenesis: insights for new treatments. Nat. Med. 2013;19(2):156–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carvalho LJ, Moreira Ada S, Daniel-Ribeiro CT, Martins YC. Vascular dysfunction as a target for adjuvant therapy in cerebral malaria. Mem. Inst. Oswaldo Cruz. 2014;109(5):577–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lovegrove FE, Tangpukdee N, Opoka RO, Lafferty EI, Rajwans N, Hawkes M, et al. Serum angiopoietin-1 and -2 levels discriminate cerebral malaria from uncomplicated malaria and predict clinical outcome in African children. PLoS One. 2009;4(3):e4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jain V, Lucchi NW, Wilson NO, Blackstock AJ, Nagpal AC, Joel PK, et al. Plasma levels of angiopoietin-1 and -2 predict cerebral malaria outcome in Central India. Malaria J. 2011;10:383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeney V, Ramos S, Bergman ML, Bechmann I, Tischer J, Ferreira A, et al. Control of disease tolerance to malaria by nitric oxide and carbon monoxide. Cell Rep. 2014;8(1):126–36. [DOI] [PubMed] [Google Scholar]
- 8.Rushworth SA, Chen XL, Mackman N, Ogborne RM, O'Connell MA. Lipopolysaccharide-induced heme oxygenase-1 expression in human monocytic cells is mediated via Nrf2 and protein kinase C. J. Immunol. 2005;175(7):4408–15. [DOI] [PubMed] [Google Scholar]
- 9.Shih RH, Yang CM. Induction of heme oxygenase-1 attenuates lipopolysaccharide-induced cyclooxygenase-2 expression in mouse brain endothelial cells. J. Neuroinflamm. 2010;7:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bouloumie A, Schini-Kerth VB, Busse R. Vascular endothelial growth factor up-regulates nitric oxide synthase expression in endothelial cells. Cardiovasc. Res. 1999;41(3):773–80. [DOI] [PubMed] [Google Scholar]
- 11.Servos S, Zachary I, Martin JF. VEGF modulates NO production: the basis of a cytoprotective effect? Cardiovasc. Res. 1999;41(3):509–10. [DOI] [PubMed] [Google Scholar]
- 12.Hunt NH, Grau GE. Cytokines: accelerators and brakes in the pathogenesis of cerebral malaria. Trends Immunol. 2003;24(9):491–9. [DOI] [PubMed] [Google Scholar]
- 13.Kwiatkowski D, Hill AV, Sambou I, Twumasi P, Castracane J, Manogue KR, et al. TNF concentration in fatal cerebral, non-fatal cerebral, and uncomplicated Plasmodium falciparum malaria. Lancet. 1990;336(8725):1201–4. [DOI] [PubMed] [Google Scholar]
- 14.Grau GE, Fajardo LF, Piguet PF, Allet B, Lambert PH, Vassalli P. Tumor necrosis factor (cachectin) as an essential mediator in murine cerebral malaria. Science. 1987;237(4819):1210–2. [DOI] [PubMed] [Google Scholar]
- 15.Grau GE, Heremans H, Piguet PF, Pointaire P, Lambert PH, Billiau A, et al. Monoclonal antibody against interferon gamma can prevent experimental cerebral malaria and its associated overproduction of tumor necrosis factor. Proc. Natl. Acad. Sci. U.S.A. 1989;86(14):5572–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller LH, Baruch DI, Marsh K, Doumbo OK. The pathogenic basis of malaria. Nature. 2002;415(6872):673–9. [DOI] [PubMed] [Google Scholar]
- 17.Amani V, Vigario AM, Belnoue E, Marussig M, Fonseca L, Mazier D, et al. Involvement of IFN-gamma receptor-medicated signaling in pathology and anti-malarial immunity induced by Plasmodium berghei infection. Eur. J. Immunol. 2000;30(6):1646–55. [DOI] [PubMed] [Google Scholar]
- 18.Lucas R, Juillard P, Decoster E, Redard M, Burger D, Donati Y, et al. Crucial role of tumor necrosis factor (TNF) receptor 2 and membrane-bound TNF in experimental cerebral malaria. Eur. J. Immunol. 1997;27(7):1719–25. [DOI] [PubMed] [Google Scholar]
- 19.Armah H, Wired EK, Dodoo AK, Adjei AA, Tettey Y, Gyasi R. Cytokines and adhesion molecules expression in the brain in human cerebral malaria. Int. J. Environ. Res. Public Health. 2005;2(1):123–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hunt NH, Ball HJ, Hansen AM, Khaw LT, Guo J, Bakmiwewa S, et al. Cerebral malaria: gamma-interferon redux. Front. Cell. Infect. Microbiol. 2014;4:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hansen DS. Inflammatory responses associated with the induction of cerebral malaria: lessons from experimental murine models. PLoS Pathog. 2012;8(12):e1003045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goncalves RM, Scopel KK, Bastos MS, Ferreira MU. Cytokine balance in human malaria: does Plasmodium vivax elicit more inflammatory responses than Plasmodium falciparum? PLoS One. 2012;7(9):e44394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Warrell DA, Looareesuwan S, Warrell MJ, Kasemsarn P, Intaraprasert R, Bunnag D, et al. Dexamethasone proves deleterious in cerebral malaria. A double-blind trial in 100 comatose patients. N. Engl. J. Med. 1982;306(6):313–9. [DOI] [PubMed] [Google Scholar]
- 24.van Hensbroek MB, Palmer A, Onyiorah E, Schneider G, Jaffar S, Dolan G, et al. The effect of a monoclonal antibody to tumor necrosis factor on survival from childhood cerebral malaria. J. Infect. Dis. 1996;174(5):1091–7. [DOI] [PubMed] [Google Scholar]
- 25.Grau GE, Frei K, Piguet PF, Fontana A, Heremans H, Billiau A, et al. Interleukin 6 production in experimental cerebral malaria: modulation by anticytokine antibodies and possible role in hypergammaglobulinemia. J. Exp. Med. 1990;172(5):1505–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Angulo I, Fresno M. Cytokines in the pathogenesis of and protection against malaria. Clin. Diagn. Lab. Immunol. 2002;9(6):1145–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitchell AJ, Hansen AM, Hee L, Ball HJ, Potter SM, Walker JC, et al. Early cytokine production is associated with protection from murine cerebral malaria. Infect. Immun. 2005;73(9):5645–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown H, Turner G, Rogerson S, Tembo M, Mwenechanya J, Molyneux M, et al. Cytokine expression in the brain in human cerebral malaria. J. Infect. Dis. 1999;180(5):1742–6. [DOI] [PubMed] [Google Scholar]
- 29.Yanez DM, Manning DD, Cooley AJ, Weidanz WP, van der Heyde HC. Participation of lymphocyte subpopulations in the pathogenesis of experimental murine cerebral malaria. J. Immunol. 1996;157(4):1620–4. [PubMed] [Google Scholar]
- 30.Tan RS, Kara AU, Feng C, Asano Y, Sinniah R. Differential interleukin-10 expression in interferon regulatory factor-1 deficient mice during Plasmodium berghei blood-stage infection. Parasite Immunol. 2000;22(9):425–35. [DOI] [PubMed] [Google Scholar]
- 31.Pamplona A, Ferreira A, Balla J, Jeney V, Balla G, Epiphanio S, et al. Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat. Med. 2007;13(6):703–10. [DOI] [PubMed] [Google Scholar]
- 32.Ziegler-Heitbrock HW. Molecular mechanism in tolerance to lipopolysaccharide. J. Inflamm. 1995;45(1):13–26. [PubMed] [Google Scholar]
- 33.Camhi SL, Alam J, Otterbein L, Sylvester SL, Choi AM. Induction of heme oxygenase-1 gene expression by lipopolysaccharide is mediated by AP-1 activation. Am. J. Respir. Cell Mol. Biol. 1995;13(4):387–98. [DOI] [PubMed] [Google Scholar]
- 34.Itaya H, Imaizumi T, Yoshida H, Koyama M, Suzuki S, Satoh K. Expression of vascular endothelial growth factor in human monocyte/macrophages stimulated with lipopolysaccharide. Thromb. Haemost. 2001;85(1):171–6. [PubMed] [Google Scholar]
- 35.Hu-Lowe DD, Zou HY, Grazzini ML, Hallin ME, Wickman GR, Amundson K, et al. Nonclinical antiangiogenesis and antitumor activities of axitinib (AG-013736), an oral, potent, and selective inhibitor of vascular endothelial growth factor receptor tyrosine kinases 1, 2, 3. Clin. Cancer Res. 2008;14(22):7272–83. [DOI] [PubMed] [Google Scholar]
- 36.Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, et al. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J. Neurosci. 1998;18(10):3659–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gown AM, Willingham MC. Improved detection of apoptotic cells in archival paraffin sections: immunohistochemistry using antibodies to cleaved caspase 3. J. Histochem. Cytochem. 2002;50(4):449–54. [DOI] [PubMed] [Google Scholar]
- 38.Rock EP, Roth EF Jr, Rojas-Corona RR, Sherwood JA, Nagel RL, Howard RJ, et al. Thrombospondin mediates the cytoadherence of Plasmodium falciparum-infected red cells to vascular endothelium in shear flow conditions. Blood. 1988;71(1):71–5. [PubMed] [Google Scholar]
- 39.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat. Med. 2003;9(6):669–76. [DOI] [PubMed] [Google Scholar]
- 40.Dulak J, Deshane J, Jozkowicz A, Agarwal A. Heme oxygenase-1 and carbon monoxide in vascular pathobiology: focus on angiogenesis. Circulation. 2008;117(2):231–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tuder RM, Flook BE, Voelkel NF. Increased gene expression for VEGF and the VEGF receptors KDR/Flk and Flt in lungs exposed to acute or to chronic hypoxia. Modulation of gene expression by nitric oxide. J. Clin. Invest. 1995;95(4):1798–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bussolati B, Ahmed A, Pemberton H, Landis RC, Di Carlo F, Haskard DO, et al. Bifunctional role for VEGF-induced heme oxygenase-1 in vivo: induction of angiogenesis and inhibition of leukocytic infiltration. Blood. 2004;103(3):761–6. [DOI] [PubMed] [Google Scholar]
- 43.Kim EH, Na HK, Surh YJ. Upregulation of VEGF by 15-deoxy-Delta12,14-prostaglandin J2 via heme oxygenase-1 and ERK1/2 signaling in MCF-7 cells. Ann. N.Y. Acad. Sci. 2006;1090:375–84. [DOI] [PubMed] [Google Scholar]
- 44.Bussolati B, Mason JC. Dual role of VEGF-induced heme-oxygenase-1 in angiogenesis. Antioxid. Redox Signal. 2006;8(7-8):1153–63. [DOI] [PubMed] [Google Scholar]
- 45.Lin HH, Chen YH, Chang PF, Lee YT, Yet SF, Chau LY. Heme oxygenase-1 promotes neovascularization in ischemic heart by coinduction of VEGF and SDF-1. J. Mol. Cell. Cardiol. 2008;45(1):44–55. [DOI] [PubMed] [Google Scholar]
- 46.Morbidelli L, Chang CH, Douglas JG, Granger HJ, Ledda F, Ziche M. Nitric oxide mediates mitogenic effect of VEGF on coronary venular endothelium. Am. J. Physiol. 1996;270(1Pt2):H411–5. [DOI] [PubMed] [Google Scholar]
- 47.Chen XL, Dodd G, Thomas S, Zhang X, Wasserman MA, Rovin BH, et al. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am. J. Physiol. Heart Circ. Physiol. 2006;290(5):H1862–70. [DOI] [PubMed] [Google Scholar]
- 48.Radaelli E, Del Piero F, Aresu L, Sciarrone F, Vicari N, Mattiello S, et al. Expression of major histocompatibility complex class II antigens in porcine leptospiral nephritis. Vet. Pathol. 2009;46(5):800–9. [DOI] [PubMed] [Google Scholar]
- 49.Tang KF, Ren H, Cao J, Zeng GL, Xie J, Chen M, et al. Decreased Dicer expression elicits DNA damage and up-regulation of MICA and MICB. J. Cell Biol. 2008;182(2):233–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Y, Hazarika S, Xie D, Pippen AM, Kontos CD, Annex BH. In mice with type 2 diabetes, a vascular endothelial growth factor (VEGF)-activating transcription factor modulates VEGF signaling and induces therapeutic angiogenesis after hindlimb ischemia. Diabetes. 2007;56(3):656–65. [DOI] [PubMed] [Google Scholar]
- 51.Clifford RL, Deacon K, Knox AJ. Novel regulation of vascular endothelial growth factor-A (VEGF-A) by transforming growth factor (beta)1: requirement for Smads, (beta)-CATENIN, AND GSK3(beta). J. Biol. Chem. 2008;283(51):35337–53. [DOI] [PubMed] [Google Scholar]
- 52.Lee YJ, Karl DL, Maduekwe UN, Rothrock C, Ryeom S, D'Amore PA, et al. Differential effects of VEGFR-1 and VEGFR-2 inhibition on tumor metastases based on host organ environment. Cancer Res. 2010;70(21):8357–67. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 53.Wakabayashi H, Takakura N, Yamauchi K, Tamura Y. Modulation of immunity-related gene expression in small intestines of mice by oral administration of lactoferrin. Clin. Vaccine Immunol. 2006;13(2):239–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takada K, Inaba M, Ichioka N, Ueda Y, Taira M, Baba S, et al. Treatment of senile osteoporosis in SAMP6 mice by intra-bone marrow injection of allogeneic bone marrow cells. Stem Cells. 2006;24(2):399–405. [DOI] [PubMed] [Google Scholar]
- 55.Xu M, Morishima N, Mizoguchi I, Chiba Y, Fujita K, Kuroda M, et al. Regulation of the development of acute hepatitis by IL-23 through IL-22 and IL-17 production. Eur. J. Immunol. 2011;41(10):2828–39. [DOI] [PubMed] [Google Scholar]
- 56.Legat A, Thomas S, Hermand P, Van Mechelen M, Goldman M, De Wit D. CD14-independent responses induced by a synthetic lipid A mimetic. Eur. J. Immunol. 2010;40(3):797–802. [DOI] [PubMed] [Google Scholar]
- 57.Longhi LN, da Silva RM, Fornazim MC, Spago MC, de Oliveira RT, Nowill AE, et al. Phenotypic and functional characterization of NK cells in human immune response against the dimorphic fungus Paracoccidioides brasiliensis. J. Immunol. 2012;189(2):935–45. [DOI] [PubMed] [Google Scholar]
- 58.Bolisetty S, Traylor AM, Kim J, Joseph R, Ricart K, Landar A, et al. Heme oxygenase-1 inhibits renal tubular macroautophagy in acute kidney injury. J. Am. Soc. Nephrol. 2010;21(10):1702–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hull TD, Bolisetty S, DeAlmeida AC, Litovsky SH, Prabhu SD, Agarwal A, et al. Heme oxygenase-1 expression protects the heart from acute injury caused by inducible Cre recombinase. Lab. Invest. 2013;93(8):868–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shih AY, Imbeault S, Barakauskas V, Erb H, Jiang L, Li P, et al. Induction of the Nrf2-driven antioxidant response confers neuroprotection during mitochondrial stress in vivo. J. Biol. Chem. 2005;280(24):22925–36. [DOI] [PubMed] [Google Scholar]
- 61.Shih AY, Erb H, Murphy TH. Dopamine activates Nrf2-regulated neuroprotective pathways in astrocytes and meningeal cells. J. Neurochem. 2007;101(1):109–19. [DOI] [PubMed] [Google Scholar]
- 62.Giulietti A, Overbergh L, Valckx D, Decallonne B, Bouillon R, Mathieu C. An overview of real-time quantitative PCR: applications to quantify cytokine gene expression. Methods. 2001;25(4):386–401. [DOI] [PubMed] [Google Scholar]
- 63.Yee CS, Yao Y, Xu Q, McCarthy B, Sun-Lin D, Tone M, et al. Enhanced production of IL-10 by dendritic cells deficient in CIITA. J. Immunol. 2005;174(3):1222–9. [DOI] [PubMed] [Google Scholar]
- 64.Myoung J, Kang HS, Hou W, Meng L, Dal Canto MC, Kim BS. Epitope-specific CD8+T cells play a differential pathogenic role in the development of a viral disease model for multiple sclerosis. J. Virol. 2012;86(24):13717–28. [DOI] [PMC free article] [PubMed] [Google Scholar]